

Abstract
Despite detailed clinical definition and refinement of neurodevelopmental disorders and neuropsychiatric conditions, the underlying genetic etiology has proved elusive. Recent genetic studies have revealed some common themes: considerable locus heterogeneity, variable expressivity for the same mutation, and a role for multiple disruptive events in the same individual affecting genes in common pathways. Recurrent copy number variation (CNV), in particular, has emphasized the importance of either de novo or essentially private mutations creating imbalances for multiple genes. CNVs have foreshadowed a model where the distinction between milder neuropsychiatric conditions from those of severe developmental impairment may be a consequence of increased mutational burden affecting more genes.
Keywords: copy number variants, variable penetrance, genomic disorders, autism, schizophrenia, intellectual disability
INTRODUCTION
Neurodevelopmental disorders are characterized by impairment of growth and development of the brain often associated with cognitive, neurological, or psychiatric dysfunction. It is an umbrella term that can traverse, to varying degrees, diverse disease classifications including intellectual disability (ID), developmental delay (DD), autism, schizophrenia, bipolar disease, etc. Despite seemingly distinct primary diagnoses, considerable clinical heterogeneity as well as overlap has been appreciated for many years. Individuals with autism, for example, can present with or without cognitive impairment or ID [Kaufman et al., 2008; Matson and Shoemaker, 2009]; this is also reflected in the observed effect of IQ on portions of the Autism Diagnostic Observation Schedule (ADOS) diagnostic criteria [Kamp-Becker et al., 2011]. Such clinical overlap has also been observed for psychiatric disorders, such as bipolar disorder and schizophrenia [Grozeva et al., 2010; Malhotra et al., 2011]. Similarly, it is well known that individuals with schizophrenia also demonstrate comorbidity with cognitive impairments of varying severities [Woodberry et al., 2008] as well as, in some cases, structural defects of the brain [Weinberger, 1995]. Epilepsy patients are more likely to develop psychoses and both ID and schizophrenia patients are more prone to seizure [Hyde and Weinberger, 1997; Cascella et al., 2009]. There are convincing epidemiological links between these diseases that support a model that at least part of the etiology may be neurodevelopmental in origin [Weinberger, 1987].
The genetics of these diverse conditions have also begun to converge. Large copy number variants (CNVs), in particular, have been implicated in these diseases to different degrees. This has included reports of overall increases in CNV burden, higher rates of de novo or sporadic mutation, and the discovery of specific recurrent CNVs observed across diverse phenotypes. With few exceptions, CNVs have been large affecting numerous genes and are extremely rare for any specific locus (<1%) but collectively common on the whole. An emerging model has been that certain CNVs disrupt the homeostasis of normal neuronal development resulting in a range of disorders as part of a neurodevelopmental continuum [Stefansson et al., 2008; The International Schizophrenia Consortium, 2008; Helbig et al., 2009; McCarthy et al., 2009; Girirajan and Eichler, 2010; Mefford et al., 2010; Mulle et al., 2010; Cooper et al., 2011; Mitchell, 2011]. In this review, we will focus on recent work in understanding the role of CNV in neurodevelopmental disorders and the implications of these results in our understanding of the classification, severity, and comorbidities of these disorders.
CNV LANDSCAPE OF NEUROPSYCHIATRIC AND NEURODEVELOPMENTAL CONDITIONS
Despite high heritability estimates for bipolar, epilepsy, schizophrenia, autism, and ID ranging from 73% to more than 90%, relatively few common single nucleotide polymorphisms (SNPs) have been convincingly associated with these diseases [Marshall et al., 2008; Stefansson et al., 2008; Wang et al., 2009; Kasperaviciūte et al., 2010; de Kovel et al., 2010; Pregelj, 2011; Psychiatric GWAS Consortium Bipolar Disorder Working Group, 2011; Shi et al., 2011; Yue et al., 2011]. This has led to a shift to the discovery of rarer genetic variation including CNV as a potential source for the missing heritability [Manolio et al., 2009]. In the past few years, studies of large cohorts have revealed several highly penetrant loci associated with neurodevelopmental disorders and a generalized increase in CNV burden compared to unaffected siblings and controls.
Two recent studies totaling to ~30,000 cases ascertained primarily for ID/DD phenotypes among pediatric cases have given us a global picture of this larger variation. This has included the confirmation as well as discovery of susceptible regions in the human genome [Cooper et al., 2011; Kaminsky et al., 2011]. Performing a systematic assessment of population frequency of CNVs at different size ranges, Cooper et al. [2011] showed a significant increase in large CNV burden in affected individuals compared to controls. Further analysis of comorbid phenotypes within the ID/multiple congenital anomalies cohort demonstrated a gradation for autosomal CNV burden, where children with developmental anomalies, such as craniofacial and cardiovascular defects showed the highest CNV burden, while those with no clinically recognized developmental issues other than autism or seizure, for example, showed the lowest (Fig. 1). Similarly, studies in large collections of idiopathic autism [1,124 Simons Simplex Collection (SSC) probands; Sanders et al., 2011] and schizophrenia (>4,500 combined cases) [The International Schizophrenia Consortium, 2008; Stefansson et al., 2008] have demonstrated significant enrichments in large rare/de novo CNV burden in cases compared to controls and unaffected siblings. Although a more subtle increase overall, the effect for schizophrenia appears more pronounced when considering CNVs which disrupt genes in neurodevelopmental pathways [Walsh et al., 2008]. More recently, the spectrum of increased CNV burden diseases has widened to include idiopathic generalized epilepsy (IGE) [de Kovel et al., 2010; Heinzen et al., 2010; Mefford et al., 2010]. The evidence for an increased CNV burden is more conflicting for bipolar disorder. Although no increase in CNV burden was detected in a study of rare CNVs (>100 kbp and <1% of the population) in 1,697 cases of bipolar disorder [Grozeva et al., 2010], a recent study that focused on rare de novo CNVs detected a significant increase in the de novo rate in a set of 199 cases [Malhotra et al., 2011]. Similarly, there is no evidence for CNV burden enrichment in dyslexia [Girirajan et al., 2011] or Tourette syndrome [Fernandez et al., 2011]. The strongest burden enrichments in all phenotypes tends to associate with large CNVs (typically defined as 500 kbp and larger), which are under purifying selection in the general population [Stefansson et al., 2008; Itsara et al., 2010; Malhotra et al., 2011], and thus represent promising candidates for pathogenicity. Additionally, burden increases are often limited to rare and de novo CNVs [Marshall et al., 2008; Pinto et al., 2010; Malhotra et al., 2011; Sanders et al., 2011]. These two parameters are tightly linked, as we have recently demonstrated that the size of a CNV is proportional to its probability of arising de novo [Cooper et al., 2011].
Figure 1.

Autosomal CNV burden across various neurodevelopmental phenotypes. Displayed are the odds ratios as a function of CNV size across various phenotypes from two recent studies. A: A comparison of multiple sample cohorts, ascertained using standard diagnostic criteria for each phenotype, profiled on the same targeted micro array CGH platform [Girirajan et al., 2011]. Odds ratios were calculated compared to 306 National Institute of Mental Health controls. B: Information obtained from 15,767 cases with a general diagnosis of ID/DD [Cooper et al., 2011]. The autism, epilepsy, and craniofacial abnormality phenotypes represent subsets of this overall ID/DD population. Odds ratios were calculated against 8,329 control samples [Cooper et al., 2011].
The common theme amongst these studies is that rare/de novo CNV burden appears at a higher odds ratio in more severe phenotypes than populations with milder phenotypes. Here, we define a more severe phenotype as one with a higher degree of cognitive impairment and/or the presence of congenital abnormalities. Three recent studies explicitly compared the autosomal CNV burdens of dyslexia, bipolar disorder, idiopathic epilepsy, autism, schizophrenia, and ID. The lowest CNV burden is observed in dyslexia and bipolar cases, with the most severe CNV burden in cases with dysmorphic features and cardiac defects, followed by ID [Cooper et al., 2011; Girirajan and Eichler, 2011; Girirajan et al., 2011]. In between these extremes are schizophrenia and autism (Fig. 1), which appear to share a similar burden of large CNVs [Cooper et al., 2011; Girirajan and Eichler, 2011; Girirajan et al., 2011; Malhotra et al., 2011; Sanders et al., 2011].
In all studies the case-enriched CNVs are typically very rare with the most common loci being hotspots where the copy number of flanking segmental duplications lead to an elevated local mutation rate [Sharp et al., 2005; Ballif et al., 2008]. In general, potentially pathogenic duplications are more enriched for the larger size range (>500 kbp) while deletions begin to manifest enrichments at a smaller size range. This fits well with a model of stronger purifying selection for haploinsufficiency when compared to increased dosage and predicts less severe outcomes for duplications. In this light, it is interesting that schizophrenia shows an enrichment for CNV duplications; similarly in autism, duplications are most significantly enriched in the 500+ kbp range, while deletions are significantly enriched in the 30–500 kbp range [The International Schizophrenia Consortium, 2008; Pinto et al., 2010]. CNV enrichment in familiar bipolar disorder is biased towards duplications [Malhotra et al., 2011]. In ID/DD, deletions are more commonly interpreted as pathogenic [Kaminsky et al., 2011] and tend to be more penetrant than duplications [Cooper et al., 2011]. In fact, reciprocal duplication events for known genomic disorders, such as Smith-Magenis and Williams syndrome, are often missed during clinical diagnosis or present as mild manifestations [Somerville et al., 2005; Potocki et al., 2007]. These data all support a stronger effect size for deletions.
