

Abstract
Pseudomonas aeruginosa (PA) secrete N-(3-oxododecanoyl)-homoserine lactone (HSL-C12) as a quorum-sensing molecule to regulate bacterial gene expression. Since HSL-C12 is membrane-permeant, multiple cell types in PA-infected airways may be exposed to HSL-C12, especially adjacent to biofilms where local [HSL-C12] may be high. Previous reports showed that HSL-C12 causes both pro- and anti-inflammatory effects. To characterize HSL-C12’s pro- and anti-inflammatory effects in host cells we measured protein synthesis, NF-κB activation and KC (mouse IL-8) and IL-6 mRNA and protein secretion in wild type (WT) mouse embryonic fibroblasts (MEF). To test the role of the ER stress inducer PERK we compared these responses in PERK−/− and PERK-corrected PERK−/− MEF. During 4 hr treatments of WT MEF, HSL-C12 potentially activated NF-κB p65 by preventing the re-synthesis of IκB and increased transcription of KC and IL-6 genes (qPCR). HSL-C12 also inhibited secretion of KC and/or IL-6 into the media (ELISA) both in control conditions and also during stimulation by TNFα. HSL-C12 also activated PERK (as shown by increased phosphorylation of eI-F2α) and inhibited protein synthesis (as measured by incorporation of 35S-methionine by MEF). Comparisons of PERK−/− and PERK-corrected MEF showed that HSL-C12’s effects were explained in part by activation of PERK → phosphorylation of eI-F2α → inhibition of protein synthesis → reduced IκBα production → activation of NF-κB → increased transcription of the KC gene but reduced translation and secretion of KC. HSL-C12 may be an important modulator of early (up to 4 hrs) inflammatory signaling in P. aeruginosa infections.
INTRODUCTION
Pseudomonas aeruginosa are gram-negative bacteria that form biofilms in the airways of patients with Cystic Fibrosis (CF) (1). P. aeruginosa coordinate the production of biofilms and virulence factors using the small molecule N-(3-oxododecanoyl)-homoserine lactone (HSL-C12) as a lipid-soluble, diffusible quorum-sensing molecule (2–4). HSL-C12 has multiple effects on mammalian cells, including inducing apoptosis and activating release of Ca2+ from endoplasmic reticulum stores (5–10). HSL-C12 has also been reported to affect inflammatory signaling, though some reports indicate an activation of pro-inflammatory signaling while others indicate a suppression of inflammatory signaling (11–17).
The goal of this study was to elucidate HSL-C12’s role in inflammatory signaling and discover associated effector molecules. To accomplish this we used mouse embryonic fibroblasts (MEF). Fibroblasts are expected to be exposed to the membrane-permeant HSL-C12 in P. aeruginosa biofilm-infected lungs. In addition, MEF are a genetically tractable system with many knockout lines available. We measured expression and secretion of KC (mouse equivalent of human IL-8) and IL-6, because these are important cytokines mediating epithelial immunity produced in response to NF-κB signaling. TNFα and IL-1β were utilized as activators of the NF-κB-proinflammatory signaling pathway. We show in the present study that both TNFα and IL-1β cause increases in KC gene transcription and KC secretion. HSL-C12 increased KC gene transcription but did not increase KC secretion, even in the presence of TNFα or IL-1β. This uncoupling of KC gene transcription from KC secretion could have resulted from an inhibition of protein synthesis resulting from HSL-C12-induced release of Ca2+ from the endoplasmic reticulum (ER) (9, 10, 18) resulting in decreased [Ca2+] in the ER, activation of ER stress and consequent inhibition of protein synthesis (19). We therefore explored the role of ER stress in the responses of MEF to HSL-C12.
We tested specifically the role of protein kinase RNA-like endoplasmic reticulum kinase (PERK), a transducer of ER stress, in HSL-C12-mediated translation inhibition. PERK, a membrane protein localized to the ER, is one of four kinases known to phosphorylate the eukaryotic translation elongation factor eI-F2α (20). PERK becomes activated when BiP chaperone proteins, which usually inhibit PERK, release from binding to PERK and are sequestered to the ER lumen due to a buildup of unfolded proteins (21). PERK is also activated by reductions in [Ca2+] in the ER. When PERK becomes active, it phosphorylates the translation elongation factor eI-F2α on serine 52 (51 in human), which causes selective inhibition of protein synthesis and induces only certain chaperones and ER stress response proteins to be translated (22). Previous studies have shown that HSL-C12 increases phosphorylation of eI-F2α in MEF (23). We therefore tested whether HSL-C12 inhibited KC secretion through its effects to activate PERK by comparing protein synthesis, NF-κB activation and KC gene transcription (mRNA production) and KC secretion by PERK−/− MEF and PERK-corrected PERK−/− MEF.
MATERIALS AND METHODS
Reagents
Unless otherwise specified, reagents and chemicals were obtained from Sigma. HSL-C12 (Cayman Chemical, Ann Arbor MI and Sigma) was dissolved in DMSO as 50 mM or 100 mM stocks, and freeze thaw cycles were limited. HSL-C12 from different suppliers displayed different activities, and therefore 50 μM or 100 μM doses were used accordingly. TNFα and IL-1β (both R&D Systems, Minneapolis, MN) were used at 10 or 20 ng/mL from 10 or 20 μg/mL stock solutions in water. The Ca2+-ATPase blocker thapsigargin (24) was prepared as a 1 mM stock in DMSO and used at 1 μM.
Cell culture of MEF
WT MEF were obtained from C. Li (Univ. Louisville). PERK−/− and corresponding PERK-corrected PERK−/− MEF cell lines were obtained from R. Kaufman (Sanford|Burnham Medical Research Institute). MEF were cultured in Dulbecco’s Modified Eagles Medium (DMEM) containing 10% FBS and 1% penicillin-streptomycin. The cells were passaged at 1:5–1:15 dilutions and the remaining cell suspension was seeded directly onto a 24-well, 12-well or 6-well tissue culture plate (BD Falcon, Bedford, MA).
ELISA and quantitative PCR for KC and IL-6
Three replicates for each of the following cells: WT, PERK−/− and PERK-corrected PERK−/− MEF were grown to confluency on 24-well plates, and experimental cells were treated for 4 hrs with HSL-C12 (50 μM), TNFα (10–20 ng/ml), IL-1β (10 ng/mL) or HSL-C12 in combination with TNFα or IL-1β. The cell culture medium was removed, the cells were washed with PBS, and samples were taken using TRIzol reagent (Life Technologies, Grand Island, NY). ELISAs were performed using R&D Systems Duo Set® kit (R&D Systems, Minneapolis, MN). Capture antibodies were incubated on Nunc MaxiSorp 96-well plates in 0.1 M sodium phosphate buffer pH 8.0 overnight at 4°C, and then the plates were blocked with DPBS with 1% BSA (PBSA) for 4 hrs at 4°C. 50 μl of culture media per well were taken from the MEF’s and applied to the plate overnight at 4°C, followed by five washes with PBS with 0.1% Tween-20 (PBST) and biotinylated capture antibodies in PBSA for 2 hrs at room temperature. Five more PBST washes were performed followed by 30 mins of streptavidin-HRP at room temperature. After five more PBST washes wells were developed for 10 min at room temperature in the dark with 1 mg/ml OPD in 0.05 M sodium phosphate / 0.02 M citrate buffer, pH 5.0 and stopped with 3 M HCl. Absorbance was read in a spectrophotometer at 490 nm.
