

Abstract
Mutations in CLN6 cause variant late-onset neuronal ceroid lipofuscinosis (vLINCL), a childhood neurodegenerative disorder resulting from aberrant neuronal cell loss and pathological accumulation of lysosomal auto-fluorescent storage material in the central nervous system. The direct function of the endoplasmic reticulum– resident protein CLN6 and how dysfunction of this protein results in vLINCL are unknown. We report that CLN6 interacts with collapsin response mediator protein-2 (CRMP-2). To further understand the significance and possible contribution to vLINCL of the CLN6–CRMP-2 interaction, we utilized the nclf mouse, which harbors mutations in CLN6. Significantly, CRMP-2 protein level was found to be reduced in the nclf mouse brain, particularly in the thalamus. Because CRMP-2 functions in growth cone collapse and is an effector protein downstream of Sema3A signaling, this pathway was examined via a dorsal root ganglion (DRG) repulsion assay. However, there were no defects in the repulsion of DRGs derived from nclf mice, indicating that the loss of CLN6 does not affect Sema3A signaling. CRMP-2 has also been implicated in controlling axon number and outgrowth, as observed in cultured hippocampal neurons. Therefore, we explored the formation and maturation of hippocampal neurons derived from nclf mice in a glial coculture system. The maturation of these neurons was reduced; by day in vitro (DIV) 8, more than 50% of nclf-derived hippocampal neurons had died. Additionally, beginning around DIV4, nclf neurons were less mature than their WT counterparts, presumably because of an inability to form mature synaptic connections. We concluded that alterations in neurite maturation resulting from a loss of CLN6–CRMP-2 interaction may contribute to neuronal dysfunction and pathology in vLINCL.
Keywords: semaphorin 3A, axon outgrowth, nclf mouse, dihydropyrimidinase-like-2 (DRP-2)
Neuronal ceroid lipofuscinoses (NCL) are lysosomal storage disorders typified by an accumulation of autofluorescent storage material in all tissues and central nervous system (CNS)–specific pathology. Mutations in the CLN6 gene result in a variant of late-onset infantile neuronal ceroid lipofuscinosis (vLINCL; Sharp et al., 2003), a childhood disease of the CNS (Lake and Cavanagh, 1978). The disease begins between 1.5 and 8 years of age with motor delay, ataxia, and seizures, ending with premature death between ages 5 and 12 (Tyynela et al., 1997). CLN6 encodes a 311-amino-acid endoplasmic reticulum (ER)–resident membrane protein that possesses seven membrane-spanning domains, with the N-terminus in the cytosol and the C-terminus in the lumen (Gao et al., 2002; Heine et al., 2004, 2007; Mole et al., 2004; Wheeler et al., 2002). How loss of this protein function precipitates vLINCL pathology is unknown, and there is no treatment or cure for this disease.
Currently, the function of CLN6 is unknown; however, some clues about the role of CLN6 in the CNS have been gained by studying mouse and sheep models of vLINCL. The Cln6 mouse model, nclf, was found as a result of a naturally occurring insertion that leads to truncation of the protein, which is a gene defect identical to that observed in a subset of human patients (Bronson et al., 1998; Gao et al., 2002). These mice exhibit progressive retinal atrophy, widespread cortical astrocytosis, accumulation of storage material, Wallerian degeneration of the spinal cord and brain stem, hind-limb paralysis, and at times terminal seizures (Bronson et al., 1998). Additionally, other naturally occurring mutations in South Hampshire and Merino sheep have provided a large animal model for CLN6, exhibiting severe neurodegeneration in the cerebral cortex, retinal atrophy, large amounts of astrocytosis, and characteristic storage material, as seen in the human disease (Broom et al., 1998; Tammen et al., 2001, 2006; Cook et al., 2002; Oswald et al., 2005, 2008). Data from human patients and animal models therefore suggest that defects in CLN6 result in massive amounts of neurodegeneration in the CNS. Therefore, it is critical to understand the pathological mechanisms behind this degeneration.
To help understand the function of CLN6 in the CNS, we screened hydrophilic regions of CLN6 against a human fetal brain library in order to identify protein– protein interactions by yeast-2-hybrid. In this article, we describe a novel interaction between CLN6 and collapsin response mediator protein-2 (CRMP-2). We also show that the CRMP-2 protein level is significantly decreased in affected brain regions of nclf mice in vivo. The CRMP-2 protein, also known as dihydropyrimidinase-like-2 (DRP-2), is involved in growth cone guidance and neuronal polarity and may contribute to synaptic neuroprotection in Wlds-mutant mice (Inagaki et al., 2001; Arimura et al., 2004; Brown et al., 2004; Wishart et al., 2007). Additionally, CRMP-2 interacts with Numb and mediates endocytosis of specific molecules such as L1 at the growth cone, suggesting it has an important role in axon growth (Nishimura et al., 2003). Through its interaction with tubulin heterodimers and the Sra-1/WAVE1–actin complex (Kawano et al., 2005), CRMP-2 promotes microtubule assembly (Fukata et al., 2002) and regulates cytoskeletal dynamics during axonal outgrowth and axon-dendrite specification (Kawano et al., 2005). Mutations that cause vLINCL lead to a loss of CLN6 function, as the severity of the disease correlates with the extent of genetic mutation (Gao et al., 2002; Wheeler et al., 2002; Sharp et al., 2003; Mole, 2008), suggesting that loss of the interaction between CLN6 and CRMP-2 may contribute to the disease pathology. To investigate the possible pathological consequences of disrupting this interaction in vLINCL, we used the nclf mouse to explore both CRMP-2-dependent semaphorin 3A (Sema3A)–induced axon repulsion and maturation and axon outgrowth in cultured hippocampal neurons, the classic model for examining neuronal polarization (Banker and Goslin, 1991). We found no effect in the repulsion of nclf dorsal root ganglion cells in the presence of Sema3A. However, we found a marked change in the maturation of hippocampal neurites derived from nclf mice. Taken together, these experiments show that CRMP-2 interacts with CLN6 in vivo and suggest that disruption of this interaction may play a role in modifying neurite maturation, contributing to vLINCL pathology.