Recurrent CNV regions have been identified across multiple disparate disorders (discussed below). Numerous hotspot and nonhotspot CNV loci have been implicated suggesting considerable locus heterogeneity with hundreds of potentially underlying genes. A minority of specific CNVs have been observed with sufficient counts to be individually detected as enriched in a case–control paradigm with multiple testing corrections. In addition to hot-spot and large CNV burden, many of these studies have demonstrated significant enrichment within the 30–500 kbp range, primarily for deletions [Itsara et al., 2010; Pinto et al., 2010; Sanders et al., 2011]. Analysis of smaller CNVs is complicated by the increasing number of rare small CNVs in the general population. Only 8% of the general population carries a CNV greater than 500 kbp (as opposed to 25% for patients with developmental delay) [Itsara et al., 2010; Cooper et al., 2011]. As the size diminishes, the number of events increases rapidly with a smaller fraction contributing to disease. Typically, tens of events (less than 10 in array studies) are seen between 100 and 500 kbp per sample, with hundreds of CNV events per sample in the 1–10 kbp range, and thousands in the 100 bp to 1 kbp range [Iafrate et al., 2004; Tuzun et al., 2005; Hormozdiari et al., 2009; Conrad et al., 2010; The 1000 Genomes Project Consortium, 2010; Alkan et al., 2011; Cooper et al., 2011]. Despite this complication, small CNVs represent a promising avenue for future studies with sufficiently powered study populations (both in sample size and resolution) and parental cases for determining de novo status. Specific examples of small CNVs are beginning to arise in genome-wide studies, such as VIPR2 duplications in schizophrenia, partial TMLHE deletions in autism, a collection of significant events in ID/DD subtypes, and events overlapping with genes identified to contain de novo mutations and indels [Boone et al., 2010; Celestino-Soper et al., 2011; Cooper et al., 2011; Kirov et al., 2011; Vacic et al., 2011].
VARIABLE EXPRESSIVITY OF SPECIFIC LOCI
Numerous CNV loci have been recurrently observed across seemingly disparate neurological phenotypes [Helbig et al., 2009; McCarthy et al., 2009; Girirajan and Eichler, 2010; Rosenfeld et al., 2010; Cooper et al., 2011; Sanders et al., 2011]. Among these are 59 pathogenic CNVs which have reached at least nominal statistical significance with respect to enrichment in cases of ID/DD [Cooper et al., 2011]. In some cases, a specific CNV is necessary and sufficient to result in a suite of characteristic features. These “syndromic CNVs” most often result in patients with moderate-to-severe ID and are by-and-large sporadic in origin. Examples include the 17q21.31 microdeletion syndrome and the Williams syndrome deletion on chromosome 7q11.23 (Fig. 2, Table I). Contrasting with these “syndromic CNVs” are CNVs which are much more variable in their outcome and more likely to be inherited. Here, we highlight this observation of variable expressivity (defined as either qualitative or quantitative phenotypic variation among individuals carrying the same CNV) by focusing on a few of these CNV loci.
Figure 2.
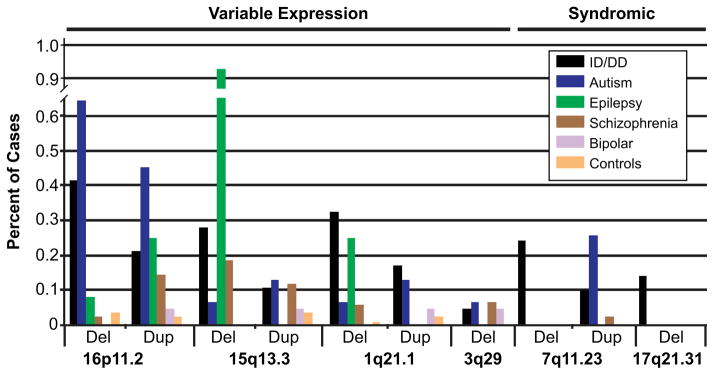
Variable expressivity of hotspot CNVs. The frequency of CNV deletions and reciprocal duplications for six genomic hotspots associated with neurological disease are shown (ID/DD, autism, epilepsy, schizophrenia, and bipolar disorders). Data sources are as follows. ID/DD: all sites n = 31,516 [Cooper et al., 2011; Kaminsky et al., 2011]. Autism: all sites n = 1,551 [Marshall et al., 2008; Sanders et al., 2011]. Epilepsy: all sites n = 399 [Mefford et al., 2010]; 15q13.3 n = 647 [Helbig et al., 2009; Mefford et al., 2010]; 16p11.2 n = 1,234 [de Kovel et al., 2010; Mefford et al., 2010]. Schizophrenia: all sites n = 4,168 [The International Schizophrenia Consortium, 2008; Malhotra et al., 2011], 15q13.3 n = 6,948 [The International Schizophrenia Consortium, 2008; Stefansson et al., 2008; Malhotra et al., 2011], 1q21.1 n = 12,117 [The International Schizophrenia Consortium, 2008; Stefansson et al., 2008; Malhotra et al., 2011], 3q29 n = 4,413 [The International Schizophrenia Consortium, 2008; Mulle et al., 2010; Malhotra et al., 2011]. Bipolar disorders: all sites n = 2,053 [Grozeva et al., 2010; Malhotra et al., 2011]. Controls: n = 8,329 [Cooper et al., 2011].
TABLE I.
Characteristics of Variably Expressive Hotspot CNVs
| CNV locus | Start (Mbp) | End (Mbp) | RefSeq genes (unique space) | Duplication inheritancea | Deletion inheritancea |
|---|---|---|---|---|---|
| 16p11.2 | 29.56 | 30.11 | 28 | 77.78% (21/27) | 36.73% (18/49) |
| 15q13.3 | 28.92 | 30.27 | 8 | 100% (11/11) | 78.79% (26/33) |
| 1q21.1 | 145.04 | 145.86 | 9 | 75.61% (31/41) | 77.78% (28/36) |
| 3q29 | 197.23 | 198.84 | 27 | — | 28% (4/14) |
| 7q11.23 | 72.38 | 73.78 | 26 | 44.44% (4/9) | 0% (0/45) |
| 17q21.31b | 16.65 | 20.42 | 10 | — | 0% (0/32) |
— Indicates no data available.
All loci (except 3q29) [Girirajan et al., unpublished], 7q11.23 deletion [Perez Jurado et al., 1996], 3q29 deletion [Willatt et al., 2005; Ballif et al., 2008], and 17q21.31 [Koolen et al., 2008].
17q21.21 does not demonstrate variable expressivity.
One locus that has received considerable attention in recent years is the 16p11.2 microdeletion/microduplication. Both the deletion and duplication have been observed in multiple conditions with significant enrichment compared to healthy controls (range: <0.01–1%).
The deletion is associated with more severe phenotypes, including cases with dysmorphic features, and is strongly enriched in ID and autism with a strong association to obesity [Walters et al., 2010]. The reciprocal duplication is not associated with any common dysmorphic features and is seen in a wider range of conditions, including clinically underweight cases mirroring the obesity phenotype for the deletion (Fig. 2, Table I) [Marshall et al., 2008; Weiss et al., 2008; Bochukova et al., 2010; Rosenfeld et al., 2010; Walters et al., 2010; Girirajan et al., 2011; Jacquemont et al., 2011; Sanders et al., 2011]. Recent work in autism has identified atypical deletions and point mutations highlighting SEZ6L2 as a strong candidate for the deletion phenotype [Kumar et al., 2008; Crepel et al., 2011; Konyukh et al., 2011]. There is also conflicting evidence that phenotypic variability at this locus may be controlled by second-site CNVs in humans. Girirajan et al. [2010], for example, noted an excess of additional larger CNVs in 9.9% of pediatric cases with ID and 16p11.2 deletions concurrent with an inheritance rate of 25.7%. However, a recent study on 16p11.2 in the context of obesity and developmental delay failed to detect evidence for an enrichment of second-hits [Jacquemont et al., 2011].
The 15q13.3 microdeletion is also detected across multiple phenotypes, with the strongest enrichment observed in cases of IGE where frequency reaches nearly 1% of cases [Dibbens et al., 2009; Helbig et al., 2009]. The microdeletion was originally discovered among patients with ID [Sharp et al., 2008; Cooper et al., 2011; Kaminsky et al., 2011]. Later, the same CNV was discovered in cases of autism, schizophrenia, and epilepsy [The International Schizophrenia Consortium, 2008; Marshall et al., 2008; Stefansson et al., 2008; Cooper et al., 2011; Kaminsky et al., 2011; Sanders et al., 2011] (Fig. 2, Table I). Atypical smaller deletions in patients with similar phenotypes have strongly implicated the nicotinamide acetylcholine receptor (CHRNA7) as the most likely candidate gene for disease especially as it relates to the seizure and ID [Shinawi et al., 2009]. While deletions of this locus have been replicated in numerous studies involving patients with idiopathic epilepsy, the deletion is not detected in focal epilepsies [Heinzen et al., 2010]. The reciprocal duplication has not been observed in cases of IGE; while it is enriched in cases of ID with respect to controls, it is more variably expressed across neurological phenotypes (ID/DD, bipolar disorder, and autism among others) and its role in pathogenicity remains unclear [Helbig et al., 2009; Shinawi et al., 2009; Szafranski et al., 2010; Cooper et al., 2011].