Apoptosis-related cell death of MEF that occurred during HSL-C12 treatments (25) could have altered measurements of KC by ELISA. We attempted to measure the magnitude of this effect by normalizing to [protein] adherent to the cell culture plate after washing. Samples were taken from organic phase from TRIzol preparation and dot-blotted onto filter paper. Filter paper was then stained with Coomassie Blue and then destained with 10% methanol 10% acetic acid. Filter paper was scanned and quantified using ImageJ. KC concentrations obtained through ELISA were divided by [protein] acquired through dot-blot. This normalization did not appreciably alter measurements of KC secretion in response to any of the treatments. KC secretion has therefore been reported in ng/ml of sampled media during four hr treatments.
For qPCR experiments three biological samples of cells were combined after each treatment. Purified RNA samples from TRIzol lysates were treated with DNase (Frementas, Glen Burnie, Maryland) and then reverse transcribed using SuperScript III (Life Technologies, Grand Island, NY) with random primers. KC gene expression level was determined by real-time PCR on triplicates carried out in 7900HT Fast Real-Time PCR System (Applied Biosystems) with SYBR mix (KAPA Biosystems, Woburn, MA) using gene specific primers. House-keeping gene Rps17 was used as normalization control throughout all experiments and all data are presented as RQ score relative to RPS17. Primers used for real-time PCR were: KC: forward: 5′-CTTGAAGGTGTTGCCCTCAG-3′ and reverse: 5′-TGGGGACACCTTTTAGCATC-3′. IL-6: forward: 5′-TCCAGTTGCCTTCTTGGGAC-3′ and reverse: 5′-GTACTCCAGAAGACCAGAGG-3′. Rps17: forward: 5′-CGCCATTATCCCCAGCAAG-3′ and reverse: 5′-TGTCGGGATCCACCTCAATG-3′
NF-κB-regulated luciferase
A recombinant adenoviral vector expressing a luciferase reporter gene driven by transcriptional activation by NF-κB (adv-NF-κB-luc) was used for studies to determine effects on NF-κB activation as described previously (26). This vector contained the luciferase gene driven by four tandem copies of the NF-κB consensus sequence and was stored in 10 mM Tris with 20% glycerol at −80°C. The virus was added to MEF at a multiplicity of infection (MOI) of 100, and they were returned to the incubator for 24 h. Cells were then washed to remove viruses and left to grow for another day. Previous experiments showed that the adenovirus elicits expression in >75% of the cells (26). WT MEF in media were left untreated or exposed to HSL-C12 (50 μM), TNFα (10 ng/ml) or HSL+TNFα for 4 h. Cells were then washed and processed with the luciferase assay system by using Reporter Lysis Buffer (Promega, Madison, WI) to measure NF-κB-mediated transcriptional induction according to the manufacturer’s protocol. Measurements of luciferase activity (relative light units) were performed in triplicate for each sample and normalized to the protein concentration (Bradford assay). Averages were then expressed relative to the average control value in the epithelial cells, which was set equal to 1.0.
Caspase 3/7 Assay
Caspase 3/7 activation was measured by cell-based homogeneous luminescent assays (Caspase-Glo, Promega, Madison, WI), in which a specific substrate that contains the tetrapeptide DEVD was cleaved by the activated cellular caspases to release aminoluciferin, which reacts with luciferase, resulting in the production of light. PERK−/− and PERK-corrected PERK−/− MEF were plated on a clear-bottom, white 96-well plate (Nunc, Penfield, NY) in 100 μl media per well and grown to confluency. Cells were treated with increasing doses of HSL-C12 in Ringer’s solution. After treatment, 100 μl of reagent mix was added to each well. The plate was incubated at room temperature for 1 hr on a shaker, and the end-point luminescence was measured in a plate-reading luminometer (LmaxII 384, Molecular Devices, Sunnyvale, CA). Data were background (blank) subtracted and averaged.
Western blotting
MEF were grown in 6-well plates to confluency and treated for up to 4 hrs with HSL-C12 or 1 hr with 1 μM thapsigargin and then lysed in M-PER mammalian protein extraction reagent (Pierce, Rockford, IL) containing 5 μg/ml leupeptin, 5 μg/ml pepstatin, 1 mM phenylmethylsulfonyl fluoride, and 50 nM calyculin A. Protein sample concentrations were determined with Bradford reagent (Bio-Rad, Hercules, CA). Immunoblot analysis was performed by first separating protein (10 to 50 μg/lane) by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferring it to nitrocellulose membranes. Individual gels with identical loading were run side by side when multiple primary antibodies were utilized. Membranes were blocked (5% nonfat dried milk) in 20 mM Tris-HCl (pH 7.5)−150 mM NaCl-0.1% Tween 20 for 1 hr and then incubated with specific antibodies overnight. Primary antibodies (diluted 1:1,000 in blocking buffer) for phosphoS51- eI-F2α (119A11), eI-F2α (9722), IκBα (L35A5), and NF-κB p65 (C22B4) were acquired from Cell Signaling (Danvers, MA). Binding of primary antibodies was visualized by enhanced chemiluminescence with horseradish peroxidase-conjugated secondary antibodies (1:2,500 in blocking buffer) and Renaissance Chemiluminescence Reagent Plus (Perkin-Elmer Life Sciences). Quantitation was performed with ImageJ (National Institutes of Health, Bethesda, MD).
35S uptake into protein
Figure 4: WT MEF were grown in 24-well plates to confluency. Wells were pretreated for 3 hrs with either 50 μM HSL-C12 or DMSO (control). At 3 hrs post-infection, medium was removed and incubated with 25 μCi/ml [35S]methionine (Perkin Elmer, Waltham, MA) in RPMI 1640 medium without methionine supplemented with 10% serum, 2 mM L-glutamine, and 50 μM HSL-C12 or DMSO. Cells were labeled for 1 hr, washed three times with cold PBS, and then lysed with radioimmunoprecipitation assay (RIPA) buffer supplemented with 2 mM Na3VO4, 1 mM PMSF, 25 mM NaF, and 1x Roche protease inhibitor mixture (no EDTA) (pH 7.2) for 10 min at 4°C. Total protein levels were measured by bicinchoninic acid assay, and equal amounts of protein were mixed with SDS sample buffer (40% glycerol, 8% SDS, 2% 2-ME, 40 mM EDTA, 0.05% bromophenol blue, and 250 mM Tris-HCl [pH 6.8]), boiled for 5 min, and then separated by SDS-PAGE. The gels were stained with Coomassie blue to show equal protein loading, dried, and exposed to a phosphor screen and visualized using a Typhoon Trio imager (GE Healthcare).
Fig. 4. HSL-C12 inhibits protein synthesis.
