

Abstract
Context:
Lung tumors are very heterogeneous histological entities. Pulmonary sarcomatoid carcinoma is a subset of tumors characterized by specific histological features. Their poor prognosis compared to other lung tumors is due to limited responses to different types of chemotherapy.
Case Report:
We report two patients with sarcomatoid tumors: A 53-year-old woman and a 46-year-old man who presented respiratory symptoms: Dyspnea, cough, associated with a deterioration of general condition.
Conclusion:
Pulmonary sarcomatoid carcinomas remained an unexplored entity, despite their poor prognosis. Based on these cases, we will discuss the histological and immunohistochemical features of these tumors, as well as report their responses to different chemotherapy regimens used in the course of treatment.
Keywords: Chemotherapy, lung, response to platinum salts, saromatoid carcinoma, surgery, targeted therapy
Introduction
Sarcomatoid carcinomas of the lung are very uncommon tumors, comprising between 0.3 and 3% of all non-small lung carcinomas.[1] It is a heterogeneous group of poorly differentiated carcinomas with two cell types: sarcomatoid or sarcomatous morphology with giant and/or spindle cells.[1] Their clinical presentation is not specific; however, this type of neoplasm is very aggressive, with an overall 5-year survival rate of approximately 20%. Few studies have examined these tumors but chemoresistance appears to be responsible for their bad prognosis. To define the types of sarcomatoid carcinomas, clarify their immunohistochemical phenotype, and examine their therapeutic features, we herein describe two cases treated at the National Institute of Oncology in Rabat.
Case Presentation
Case 1
A 53-year-old female presented in January 2010 with a cervical mass of gradually increasing volume that was associated with dyspnea and dry cough. Chest computed tomography (CT) scan revealed a cervico-mediastino-pulmonary lesional process of 120/80, 70/70, and 50 mm sheathing cervical and intrathoracic vascular axes and displacing the mediastinum. Pleural effusion was present [Figure 1]. A biopsy was performed and the histological examination revealed diffuse cellular water with large cells and hypertrophic irregular macro-nucleoli and macro-nuclei. We observed many mitoses, and the stroma was scant and fibro-inflammatory. Immunohistochemically, the tumor cells were positive for vimentin. However, they were negative for thyroid transcription factor-1 (TTF-1), epithelial membrane antigen (EMA), muscle actin (HHF35), cytokeratins 7, 20, and 5/6, PS100, actin, and calretinin. Abdominal CT scans and bone scintigraphy were normal. The tumor was classified as stage IV (T4N0M1) and the patient received the following chemotherapy-based platinum salts: navelbine (25 mg/m2 J1, J8) and cisplatin (80 mg/m2 J1). Toxicity was marked by nausea with grade II anemia. Evaluation after the third and sixth treatment regimens indicated stable disease.
Figure 1.
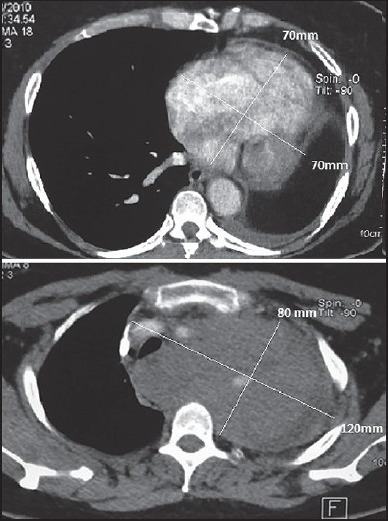
Chest CT scan showing a right cervicothoracic mass with vascular sheathing
After 18 months with the stable disease, the patient clinically progressed with the onset of chest pain. A chest CT examination showed increased tumor volume (from 120 to 160 mm). Docetaxel (75 mg/m2) was administered as second-line therapy. The observed toxicity was predominantly hematological, with neutropenia and grade I anemia. Evaluation after the third and sixth courses showed significant clinical benefit with radiographic disease stabilization. The condition of the patient remained stable for 4 months and is currently being monitored routinely.