The 3q29 microdeletion is particularly rare (<1/1,000) and has been associated with severe schizophrenia [Mulle et al., 2010], ID, and autistic features with mild dysmorphism present in the majority of cases [Willatt et al., 2005; Ballif et al., 2008]. Recently, Carroll et al., [2011] identified rare mutations in one gene (DLG1) highlighting its potential involvement in the schizophrenia phenotype. The variability in expression is further supported by a case report of a child with autistic features, an elongated face, and normal IQ [Cobb et al., 2010]. Pathogenicity of the reciprocal duplication remains uncertain with the loci failing to reach significance in ID/DD and the observation that a family carrying the duplication also carries a second CNV that may explain the clinical features [Ballif et al., 2008; Cooper et al., 2011].
Finally, 1q21.1 deletions and duplications demonstrate some of the most pronounced variability in phenotypic outcome with the highest rates of deletions observed among ID and IGE patients [Mefford et al., 2010; Cooper et al., 2011; Kaminsky et al., 2011] and the highest rates of duplications found in ID and autism [Marshall et al., 2008; Cooper et al., 2011; Kaminsky et al., 2011; Sanders et al., 2011]. The variable expressivity between cases has been explained by secondary insults including environmental exposure, stochastic variation during development, or differences in the genetic backgrounds. Studies involving large panels of patients with the deletion [Mefford et al., 2008] suggest a relatively finite number of “sub-syndromic” outcomes including mild-to-moderate ID, micro-cephaly, cardiac abnormalities, and cataracts. The commonalities and differences between duplication and deletion phenotypes may be explained, in part, by the variable dosage sensitivity of the genes contained in the CNV region. To this end, Harvard et al. [2011] performed functional analyses on two genes with a strong correlation between expression levels and copy number state. In a cell model the authors observed that one gene (CHD1L) demonstrated a functional deficit under both over-and underexpression conditions, while another gene (PRKAB2) only affected its associated functions in a deletion state. The complex comorbidities of these loci mirror the complex interactions of the clinical phenotypes and highlight not just locus heterogeneity but variable expressivity and overlap in neurodevelopmental conditions [Matson and Shoemaker, 2009; Girirajan and Eichler, 2010; Rosenfeld et al., 2010; Auerbach et al., 2011]. While deletions and duplications of specific loci may yield drastically different phenotypes, the contradictory observation of both deletions and duplications leading to similar phenotypes is also common. This may be explained by the general sensitivity of certain cellular functions to dosage imbalance, such as that observed for the 1q21.1 region [Harvard et al., 2011]. This model is also supported in an alternative gene context by Auerbach et al. [2011], who recently demonstrated that fragile X and tuberous sclerosis mouse models affect the same pathway (glutamate receptor 5) in opposite directions with both disruptions resulting in syndromic autism and ID features. This complex variability in phenotypic expression is not limited to the hotspots discussed here but has been observed for many CNV loci, and variable expressivity has been explained by both variable breakpoints and the interaction of these CNVs with secondary mutation events. This model is particularly striking for 16p12.1 deletions where phenotypic severity is strongly associated with additional CNVs and appears to be most strongly correlated with phenotypic modification of inherited CNV sites [Girirajan et al., 2010]. Additional evidence has suggested that variable expressivity can be the result of genetic background and additional subclinical phenotypes, only obvious in studies of large populations with detailed clinical information, may exist in cases defined as normal or with other assigned phenotypes [Mefford et al., 2008].
CNV BURDEN AND PHENOTYPE SEVERITY
Taken together, the results of recent genome-wide screens of CNVs of neurodevelopmental disorders reveal a striking correlation of effect size and the number of genes affected (both by large CNVs and multiple loci) [Itsara et al., 2010; Cooper et al., 2011; Girirajan and Eichler, 2011; Malhotra et al., 2011]. A simple additive model may be that these diseases are part of a neurodevelopmental continuum where more rare and disruptive mutations in an individual lead to increasing severity. In this model, larger CNVs create imbalance for more genes during neurodevelopment leading to a more severe outcome. In the study by Girirajan et al. [2011], we noted an increased CNV burden in cases of autism with ID compared to cases without ID; this is further supported by the increased CNV burden in the autism cohorts highlighted in Figure 1. To further investigate the relationship between CNV burden and effect size, we performed a meta-analysis of rare CNVs from a study with a quantitative measurement of severity. In the SSC autism cohort (ascertained by multiple criteria described by Risi et al. [2006]) studied by Sanders et al. [2011], each sample has an associated full-scale IQ score. We reanalyzed CNVs using the algorithm described by Itsara et al. [2009] to include CNVs that would be excluded due to the stringent size filtering in Sanders study and filtered CNVs by their frequency in both siblings (<0.1%) and an independent cohort of 2,090 controls (<0.1%) also profiled on Illumina SNP arrays with similar density [Cooper et al., 2011]. CNV calls were filtered to exclude those not detectable on all three ~1 M Illumina platforms represented. This resulted in 2,227 rare CNV calls in 841 probands with no events detected in >1% of probands. Although the individual level correlation between the largest rare CNV carried and full-scale IQ (Fig. 3a) is relatively weak, we note that a general trend does appear to indicate reduced IQ for cases with larger CNVs similar to the observation of Sanders et al. [2011]. When we examine the median IQ for populations of cases with increasing minimum CNV size cutoffs, we note an emerging trend (Spearman’s r = −0.7894, permutation P = 0.074) with the effect appearing strongest past ~500 kbp (Fig. 3b). Filtering of cases with CNVs smaller than 500 kbp resulted in a striking correlation of minimum CNV size and median IQ (Spearman’s r = −0.97, P = 0.0009). To confirm that the effect is not solely due to large outliers, we repeated the analysis excluding very large CNVs (>3 Mbp) and noted that the correlation remained significant (Spearman’s r = −0.96, P = 0.0056). We also note that a statistically significant reduction in median IQ compared to the general SSC cohort is reached at a cutoff of 670 kbp (P = 0.0321). Thus, within this limited cohort we can confirm that large CNVs have an increasing effect size on quantifiable phenotypic severity. To further investigate the role of rare and de novo CNVs on phenotypic severity and the potential for multiple rare CNVs in a single case [Girirajan et al., 2010; Sanders et al., 2011], we repeated the analysis using the number of genes affected by rare CNVs and observed a striking downward trend in IQ with an increasing number of genes, with the median IQ crossing the threshold for ID at ≥18 genes affected (P = 0.00225). These data support a significant role for rare CNVs in phenotypic severity with both individual large CNVs and the combined effect of multiple rare CNVs leading to an increased effect size.
Figure 3.

Phenotypic severity as an outcome of CNV burden. Rare CNVs (defined as present in <0.1% of controls and unaffected siblings) were analyzed in the context of full-scale IQ for 841 Simons Simplex autism probands. A: Full-scale IQ weakly correlates with the largest rare CNV carried by an individual (Pearson’s r = −0.11, >500 kbp, r = −0.36); however, when we split the population at increasing CNV size cutoffs (B), we observe a striking drop in median IQ, most noticeable past 500 kbp. When we filter CNVs <500 kbp, we observe a striking correlation between median IQ and CNV size (Spearman’s r = −0.97, P = 0.0009). The calculation of P-values was performed by computing 10,000 random permutations of IQ across samples and repeating the median IQ calculations. The P-value represents the fraction of permutations with a more extreme correlation than that observed for the raw data.
SUMMARY
The neurodevelopmental conditions of bipolar disorder, epilepsy, schizophrenia, autism, and ID share a highly similar large CNV landscape with pathogenicity ascribed to a plethora of individually rare, collectively common CNVs that are both restricted to and shared between these and other phenotypes. The most significant enrichment at a single locus in all conditions is predominantly associated with segmental duplication mediated hotspots [Bailey et al., 2002], such as 16p11.2 deletions in autism and 15q13.3 deletions in IGE, which have both been observed in up to 1% of cases for these specific phenotypes [Helbig et al., 2009; Sanders et al., 2011]. Analysis of hotspot loci has demonstrated remarkable heterogeneity of clinical features supporting a similar mechanism of neurodevelopmental impairment across neurological conditions, particularly those with an increased CNV burden compared to unaffected controls (Figs. 1 and 2). In most conditions discussed here, an increased burden of large rare/de novo CNVs has been reported, although evidence is conflicting for bipolar disorders, which demonstrate the lowest burden increase. Larger CNVs are associated with more severe IQ deficit and the disruption of an increasing number of genes by both single large and multiple small rare CNVs leading to a reduction in median IQ. This effect is also supported by the increasing burden of large CNVs in more severe phenotypes [Cooper et al., 2011; Girirajan and Eichler, 2011; Girirajan et al., 2011].