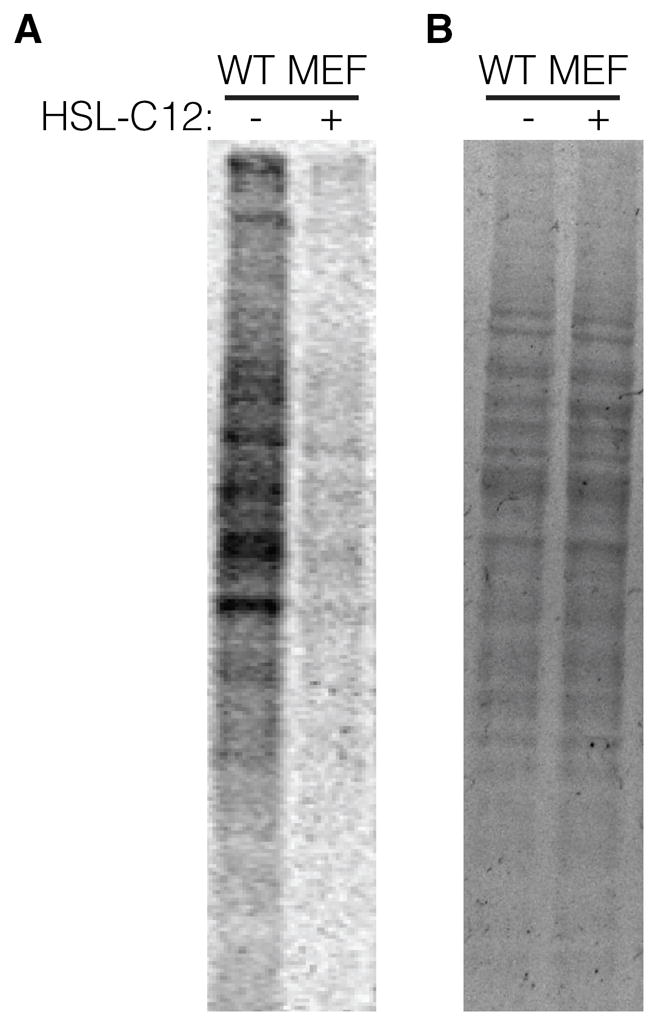
(A) 35S methionine radio-labeling of bulk protein from WT MEF treated with 50 μM HSL-C12 or DMSO (mock-treated) and then imaged with a phosphor screen. HSL-C12 treatments were for 4 hrs total with radio-labeling performed during final hr. (B) Coomassie-stained SDS-PAGE gel from same experiment. Results are representative of two experiments.
Figure 7: Same as Figure 4, with treatments of 100uM HSL-C12 combined with 10ng/ml TNFα for 3 hours pre-labeling. Phosphor and coomassie images were quantified using ImageJ (NIH, Bethesda, MD).
Fig. 7. HSL-C12’s inhibition of protein synthesis is PERK-dependent.
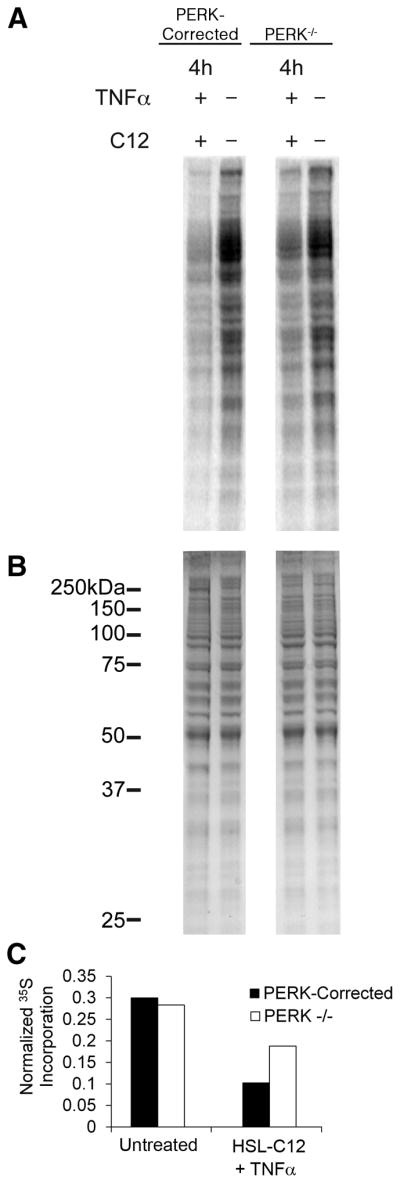
(A) 35S methionine radio-labeling of bulk protein from PERK−/− and PERK-corrected MEF untreated or treated with 100 μM HSL-C12 + 10 ng/mL TNFα imaged with a phosphor screen. MEF were treated with HSL-C12 + TNFα for 4 hrs, with radio-labeling performed during final hr. (B) Coomassie-stained SDS-PAGE gel from same experiment. (C) Graph of phosphor images from (A) were quantified and normalized to Coomassie image from (B). Normalized 35S incorporation was calculated from the entire lane for PERK-corrected and PERK−/− MEF, untreated or treated with HSL-C12+TNFα. Results typical of two similar experiments.
Statistics
Significance was tested using the two tailed students T-test with equal or unequal distribution as mentioned in the figure legends. p values < 0.05 were considered significant. Error bars in graphs indicate standard errors (SE). Symbols *, # and % in figures denote significant differences between different treatments as stated in legends.
RESULTS
HSL-C12 increases proinflammatory cytokine gene transcription but reduces cytokine secretion
We used 50–100 μM HSL-C12 for studies testing inflammatory responses in MEF. These concentrations were based on previous studies showing that HSL-C12 activated apoptosis in MEF with a threshold of 1–10 μM and maximal effects at 50–100 μM (25). Similar concentration-dependence has been observed for HSL-C12 activation of Cl− secretion (9) and apoptosis (10) by airway epithelial cells.
In order to determine HSL-C12’s effects on inflammatory signaling in MEF we measured both transcription of the proinflammatory gene KC (mouse equivalent of IL-8) into mRNA using qPCR and secretion of KC protein product into the cell medium using ELISA. MEF were treated for 4 hrs with HSL-C12 (50 μM), TNFα (10 ng/ml) or a combination of the two. Results have been summarized in Fig. 1. Compared to untreated MEF, TNFα caused expected increases in KC gene transcription (Fig. 1A) and KC secretion (Fig. 1B). In contrast, treatment with HSL-C12 caused increased transcription of KC mRNA (Fig. 1A) but a decrease in secretion of KC (Fig. 1B). Similarly, exposure of MEF to TNFα + HSL-C12 caused increased KC gene transcription that was similar to that elicited by TNFα-treated MEF (Fig. 1A), but secretion of KC was less than that stimulated by TNFα and roughly equal to that of untreated, control MEF (Fig. 1B). Similar results were obtained when IL-6 mRNA transcription and secretion were measured in response to TNFα and HSL-C12 (Figs 2A, B). These data showed that TNFα caused predictable increases in transcription of KC and IL-6 mRNA and secretion of KC and IL-6. In contrast, C12-HSL, both on its own and in the presence of TNFα, caused increases in transcription of KC or IL-6 mRNA, but secretion of KC or IL-6 was either unaffected or inhibited.
Fig. 1. HSL-C12 and TNFα stimulate KC gene transcription, but C12-HSL inhibits KC secretion.
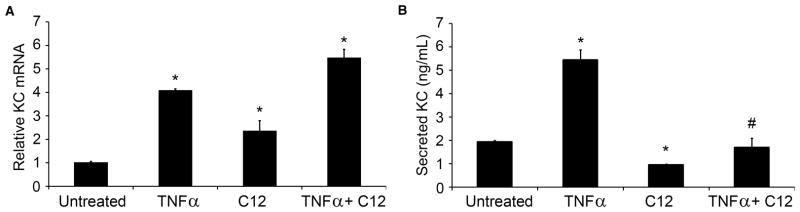
WT MEF were rinsed with fresh media then left untreated or treated with TNFα (10 ng/ml), HSL-C12 (50 μM) or TNFα+HSL-C12 for 4 hrs. Samples were taken from cell media at t = 4 hrs, and KC contents were assayed by ELISA. At the end of the experiment, RNA was isolated, and cDNA was formed for qPCR assay. (A) Quantitative PCR data for KC mRNA are given as RQ score normalized to RPS17 cDNA. Averages displayed with min and max. TNFα and HSL-C12 each increased production of KC mRNA, and treatment with TNFα+HSL-C12 caused an even larger increase than during treatment with either agonist alone. * comparison to control. (B) KC secretion (in ng/ml) for the same treatments. TNFα increased KC secretion, but HSL-C12 decreased KC secretion. Averages +/− Std. Error. N=3 biological replicates for all conditions. * comparison to control; # comparison to TNFα.