Materials and Methods
Yeast Two-Hybrid
CLN6 has a predicted five-transmembrane topology, with the N-terminus facing the cytosol, two luminal loops, two cytosolic loops, and a luminal C-terminus. Based on this topology, the hydrophilic regions of CLN6 (amino acids 1– 15, 72–110, 201–229, 130–173, and 282–311) were screened against a human fetal brain library using the Cytotrap yeast two-hybrid system (Y2H; Stratagene, La Jolla, CA). The cytosolic loop between membrane regions 4 and 5 was omitted from the screen because of hydrophobicity. Briefly, this Y2H system utilizes the temperature-sensitive yeast strain Cdc25H, which harbors a mutation in the homologue of the human Sos (hSos) protein, Cdc25p. The CLN6 fragments are fused with hSos and screened against the cDNA library in the pMyr vector, which myristoylates the library proteins, anchoring them in the plasma membrane. In the Cdc25H yeast strain, when the hSos–CLN6 fragment fusion protein is brought to the plasma membrane through an interaction with the myristoylated library protein, growth at 37°C is restored. A positive candidate from the library was verified by rescreening through the yeast two-hybrid and also sequenced to determine its identity.
cDNA Constructs
For the yeast two-hybrid, the CLN6 nucleotides corresponding to the N-terminal amino acids (1–15) of the CLN6 protein were amplified using primers 5′-GGATCCCC′AT GGAGGCGACGCGGAGGC-3′ and 5′-ACGCGTGC′CG CCCGTCGCTCCCAG-3′ specifically for the interacting region of CLN6 described here. The product was cloned into the pSos vector using the BamH1 and Mlu1 sites.
For validation of the interaction, full-length CLN6 was subcloned into the CMV multiple cloning site in the pBudCE4 vector (Invitrogen, Carlsbad, CA) following amplification of CLN6 (using primers 5′-ATGGAGGCGACGCG GAGGC-3′ and 5′-GGATCCGTGCCGACTGCTGACGT GAAG-3′), cloning the cDNA into the pCR blunt shuttle vector (Invitrogen, Carlsbad, CA), and digestion by BamH1. Full-length CRMP-2 was subcloned into the EF-1α multiple cloning site in the CLN6-containing pBudCE4 vector following amplification of CRMP-2 (using primers 5′-AAGCGGC CGCGAGATGTCTTATCAGGGGAAGAAAAATATTC-3′ and 5′- AAGCGGCCGCCCAGGCAGGTGATGTTG-3′), cloning the cDNA into the pCR blunt shuttle vector, and digestion by Not1.
Coimmunoprecipitation and Western Blotting
CLN6-myc/CRMP-2-V5 or CRMP-2-V5 only containing the pBudCE4 vector was transfected into NIH/3T3 fibroblasts using Lipofectamine 2000 per the manufacturer's protocol (Invitrogen, Carlsbad, CA). Protein was expressed for 36 hr and then extracted by incubating the cell pellet in ice-cold nondenaturing lysis buffer [50 mM Tris-Cl (pH 7.5), 300 mM NaCl, 5 mM EDTA, 1% Triton X-100) for 30 min. Cell debris was pelleted, and the supernatant was isolated and precleared using protein A–conjugated agarose beads. Cell lysates were incubated with anti-c-myc antibody (Cell Signaling, Danvers, MA) overnight at 4°C. Lysates were then incubated with protein A–conjugated agarose beads for 1 hr and pelleted, and the supernatant was removed. The immunoprecipitate was washed with ice-cold nondenaturing wash buffer [50 mM Tris-Cl (pH 7.5), 300 mM NaCl, 5 mM EDTA, 0.01% Triton X-100], pelleted, resuspended in Laemmli loading buffer, and boiled for 3 min. Immunoprecipitates and lysates were run on a 12% SDS-PAGE and transferred onto a nitrocellulose membrane. The membranes were Western-blotted using either an anti-V5 antibody (Invitrogen, Carlsbad, CA) or an anti-myc antibody (Cell Signaling, Danvers, MA). Membranes were washed with TBS-T and incubated with the appropriate HRP-conjugated secondary antibody, and ECL detection (Amersham, Piscataway, NJ) was used to visualize the protein.
Quantitative Western Blotting
Brains from 90-day-old nclf mice and controls (n = 3) were freshly dissected into regions containing the thalamus, cortex, and cerebellum. Protein was extracted as described previously (Wishart et al., 2007). Thirty-five micrograms of total protein from each brain region was separated by SDS/polyacrylamide gel electrophoresis on 4%–20% precast NuPage Bis Tris gradient gels (Invitrogen) and then transferred to a PVDF membrane overnight. The membranes were then blocked using Odyssey blocking buffer (Li-COR) and incubated with primary antibodies as per manufacturers' instructions (beta tubulin, Abcam, Cambridge, MA; VDAC2, GeneTex; CRMP-2 and phosphorylated CRMP-2, a kind gift from Dr. Calum Sutherland, University of Dundee). Odyssey secondary antibodies were added according to manufacturers' instructions (donkey antisheep IRDye 680 and goat antimouse IRDye 800). Blots were imaged using an Odyssey Infrared Imaging System (Li-COR Biosciences). Scan resolution of this instrument ranges from 21–339 μm, and in this study blots were imaged at 169 lm. Quantification was performed on single channels with the analysis software provided, as described previously (Wishart et al., 2007).
Immunofluorescence
A low-density NIH/3T3 culture grown on a poly-d-lysine-coated cover glass was transfected with CLN6-myc/CRMP-2-V5 or CRMP-2-V5 containing pBudCE4 vector using Lipofectamine 2000. Protein was expressed for 36 hr, and the cover glass was washed with PBS and fixed for 30 min at room temperature [4% PFA/4% sucrose phosphate buffer (pH7.5)]. Cells were washed with PBS, permeabilized (PBS, 0.1% Triton X-100), and blocked in Blotto (4% nonfat dried milk, PBS, 0.1% Triton X-100). Anti-V5 (Invitrogen, Carlsbad, CA) and anti-myc (Cell Signaling, Danvers, MA) antibodies were diluted in Blotto and incubated overnight at 4°C. Cells were washed and incubated in the appropriate Alexa-conjugated secondary antibodies (Molecular Probes, Carlsbad, CA). Cells were washed, mounted, and then visualized using confocal microscopy.
For hippocampal neuron cultures, the same preparative protocol was used, and the primary antibodies used were anti– neuronal class III β-tubulin (Covance Research Products, Berkeley, CA) and anti-TOAD64 (BD PharMingen, Mississauga, Ontario, Canada).