In the phenotypes with fewer large CNVs, whole-genome or exome studies are very likely to yield fruitful results specifying particular candidate genes. Proving pathogenicity will be challenging as the equivalent of genomic hot-spots prone to recurrent mutation are not known to exist for most smaller mutational events. Current analyses indicate that the burden of point mutations and indels is only subtly enriched in schizophrenia and autism versus controls [Awadalla et al., 2010; Girard et al., 2011; O’Roak et al., 2011; Xu et al., 2011]. It is thus likely that a similar model will apply to smaller CNVs where the effect is not simply the number of events but the specific events observed, such as the type of genes or the nature of the copy number event (e.g., gene-disruptive or leading to a gene fusion). Given the current estimates of locus heterogeneity (~1,000), this will necessitate the screening of extremely larger numbers of cases and controls or detailed family studies where additional evidence from linkage can be leveraged to increase the likelihood of discovering the pathogenic mutation. Large CNVs are expected to have a significant effect size as they perturb dosage of many genes, while small events will need to either individually hit a critical gene or combine to have a significant effect. The extent of locus heterogeneity is supported by observations of 50 distinct and novel gene mutations observed in 136 consanguineous families with recessive ID [Najmabadi et al., 2011]. The notion of multiple hits has been demonstrated in the context of idiopathic high-functioning autism where multiple inherited and de novo mutations have been demonstrated to lead to a clinical phenotype, while select single-gene events can lead to syndromic autism [Schaaf et al., 2011]. In addition, evidence supports a combination of these models in autism with both de novo and rare point mutations/indels and structural variants coming together in specific individuals to converge on particular molecular pathways related to disease [O’Roak et al., 2011, 2012].
Together these data support an oligogenic model for phenotypic expression of CNVs. As more, or larger, rare de novo genic CNVs are present, severity markedly increases with bipolar disorders demonstrating the least CNV burden and ID demonstrating the most significant burden (Fig. 4). Importantly, these mutations are very rare, they are gene disruptive, and they exist in the heterozygous state but collectively different mutations or imbalances converging on common pathways lead to disease. If this model holds, exome and whole-genome sequencing should identify hundreds of genes underlying neuropsychiatric and neurodevelopement disease as both disruptive point mutations as well as smaller CNVs are systematically discovered [Hardenbol et al., 2003; Porreca et al., 2007; Turner et al., 2009]. The prospects for understanding the genetic etiology and the biological pathways underlying the development of the human brain have never been better.
Figure 4.
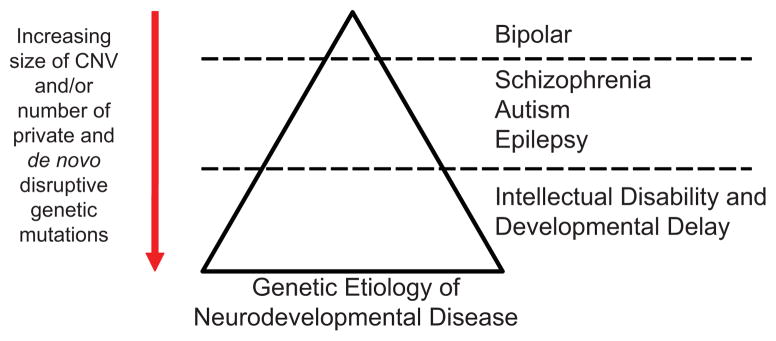
An oligogenic model for neurodevelopmental phenotypes. Presented is a model where an increasing number of genes disrupted by rare CNVs correlates with an increase in phenotypic severity (defined as increasing comorbidity of ID and congenital abnormalities).
Biographies
Santhosh Girirajan is a postdoctoral fellow in Evan Eichler’s group in the Department of Genome Sciences at the University of Washington School of Medicine. His research interest involves unraveling the genetic basis of variable phenotypes associated with pathogenic CNVs (i.e., genomic disorders).
Evan E. Eichler is a professor and Howard Hughes Medical Institute investigator in the Department of Genome Sciences at the University of Washington School of Medicine. He received his Ph.D. in 1995 from the Department of Molecular and Human Genetics at Baylor College of Medicine in Houston, Texas, USA. He is a fellow of the American Association for the Advancement of Science and was a recipient of the American Society of Human Genetics Curt Stern Award in 2008. His research group develops experimental and computational methods for studying genome structural variation in evolution and human disease.
Bradley P. Coe is a postdoctoral fellow in Evan Eichler’s group in the Department of Genome Sciences at the University of Washington School of Medicine, where he is supported by a fellowship from the Canadian Institutes of Health Research. His current work focuses on the identification and study of rare copy number variation in intellectual disability and developmental delay.
References
- Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12:363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480:63–68. doi: 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awadalla P, Gauthier J, Myers RA, Casals F, Hamdan FF, Griffing AR, Cote M, Henrion E, Spiegelman D, Tarabeux J, Piton A, Yang Y, Boyko A, Bustamante C, Xiong L, Rapoport JL, Addington AM, DeLisi JL, Krebs MO, Joober R, Millet B, Fombonne E, Mottron L, Zilversmit M, Keebler J, Daoud H, Marineau C, Roy-Gagnon MH, Dubé MP, Eyre-Walker A, Drapeau P, Stone EA, Lafrenière RG, Rouleau GA. Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am J Hum Genet. 2010;87:316–324. doi: 10.1016/j.ajhg.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- Ballif BC, Theisen A, Coppinger J, Gowans GC, Hersh JH, Madan-Khetarpal S, Schmidt KR, Tervo R, Escobar LF, Friedrich CA, McDonald M, Campbell L, Ming JE, Zackai EH, Bejjani BA, Shaffer LG. Expanding the clinical phenotype of the 3q29 microdeletion syndrome and characterization of the reciprocal microduplication. Mol Cytogenet. 2008;1:8. doi: 10.1186/1755-8166-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochukova EG, Huang N, Keogh J, Henning E, Purmann C, Blaszczyk K, Saeed S, Hamilton-Shield J, Clayton-Smith J, O’Rahilly S, Hurles ME, Farooqi IS. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature. 2010;463:666–670. doi: 10.1038/nature08689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone PM, Bacino CA, Shaw CA, Eng PA, Hixson PM, Pursley AN, Kang SH, Yang Y, Wiszniewska J, Nowakowska BA, del Gaudio D, Xia Z, Simpson-Patel G, Immken LL, Gibson JB, Tsai AC, Bowers JA, Reimschisel TE, Schaaf CP, Potocki L, Scaglia F, Gambin T, Sykulski M, Bartnik M, Derwinska K, Wisniowiecka-Kowalnik B, Lalani SR, Probst FJ, Bi W, Beaudet AL, Patel A, Lupski JR, Cheung SW, Stankiewicz P. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat. 2010;31:1326–1342. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll LS, Williams HJ, Walters J, Kirov G, O’Donovan MC, Owen MJ. Mutation screening of the 3q29 microdeletion syndrome candidate genes DLG1 and PAK2 in schizophrenia. Am J Med Genet Part B. 2011;156B:844–849. doi: 10.1002/ajmg.b.31231. [DOI] [PubMed] [Google Scholar]