Fig. 2. Effects of HSL-C12 and TNFα on IL-6 gene transcription and IL-6 secretion.
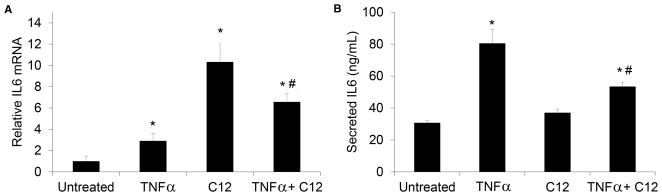
WT MEF were treated with TNFα (10 ng/ml), HSL-C12 (50 μM) or TNFα+HSL-C12 for 4 hrs, and ELISA and qPCR were performed as in Fig. 1. (A) Quantitative PCR data for IL-6 mRNA are given as RQ score normalized to RPS17 cDNA. Averages displayed with min and max. * comparison to control; # comparison to TNFα. (B) IL-6 secretion (in ng/ml) for conditions shown. Averages +/− Std. Error, n=3 biological replicates for all conditions. *p comparison to control; # comparison to TNFα.
HSL-C12 inhibits protein synthesis
The apparently contradictory effects of HSL-C12 (alone and in combination with TNFα) to stimulate KC/IL-6 gene transcription but inhibit KC/IL-6 secretion could have resulted from an inhibition of secretion of the cytokines or from an inhibition of synthesis of these two proteins. Inhibition of protein synthesis could also explain the discrepancy between measured levels of mRNA induced in response to HSL-C12 (Figs 1 and 2) and levels of NF-κB-regulated luciferase induced by HSL-C12 (Fig 3). The NF-κB-regulated luciferase method involves transfecting cells with a plasmid that expresses luciferase driven by an NF-κB-regulated promoter. When NF-κB signaling becomes activated, transcription of the luciferase gene also becomes activated, and luciferase protein is produced by the cells, which are then assayed after 4 hrs of incubation. If HSL-C12 were inhibiting protein synthesis, this could prevent increases in luciferase expression, as observed here (Fig 3) and in previous experiments (23).
Fig. 3. HSL-C12 reduces activation of NF-κB luciferase.
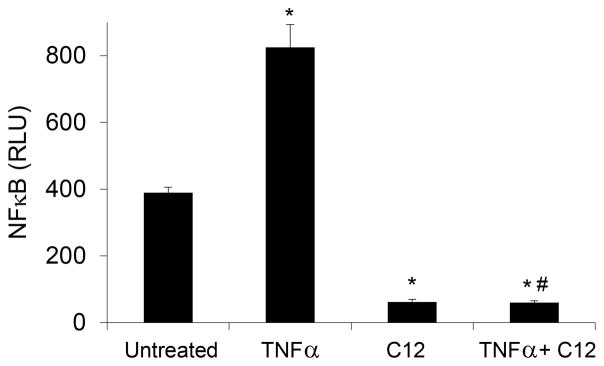
TNFα (10 ng/ml) stimulated NF-κB luciferase activity. Compared to control or TNFα-treated MEF, HSL-C12 (50 μM) decreased NF-κB luciferase activity. Data are averages (+/-SD) of three biological replicates, each performed in triplicate. * comparsion to control; # comparison to TNFα
To test for potential HSL-C12-induced inhibition of protein synthesis, we utilized a 35S-methionine metabolic labeling approach to measure global translation levels in MEF after treatment with HSL-C12 (Fig. 4). During the final hour of HSL-C12 or DMSO 4 hr treatments on WT MEF, the media were replaced with media in which the only source of methionine was 35S-methionine. Any new proteins synthesized during this time would incorporate radioactive methionine, and the level of incorporation would be directly proportional to the amount of translation occurring in those cells. When protein samples were taken, run on an SDS-PAGE gel and exposed on a phosphor screen, WT MEF samples treated with only DMSO contained radio-labeled proteins over a wide range of molecular weights (Fig. 4A). In contrast, MEF treated with HSL-C12 showed very little 35S-labeling, indicating that HSL-C12 was inhibiting protein synthesis. Coomassie labeling of the SDS-PAGE gel revealed equal protein loading between lanes (Fig. 4B), indicating that the difference in radio-labeling in response to HSL-C12 was not caused by differential loading. These results suggested that the HSL-C12-induced decoupling of increased KC gene transcription with no increases in KC secretion in MEF may have resulted at least in part from a global reduction in protein synthesis.
ER stress transducer PERK plays a role in HSL-C12-mediated inhibition of protein translation
To assay the role of PERK in HSL-C12-mediated inhibition of translation, we compared HSL-C12-induced responses in PERK−/− and corresponding PERK-corrected PERK−/− (termed PERK-corrected from here on) MEF cell lines. Western blot analysis indicated that PERK levels were similar in the WT and PERK-corrected MEF, and PERK was absent in PERK−/− MEF (Figs. 5A, B). Western blots of PERK from WT and PERK-corrected MEF displayed similar gel shifts when treated with HSL-C12, consistent with phosphorylation and activation of PERK during treatment with HSL-C12 (Fig. 5A).
Fig. 5. Characterization of PERK−/− and PERK-corrected MEF.
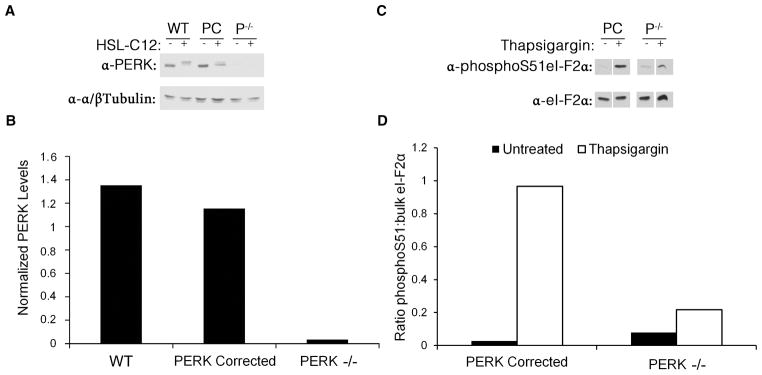
(A) PERK-corrected PERK−/− (PC) and PERK−/− (P−/−) MEF were either untreated or treated for 1 hr with 100 μM HSL-C12. Protein samples were taken, run at equal concentrations on an SDS-PAGE gel, and western blots were performed using anti-PERK and anti-α/βTubulin antibodies. (B) Results from untreated lanes from (A) quantified and displayed as ratio PERK:α/βTubulin. Results typical of two experiments. (C) PERK-corrected PERK−/− (PC) and PERK−/− (P−/−) MEF were either untreated or treated for 1 hr with 1 μM thapsigargin to activate ER stress. Protein samples were taken, run at equal concentrations on an SDS-PAGE gel, and western blots were performed using anti-phosphoS51-eI-F2α and anti-eI-F2α antibodies. (D) Results from (C) quantified and displayed as ratio phosphoS51-eI-F2α to bulk eI-F2α. Thapsigargin increased phosphorylation of eI-F-2α in PERK-corrected but much less in PERK−/− MEF. Results typical of two experiments.