Dorsal Root Ganglion Repulsion Assay
Dorsal root ganglion (DRG) neurons were removed from embryonic day 14.5 nclf and C57BL6/J mice (n = 3). HEK293 human fibroblasts were transfected with either semaphorin 3F (Sem3F) or semaphorin 3A (Sema3A) expression constructs. Cells were incubated at 37°C for 24 hr to guarantee protein expression, which was assayed by detecting the presence of released Sem3F and Sema3A in the media. The HEK293 cells were centrifuged and embedded in collagen. To plate the DRGs and HEK293 cells, a drop of collagen was placed in a culture dish. Once hardened, collagen-embedded HEK293 cells were placed adjacent to DRG neurons, and these were secured by overlaying collagen. Cells were incubated for 48 hr at 37°C to allow ample time for axon outgrowth. The cells were fixed with fresh ice-cold 4% PFA, washed, and blocked using PHT (PBS, 1% horse serum, 0.1% Triton-X 100). Cells were incubated in anti–neurofilament 2H3 antibody (Developmental Studies Hybridoma Bank, Iowa City, IA) diluted in PHT for 24 hr at 4°C, washed, incubated in HRP-conjugated antimouse IgG at 4°C for 24 hr, washed, and developed using an ABC kit per the manufacturer's protocol (Vector Labs, Burlingame, CA). Cells were visualized by light microscopy, and images were collected using a SPOT camera.
Hippocampal Neuron Cultures
The hippocampi were removed from embryonic day 16.5 nclf and C57BL6/J mice (age- and sex-matched wild-type control strain, n = 3) to prepare primary neuronal cultures as described by Banker and Goslin (1991). Briefly, dissected hippocampi in balanced salt solution were digested in trypsin and mechanically dissociated using a Pasteur pipette. The dissociated neurons were plated at 50 cells/mm2 on a poly-l-lysine-coated cover glass in MEM with 10% horse serum for 3 hr. Coverslips (with paraffin feet) were flipped and cocultured over astrocytes. These astrocytes were isolated from postnatal day 1 rat brains and cultured in Neurobasal medium (Invitrogen) supplemented with B27 and Glutamax for 1 week prior to coculture. The medium was replaced 1 day before the neuronal coculture was set up to ensure that it was conditioned. Two days following the coculture set up, Ara-C (5 μM) was added to prevent glial proliferation on the cover glass.
Results
CLN6 Interacts with CRMP-2
The CLN6 membrane protein has been localized to the ER (Mole et al., 2004), but the functions of this protein remain to be determined. Therefore, we employed Stratagene's Cytotrap yeast two-hybrid (Y2H) system to identify protein interactions using hydrophilic regions of CLN6, which were screened against a human fetal brain cDNA library. This Y2H screen identified collapsin response mediator protein 2 (CRMP-2) as an interactor with the cystosolic N-terminus of CLN6 (Fig. 1A). For validation of this identified interaction, CLN6 and CRMP-2 were coexpressed and coimmunoprecipitated in NIH/3T3 fibroblasts in order to verify the interaction in an alternate mammalian cell system, using the full-length CLN6 protein. NIH/3T3 fibroblasts were transfected with pBudCE4 vector coexpressing CLN6 with a c-terminal myc tag and CRMP-2 with a c-terminal V5 tag or with pBudCE4 containing CRMP-2-V5 alone as a negative control. Lysates containing the coexpressed proteins were immunoprecipitated with an anti-myc antibody, separated on an SDS-PAGE, and Western-blotted, which revealed copurification of CLN6-myc and CRMP-V5 (Fig. 1B,C). Endogenous coimmunoprecipitation of these two proteins from mouse tissue homogenates with a CLN6 antibody was not possible using the available antibodies to CLN6. Also, following the transfection of NIH/3T3 fibroblasts with the CLN6-myc/CRMP-2-V5 construct and staining the cells with anti-c-myc and anti-V5 antibodies, we found that the two proteins colocalized not only in the cell soma but also at the distal extremities of cellular processes (Fig. 1D). Taken together, these data show that CRMP-2 interacts with CLN6.
Fig. 1.
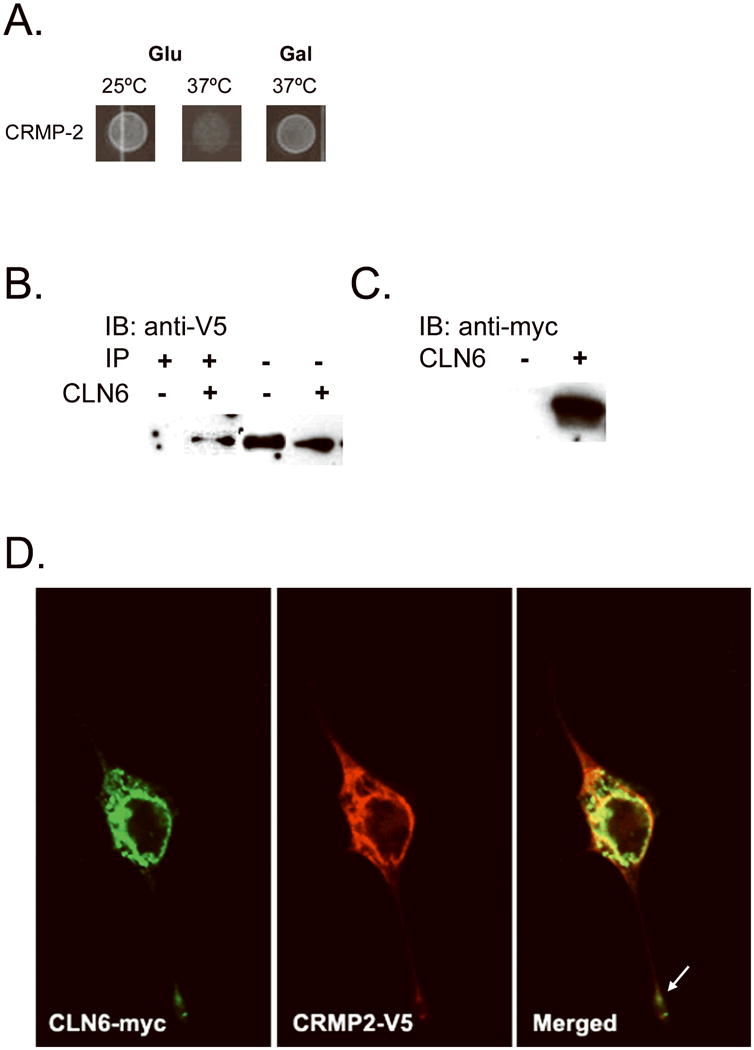
CLN6 interacts with collapsin response mediator protein-2 (CRMP-2). CLN6 hydrophilic fragments fused to the human Sos protein were screened using the Cytotrap yeast two-hybrid (Y2H) method against a human fetal brain library to identify interacting partners. The library proteins were expressed when grown on galactose media; thus, positive interaction was achieved when there is growth on galactose at the permissive temperature of 37°C. We found that CLN6 interacted with CRMP-2 (A). To validate the CRMP-2 interaction, CLN6-myc and CRMP-2-V5 were coexpressed or CRMP-2 alone was expressed in NIH/3T3 fibroblasts, and protein lysates were immunoprecipitated with anti-myc antibody. The immunoprecipitate (B, left) and the lysate (B, right) were probed with an anti-V5 antibody. This showed a specific CRMP-2 band in the immunoprecipitation only in the presence of CLN6. The lysate was probed with an anti-myc antibody to verify CLN6 expression (C). To visualize intracellular localization, CLN6-myc and CRMP-2-V5 were transfected into NIH/3T3 fibroblasts grown on poly-d-lysine-treated cover glass. Anti-myc and Anti-V5 antibodies were used to stain CLN6-myc (D, left) and CRMP-2-V5 (D, middle), respectively. Alexa-fluor antibodies were used to visualize the proteins. Confocal microscopy was used to image the cells. Colocalization of the two proteins is shown by the yellow color in the merged image (D, right). The white arrow indicates costaining down a process (magnification 40×).