Case 2
In February 2013, a 46-year-old male with a history of cigarette smoking for 20 years (30 cigarettes per day) presented with dyspnea stage II of the New York Heart Association classification, (NYHA), chest pain, and anorexia. Symptomatology evolved in a context of progressive deterioration of the general state and poor general condition. Upon physical examination, he appeared weak (stage II of WHO score), with a body temperature of 36.2°C. His lungs, heart, and abdomen were normal. Chest CT scans showed a large mass in the right lower lobe with spiculated contours that measured 110 mm. The mass was associated with latero-tracheal, anterior mediastinal, and right hilar lymphadenopathy. Further staging showed multiple lesions of the spine (D8), right sacroiliac joint, 4th right odds. However, no visceral lesion was observed after abdominal ultrasonographic examination. Using a scan-guided biopsy, histological and immunohistochemical evidences supported a sarcomatoid carcinoma, with carcinomatous proliferation of pleomorphic spindle and globoid cells [Figure 2a]. Tumor cells were positive for vimentin [Figure 2b], cytokeratin, and TTF1 [Figure 2c]. They did not express cytokeratins 7, 20, or 5/6. In addition, they did not express p63. The patient received cisplatin and navelbine, with a partial response after the third cycle (70 mm versus 110 mm). We decided to continue the same chemotherapy after the good clinical and radiological response; however, the patient presented with an acute kidney injury after the fourth cycle, with an estimated clearance of 25 ml/min according to the Cockroft-Gault formula. The decision was made to switch to carboplatin (AUC 5) and paclitaxel (175 mg/m2). Chest CT was performed again and showed the appearance of a lesion process in the right posterior of 7.5 cm, associated with two left hilar lymphadenopathies (20 mm and 26 mm, respectively). Therefore, we moved to a second-line therapy of docetaxel (75 mg/m2). The patient received 3 courses, and the period between these cycles of chemotherapy was marked by neuropathy grade I. A chest thoracic scan showed an increase in the size of the right posterior parietal lesion process, measuring 105/77, 5 mm vs. 75.5/36 mm. The process extended behind the 4th, 5th, and 6th dimensions, with invasion of the erector spinae muscles, trapezius invasion, and root canal. The mediastinal lymphadenopathy kept the same dimensions. We decided to go to the palliative care. The patient died a few days after.
Figure 2.
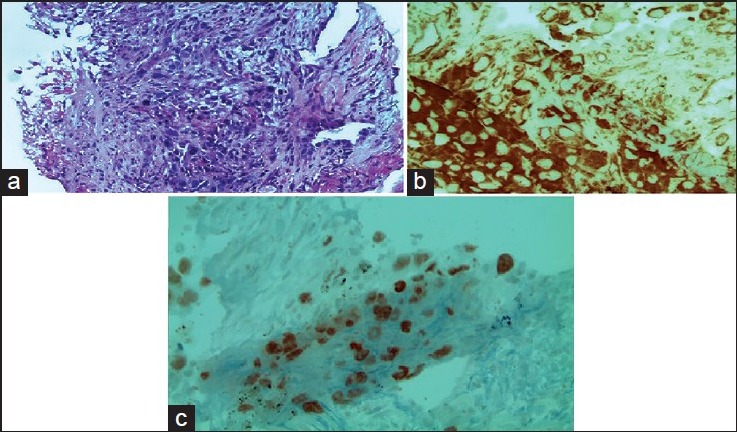
(a) H&E staining G× 200 carcinomatous proliferation of pleomorphic spindle and globoid cells (b) H&E staining G× 400: Tumor cells expressing the vimentin antibody (clone V9, IMMUNOTEC) (c) H&E staining G× 400: Tumor cells expressing the CK (clone AE1/AE3, DAKO) (+) and thyroid transcription factor-1 (TTF1) (clone 8G7G3 / 1 DAKO (flex). Nuclear staining (+)
Discussion
Sarcomatoid carcinoma is a rare tumor that comprises approximately 1% of all lung malignancies. They are more common among men than women with a gender ratio of 4/1. The mean age at diagnosis is between 65-75 years. The patients are usually heavy smokers or have a history of chronic smoking.[1,2] Some cases have been reported related to asbestos exposure. In a study by Fisbak, 3% patients with sarcomatoid carcinoma were exposed to asbestos. Thus, there could be a direct mechanism between toxicity and emergence of a sarcomatoid component.[2,3]
This tumor is frequent with the local advancement and has high rates of recurrence. However, there is no specific clinical presentation. Patients generally have several symptoms. Dyspnea and cough were the first symptoms to appear in our patients, which were followed by generally rapid physical deterioration. Other symptoms may occur including weakness, shortness of breath, and fever. For proximal tumors, hemoptysis is present in 50% of cases, whereas for peripheral tumors, chest pain is frequently described.[4] Radiologically, sarcomatoid carcinoma is usually a single lesion, with a large diameter (from 40 to 180 mm), and central or peripheral location in the upper lobes. The tumor is usually and locally advanced at the time of diagnosis with a large proportion of pleural invasion, either vascular or parietal (40-70% of cases).[1,3] Our patients had large tumors (160 and 110 mm, respectively) with vascular and nervous axis invasion (Case 1).