We tested the functional PERK activity of these cell lines by activating ER stress using the common ER stress inducer thapsigargin, which, like HSL-C12, causes release of Ca2+ from the ER (9, 10). As measured in western blots, thapsigargin caused only a small increase in eI-F2α phosphorylation in PERK−/− cells, while PERK-corrected cells displayed much more phosphorylation (Figs 5C, D).
50 μM HSL-C12 also increased phosphorylation of eI-F2α to high levels in PERK-corrected cells but much less in PERK−/− cells, particularly during the first one-two hrs of treatment (Figs 6A, B). A summary of eI-F2α from three experiments using 100 μM HSL-C12 is shown in Fig. 6C. The higher dose of HSL-C12 resulted in an even greater difference in the response of PERK+/+ and PERK−/− cells. HSL-C12 also increased phosphorylation of eI-F2α in WT MEF (data not shown). These results indicated that HSL-C12 would induce a more pronounced inhibition of protein synthesis in PERK-corrected cells compared to PERK−/− cells. Measurements of protein synthesis using 35S labeling showed that when cells were treated with HSL-C12+TNFα there was greater inhibition of protein synthesis in PERK-corrected than PERK−/− cells (Fig. 7). Similar PERK-dependent inhibition of 35S labeling was obtained during treatment with HSL-C12 alone (data not shown). Together these results indicated that a portion of HSL-C12’s inhibitory effects on protein synthesis resulted from activation of PERK and the phosphorylation of eI-F2α.
Fig. 6. HSL-C12 increases phosphorylation of el-F2α.

(A) PERK−/− and PERK-corrected MEF were treated with 50 μM HSL-C12 for 0, 1, 2, 3 or 4 hrs. Protein samples were taken, and run at equal concentrations on an SDS-PAGE gel; western blots were performed using anti-phosphoS51-eI-F2α and anti-eI-F2α antibodies. Results typical of three similar experiments. (B) Results from (A) were quantified and displayed as the ratio of phosphoS51-eI-F2α to bulk eI-F2α. (C) Average induction (+/− std errors) of eI-F2α phosphorylation of WT, PERK−/− and PERK-corrected MEF in response to treatment with 100 μM HSL-C12 for 1 hr from two further experiments. * comparison PERK−/− to PERK-corrected, n= 3 experiments.
HSL-C12’s effects on KC gene transcription and secretion in MEF’s are PERK-dependent
The data to this point suggested that HSL-C12 caused a PERK-dependent block in host mRNA-to-protein translation that could have inhibited the production of KC. Therefore, we explored PERK’s role in KC responses to TNFα and IL-1β (a more potent activator than TNFα of the NF-κB pathway in MEF) in combination with HSL-C12. Results from these experiments have been summarized in Figs 8A–D. Similar to WT MEF (Fig. 1), PERK−/− and PERK-corrected MEF both responded to treatment with TNFα or IL-1β with increases in KC gene transcription (Figs 8A and C) and KC secretion (Figs 8B and D).
Fig. 8. HSL-C12 effects on KC gene expression and secretion are PERK-dependent.

PERK−/− and PERK-corrected MEF were rinsed with fresh media then treated for 4 hours with TNFα (20 ng/ml), HSL-C12 (100 μM) or TNFα + HSL-C12 (A and B) or with IL-1β (10 nM), HSL-C12 (100 μM) or IL-1β + HSL-C12 (C and D). Samples were taken from the media after 4 hr for measurement of KC secretion (in ng/ml of medium) by ELISA (A and C). Cells were then treated with Trizol and cDNA was prepared for qPCR analysis, where results are given as RQ score normalized to RPS17 cDNA (B and D). Averages +/− Std error; n = 3 biological replicates for all conditions, except KC secretion by PERK−/− MEF in response to IL-1β+HSL-C12, where n = 2. For experiments on PERK-corrected MEF: * comparisons to control; # comparison to TNFα (A and B) or IL-1β (C and D). For experiments on PERK−/− MEF, $ comparison to control; % comparison to TNFα (A and B) or IL-1β (C and D).
In PERK-corrected cells, HSL-C12 elicited modest increases in KC gene transcription (Figs 8A and C) but inhibited KC secretion (Figs 8B and D). In the presence of either TNFα or IL-1β, HSL-C12 increased KC gene transcription (Figs 8A and C), but KC secretion decreased markedly (i.e., compared to treatment with TNFα or IL-1β alone: Figs 8B and D). These results were similar to those obtained in wild type MEF (Figs 1 and 2). Results were different in PERK−/− MEF: HSL-C12 caused only small changes in either KC gene transcription Figs 8A and C) or KC secretion (Figs 8B and D). In the presence of either TNFα or IL-1β, HSL-C12 had only small effects on KC gene transcription (i.e., compared to TNFα or IL-1β alone: Figs 8A and C), and KC secretion was either not affected or was modestly inhibited (i.e., compared to TNFα or IL-1β alone: Figs 8B and D). These data were consistent with HSL-C12 triggering PERK-dependent increases in KC gene expression while inhibiting KC secretion. These PERK-dependent effects of HSL-C12 were particularly evident when HSL-C12 was added in combination with TNFα or IL-1β: in PERK-corrected MEF treated with either TNFα or IL-1β, HSL-C12 caused large increases in KC gene transcription but inhibitions of KC secretion, while in PERK−/− MEF treated with either TNFα or IL-1β, HSL-C12 caused only small stimulations of KC gene transcription and KC secretion remained elevated.
HSL-C12 activates NF-κB: PERK-dependence
The ability of HSL-C12 to stimulate KC gene transcription on its own and to synergize with TNFα or IL-1β in stimulating KC gene expression in PERK-corrected (but not PERK−/−) MEF (Fig. 8) may be explained by the inhibition of protein synthesis. IκB, the repressor of NF-κB, is a high turnover protein (27). Treatment with an activator of the NF-κB pathway (e.g., TNFα or IL-1β) causes phosphorylation and increased degradation of IκBα (27). It has been reported that activators of PERK, including UV-light, can also lead to NF-κB signaling by inhibiting protein synthesis, thereby decreasing the available pool of IκBα (28). We measured IκBα and p65 (a subunit of NF-κB) levels and found that over the course of a 4 hr treatment HSL-C12 caused IκBα levels to decrease more in PERK-corrected than PERK−/− cells, particularly during the first two hrs of treatment (Fig. 9A). Levels of p65 were similar between the two lines, so the ratio of p65 to IκBα, and therefore potential NF-κB activity, was greater in PERK-corrected cells during the first two hrs of treatment (Fig. 9B). These results indicated that PERK was responsible for a major part of the effects of HSL-C12 to potentially activate NF-κB (Fig. 9) and thereby increase KC gene transcription (Figs 8A and C). By inhibiting protein synthesis (Figs 4 and 7), PERK may have prevented the production and secretion of KC (Figs 8B and D), while synergistically increasing KC gene transcription when co-treated with IL-1β (Figs 8A and C) by causing reduced re-synthesis of the NF-κB repressor, IκB.
Fig. 9. HSL-C12-induced reduction in IκBα is PERK-dependent.