CRMP-2 Protein Levels Are Selectively Decreased in Thalamus of nclf Mice
We next investigated whether we could detect any dysregulation of CRMP-2 levels resulting from a disrupted interaction with CLN6 protein in the nclf mouse, which harbors a truncation mutation in Cln6. Total CRMP-2 and phosphorylated CRMP-2 protein levels were determined in 90-day-old nclf mice by quantitative Western blot (Fig. 2). A significant decrease (P < 0.0001; unpaired two-tailed t test) in total CRMP-2 protein level was found in the thalami of nclf mice (Fig. 2A). Similarly, levels of phosphorylated CRMP-2 (measured using antibodies specific for phosphorylation of CRMP-2 at Thr514/509 residues) were significantly decreased in the thalami of nclf mice (P < 0.001; Fig. 2A). For control purposes, protein expression of beta-tubulin and mitochondrial voltage-dependent anion channel-2 (VDAC2) protein was determined and compared, with no significant differences observed for either protein (Fig. 2A).
Fig. 2.
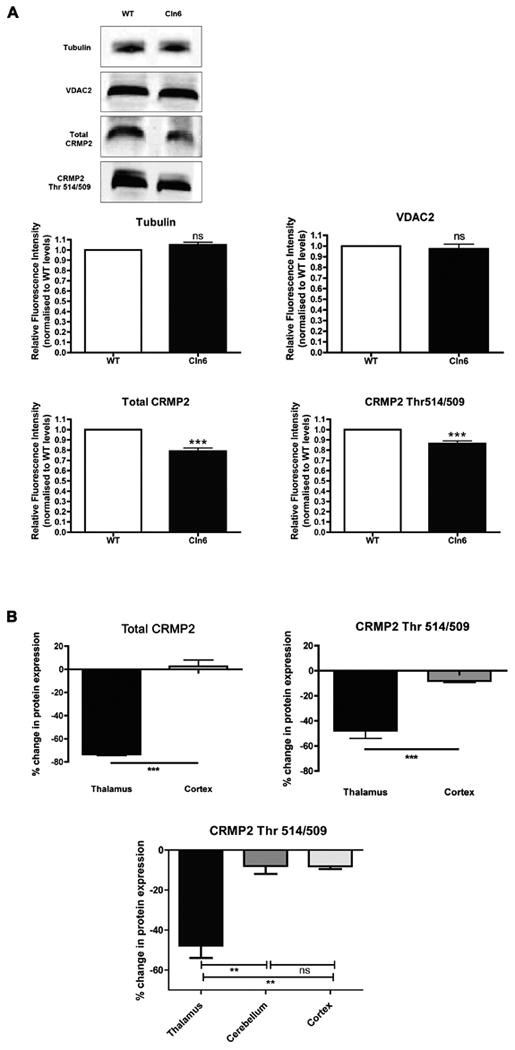
Altered CRMP-2 protein levels in the thalamus of nclf mice. CRMP-2 protein levels in the brains of 90-day-old nclf mice were examined by quantitative Western blotting (n = 3). A: Levels of both total CRMP-2 and phosphorylated CRMP-2 (Thr514/509) were significantly reduced in the thalami of nclf mice (P < 0.0001 for both; unpaired two-tailed t test), whereas levels of a control protein (tubulin and VDAC2) remained unchanged. B: Significant reductions in CRMP-2 protein levels were observed in the thalamus, but not in the cortex, suggesting a region-specific modulation of CRMP-2 in nclf mice.
As the thalamus is regarded as a primary target in the brain across a range of NCLs (Pontikis et al., 2005; Autti et al., 2007a, 2007b), we also asked whether reduced CRMP-2 levels were also detectable in the cortex. Total CRMP-2 levels were significantly reduced in the thalami, but not the cortices, of nclf mice (Fig. 2B). Similarly, the phosphorylated form of CRMP-2 showed a statistically significant decrease in expression in the thalamus, whereas it was only modestly reduced in the cortex (Fig. 2B). Thus, significant reductions in CRMP-2 levels appear to be specific to the thalamus.
Sema3A-Induced Growth Cone Collapse in DRG Neurons from nclf Mice
We have shown that CLN6 physically interacts with CRMP-2 and that CRMP-2 protein levels are modified in the thalami of nclf mice; however, the pathological implications of losing this interaction in vLINCL are not clear. This led us to explore possible CRMP-2-associated phenotypes in the nclf mouse. CRMP-2 plays a role in growth cone collapse in response to the binding of the extracellular chemorepellent semaphorin 3A to its receptor, the neuropilin-1/plexin-A complex (Takahashi et al., 1999). This binding event triggers a signaling pathway that eventually results in phosphorylation of CRMP-2 by GSK-3β, which causes inactivation of CRMP-2 and growth cone collapse (Brown et al., 2004). This function of CRMP-2 can be assayed by observing the outgrowth of axons within DRG neurons derived from the spinal cord of embryonic mice. If the CRMP-2 pathway functions properly, outgrowth of DRG neurons cultured in the presence of Sema3A would be repelled. However, if CRMP-2 function is impaired, there would be abnormal outgrowth of DRG neurites toward the source of Sema3A. Therefore, we examined whether there is a defect in the outgrowth of DRG neurites derived from nclf mice. We cultured DRG neurons derived from nclf and WT control mice in the presence of either Sema3A, to induce CRMP-2-dependent inhibition of outgrowth, or Sem3F, which induces inhibition of neurite outgrowth in a CRMP-2-independent manner. We found no difference in the repulsion of neurite outgrowth between the nclf and WT mice in response to either molecule (Fig. 3). This indicates there was no Sema3A-related CRMP-2-dependent phenotype in the DRG neurons from nclf mice.