It is difficult to distinguish pulmonary sarcomatoid carcinomas from true sarcomas. They are defined in the World Health Organization classification as “poorly differentiated non-small cell lung carcinomas (NSCLCs) containing a sarcoma-like element (malignant spindle or giant cells) or sarcomatous component.[1] We can define five types of carcinomas based on specific histological criteria: Giant cell carcinoma, pleomorphic carcinoma, carcinosarcoma, spindle cell carcinoma, and pulmonary blastoma.[1]
Pleomorphic carcinoma is formed by malignant giant and/or spindle cells plus epithelial components, such as squamous cells. The spindle and/or giant cells appear as cohesive aggregates of the tumor cell, without glandular or squamous differentiation. The neoplastic spindle cells are isolated or in loose clusters, and are usually very polymorphic and elongated. Nucleoli are prominent, and nuclei are solitary, large, and spindled. Additionally, the nuclear-cytoplasmic ratio is high.[1] In some cases, multiple keratin antibodies and EMA are necessary to demonstrate epithelial differentiation in the sarcomatoid component. The tumor cells often co-express cytokeratin, vimentin, carcinoembryonic antigen, and smooth muscle markers. The TTF-1 may be positive in giant cell carcinomas. The differential diagnosis for pleomorphic carcinoma consists of other tumors as both primary and metastatic sarcomas.
Epidermal growth factor receptor (EGFR) is a membrane of tyrosine kinase receptor that becomes oncogenic by acquiring a mutation within its catalytic domain. A study by Italiano suggested that a role for EGFR in SC. Indeed, in 22 surgically treated patients, EGFR expression was analyzed and EGFR gene mutations were observed. EGFR overexpression was found in all cases, with mutation analyses for EGFR exons 18, 19, 20, and 21 were obtained in 19 cases. No results could be obtained in three cases, but no EGFR mutations were detected.[6] Recently, Chang et al., reported results that confirm the existence of carcinomas with pleomorphic EGFR mutations in a Taiwanese population.[7] Sarcomatoid carcinomas have also evaluated for their K-ras status. Pelosi et al., reported 22% of patients had K-ras mutations at codon 12.[8]
For localized tumors, surgery remains adequate for treatment and appears to provide adequate local control.[3] However, for metastatic disease, no data are currently available and sarcomatoid carcinomas are usually treated as non-small cell lung cancer (NSCLCs).[9] Cytotoxic agents used for the treatment of sarcomatoid carcinomas are the same used as those used for NSCLCs. In the series of Vieira et al., study 97 patients with metastatic disease were included, of which 73% of them received chemotherapy-based platinum salts. The difference in progression-free survival (PFS) was not statistically significant between patients receiving or not a platinum-based chemotherapy, no statistically significant difference in overall survival was observed (7months with platinum versus 5.3 months without, P = 0.096).[5]
Our two patients received platinum-based doublets for first-line therapy but their responses varied between stabilization and progression, but these elements remain insufficient to conclude that platinum salts possess an activity on this type of tumor or not. Other protocols have been used, such as CAV (cyclophosphamide, adriamycin, and vincristine), to target the epithelial and sarcomatous component. However, the objective response rates remain poor.[2,3] One study evaluated the short-term outcomes of using doxorubicin and ifosfamide in the treatment of metastatic sarcomatoid carcinoma. Among 15 patients, 50% showed partial responses to short-term chemotherapy with doxorubicin and ifosfamide. However, all patients experienced disease progression and died despite continuous treatment.[10] For targeted therapies, the study by Italiano showed that patients with lung sarcomatoid carcinoma did not benefit from anti-EGFR treatment.[6]