PERK−/− and PERK-corrected MEF were treated with 100 μM HSL-C12. (A) Protein samples were taken from cells at times shown, and run at equal concentrations on an SDS-PAGE gel; western blots were performed using anti-IκBα and anti-p65 antibodies. (B) Results from (A) quantified and displayed as ratio p65 to bulk IκBα. High ratios indicate high potential for NF-κB p65-driven transcription. Results typical of two experiments.
We also tested if the ER stress pathway that is based on IRE1α affected the HSL-C12-mediated transcriptional regulation of KC in IRE1α +/+ and IRE1α −/− MEF cells. Data shown in the Supplemental Data, Fig. S1 indicate that C12-mediated activation of KC transcription was not dependent on IRE1α since increased KC mRNA levels were observed upon C12 treatment in IRE1α −/− MEF cells.
Role for PERK in HSL-C12-triggered apoptosis?
Given the apparent role for PERK in HSL-C12 inflammatory phenotypes, we also tested whether PERK might play a role in other HSL-C12-triggered responses. PERK and other members of the unfolded protein response are known to produce pro-apoptotic signals under certain conditions (29–31), and 50 μM HSL-C12 increased caspase 3/7 activity in WT MEF (25). In the present experiments, HSL-C12 caused equivalent concentration-dependent activation of caspase 3/7 in PERK-corrected and PERK−/− MEF (Fig. 10). HSL-C12 has also been reported to cause depolarization of mitochondrial membrane potential (10), but PERK-corrected and PERK−/− cells showed similar HSL-C12-induced rates of depolarization of mitochondrial membrane potential as measured using the dye JC-1 (data not shown). Together, these results indicated that HSL-C12 affected KC signaling and apoptosis through different pathways.
Fig. 10. HSL-C12 induced apoptosis is PERK-independent.

PERK−/− and PERK-corrected MEF were treated with increasing doses of HSL-C12 for four hrs, and caspase 3/7 activation was measured. Data are averages +/− SE (n = 3 biological replicates).
DISCUSSION
Results from this study showed that, depending on the assay, HSL-C12 had both pro-inflammatory and anti-inflammatory effects on WT MEF, but these responses appeared to be mediated at least in part through a common molecular mechanism. HSL-C12 caused increased transcription of KC and IL-6 genes, but the expected increase in KC and IL-6 secretion did not occur - secretion of these two pro-inflammatory cytokines was either reduced (KC) or remained unaffected (IL-6). Because HSL-C12 also increased the phosphorylation of eI-F2α and inhibited protein synthesis, it seems likely that at least part of the inhibitory effect of HSL-C12 on pro-inflammatory cytokine secretion in the face of increases in gene transcription resulted from HSL-C12 inhibiting synthesis of these cytokines. It also seems likely that similar effects contributed to HSL-C12-triggered increases in TNFα- or IL-1β-stimulated KC or IL-6 gene expression but decreases in TNFα-stimulated secretion of KC or IL-6. These apparently contradictory effects of HSL-C12 to elicit both pro-inflammatory (increased gene transcription) and anti-inflammatory (reduced activation of NF-κB-luciferase and reduced secretion of KC and IL-6) may explain some of the apparent contradictions surrounding previously reported pro-inflammatory (11, 12, 15) vs. anti-inflammatory (16, 23) effects of HSL-C12 on host cells.
Many of HSL-C12’s effects to alter inflammatory signaling and responses appeared to be mediated largely through HSL-C12-induced activation of PERK. This conclusion is based on the following observations: (i) HSL-C12 caused larger increases in eI-F2α phosphorylation, greater inhibition of protein synthesis and larger degradation of IκBα in PERK-corrected MEF compared with PERK−/− MEF. (ii) Compared to treatments with IL-1β or TNFα alone, treatments with IL-1β or TNFα + HSL-C12 caused larger increases in KC gene transcription and less KC secretion in PERK-corrected MEF compared with PERK−/− MEF. These data are consistent with the interpretation that HSL-C12 acts through PERK and eI-F2α to inhibit protein synthesis, leading to reduced production of the high turnover protein IκBα, increased activation of NF-κB and increased KC gene transcription but decreased KC synthesis and secretion, particularly in the presence of the pro-inflammatory mediators IL-1β and TNFα.
A flow chart summarizing these conclusions about the effects of HSL-C12 is presented in Fig. 11. HSL-C12 activates PERK and the subsequent phosphorylation of eI-F2α and inhibition of protein synthesis. Since HSL-C12 causes the release of Ca2+ stores from the ER and many of the ER chaperones require Ca2+, it is possible that reducing Ca2+ levels in the ER activates the unfolded protein response and PERK (20, 29). This idea is supported by the fact that HSL-C12 and thapsigargin elicited similar phosphorylation of eI-F2α in PERK-corrected MEF, but much less in PERK−/− MEF. Once eI-F2α becomes phosphorylated, protein synthesis, including re-synthesis of the high turnover protein IκBα, the NF-κB inhibitor (28), will be inhibited. As IκBα levels fall, NF-κB will be released to enter the nucleus and induce transcription of pro-inflammatory gene products like KC. [HSL-C12 causes p65 to migrate from the cytosol to the nucleus within 30–60 mins in the airway epithelial cell line JME (Schwarzer and Machen, unpublished observations).] However, because protein synthesis has been inhibited, new synthesis and secretion of KC will be inhibited, even in the presence of ligands like TNFα and IL-1β that normally induce inflammatory secretion. Of course, this proposal requires that p65 that enters the nucleus will be the phosphorylated, active version that serves as the potent proinflammatory transcription factor, and it has been argued that p65 that enters the nucleus of HSL-C12-treated cells remains unphosphorylated (29). Further experiments testing the mechanism by which HSL-C12 activates proinflammatory gene transcription will help clarify the specific roles of PERK, eI-F2α and NF-κB p65 in responses to HSL-C12.
Fig. 11. Proposed flowchart showing the inflammatory phenotypes during HSL-C12 treatment based on the present study.

See text for details.
Although the present data indicate that HSL-C12-triggered effects on inflammatory signaling and responses in MEF were mediated largely through activation of PERK, it is also clear that HSL-C12 had effects that were mediated through other effector pathways. PERK was not required for HSL-C12 to activate caspase 3/7, indicating that PERK was not involved in mediating HSL-C12-triggered apoptosis. Instead, HSL-C12-triggered apoptosis (10, 25) seems to require activation of another ER stress pathway mediated by IRE1α and XBP-1 (31). In addition, HSL-C12 elicited a less than maximal but still detectable increase in eI-F2α phosphorylation, inhibition of protein synthesis, and degradation of IκB in PERK−/− MEF. All of these effects likely resulted from HSL-C12 activating other effector pathways besides PERK-eI-F2α. One potential PERK-independent molecule that could become activated by HSL-C12 is the eI-F2α kinase GCN2, which is activated by metabolic stress through the buildup of unloaded tRNAs (31). HSL-C12’s effects on the mitochondria (10, 14) may lead to metabolic stress and subsequent GCN2 activation. Previous work showed that HSL-C12 activates both p-38 and c-Jun in MEF (31), so these signaling pathways may also be involved in HSL-C12-triggered responses. The newly identified, selective inhibitors of the anti-inflammatory and pro-apoptotic effects of HSL-C12 on MEF (triazolo[4,3-a]quinolones) should help in testing for alternative signaling pathways affected by HSL-C12 (23).