Fig. 3.
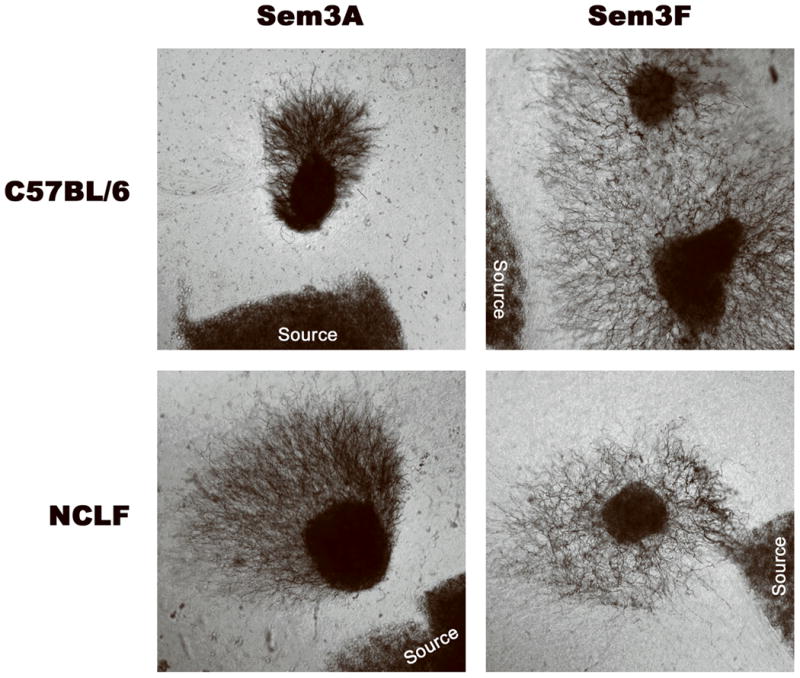
Repulsion of DRG neurons derived from nclf mice was normal when exposed semaphorin 3A. Dorsal root ganglion (DRG) neurons were removed from either E14.5 C57BL/6 (wild-type) or nclf mice (n = 3). DRGs were cultured adjacent to collagen-embedded HEK293 fibroblasts expressing either semaphorin 3A (Sema3A) or 3F (Sem3F). These fibroblasts then release these factors into the media. After incubation at 37°C for 48 hr, images were obtained looking for repulsion of outgrowth. Sema3A signaling is one of many pathways in which CRMP-2 is downstream. If there is a defect in CRMP-2 signaling through stimulation by Sema3A, no repulsion would be expected. In contrast, Sem3F functions in a CRMP-2 independent pathway, therefore acting as a negative control. We observed no differences in CRMP-2-dependent Sema3A signaling between nclf mouse and wild-type-derived DRGs (magnification 4×).
Defects in Hippocampal Neuron Development In Vitro
Hippocampal neuron cultures generated by dissociation from embryonic hippocampi have become a classic model for examining polarization of neurons, as these cultures develop dendritic processes and a single axonal projection. Over several days of coculture, these cells undergo dramatic changes in morphology because of this polarization and are typically considered to be polarized and mature by 7 days in vitro (Dotti et al., 1988). Functional roles of CRMP-2 in neurite outgrowth and neuronal polarity have been described in dissociated hippocampal cultures. CRMP-2 inhibition causes a decrease in neurite outgrowth, whereas CRMP-2 overexpression leads to aberrant formation of multiple axons (Inagaki et al., 2001). Additionally, in vivo, CRMP-2 is downstream of a trophic factor that signals in the developing hippocampus, which aids in axon guidance (Arimura et al., 2005). Therefore, we hypothesized that the loss of the CLN6–CRMP-2 interaction in nclf mice would cause CRMP-2-associated phenotypes in hippocampal neurons. We isolated hippocampi from E16.5 nclf and C57BL6/J mice and grew cocultures of dissociated neurons above a glial feeder layer derived from postnatal day 1 rats. We did not observe any obvious differences in axon outgrowth in the early stages of in vitro development. However, on day in vitro (DIV) 4, we observed a change in the morphology of the nclf neurons (Fig. 4). Cellular processes from nclf neurons were not as developed and did not appear to make as many putative synaptic connections as did neurons from control mice. In DIV8 cultures, there was a marked change in the morphology of the nclf neurons, whose processes were much thinner and less elaborate. Additionally, although the same number of neurons was plated for both genotypes and equal amounts remained after DIV4, we observed about a 50% decrease in the number of cells remaining in the nclf mouse cultures at DIV8 (Fig. 4). This suggests that abnormal development and/or maturation of nclf hippocampal neurons leads to greater cell death in culture.
Fig. 4.