Conclusion
Sarcomatoid carcinoma of the lung remains poorly explored. The histological and immunohistochemical characteristics of this tumor type are specific and very different from non-small cell lung carcinoma, resulting in an aggressive entity. Conventional chemotherapy did not show satisfactory results, and future studies are needed to explore the molecular profile of these tumors in order to determine the best therapeutic approach.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Travis WD, Brambilla E, Müller-Hermelink K, Harris C, Kleihues C, Sobin P. World Health Organization Classification of Tumours. Lyon: IARC Press; 2004. Pathology and genetics of tumors of the lung, pleura, thymus, and heart; pp. 53–58. http://www.iarc.fr/en/publications/pdfs-online/pat-gen/bb10/BB10 . [Google Scholar]
- 2.Ishida T, Tateishi M, Kaneko S, Yano T, Mitsudomi T, Sugimachi K, et al. Carcinosarcoma and spindle cell carcinoma of the lung. Clinicopathologic and immunohistochemical studies. J Thorac Cardiovasc Surg. 1990;100:844–52. [PubMed] [Google Scholar]
- 3.Davis MP, Eagan RT, Weiland LH, Pairolero PC. Carcinosarcoma of the lung: Mayo Clinic experience and response to chemotherapy. Mayo Clin Proc. 1984;59:598–603. doi: 10.1016/s0025-6196(12)62410-0. [DOI] [PubMed] [Google Scholar]
- 4.Yoshino N, Kubokura H, Yamauchi S, Ohaki Y, Koizumi K, Shimizu K. A true pulmonary carcinosarcoma that required diagnostic differentiation from a pleomorphic adenoma: A case report. Ann Thorac Cardiovasc Surg. 2009;15:42–5. [PubMed] [Google Scholar]
- 5.Vieira T, Girard N, Ung M, Monnet I, Cazes A, Bonnette P, et al. Efficacy of first-line chemotherapy in patients with advanced lung sarcomatoid carcinoma. J Thorac Oncol. 2013;8:1574–7. doi: 10.1097/01.JTO.0000437008.00554.90. [DOI] [PubMed] [Google Scholar]
- 6.Italiano A, Cortot AB, Ilie M, Martel-Planche G, Fabas T, Pop D, et al. EGFR and K-RAS status of primary sarcomatoid carcinomas of the lung: Implications for anti-EGFR treatment of a rare lung malignancy. Int J Cancer. 2009;125:2479–82. doi: 10.1002/ijc.24610. [DOI] [PubMed] [Google Scholar]
- 7.Chang YL, Wu CT, Shih JY, Lee YC. EGFR and p53 status of pulmonary pleomorphic carcinoma: Implications for EGFR tyrosine kinase inhibitors therapy of an aggressive lung malignancy. Ann Surg Oncol. 2011;18:2952–60. doi: 10.1245/s10434-011-1621-7. [DOI] [PubMed] [Google Scholar]
- 8.Pelosi G, Scarpa A, Manzotti M, Veronesi G, Spaggiari L, Fraggetta F, et al. K-ras gene mutational analysis supports a monoclonal origin of biphasic pleomorphic carcinoma of the lung. Mod Pathol. 2004;17:538–46. doi: 10.1038/modpathol.3800058. [DOI] [PubMed] [Google Scholar]
- 9.Mason RJ, Broaddus VC, Martin T, King T, Jr, Schraufnagel D, John F. Rare malignant primary pulmonary epithelial tumors. 5th ed. Ch. 48, Part 4. Primary Pulmonary Sarcomas; 2010. Murray and Nadel's Textbook of Respiratory Medicine. http://www.mdconsult.com/books/ [Google Scholar]
- 10.Staddon AP, Crawford EA, Hartner L, Lackman RD, Ogilvie MC. Treatment of metastatic sarcomatoid carcinoma with doxorubicin and ifosfamide. J Clin Oncol. 2008;26:21518. [Google Scholar]