

To the Editor
A surprise revealed by the success of the human genome project was the lower-than-anticipated number of genes identified: ~20,300, rather than the ~100,000 estimated1. This finding led to the recognition that much of the complexity afforded by our biological machinery is at the level of protein variation rather than due to a high number of distinct genes2. The divergences among highly related, but chemically different, protein molecules arise from variation within populations, cell and tissue types and subcellular localization. On the DNA, RNA and protein levels, complexity can arise from allelic variations, from alternative splicing of RNA transcripts and from many post-translational modifications, respectively. These events create distinct protein molecules that modulate a wide variety of biological processes, from cell signaling inside or between cells to gene regulation and activation of protein complexes.
Although the complexity of protein forms was first revealed by two-dimensional gel electrophoresis, newer proteomic technologies can provide the precise compositions of whole protein molecules. Mass spectrometry has emerged as a key platform for proteomic analyses, with two contrasting approaches referred to as ‘bottom-up’ and ‘top-down’ proteomics. In the bottom-up approach, proteins are digested into peptides using trypsin or other proteases and are then identified by liquid chromatography and tandem mass spectrometry. In top-down proteomics, digestion into peptides is avoided, and protein identification is obtained directly from fragmentation of the intact protein. When available, the top-down approach provides the richest data for both precise identification (that is, the specific gene in a higher eukaryote that encodes the protein measured)3 and full characterization of molecular composition. However, it is considerably more challenging to execute than the bottom-up approach because of the complexity of the data generated and various technical limitations.
Given the importance of capturing this protein variation in basic and translational research, and that technologies now exist to reveal it, we point out an ongoing problem in nomenclature regarding what to call it. In the literature, one finds the following terms: “protein forms”, “protein isoforms”, “protein species”2 and “protein variants.” Protein “mod forms” was also recently introduced4. None of these terms is very satisfactory. The term “isoform” is widely used, but often incorrectly according to the International Union of Pure and Applied Chemistry (IUPAC) definition, which refers only to genetic differences and not to variation at the protein level2. The term “protein species” was proposed in 2009 (ref. 2) but does not distinguish between proteins originating from different genes and those originating from a single gene, and thus we find it confusing. A similar issue arises with the term “protein variants.”
The UniProt Knowledgebase (a definitive, gene-centric protein database)5 uses the term “isoform” in yet a different manner, one that denotes related forms of protein molecules arising from the same gene by alternative splicing or variable promoter usage (Fig. 1). Such events create a variable set of protein sequences that significantly change the numbering of amino acids for the protein as compared to the canonical sequence. These changes to the base primary sequence are referred to by some as “isoforms” and are denoted in UniProt by a −1, −2 and so on following the accession number (Fig. 1). However, genetic changes (for example, mutations and polymorphisms) are not covered by this terminology and create a conflict with the IUPAC definition of isoform2. Differences in IUPAC and UniProt definitions notwithstanding, the terms “variants” and “iso-forms” were intended to describe proteins derived from distinct DNA or RNA; their use to describe modified proteins is confusing.
Figure 1.
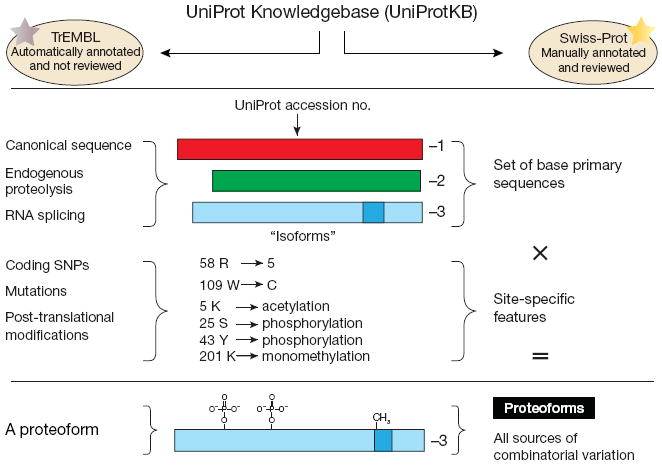
The origins of the proteoform terminology to cleanly describe biological variability at the level of protein primary structure. UniProt is a gene-centric database, meaning that it strives to have a single accession number for each gene. There are occasional deviations due to multiple genes (in one species) producing precisely the same primary protein sequence in higher eukaryotes. SNP, single-nucleotide polymorphism.
Accordingly, we propose that the term ‘proteoform’ be used to designate all of the different molecular forms in which the protein product of a single gene can be found, including changes due to genetic variations, alternatively spliced RNA transcripts and post-translational modifications (Fig. 1). Any gene or protein processing events such as those using inteins or RNA-editing mechanisms are now covered cleanly by the term ‘proteoform’. The term should include all post-translational modifications in the PSI-MOD ontology except those classified as reagent-derivatized or isotope-labeled residues (see the Supplementary Note for a precise definition). Products of multigene families should continue to be categorized on the basis of sequence identity (for example, >90%, >99% and so on). The term is compatible with a gene-centric approach for referring to proteins, which we support, because grouping related forms of proteins together even though they are the products of different genes leads to imprecision in protein identification5.
We have begun to use the term ‘proteoform’ in our own writing and presentations, and we find it to be intuitive and readily grasped by readers and audiences. It has an aesthetic appeal, as the simple protein analog of the genetic term ‘isoform’. It is a single word rather than a pair of words, and it does not present the ambiguity of a half-dozen alternative terms, many with historical uses. We have already found its use helpful to us, and the adoption of the term by UniProt5, the Protein Ontology6 and the wider community will improve readability and comprehension of the often technically dense publications that characterize the proteomics field.
Supplementary Material
Acknowledgments
The authors acknowledge the support of the US National Institutes of Health (1R01GM067193 to N.L.K., 1P50HG004952 to L.M.S.) and a grant to N.L.K. from the Chicago Biomedical Consortium, which is supported by the Searle Funds at The Chicago Community Trust. This paper is endorsed by the Consortium for Top Down Proteomics, whose members, including ad hoc members (indicated by an asterisk), at the March 2012 meeting were: Michal Linial, David Goodlett, Pat Langridge-Smith, Young Ah Goo, George Safford, Leo Bonilla*, George Kruppa, Roman Zubarev, Jon Rontree, Julia Chamot-Rooke, John Garavelli, Albert Heck, Joseph Loo, Deborah Penque, Martin Hornshaw, Christopher Hendrickson, Ljiljana Pasa-Tolic, Christoph Borchers, Dominic Chan, Nicholas Young*, Jeffrey Agar, Christophe Masselon, Michael Gross*, Fred McLafferty, Yury Tsybin, Ying Ge, Ian Sanders*, James Langridge, Julian Whitelegge* and Alan Marshall.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Supplementary information is available at http://www.nature.com/doifinder/10.1038/nmeth.2369
References
- 1.Pruitt KD, Tatusova T, Maglott DR. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schlüter H, Apweiler R, Holzhütter HG, Jungblut PR. Chem Cent J. 2009;3:11. doi: 10.1186/1752-153X-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tran JC, et al. Nature. 2011;480:254–258. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prabakaran S, Lippens G, Steen H, Gunawardena J. Wiley Interdiscip Rev Syst Biol Med. 2012;4:565–583. doi: 10.1002/wsbm.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The UniProt Consortium. Nucleic Acids Res. 2012;40:D71–D75. [Google Scholar]
- 6.Natale DA, et al. Nucleic Acids Res. 2011;39:D539–D545. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.