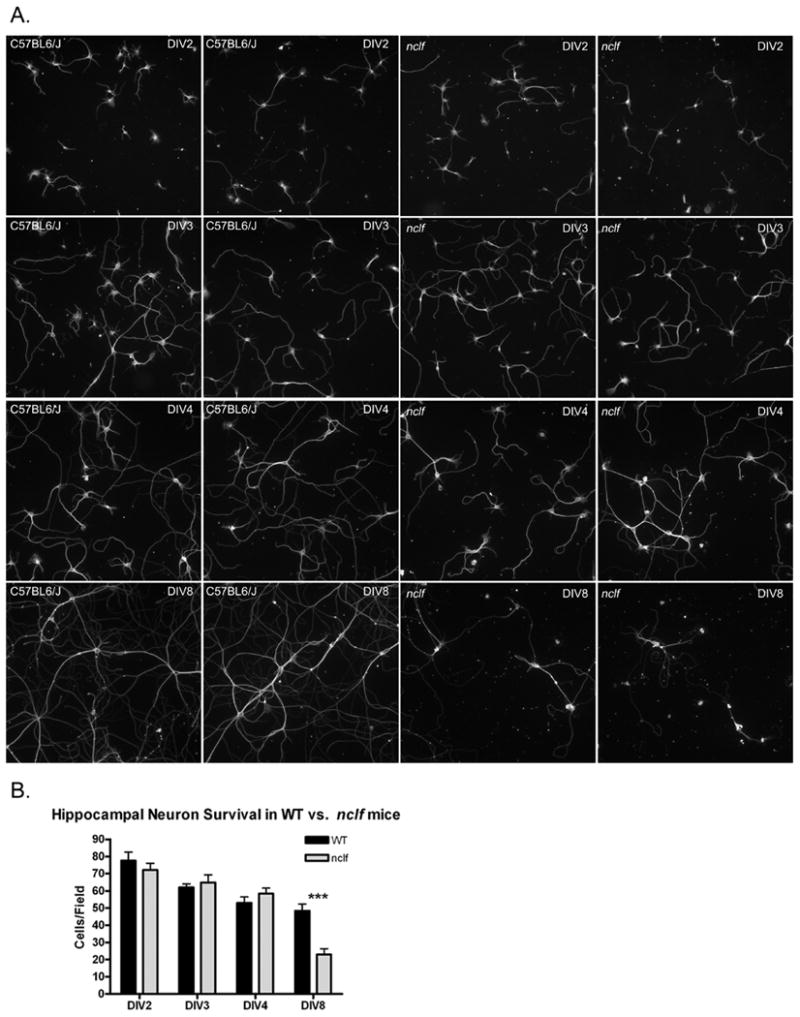
nclf Mice exhibited defects in the maturation and viability of hippocampal neurons. Hippocampal neurons isolated from embryonic day 16.5 nclf and C57BL6/J (wild-type) mice (n = 3) were plated at the same density on a poly-l-lysine-coated cover glass as a dissociated coculture and were grown suspended above a rat glial layer. To observe the development of these neurons, we fixed and stained the cells with an antitubulin antibody at four times: day in vitro (DIV) 2, DIV3, DIV4, and DIV8 (A). Two fields are represented for each genotype at each time, which are representative of two independent experiments. We did not observe any differences between the two genotypes in the early stages, DIV2 (first row) and DIV3 (second row). However, starting on DIV4 (third row), the nclf neurons appeared to be immature. Additionally, by DIV8 (fourth row), the cells were immature compared with those of the wild type, rarely make synaptic connections with neighboring cells, and appeared to have fewer cells. Therefore, the cell nuclei were stained with DAPI, and the number of cells was counted. There was no difference in cell number on DIV2, DIV3, and DIV4, but we observed a significant decrease of 50% in nclf cell number (★★★P < 0.001, two-way ANOVA, Bonferroni's post hoc test; magnification 10×) (B).
Discussion
Variant late-onset NCL (vLINCL) is a rapidly progressing NCL with no known treatment or cure. vLINCL is known to be caused by at least 29 distinct mutations in the CLN6 gene (Mole, 2008). Currently, however, the function of CLN6 is unknown. In this study, we have provided evidence of a novel CLN6 interaction that provides clues to CLN6 function. Using the yeast two-hybrid, we identified collapsin mediator response protein-2 (CRMP-2) as a protein that interacts with the N-terminus of CLN6. The nclf mouse is homozygous for a 1-bp insertion (c316insC), which causes a frameshift after P105 (Wheeler et al., 2002). This same mutation has been found in vLINCL patients, and the nclf mouse recapitulates many of the features seen in vLINCL. In the nclf mouse, we observed a significant decrease in total CRMP-2 protein level in the thalamus and a comparable decrease in phosphorylated CRMP-2 in this region. This suggests that CRMP-2 level is modified in the absence of CLN6 in the thalamus. However, modified expression of CRMP-2 was not ubiquitous throughout the brain. For example, we could find no evidence of a decrease in CRMP-2 level in the cortex (nor in the cerebellum; data not shown) of the nclf mouse, although neuronal populations in the cortex are known to be affected in vLINCL and in the South Hampshire sheep model, which harbors mutations in OCL6, the gene homologous to CLN6 (Jolly and Walkley, 1999; Heine et al., 2003; Oswald et al., 2008). Specifically, GABAergic interneurons positive for parvalbumin immunoreactivity in the cerebral cortex and hippocampus are lost as the disease progresses (Oswald et al., 2005, 2008). This suggests that any relationship between the pathological progression of vLINCL and the CLN6–CRMP-2 interaction is likely to be complex.
To understand the CLN6–CRMP-2 interaction further in the nclf mouse, we assayed phenotypes associated with CRMP-2 function in order to reveal a primary function of this interaction, a consequence of CLN6 loss or by extension a contributing factor to vLINCL pathology. CRMP-2 is highly expressed in the developing CNS, where it plays a crucial role in many cellular pathways in vivo. For example, CRMP-2 plays a role in microtubule dynamics through its interaction with tubulin heterodimers and its interaction with the microtubule binding protein kinesin-1 (Fukata et al., 2002; Kimura et al., 2005). CRMP-2 activity can be modulated through various phosphorylation events, which inactivate CRMP-2 function. Sema3A binds to the neuropilin-1/plexin-A complex and activates a signal cascade that causes axonal repulsion through growth cone collapse, resulting from inactivation of CRMP-2 by phosphorylation (Takahashi et al., 1999). We examined this pathway in dorsal root ganglion neurons from nclf mice. Sema3A induced growth cone collapse in DRG neurons in a CRMP-2 dependent manner; however, we found that Sema3A-induced growth cone collapse was not defective in the nclf mice, indicating that CRMP-2 dependent axon guidance is not affected.
Although the CRMP-2-dependent Sema3A pathway was not affected by the loss of CLN6 in DRGs, other CRMP-2 associated pathways may be affected in vLINCL. For example, CRMP-2 activity in regulating neurite outgrowth and neuron polarity in cultured hippocampal neurons have been well studied. Overexpression of CRMP-2 causes abnormal outgrowth of multiple axons and elongation of the primary axon (Inagaki et al., 2001; Cole et al., 2004). Alternatively, the use of dominant-negative CRMP-2 or the use of RNA interference to knock down CRMP-2 inhibits the formation of axons. Based on these studies, we examined neuronal morphology and neurite outgrowth in dissociated hippocampal neurons from nclf mice. Initially, we did not observe any differences in axon outgrowth or neuronal morphology; however, after the inception of stage 4 (around DIV4), when dendrites begin to mature, the development of nclf neurons arrested. In fact, by DIV8, a time when WT neurons are mature, the nclf neurons were still immature, having much less secondary and tertiary neurite branching and also an abnormal morphology with thinner processes. Overall, we observed an approximately 50% decrease in the number of nclf neurons surviving in culture. We did not observe a multiple axon phenotype typical of overactivity of CRMP-2 by staining for either B-tubulin (Fig. 4) or Tau (data not shown), nor did we observe a delay in axon formation, which could indicate decreased CRMP-2 activity. Our findings reveal an inherent and significant abnormality in nclf hippocampal neurons in coculture, which could be attributable to CRMP-2-related modifications in neurite morphology and outgrowth.