Although it has not been fully established that HSL-C12 is an important P. aeruginosa virulence factor in vivo, the present results indicate that HSL-C12’s effects on both inflammatory signaling and apoptosis might affect the course of P. aeruginosa lung infections. HSL-C12 secreted by P. aeruginosa would have the short term effects of suppressing secretion of pro-inflammatory mediators (present work) and activating anti-inflammatory mediators (17). Consistent with this idea, we have found that Calu-3 airway epithelial cells secreted more IL-8 during exposure to planktonic P. aeruginosa compared to exposure to biofilm P. aeruginosa, which are expected to produce higher [HSL-C12] compared to planktonic P. aeruginosa (Supplemental Data, Fig. S2). The apparent inhibitory effect of biofilm (i.e., compared to planktonic) P. aeruginosa on IL-8 secretion appeared to be mediated in part by HSL-C12 because the inhibitory effect of the biofilm PAO1 was reduced by biofilm PAO1 that were defective in expression of lasI, the enzyme responsible for producing HSL-C12 (Supplemental Data, Fig. S2). HSL-C12 is also expected to create gaps in epithelia resulting from its pro-apoptotic effects (5–7, 10), allowing P. aeruginosa access to the basolateral membrane, an important factor for virulence (32, 33).
The roles of PERK in modulating host cell inflammatory responses to HSL-C12 (Fig. 11) and of IRE1α and XBP1 in mediating the apoptosis response to HSL-C12 (30) suggest ties to phenotypes observed in cells from CF patients. CF airway cells are characterized by increased ER volume and a chronically active unfolded protein response (34), perhaps indicating prolonged exposure to ER stress inducers like HSL-C12. However, these conclusions are mostly based on results from our short-term (up to 4 hr) experiments, and CF patients may be chronically exposed to P. aeruginosa biofilms and HSL-C12. The experimental conditions applied in our current study did not allow us to monitor long term effects of HSL-C12 since increased cell death dominated over and masked other cellular responses. Further studies of HSL-C12 effects on airway and other epithelia during exposures longer than the 4 hr treatments and with lower doses used here are therefore warranted. Previous work on the intestinal cell line Caco-2 has shown that HSL-C12-triggered degradation of barrier function and tight junction structure is largely reversed during 24 hr exposures (35, 36), and it will be important to test whether there is a similar reversal in inflammatory and apoptotic responses of airway epithelia during 24 hr treatments with HSL-C12. Despite the uncertainty associated with making conclusions about chronic conditions in vivo based on results from short term experiments in vitro, accumulating evidence indicates that HSL-C12 is an important virulence factor in P. aeruginosa infections not only because it regulates expression of key bacterial genes but also because it has direct, pathological effects on host cells.
Supplementary Material
Acknowledgments
We thank C. Li (U. Louisville) for providing WT MEF and R. Kaufman (Sanford|Burnham Medical Research Institute) for PERK−/− and PERK-corrected MEF. We are grateful to Russell Vance (UC Berkeley) for discussions.
Abbreviations
- HSL-C12
N-(3-oxododecanoyl)-homoserine lactone
- MEF
Mouse embryo fibroblast
- PERK
Protein kinase RNA-like Endoplasmic Reticulum Kinase, the ER kinase activated by ER stress
- eI-F2α
eukaryotic initiation factor 2
Footnotes
This research was funded by grants from the NIH (2PNE Y016241) and Cystic Fibrosis Research, Inc (New Horizons).
CONFLICTS OF INTEREST
The authors have no conflicts of interest.
References
- 1.Hoiby N, Flensborg EW, Beck B, Friis B, Jacobsen SV, Jacobsen L. Pseudomonas aeruginosa infection in cystic fibrosis. Diagnostic and prognostic significance of Pseudomonas aeruginosa precipitins determined by means of crossed immunoelectrophoresis. Scandinavian journal of respiratory diseases. 1977;58:65–79. [PubMed] [Google Scholar]
- 2.Schuster M, Peter Greenberg E. A network of networks: Quorum-sensing gene regulation in Pseudomonas aeruginosa. International journal of medical microbiology. 2006;296:73–81. doi: 10.1016/j.ijmm.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 3.Rumbaugh KP. Convergence of hormones and autoinducers at the host/pathogen interface. Analytical and bioanalytical chemistry. 2007;387:425–435. doi: 10.1007/s00216-006-0694-9. [DOI] [PubMed] [Google Scholar]
- 4.Irie Y, Parsek M. Bacterial biofilms. Springer; 2008. Quorum sensing and microbial biofilms; pp. 67–84. [DOI] [PubMed] [Google Scholar]
- 5.Tateda K, Ishii Y, Horikawa M, Matsumoto T, Miyairi S, Pechere JC, Standiford TJ, Ishiguro M, Yamaguchi K. The Pseudomonas aeruginosa autoinducer N-3-oxododecanoyl homoserine lactone accelerates apoptosis in macrophages and neutrophils. Infection and immunity. 2003;71:5785–5793. doi: 10.1128/IAI.71.10.5785-5793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li L, Hooi D, Chhabra SR, Pritchard D, Shaw PE. Bacterial N-acylhomoserine lactone-induced apoptosis in breast carcinoma cells correlated with down-modulation of STAT3. Oncogene. 2004;23:4894–4902. doi: 10.1038/sj.onc.1207612. [DOI] [PubMed] [Google Scholar]
- 7.Jacobi CA, Schiffner F, Henkel M, Waibel M, Stork B, Daubrawa M, Eberl L, Gregor M, Wesselborg S. Effects of bacterial N-acyl homoserine lactones on human Jurkat T lymphocytes-OdDHL induces apoptosis via the mitochondrial pathway. International Journal of Medical Microbiology. 2009;299:509–519. doi: 10.1016/j.ijmm.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 8.Li H, Wang L, Ye L, Mao Y, Xie X, Xia C, Chen J, Lu Z, Song J. Influence of Pseudomonas aeruginosa quorum sensing signal molecule N-(3-oxododecanoyl) homoserine lactone on mast cells. Medical microbiology and immunology. 2009;198:113–121. doi: 10.1007/s00430-009-0111-z. [DOI] [PubMed] [Google Scholar]
- 9.Schwarzer C, Wong S, Shi J, Matthes E, Illek B, Ianowski JP, Arant RJ, Isacoff E, Vais H, Foskett JK. Pseudomonas aeruginosa homoserine lactone activates store-operated cAMP and cystic fibrosis transmembrane regulator-dependent Cl– secretion by human airway epithelia. Journal of Biological Chemistry. 2010;285:34850–34863. doi: 10.1074/jbc.M110.167668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwarzer C, Fu Z, Patanwala M, Hum L, Lopez-Guzman M, Illek B, Kong W, Lynch SV, Machen TE. Pseudomonas aeruginosa biofilm-associated homoserine lactone C12 rapidly activates apoptosis in airway epithelia. Cellular microbiology. 2012;14:698–709. doi: 10.1111/j.1462-5822.2012.01753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Telford G, Wheeler D, Williams P, Tomkins P, Appleby P, Sewell H, Stewart GS, Bycroft BW, Pritchard DI. The Pseudomonas aeruginosaQuorum-Sensing Signal MoleculeN-(3-Oxododecanoyl)-l-Homoserine Lactone Has Immunomodulatory Activity. Infection and Immunity. 1998;66:36–42. doi: 10.1128/iai.66.1.36-42.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith RS, Fedyk ER, Springer T, Mukaida N, Iglewski BH, Phipps RP. IL-8 production in human lung fibroblasts and epithelial cells activated by the Pseudomonas autoinducer N-3-oxododecanoyl homoserine lactone is transcriptionally regulated by NF-κB and activator protein-2. The Journal of Immunology. 2001;167:366–374. doi: 10.4049/jimmunol.167.1.366. [DOI] [PubMed] [Google Scholar]