- Cascella NG, Schretlen DJ, Sawa A. Schizophrenia and epilepsy: Is there a shared susceptibility? Neurosci Res. 2009;63:227–235. doi: 10.1016/j.neures.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celestino-Soper PB, Shaw CA, Sanders SJ, Li J, Murtha MT, Ercan-Sencicek AG, Davis L, Thomson S, Gambin T, Chinault AC, Ou Z, German JR, Milosavljevic A, Sutcliffe JS, Cook EH, Jr, Stankiewicz P, State MW, Beaud AL. Use of array CGH to detect exonic copy number variants throughout the genome in autism families detects a novel deletion in TMLHE. Hum Mol Genet. 2011;20:4360–4370. doi: 10.1093/hmg/ddr363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb W, Anderson A, Turner C, Hoffman RD, Schonberg S, Levin SW. 1.3 Mb de novo deletion in chromosome band 3q29 associated with normal intelligence in a child. Eur J Med Genet. 2010;53:415–418. doi: 10.1016/j.ejmg.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Bird C, Blackburne B, Lindsay S, Mamanova L, Lee C, Turner DJ, Hurles ME. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat Genet. 2010;42:385–391. doi: 10.1038/ng.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, Abdel-Hamid H, Bader P, McCracken E, Niyazov D, Leppig K, Thiese H, Hummel M, Alexander N, Gorski J, Kussmann J, Shashi V, Johnson K, Rehder C, Ballif BC, Shaffer LG, Eichler EE. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepel A, Steyaert J, De la Marche W, De Wolf V, Fryns JP, Noens I, Devriendt K, Peeters H. Narrowing the critical deletion region for autism spectrum disorders on 16p11.2. Am J Med Genet Part B. 2011;156B:243–245. doi: 10.1002/ajmg.b.31163. [DOI] [PubMed] [Google Scholar]
- de Kovel CG, Trucks H, Helbig I, Mefford HC, Baker C, Leu C, Kluck C, Muhle H, von Spiczak S, Ostertag P, Obermeier T, Kleefuss-Lie AA, Hallmann K, Steffens M, Gaus V, Klein KM, Hamer HM, Rosenow F, Brilstra EH, Trenité DK, Swinkels ME, Weber YG, Unterberger I, Zimprich F, Urak L, Feucht M, Fuchs K, Møller RS, Hjalgrim H, De Jonghe P, Suls A, Rückert IM, Wichmann HE, Franke A, Schreiber S, Nürnberg P, Elger CE, Lerche H, Stephani U, Koeleman BP, Lindhout D, Eichler EE, Sander T. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010;133:23–32. doi: 10.1093/brain/awp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibbens LM, Mullen S, Helbig I, Mefford HC, Bayly MA, Bellows S, Leu C, Trucks H, Obermeier T, Wittig M, Franke A, Caglayan H, Yapici Z, Sander T, Eichler EE, Scheffer IE, Mulley JC, Berkovic SF EPICURE Consortium. Familial and sporadic 15q13.3 microdeletions in idiopathic generalized epilepsy: Precedent for disorders with complex inheritance. Hum Mol Genet. 2009;18:3626–3631. doi: 10.1093/hmg/ddp311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez TV, Sanders SJ, Yurkiewicz IR, Ercan-Sencicek AG, Kim YS, Fishman DO, Raubeson MJ, Song Y, Yasuno K, Ho WS, Bilguvar K, Glessner J, Chu SH, Leckman JF, King RA, Gilbert DL, Heiman GA, Tischfield JA, Hoekstra PJ, Devlin B, Hakonarson H, Mane SM, Günel M, State MW. Rare copy number variants in Tourette syndrome disrupt genes in histaminergic pathways and overlap with autism. Biol Psychiatry. 2012;71:392–402. doi: 10.1016/j.biopsych.2011.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard SL, Gauthier J, Noreau A, Xiong L, Zhou S, Jouan L, Dionne-Laporte A, Spiegelman D, Henrion E, Diallo O, Thibodeau P, Bachand I, Bao JY, Tong AH, Lin CH, Millet B, Jaafari N, Joober R, Dion PA, Lok S, Krebs MO, Rouleau GA. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat Genet. 2011;43:860–863. doi: 10.1038/ng.886. [DOI] [PubMed] [Google Scholar]
- Girirajan S, Eichler EE. Phenotypic variability and genetic susceptibility to genomic disorders. Hum Mol Genet. 2010;19:R176–R187. doi: 10.1093/hmg/ddq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Eichler EE. De novo CNVs in bipolar disorder: recurrent themes or new directions? Neuron. 2011;72:885–887. doi: 10.1016/j.neuron.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, Vives L, Walsh T, McCarthy SE, Baker C, Mefford HC, Kidd JM, Browning SR, Browning BL, Dickel DE, Levy DL, Ballif BC, Platky K, Farber DM, Gowans GC, Wetherbee JJ, Asamoah A, Weaver DD, Mark PR, Dickerson J, Garg BP, Ellingwood SA, Smith R, Banks VC, Smith W, McDonald MT, Hoo JJ, French BN, Hudson C, Johnson JP, Ozmore JR, Moeschler JB, Surti U, Escobar LF, El-Khechen D, Gorski JL, Kussmann J, Salbert B, Lacassie Y, Biser A, McDonald-McGinn DM, Zackai EH, Deardorff MA, Shaikh TH, Haan E, Friend KL, Fichera M, Romano C, Gécz J, DeLisi LE, Sebat J, King MC, Shaffer LG, Eichler EE. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42:203–209. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Brkanac Z, Coe BP, Baker C, Vives L, Vu TH, Shafer N, Bernier R, Ferrero GB, Silengo M, Warren ST, Moreno CS, Fichera M, Romano C, Raskind WH, Eichler EE. Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet. 2011;7:e1002334. doi: 10.1371/journal.pgen.1002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozeva D, Kirov G, Ivanov D, Jones IR, Jones L, Green EK, St Clair DM, Young AH, Ferrier N, Farmer AE, McGuffin P, Holmans PA, Owen MJ, O’Donovan MC, Craddock N Wellcome Trust Case Control Consortium. Rare copy number variants: A point of rarity in genetic risk for bipolar disorder and schizophrenia. Arch Gen Psychiatry. 2010;67:318–327. doi: 10.1001/archgenpsychiatry.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardenbol P, Baner J, Jain M, Nilsson M, Namsaraev EA, Karlin-Neumann GA, Fakhrai-Rad H, Ronaghi M, Willis TD, Landegren U, Davis RW. Multiplexed genotyping with sequence-tagged molecular inversion probes. Nat Biotechnol. 2003;21:673–678. doi: 10.1038/nbt821. [DOI] [PubMed] [Google Scholar]
- Harvard C, Strong E, Mercier E, Colnaghi R, Alcantara D, Chow E, Martell S, Tyson C, Hrynchak M, McGillivray B, Hamilton S, Marles S, Mhanni A, Dawson AJ, Pavlidis P, Qiao Y, Holden JJ, Lewis SM, O’Driscoll M, Rajcan-Separovic E. Understanding the impact of 1q21.1 copy number variant. Orphanet J Rare Dis. 2011;6:54. doi: 10.1186/1750-1172-6-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzen EL, Radtke RA, Urban TJ, Cavalleri GL, Depondt C, Need AC, Walley NM, Nicoletti P, Ge D, Catarino CB, Duncan JS, Kasperaviciūte D, Tate SK, Caboclo LO, Sander JW, Clayton L, Linney KN, Shianna KV, Gumbs CE, Smith J, Cronin KD, Maia JM, Doherty CP, Pandolfo M, Leppert D, Middleton LT, Gibson RA, Johnson MR, Matthews PM, Hosford D, Kälviäinen R, Eriksson K, Kantanen AM, Dorn T, Hansen J, Krämer G, Steinhoff BJ, Wieser HG, Zumsteg D, Ortega M, Wood NW, Huxley-Jones J, Mikati M, Gallentine WB, Husain AM, Buckley PG, Stallings RL, Podgoreanu MV, Delanty N, Sisodiya SM, Goldstein DB. Rare deletions at 16p13.11 predispose to a diverse spectrum of sporadic epilepsy syndromes. Am J Hum Genet. 2010;86:707–718. doi: 10.1016/j.ajhg.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, Franke A, Muhle H, de Kovel C, Baker C, von Spiczak S, Kron KL, Steinich I, Kleefuss-Lie AA, Leu C, Gaus V, Schmitz B, Klein KM, Reif PS, Rosenow F, Weber Y, Lerche H, Zimprich F, Urak L, Fuchs K, Feucht M, Genton P, Thomas P, Visscher F, de Haan GJ, Møller RS, Hjalgrim H, Luciano D, Wittig M, Nothnagel M, Elger CE, Nürnberg P, Romano C, Malafosse A, Koeleman BP, Lindhout D, Stephani U, Schreiber S, Eichler EE, Sander T. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009;41:160–162. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F, Alkan C, Eichler EE, Sahinalp SC. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 2009;19:1270–1278. doi: 10.1101/gr.088633.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde TM, Weinberger DR. Seizures and schizophrenia. Schizophr Bull. 1997;23:611–622. doi: 10.1093/schbul/23.4.611. [DOI] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, Krauss RM, Myers RM, Ridker PM, Chasman DI, Mefford H, Ying P, Nickerson DA, Eichler EE. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itsara A, Wu H, Smith JD, Nickerson DA, Romieu I, London SJ, Eichler EE. De novo rates and selection of large copy number variation. Genome Res. 2010;20:1469–1481. doi: 10.1101/gr.107680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Reymond A, Zufferey F, Harewood L, Walters RG, Kutalik Z, Martinet D, Shen Y, Valsesia A, Beckmann ND, Thorleifsson G, Belfiore M, Bouquillon S, Campion D, de Leeuw N, de Vries BB, Esko T, Fernandez BA, Fernández-Aranda F, Fernández-Real JM, Gratacòs M, Guilmatre A, Hoyer J, Jarvelin MR, Kooy RF, Kurg A, Le Caignec C, Männik K, Platt OS, Sanlaville D, Van Haelst MM, Villatoro Gomez S, Walha F, Wu BL, Yu Y, Aboura A, Addor MC, Alembik Y, Antonarakis SE, Arveiler B, Barth M, Bednarek N, Béna F, Bergmann S, Beri M, Bernardini L, Blaumeiser B, Bonneau D, Bottani A, Boute O, Brunner HG, Cailley D, Callier P, Chiesa J, Chrast J, Coin L, Coutton C, Cuisset JM, Cuvellier JC, David A, de Freminville B, Delobel B, Delrue MA, Demeer B, Descamps D, Didelot G, Dieterich K, Disciglio V, Doco-Fenzy M, Drunat S, Duban-Bedu B, Dubourg C, El-Sayed Moustafa JS, Elliott P, Faas BH, Faivre L, Faudet A, Fellmann F, Ferrarini A, Fisher R, Flori E, Forer L, Gaillard D, Gerard M, Gieger C, Gimelli S, Gimelli G, Grabe HJ, Guichet A, Guillin O, Hartikainen AL, Heron D, Hippolyte L, Holder M, Homuth G, Isidor B, Jaillard S, Jaros Z, Jiménez-Murcia S, Helas GJ, Jonveaux P, Kaksonen S, Keren B, Kloss-Brandstätter A, Knoers NV, Koolen DA, Kroisel PM, Kronenberg F, Labalme A, Landais E, Lapi E, Layet V, Legallic S, Leheup B, Leube B, Lewis S, Lucas J, MacDermot KD, Magnusson P, Marshall C, Mathieu-Dramard M, McCarthy MI, Meitinger T, Mencarelli MA, Merla G, Moerman A, Mooser V, Morice-Picard F, Mucciolo M, Nauck M, Ndiaye NC, Nordgren A, Pasquier L, Petit F, Pfundt R, Plessis G, Rajcan-Separovic E, Ramelli GP, Rauch A, Ravazzolo R, Reis A, Renieri A, Richart C, Ried JS, Rieubland C, Roberts W, Roetzer KM, Rooryck C, Rossi M, Saemundsen E, Satre V, Schurmann C, Sigurdsson E, Stavropoulos DJ, Stefansson H, Tengström C, Thorsteinsdóttir U, Tinahones FJ, Touraine R, Vallée L, van Binsbergen E, Van der Aa N, Vincent-Delorme C, Visvikis-Siest S, Vollenweider P, Völzke H, Vulto-van Silfhout AT, Waeber G, Wallgren-Pettersson C, Witwicki RM, Zwolinksi S, Andrieux J, Estivill X, Gusella JF, Gustafsson O, Metspalu A, Scherer SW, Stefansson K, Blakemore AI, Beckmann JS, Froguel P. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature. 2011;478:97–102. doi: 10.1038/nature10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, Moreno-De-Luca D, Moreno-De-Luca A, Mulle JG, Warren ST, Richard G, Compton JG, Fuller AE, Gliem TJ, Huang S, Collinson MN, Beal SJ, Ackley T, Pickering DL, Golden DM, Aston E, Whitby H, Shetty S, Rossi MR, Rudd MK, South ST, Brothman AR, Sanger WG, Iyer RK, Crolla JA, Thorland EC, Aradhya S, Ledbetter DH, Martin CL. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med. 2011;13:777–784. doi: 10.1097/GIM.0b013e31822c79f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp-Becker I, Ghahreman M, Heinzel-Gutenbrunner M, Peters M, Remschmidt H, Becker K. Evaluation of the revised algorithm of Autism Diagnostic Observation Schedule (ADOS) in the diagnostic investigation of high-functioning children and adolescents with autism spectrum disorders. Autism. 2011 May 24; doi: 10.1177/1362361311408932. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Kasperaviciūte D, Catarino CB, Heinzen EL, Depondt C, Cavalleri GL, Caboclo LO, Tate SK, Jamnadas-Khoda J, Chinthapalli K, Clayton LM, Shianna KV, Radtke RA, Mikati MA, Gallentine WB, Husain AM, Alhusaini S, Leppert D, Middleton LT, Gibson RA, Johnson MR, Matthews PM, Hosford D, Heuser K, Amos L, Ortega M, Zumsteg D, Wieser HG, Steinhoff BJ, Krämer G, Hansen J, Dorn T, Kantanen AM, Gjerstad L, Peuralinna T, Hernandez DG, Eriksson KJ, Kälviäinen RK, Doherty CP, Wood NW, Pandolfo M, Duncan JS, Sander JW, Delanty N, Goldstein DB, Sisodiya SM. Common genetic variation and susceptibility to partial epilepsies: A genome-wide association study. Brain. 2010;133:2136–2147. doi: 10.1093/brain/awq130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman WE, Capone GT, Clarke M, Budimirovic DB. Chapter 4: Autism in genetic intellectual disability. In: Zimmerman AW, editor. Autism: Current theories and evidence. Totowa, NJ: Humana Press; 2008. pp. 81–107. [Google Scholar]
- Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, Moran J, Chambert K, Toncheva D, Georgieva L, Grozeva D, Fjodorova M, Wollerton R, Rees E, Nikolov I, van de Lagemaat LN, Bayés A, Fernandez E, Olason PI, Böttcher Y, Komiyama NH, Collins MO, Choudhary J, Stefansson K, Stefansson H, Grant SG, Purcell S, Sklar P, O’Donovan MC, Owen MJ. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2012;17:142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konyukh M, Delorme R, Chaste P, Leblond C, Lemiere N, Nygren G, Anckarsater H, Rastam M, Stahlberg O, Amsellem F, Gillberg IC, Mouren-Simeoni MC, Herbrecht E, Fauchereau F, Toro R, Gillberg C, Leboyer M, Bourgeron T. Variations of the candidate SEZ6L2 gene on chromosome 16p11.2 in patients with autism spectrum disorders and in human populations. PLoS ONE. 2011;6:e17289. doi: 10.1371/journal.pone.0017289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolen DA, Sharp AJ, Hurst JA, Firth HV, Knight SJ, Goldenberg A, Saugier-Veber P, Pfundt R, Vissers LE, Destree A, Grisart B, Rooms L, Van der Aa N, Field M, Hackett A, Bell K, Nowaczyk MJ, Mancini GM, Poddighe PJ, Schwartz CE, Rossi E, De Gregori M, Antonacci-Fulton LL, McLellan MD, II, Garrett JM, Wiechert MA, Miner TL, Crosby S, Ciccone R, Willatt L, Rauch A, Zenker M, Aradhya S, Manning MA, Strom TM, Wagenstaller J, Krepischi-Santos AC, Vianna-Morgante AM, Rosenberg C, Price SM, Stewart H, Shaw-Smith C, Brunner HG, Wilkie AO, Veltman JA, Zuffardi O, Eichler EE, de Vries BB. Clinical and molecular delineation of the 17q21.31 microdeletion syndrome. J Med Genet. 2008;45:710–720. doi: 10.1136/jmg.2008.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH, Jr, Dobyns WB, Christian SL. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- Malhotra D, McCarthy S, Michaelson JJ, Vacic V, Burdick KE, Yoon S, Cichon S, Corvin A, Gary S, Gershon ES, Gill M, Karayiorgou M, Kelsoe JR, Krastoshevsky O, Krause V, Leibenluft E, Levy DL, Makarov V, Bhandari A, Malhotra AK, McMahon FJ, Nöthen MM, Potash JB, Rietschel M, Schulze TG, Sebat J. High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron. 2011;72:951–963. doi: 10.1016/j.neuron.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, Thiruvahindrapduram B, Fiebig A, Schreiber S, Friedman J, Ketelaars CE, Vos YJ, Ficicioglu C, Kirkpatrick S, Nicolson R, Sloman L, Summers A, Gibbons CA, Teebi A, Chitayat D, Weksberg R, Thompson A, Vardy C, Crosbie V, Luscombe S, Baatjes R, Zwaigenbaum L, Roberts W, Fernandez B, Szatmari P, Scherer SW. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson JL, Shoemaker M. Intellectual disability and its relationship to autism spectrum disorders. Res Dev Disabil. 2009;30:1107–1114. doi: 10.1016/j.ridd.2009.06.003. [DOI] [PubMed] [Google Scholar]
- McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, Perkins DO, Dickel DE, Kusenda M, Krastoshevsky O, Krause V, Kumar RA, Grozeva D, Malhotra D, Walsh T, Zackai EH, Kaplan P, Ganesh J, Krantz ID, Spinner NB, Roccanova P, Bhandari A, Pavon K, Lakshmi B, Leotta A, Kendall J, Lee YH, Vacic V, Gary S, Iakoucheva LM, Crow TJ, Christian SL, Lieberman JA, Stroup TS, Lehtimäki T, Puura K, Haldeman-Englert C, Pearl J, Goodell M, Willour VL, Derosse P, Steele J, Kassem L, Wolff J, Chitkara N, McMahon FJ, Malhotra AK, Potash JB, Schulze TG, Nöthen MM, Cichon S, Rietschel M, Leibenluft E, Kustanovich V, Lajonchere CM, Sutcliffe JS, Skuse D, Gill M, Gallagher L, Mendell NR, Craddock N, Owen MJ, O’Donovan MC, Shaikh TH, Susser E, Delisi LE, Sullivan PF, Deutsch CK, Rapoport J, Levy DL, King MC, Sebat J Wellcome Trust Case Control Consortium. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, Huang S, Maloney VK, Crolla JA, Baralle D, Collins A, Mercer C, Norga K, de Ravel T, Devriendt K, Bongers EM, de Leeuw N, Reardon W, Gimelli S, Bena F, Hennekam RC, Male A, Gaunt L, Clayton-Smith J, Simonic I, Park SM, Mehta SG, Nik-Zainal S, Woods CG, Firth HV, Parkin G, Fichera M, Reitano S, Lo Giudice M, Li KE, Casuga I, Broomer A, Conrad B, Schwerzmann M, Räber L, Gallati S, Striano P, Coppola A, Tolmie JL, Tobias ES, Lilley C, Armengol L, Spysschaert Y, Verloo P, De Coene A, Goossens L, Mortier G, Speleman F, van Binsbergen E, Nelen MR, Hochstenbach R, Poot M, Gallagher L, Gill M, McClellan J, King MC, Regan R, Skinner C, Stevenson RE, Antonarakis SE, Chen C, Estivill X, Menten B, Gimelli G, Gribble S, Schwartz S, Sutcliffe JS, Walsh T, Knight SJ, Sebat J, Romano C, Schwartz CE, Veltman JA, de Vries BB, Vermeesch JR, Barber JC, Willatt L, Tassabehji M, Eichler EE. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Muhle H, Ostertag P, von Spiczak S, Buysse K, Baker C, Franke A, Malafosse A, Genton P, Thomas P, Gurnett CA, Schreiber S, Bassuk AG, Guipponi M, Stephani U, Helbig I, Eichler EE. Genome-wide copy number variation in epilepsy: Novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6:e1000962. doi: 10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell KJ. The genetics of neurodevelopmental disease. Curr Opin Neurobiol. 2011;21:197–203. doi: 10.1016/j.conb.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Mulle JG, Dodd AF, McGrath JA, Wolyniec PS, Mitchell AA, Shetty AC, Sobreira NL, Valle D, Rudd MK, Satten G, Cutler DJ, Pulver AE, Warren ST. Microdeletions of 3q29 confer high risk for schizophrenia. Am J Hum Genet. 2010;87:229–236. doi: 10.1016/j.ajhg.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, Hosseini M, Behjati F, Haas S, Jamali P, Zecha A, Mohseni M, Püttmann L, Vahid LN, Jensen C, Moheb LA, Bienek M, Larti F, Mueller I, Weissmann R, Darvish H, Wrogemann K, Hadavi V, Lipkowitz B, Esmaeeli-Nieh S, Wieczorek D, Kariminejad R, Firouzabadi SG, Cohen M, Fattahi Z, Rost I, Mojahedi F, Hertzberg C, Dehghan A, Rajab A, Banavandi MJ, Hoffer J, Falah M, Musante L, Kalscheuer V, Ullmann R, Kuss AW, Tzschach A, Kahrizi K, Ropers HH. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, Rieder MJ, Nickerson DA, Bernier R, Fisher SE, Shendure J, Eichler EE. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez Jurado LA, Peoples R, Kaplan P, Hamel BC, Francke U. Molecular definition of the chromosome 7 deletion in Williams syndrome and parent-of-origin effects on growth. Am J Hum Genet. 1996;59:781–792. [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bölte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Bryson SE, Carson AR, Casallo G, Casey J, Chung BH, Cochrane L, Corsello C, Crawford EL, Crossett A, Cytrynbaum C, Dawson G, de Jonge M, Delorme R, Drmic I, Duketis E, Duque F, Estes A, Farrar P, Fernandez BA, Folstein SE, Fombonne E, Freitag CM, Gilbert J, Gillberg C, Glessner JT, Goldberg J, Green A, Green J, Guter SJ, Hakonarson H, Heron EA, Hill M, Holt R, Howe JL, Hughes G, Hus V, Igliozzi R, Kim C, Klauck SM, Kolevzon A, Korvatska O, Kustanovich V, Lajonchere CM, Lamb JA, Laskawiec M, Leboyer M, Le Couteur A, Leventhal BL, Lionel AC, Liu XQ, Lord C, Lotspeich L, Lund SC, Maestrini E, Mahoney W, Mantoulan C, Marshall CR, McConachie H, McDougle CJ, McGrath J, McMahon WM, Merikangas A, Migita O, Minshew NJ, Mirza GK, Munson J, Nelson SF, Noakes C, Noor A, Nygren G, Oliveira G, Papanikolaou K, Parr JR, Parrini B, Paton T, Pickles A, Pilorge M, Piven J, Ponting CP, Posey DJ, Poustka A, Poustka F, Prasad A, Ragoussis J, Renshaw K, Rickaby J, Roberts W, Roeder K, Roge B, Rutter ML, Bierut LJ, Rice JP, Salt J, Sansom K, Sato D, Segurado R, Sequeira AF, Senman L, Shah N, Sheffield VC, Soorya L, Sousa I, Stein O, Sykes N, Stoppioni V, Strawbridge C, Tancredi R, Tansey K, Thiruvahindrapduram B, Thompson AP, Thomson S, Tryfon A, Tsiantis J, Van Engeland H, Vincent JB, Volkmar F, Wallace S, Wang K, Wang Z, Wassink TH, Webber C, Weksberg R, Wing K, Wittemeyer K, Wood S, Wu J, Yaspan BL, Zurawiecki D, Zwaigenbaum L, Buxbaum JD, Cantor RM, Cook EH, Coon H, Cuccaro ML, Devlin B, Ennis S, Gallagher L, Geschwind DH, Gill M, Haines JL, Hallmayer J, Miller J, Monaco AP, Nurnberger JI, Jr, Paterson AD, Pericak-Vance MA, Schellenberg GD, Szatmari P, Vicente AM, Vieland VJ, Wijsman EM, Scherer SW, Sutcliffe JS, Betancur C. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porreca GJ, Zhang K, Li JB, Xie B, Austin D, Vassallo SL, LeProust EM, Peck BJ, Emig CJ, Dahl F, Gao Y, Church GM, Shendure J. Multiplex amplification of large sets of human exons. Nat Methods. 2007;4:931–936. doi: 10.1038/nmeth1110. [DOI] [PubMed] [Google Scholar]
- Potocki L, Bi W, Treadwell-Deering D, Carvalho CM, Eifert A, Friedman EM, Glaze D, Krull K, Lee JA, Lewis RA, Mendoza-Londono R, Robbins-Furman P, Shaw C, Shi X, Weissenberger G, Withers M, Yatsenko SA, Zackai EH, Stankiewicz P, Lupski JR. Characterization of Potocki–Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet. 2007;80:633–649. doi: 10.1086/512864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pregelj P. Gene environment interactions in bipolar disorder. Psychiatr Danub. 2011;23:S91–S93. [PubMed] [Google Scholar]
- Psychiatric GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43:977–983. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risi S, Lord C, Gotham K, Corsello C, Chrysler C, Szatmari P, Cook EH, Jr, Leventhal BL, Pickles A. Combining information from multiple sources in the diagnosis of autism spectrum disorders. J Am Acad Child Adolesc Psychiatry. 2006;45:1094–1103. doi: 10.1097/01.chi.0000227880.42780.0e. [DOI] [PubMed] [Google Scholar]
- Rosenfeld JA, Coppinger J, Bejjani BA, Girirajan S, Eichler EE, Shaffer LG, Ballif BC. Speech delays and behavioral problems are the predominant features in individuals with developmental delays and 16p11.2 microdeletions and microduplications. J Neurodev Disord. 2010;2:26–38. doi: 10.1007/s11689-009-9037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, Mason CE, Bilguvar K, Celestino-Soper PB, Choi M, Crawford EL, Davis L, Wright NR, Dhodapkar RM, DiCola M, DiLullo NM, Fernandez TV, Fielding-Singh V, Fishman DO, Frahm S, Garagaloyan R, Goh GS, Kammela S, Klei L, Lowe JK, Lund SC, McGrew AD, Meyer KA, Moffat WJ, Murdoch JD, O’Roak BJ, Ober GT, Pottenger RS, Raubeson MJ, Song Y, Wang Q, Yaspan BL, Yu TW, Yurkiewicz IR, Beaudet AL, Cantor RM, Curland M, Grice DE, Günel M, Lifton RP, Mane SM, Martin DM, Shaw CA, Sheldon M, Tischfield JA, Walsh CA, Morrow EM, Ledbetter DH, Fombonne E, Lord C, Martin CL, Brooks AI, Sutcliffe JS, Cook EH, Jr, Geschwind D, Roeder K, Devlin B, State MW. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CP, Sabo A, Sakai Y, Crosby J, Muzny D, Hawes A, Lewis L, Akbar H, Varghese R, Boerwinkle E, Gibbs RA, Zoghbi HY. Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum Mol Genet. 2011;20:3366–3375. doi: 10.1093/hmg/ddr243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, Oseroff VV, Albertson DG, Pinkel D, Eichler EE. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Mefford HC, Li K, Baker C, Skinner C, Stevenson RE, Schroer RJ, Novara F, De Gregori M, Ciccone R, Broomer A, Casuga I, Wang Y, Xiao C, Barbacioru C, Gimelli G, Bernardina BD, Torniero C, Giorda R, Regan R, Murday V, Mansour S, Fichera M, Castiglia L, Failla P, Ventura M, Jiang Z, Cooper GM, Knight SJ, Romano C, Zuffardi O, Chen C, Schwartz CE, Eichler EE. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Li Z, Xu Q, Wang T, Li T, Shen J, Zhang F, Chen J, Zhou G, Ji W, Li B, Xu Y, Liu D, Wang P, Yang P, Liu B, Sun W, Wan C, Qin S, He G, Steinberg S, Cichon S, Werge T, Sigurdsson E, Tosato S, Palotie A, Nöthen MM, Rietschel M, Ophoff RA, Collier DA, Rujescu D, Clair DS, Stefansson H, Stefansson K, Ji J, Wang Q, Li W, Zheng L, Zhang H, Feng G, He L. Common variants on 8p12 and 1q24.2 confer risk of schizophrenia. Nat Genet. 2011;43:1224–1227. doi: 10.1038/ng.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinawi M, Schaaf CP, Bhatt SS, Xia Z, Patel A, Cheung SW, Lanpher B, Nagl S, Herding HS, Nevinny-Stickel C, Immken LL, Patel GS, German JR, Beaudet AL, Stankiewicz P. A small recurrent deletion within 15q13.3 is associated with a range of neurodevelopmental phenotypes. Nat Genet. 2009;41:1269–1271. doi: 10.1038/ng.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerville MJ, Mervis CB, Young EJ, Seo EJ, del Campo M, Bamforth S, Peregrine E, Loo W, Lilley M, Perez-Jurado LA, Morris CA, Scherer SW, Osborne LR. Severe expressive-language delay related to duplication of the Williams–Beuren locus. N Engl J Med. 2005;353:1694–1701. doi: 10.1056/NEJMoa051962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, Jonasdottir A, Jonasdottir A, Bjornsson A, Mattiasdottir S, Blondal T, Haraldsson M, Magnusdottir BB, Giegling I, Möller HJ, Hartmann A, Shianna KV, Ge D, Need AC, Crombie C, Fraser G, Walker N, Lonnqvist J, Suvisaari J, Tuulio-Henriksson A, Paunio T, Toulopoulou T, Bramon E, Di Forti M, Murray R, Ruggeri M, Vassos E, Tosato S, Walshe M, Li T, Vasilescu C, Mühleisen TW, Wang AG, Ullum H, Djurovic S, Melle I, Olesen J, Kiemeney LA, Franke B, Sabatti C, Freimer NB, Gulcher JR, Thorsteinsdottir U, Kong A, Andreassen OA, Ophoff RA, Georgi A, Rietschel M, Werge T, Petursson H, Goldstein DB, Nöthen MM, Peltonen L, Collier DA, St Clair D, Stefansson K GROUP. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szafranski P, Schaaf CP, Person RE, Gibson IB, Xia Z, Mahadevan S, Wiszniewska J, Bacino CA, Lalani S, Potocki L, Kang SH, Patel A, Cheung SW, Probst FJ, Graham BH, Shinawi M, Beaudet AL, Stankiewicz P. Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRN A7: Benign or pathological? Hum Mutat. 2010;31:840–850. doi: 10.1002/humu.21284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. 2010. A map of human genome variation from population-scale sequencing. Nature. 467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives HF, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Reider MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012 doi: 10.1038/nature10989. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner EH, Lee C, Ng SB, Nickerson DA, Shendure J. Massively parallel exon capture and library-free resequencing across 16 genomes. Nat Methods. 2009;6:315–316. doi: 10.1038/nmeth.f.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, Olson MV, Eichler EE. Fine-scale structural variation of the human genome. Nat Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- Vacic V, McCarthy S, Malhotra D, Murray F, Chou HH, Peoples A, Makarov V, Yoon S, Bhandari A, Corominas R, Iakoucheva LM, Krastoshevsky O, Krause V, Larach-Walters V, Welsh DK, Craig D, Kelsoe JR, Gershon ES, Leal SM, Dell Aquila M, Morris DW, Gill M, Corvin A, Insel PA, McClellan J, King MC, Karayiorgou M, Levy DL, DeLisi LE, Sebat J. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature. 2011;471:499–503. doi: 10.1038/nature09884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, Stray SM, Rippey CF, Roccanova P, Makarov V, Lakshmi B, Findling RL, Sikich L, Stromberg T, Merriman B, Gogtay N, Butler P, Eckstrand K, Noory L, Gochman P, Long R, Chen Z, Davis S, Baker C, Eichler EE, Meltzer PS, Nelson SF, Singleton AB, Lee MK, Rapoport JL, King MC, Sebat J. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- Walters RG, Jacquemont S, Valsesia A, de Smith AJ, Martinet D, Andersson J, Falchi M, Chen F, Andrieux J, Lobbens S, Delobel B, Stutzmann F, El-Sayed Moustafa JS, Chèvre JC, Lecoeur C, Vatin V, Bouquillon S, Buxton JL, Boute O, Holder-Espinasse M, Cuisset JM, Lemaitre MP, Ambresin AE, Brioschi A, Gaillard M, Giusti V, Fellmann F, Ferrarini A, Hadjikhani N, Campion D, Guilmatre A, Goldenberg A, Calmels N, Mandel JL, Le Caignec C, David A, Isidor B, Cordier MP, Dupuis-Girod S, Labalme A, Sanlaville D, Béri-Dexheimer M, Jonveaux P, Leheup B, Ounap K, Bochukova EG, Henning E, Keogh J, Ellis RJ, Macdermot KD, van Haelst MM, Vincent-Delorme C, Plessis G, Touraine R, Philippe A, Malan V, Mathieu-Dramard M, Chiesa J, Blaumeiser B, Kooy RF, Caiazzo R, Pigeyre M, Balkau B, Sladek R, Bergmann S, Mooser V, Waterworth D, Reymond A, Vollenweider P, Waeber G, Kurg A, Palta P, Esko T, Metspalu A, Nelis M, Elliott P, Hartikainen AL, McCarthy MI, Peltonen L, Carlsson L, Jacobson P, Sjöström L, Huang N, Hurles ME, O’Rahilly S, Farooqi IS, Männik K, Jarvelin MR, Pattou F, Meyre D, Walley AJ, Coin LJ, Blakemore AI, Froguel P, Beckmann JS. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;463:671–675. doi: 10.1038/nature08727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PM, Kim CE, Hou C, Frackelton E, Chiavacci R, Takahashi N, Sakurai T, Rappaport E, Lajonchere CM, Munson J, Estes A, Korvatska O, Piven J, Sonnenblick LI, Alvarez Retuerto AI, Herman EI, Dong H, Hutman T, Sigman M, Ozonoff S, Klin A, Owley T, Sweeney JA, Brune CW, Cantor RM, Bernier R, Gilbert JR, Cuccaro ML, McMahon WM, Miller J, State MW, Wassink TH, Coon H, Levy SE, Schultz RT, Nurnberger JI, Haines JL, Sutcliffe JS, Cook EH, Minshew NJ, Buxbaum JD, Dawson G, Grant SF, Geschwind DH, Pericak-Vance MA, Schellenberg GD, Hakonarson H. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44:660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- Weinberger DR. From neuropathology to neurodevelopment. Lancet. 1995;346:552–557. doi: 10.1016/s0140-6736(95)91386-6. [DOI] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, Platt OS, Ruderfer DM, Walsh CA, Altshuler D, Chakravarti A, Tanzi RE, Stefansson K, Santangelo SL, Gusella JF, Sklar P, Wu BL, Daly MJ Autism Consortium. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Willatt L, Cox J, Barber J, Cabanas ED, Collins A, Donnai D, FitzPatrick DR, Maher E, Martin H, Parnau J, Pindar L, Ramsay J, Shaw-Smith C, Sistermans EA, Tettenborn M, Trump D, de Vries BB, Walker K, Raymond FL. 3q29 Microdeletion syndrome: Clinical and molecular characterization of a new syndrome. Am J Hum Genet. 2005;77:154–160. doi: 10.1086/431653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodberry KA, Giuliano AJ, Seidman LJ. Premorbid IQ in schizophrenia: A meta-analytic review. Am J Psychiatry. 2008;165:579–587. doi: 10.1176/appi.ajp.2008.07081242. [DOI] [PubMed] [Google Scholar]
- Xu B, Roos JL, Dexheimer P, Boone B, Plummer B, Levy S, Gogos JA, Karayiorgou M. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat Genet. 2011;43:864–868. doi: 10.1038/ng.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue WH, Wang HF, Sun LD, Tang FL, Liu ZH, Zhang HX, Li WQ, Zhang YL, Zhang Y, Ma CC, Du B, Wang LF, Ren YQ, Yang YF, Hu XF, Wang Y, Deng W, Tan LW, Tan YL, Chen Q, Xu GM, Yang GG, Zuo XB, Yan H, Ruan YY, Lu TL, Han X, Ma XH, Wang Y, Cai LW, Jin C, Zhang HY, Yan J, Mi WF, Yin XY, Ma WB, Liu Q, Kang L, Sun W, Pan CY, Shuang M, Yang FD, Wang CY, Yang JL, Li KQ, Ma X, Li LJ, Yu X, Li QZ, Huang X, Lv LX, Li T, Zhao GP, Huang W, Zhang XJ, Zhang D. Genome-wide association study identifies a susceptibility locus for schizophrenia in Han Chinese at 11p11.2. Nat Genet. 2011;43:1228–1231. doi: 10.1038/ng.979. [DOI] [PubMed] [Google Scholar]