Although this coculture system provides the best system to study these cells in close to in vivo–like conditions because of the presence of glial support cells, it still does not contain conditions identical to those in vivo. A number of other cell types are present in the developing CNS that signal the maturation and growth of nascent cells. Because there is not massive cell loss early in the development of the nclf hippocampus, it is likely that there are compensatory or additional pathways that correct for the underlying defect resulting from the loss of Cln6 function revealed in vitro, which would explain why the hippocampi remain largely intact in nclf mice in vivo.
Understanding the pathological mechanisms that lead to vLINCL is an arduous task, but as we begin to grasp the function of CLN6, it will become clear how a deficit in this protein can lead to neurodegeneration. Taken together, we have described an interaction of the ER-resident transmembrane protein CLN6 with CRMP-2 that may have implications in neuronal maturation or integrity in vLINCL. As CLN6 is highly expressed in the hippocampus, and patients experience a marked decrease in learning ability, it is reasonable to hypothesize that CLN6 plays a role in neuronal maturation in this region. This study provides the first evidence of a direct molecular interaction of CLN6 with CRMP-2 and highlights possible functional consequences of losing this interaction.
Acknowledgments
The authors thank Rita Giuliano for help with neuronal cultures, Jill Weimer for help with the yeast two-hybrid, Alice Palmer for assistance with some of the quantitative Western blotting experiments, and Dr. Calum Sutherland (University of Dundee, UK) for the kind gift of CRMP-2 antibodies. This work was supported in part by NIH R01 NS43310 (to D.A.P.), BBSRC grant BB/D001722/1 (to T.H.G.), and the Batten Disease Support and Research Association fellowship (to J.W.B.).
Contract grant sponsor: NIH; Contract grant number: R01 NS43310 (to D.A.P.); Contract grant sponsor: BBSRC; Contract grant number: BB/D001722/1 (to T.H.G.); Contract grant sponsor: Batten Disease Support and Research Association fellowship (to J.W.B.).
References
- Arimura N, Menager C, Fukata Y, Kaibuchi K. Role of CRMP-2 in neuronal polarity. J Neurobiol. 2004;58:34–47. doi: 10.1002/neu.10269. [DOI] [PubMed] [Google Scholar]
- Arimura N, Menager C, Kawano Y, Yoshimura T, Kawabata S, Hattori A, Fukata Y, Amano M, Goshima Y, Inagaki M, Morone N, Usukura J, Kaibuchi K. Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Mol Cell Biol. 2005;25:9973–9984. doi: 10.1128/MCB.25.22.9973-9984.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autti T, Hamalainen J, Aberg L, Lauronen L, Tyynela J, Van Leemput K. Thalami and corona radiata in juvenile NCL (CLN3): a voxel-based morphometric study. Eur J Neurol. 2007a;14:447–450. doi: 10.1111/j.1468-1331.2007.01692.x. [DOI] [PubMed] [Google Scholar]
- Autti T, Joensuu R, Aberg L. Decreased T2 signal in the thalami may be a sign of lysosomal storage disease. Neuroradiology. 2007b;49:571–578. doi: 10.1007/s00234-007-0220-6. [DOI] [PubMed] [Google Scholar]
- Banker G, Goslin K. In: Rat Hippocampal Neurons in Low-Density Culture. Banker G, Goslin K, editors. Cambridge, MA: MIT Press; 1991. [Google Scholar]
- Bronson RT, Donahue LR, Johnson KR, Tanner A, Lane PW, Faust JR. Neuronal ceroid lipofuscinosis (nclf), a new disorder of the mouse linked to chromosome 9. Am J Med Genet. 1998;77:289–297. doi: 10.1002/(sici)1096-8628(19980526)77:4<289::aid-ajmg8>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Broom MF, Zhou C, Broom JE, Barwell KJ, Jolly RD, Hill DF. Ovine neuronal ceroid lipofuscinosis: a large animal model syntenic with the human neuronal ceroid lipofuscinosis variant CLN6. J Med Genet. 1998;35:717–721. doi: 10.1136/jmg.35.9.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M, Jacobs T, Eickholt B, Ferrari G, Teo M, Monfries C, Qi RZ, Leung T, Lim L, Hall C. Alpha2-chimaerin, cyclin-dependent Kinase 5/p35, and its target collapsin response mediator protein-2 are essential components in semaphorin 3A–induced growth-cone collapse. J Neurosci. 2004;24:8994–9004. doi: 10.1523/JNEUROSCI.3184-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole AR, Knebel A, Morrice NA, Robertson LA, Irving AJ, Connolly CN, Sutherland C. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J Biol Chem. 2004;279:50176–50180. doi: 10.1074/jbc.C400412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook RW, Jolly RD, Palmer DN, Tammen I, Broom MF, McKinnon R. Neuronal ceroid lipofuscinosis in Merino sheep. Aust Vet J. 2002;80:292–297. doi: 10.1111/j.1751-0813.2002.tb10847.x. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Itoh TJ, Kimura T, Menager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H, Kaibuchi K. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol. 2002;4:583–591. doi: 10.1038/ncb825. [DOI] [PubMed] [Google Scholar]
- Gao H, Boustany RM, Espinola JA, Cotman SL, Srinidhi L, Antonellis KA, Gillis T, Qin X, Liu S, Donahue LR, Bronson RT, Faust JR, Stout D, Haines JL, Lerner TJ, MacDonald ME. Mutations in a novel CLN6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man and mouse. Am J Hum Genet. 2002;70:324–335. doi: 10.1086/338190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine C, Koch B, Storch S, Kohlschutter A, Palmer DN, Braulke T. Defective endoplasmic reticulum-resident membrane protein CLN6 affects lysosomal degradation of endocytosed arylsulfatase A. J Biol Chem. 2004;279:22347–22352. doi: 10.1074/jbc.M400643200. [DOI] [PubMed] [Google Scholar]