- 13.Smith RS, Kelly R, Iglewski BH, Phipps RP. The Pseudomonas autoinducer N-(3-oxododecanoyl) homoserine lactone induces cyclooxygenase-2 and prostaglandin E2 production in human lung fibroblasts: implications for inflammation. The Journal of Immunology. 2002;169:2636–2642. doi: 10.4049/jimmunol.169.5.2636. [DOI] [PubMed] [Google Scholar]
- 14.Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Wood MR, Brogan AP, Lehmann M, Mee JM, Iwata K. N-(3-oxo-acyl) homoserine lactones signal cell activation through a mechanism distinct from the canonical pathogen-associated molecular pattern recognition receptor pathways. Journal of Biological Chemistry. 2006;281:28822–28830. doi: 10.1074/jbc.M606613200. [DOI] [PubMed] [Google Scholar]
- 15.Jahoor A, Patel R, Bryan A, Do C, Krier J, Watters C, Wahli W, Li G, Williams SC, Rumbaugh KP. Peroxisome proliferator-activated receptors mediate host cell proinflammatory responses to Pseudomonas aeruginosa autoinducer. Journal of bacteriology. 2008;190:4408–4415. doi: 10.1128/JB.01444-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Grauer DC, Lehmann M, Meijler MM, Janda KD, Ulevitch RJ. Modulation of gene expression via disruption of NF-κB signaling by a bacterial small molecule. Science. 2008;321:259–263. doi: 10.1126/science.1156499. [DOI] [PubMed] [Google Scholar]
- 17.Glucksam-Galnoy Y, Sananes R, Silberstein N, Krief P, Kravchenko VV, Meijler MM, Zor T. The bacterial quorum-sensing signal molecule N-3-oxo-dodecanoyl-L-homoserine lactone reciprocally modulates pro-and anti-inflammatory cytokines in activated macrophages. The Journal of Immunology. 2013;191:337–344. doi: 10.4049/jimmunol.1300368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shiner E, Terentyev D, Bryan A, Sennoune S, Martinez-Zaguilan R, Li G, Gyorke S, Williams S, Rumbaugh K. Pseudomonas aeruginosa autoinducer modulates host cell responses through calcium signalling. Cellular microbiology. 2006;8:1601–1610. doi: 10.1111/j.1462-5822.2006.00734.x. [DOI] [PubMed] [Google Scholar]
- 19.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes & Development. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 20.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 21.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 22.Hinnebusch AG. Seminars in cell biology. Elsevier; 1994. The eIF-2α kinases: regulators of protein synthesis in starvation and stress; pp. 417–426. [DOI] [PubMed] [Google Scholar]
- 23.Valentine CD, Zhang H, Phuan PW, Nguyen J, Verkman AS, Haggie PM. Small molecule screen yields inhibitors of pseudomonas homoserine lactone-induced host responses. Cell Microbiol. 2013 Aug 2; doi: 10.1111/cmi.12176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christensen SB, Andersen A, Poulsen JCJ, Treiman M. Derivatives of thapsigargin as probes of its binding site on endoplasmic reticulum Ca2+ ATPase: Stereoselectivity and important functional groups. FEBS Letters. 1993;335:345–348. doi: 10.1016/0014-5793(93)80416-r. [DOI] [PubMed] [Google Scholar]
- 25.Schwarzer C, Fu Z, Shuai S, Babbar S, Zhao G, Li C, Machen TE. Pseudomonas aeruginosa Homoserine Lactone Triggers Apoptosis and Bak/Bax-Independent Release of Mitochondrial Cytochrome C in Fibroblasts. Cell Microbiol. 2014 Jan 20; doi: 10.1111/cmi.12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hybiske K, Fu Z, Schwarzer C, Tseng J, Do J, Huang N, Machen TE. Effects of cystic fibrosis transmembrane conductance regulator and DeltaF508CFTR on inflammatory response, ER stress, and Ca2+ of airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1250–60. doi: 10.1152/ajplung.00231.2007. [DOI] [PubMed] [Google Scholar]
- 27.Krappmann D, Scheidereit C. Regulation of NF-κB activity by IκBα and IκBβ stability. Immunobiology. 1997;198:3–13. doi: 10.1016/s0171-2985(97)80022-8. [DOI] [PubMed] [Google Scholar]
- 28.Wu S, Tan M, Hu Y, Wang JL, Scheuner D, Kaufman RJ. Ultraviolet light activates NFκB through translational inhibition of IκBα synthesis. Journal of Biological Chemistry. 2004;279:34898–34902. doi: 10.1074/jbc.M405616200. [DOI] [PubMed] [Google Scholar]
- 29.Jordan R, Wang L, Graczyk TM, Block TM, Romano PR. Replication of a cytopathic strain of bovine viral diarrhea virus activates PERK and induces endoplasmic reticulum stress-mediated apoptosis of MDBK cells. Journal of virology. 2002;76:9588–9599. doi: 10.1128/JVI.76.19.9588-9599.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kravchenko VV, Kaufmann GF. Bacterial inhibition of inflammatory responses via TLR-independent mechanisms. Cell Microbiol. 2013;15:527–36. doi: 10.1111/cmi.12109. [DOI] [PubMed] [Google Scholar]
- 31.Valentine CD, Anderson MO, Papa FR, Haggie PM. X-box binding protein 1 (XBP1s) is a critical determinant of Pseudomonas aeruginosa homoserine lactone-mediated apoptosis. PLoS Pathog. 2013;9:e1003576. doi: 10.1371/journal.ppat.1003576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hybiske K, Ichikawa JK, Huang V, Lory SJ, Machen TE. Cystic fibrosis airway epithelial cell polarity and bacterial flagellin determine host response to Pseudomonas aeruginosa. Cellular microbiology. 2004;6:49–63. doi: 10.1046/j.1462-5822.2003.00342.x. [DOI] [PubMed] [Google Scholar]
- 33.Tseng J, Do J, Widdicombe JH, Machen TE. Innate immune responses of human tracheal epithelium to Pseudomonas aeruginosa flagellin, TNF-α, and IL-1β. American Journal of Physiology-Cell Physiology. 2006;290:C678–C690. doi: 10.1152/ajpcell.00166.2005. [DOI] [PubMed] [Google Scholar]
- 34.Martino ME, Olsen JC, Fulcher NB, Wolfgang MC, O’Neal WK, Ribeiro CM. Airway epithelial inflammation-induced endoplasmic reticulum Ca2+ store expansion is mediated by X-box binding protein-1. Journal of Biological Chemistry. 2009;284:14904–14913. doi: 10.1074/jbc.M809180200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vikström E, Tafazoli F, Magnusson KE. Pseudomonas aeruginosa quorum sensing molecule N-(3 oxododecanoyl)-l-homoserine lactone disrupts epithelial barrier integrity of Caco-2 cells. FEBS letters. 2006;580:6921–6928. doi: 10.1016/j.febslet.2006.11.057. [DOI] [PubMed] [Google Scholar]
- 36.Vikström E, Bui L, Konradsson P, Magnusson KE. The junctional integrity of epithelial cells is modulated by Pseudomonas aeruginosa quorum sensing molecule through phosphorylation-dependent mechanisms. Exp Cell Res. 2009;315:313–26. doi: 10.1016/j.yexcr.2008.10.044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.