- Heine C, Quitsch A, Storch S, Martin Y, Lonka L, Lehesjoki AE, Mole SE, Braulke T. Topology and endoplasmic reticulum retention signals of the lysosomal storage disease-related membrane protein CLN6. Mol Membr Biol. 2007;24:74–87. doi: 10.1080/09687860600967317. [DOI] [PubMed] [Google Scholar]
- Heine C, Tyynela J, Cooper JD, Palmer DN, Elleder M, Kohlschutter A, Braulke T. Enhanced expression of manganese-dependent superoxide dismutase in human and sheep CLN6 tissues. Biochem J. 2003;376:369–376. doi: 10.1042/BJ20030598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Chihara K, Arimura N, Menager C, Kawano Y, Matsuo N, Nishimura T, Amano M, Kaibuchi K. CRMP-2 induces axons in cultured hippocampal neurons. Nat Neurosci. 2001;4:781–782. doi: 10.1038/90476. [DOI] [PubMed] [Google Scholar]
- Jolly RD, Walkley SU. Ovine ceroid lipofuscinosis (OCL6): postulated mechanism of neurodegeneration. Mol Genet Metab. 1999;66:376–380. doi: 10.1006/mgme.1999.2821. [DOI] [PubMed] [Google Scholar]
- Kawano Y, Yoshimura T, Tsuboi D, Kawabata S, Kaneko-Kawano T, Shirataki H, Takenawa T, Kaibuchi K. CRMP-2 is involved in kinesin-1-dependent transport of the Sra-1/WAVE1 complex and axon formation. Mol Cell Biol. 2005;25:9920–9935. doi: 10.1128/MCB.25.22.9920-9935.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Watanabe H, Iwamatsu A, Kaibuchi K. Tubulin and CRMP-2 complex is transported via Kinesin-1. J Neurochem. 2005;93:1371–1382. doi: 10.1111/j.1471-4159.2005.03063.x. [DOI] [PubMed] [Google Scholar]
- Lake BD, Cavanagh NP. Early-juvenile Batten's disease—a recognisable sub-group distinct from other forms of Batten's disease. Analysis of 5 patients. J Neurol Sci. 1978;36:265–271. doi: 10.1016/0022-510x(78)90087-4. [DOI] [PubMed] [Google Scholar]
- Mole SE, Michaux G, Codlin S, Wheeler RB, Sharp JD, Cutler DF. CLN6, which is associated with a lysosomal storage disease, is an endoplasmic reticulum protein. Exp Cell Res. 2004;298:399–406. doi: 10.1016/j.yexcr.2004.04.042. [DOI] [PubMed] [Google Scholar]
- Mole SEea. NCL Resource: A Gateway for Batten Disease. 2008 Available at: http://www.ucl.ac.uk/ncl/cln6.shtml.
- Nishimura T, Fukata Y, Kato K, Yamaguchi T, Matsuura Y, Kamiguchi H, Kaibuchi K. CRMP-2 regulates polarized Numb-mediated endocytosis for axon growth. Nat Cell Biol. 2003;5:819–826. doi: 10.1038/ncb1039. [DOI] [PubMed] [Google Scholar]
- Oswald MJ, Palmer DN, Kay GW, Barwell KJ, Cooper JD. Location and connectivity determine GABAergic interneuron survival in the brains of South Hampshire sheep with CLN6 neuronal ceroid lipofuscinosis. Neurobiol Dis. 2008 doi: 10.1016/j.nbd.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald MJ, Palmer DN, Kay GW, Shemilt SJ, Rezaie P, Cooper JD. Glial activation spreads from specific cerebral foci and precedes neurodegeneration in presymptomatic ovine neuronal ceroid lipofuscinosis (CLN6) Neurobiol Dis. 2005;20:49–63. doi: 10.1016/j.nbd.2005.01.025. [DOI] [PubMed] [Google Scholar]
- Pontikis CC, Cotman SL, MacDonald ME, Cooper JD. Thalamocortical neuron loss and localized astrocytosis in the Cln3Deltaex7/8 knock-in mouse model of Batten disease. Neurobiol Dis. 2005;20:823–836. doi: 10.1016/j.nbd.2005.05.018. [DOI] [PubMed] [Google Scholar]
- Sharp JD, Wheeler RB, Parker KA, Gardiner RM, Williams RE, Mole SE. Spectrum of CLN6 mutations in variant late infantile neuronal ceroid lipofuscinosis. Hum Mutat. 2003;22:35–42. doi: 10.1002/humu.10227. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Fournier A, Nakamura F, Wang LH, Murakami Y, Kalb RG, Fujisawa H, Strittmatter SM. Plexin-neuropilin-1 complexes form functional semaphorin-3A receptors. Cell. 1999;99:59–69. doi: 10.1016/s0092-8674(00)80062-8. [DOI] [PubMed] [Google Scholar]
- Tammen I, Cook RW, Nicholas FW, Raadsma HW. Neuronal ceroid lipofuscinosis in Australian Merino sheep: a new animal model. Eur J Paediatr Neurol. 2001;5:37–41. doi: 10.1053/ejpn.2000.0432. [DOI] [PubMed] [Google Scholar]
- Tammen I, Houweling PJ, Frugier T, Mitchell NL, Kay GW, Cavanagh JA, Cook RW, Raadsma HW, Palmer DN. A missense mutation (c.184C>T) in ovine CLN6 causes neuronal ceroid lipofuscinosis in Merino sheep whereas affected South Hampshire sheep have reduced levels of CLN6 mRNA. Biochim Biophys Acta. 2006;1762:898–905. doi: 10.1016/j.bbadis.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Tyynela J, Suopanki J, Santavuori P, Baumann M, Haltia M. Variant late infantile neuronal ceroid-lipofuscinosis: pathology and biochemistry. J Neuropathol Exp Neurol. 1997;56:369–375. doi: 10.1097/00005072-199704000-00005. [DOI] [PubMed] [Google Scholar]
- Wheeler RB, Sharp JD, Schultz RA, Joslin JM, Williams RE, Mole SE. The gene mutated in variant late-infantile neuronal ceroid lipofuscinosis (CLN6) and in nclf mutant mice encodes a novel predicted transmembrane protein. Am J Hum Genet. 2002;70:537–542. doi: 10.1086/338708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart TM, Paterson JM, Short DM, Meredith S, Robertson KA, Sutherland C, Cousin MA, Dutia MB, Gillingwater TH. Differential proteomics analysis of synaptic proteins identifies potential cellular targets and protein mediators of synaptic neuroprotection conferred by the slow Wallerian degeneration (Wlds) gene. Mol Cell Proteomics. 2007;6:1318–1330. doi: 10.1074/mcp.M600457-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]