

Abstract
Mitochondrial dysfunction is becoming a pivotal target for neuroprotective strategies following contusion spinal cord injury (SCI) and the pharmacological compounds that maintain mitochondrial function confer neuroprotection and improve long-term hindlimb function after injury. In the current study we evaluated the efficacy of cell-permeating thiol, N-acetylcysteineamide (NACA), a precursor of endogenous antioxidant glutathione (GSH), on mitochondrial function acutely, and long-term tissue sparing and hindlimb locomotor recovery following upper lumbar contusion SCI. Some designated injured adult female Sprague-Dawley rats (n=120) received either Vehicle or NACA (75, 150, 300 or 600 mg/kg) at 15min and 6hrs post-injury. After 24hr the total, synaptic, and non-synaptic mitochondrial populations were isolated from a single 1.5cm spinal cord segment (centered at injury site) and assessed for mitochondrial bioenergetics. Results showed compromised total mitochondrial bioenergetics following acute SCI that was significantly improved with NACA treatment in a dose-dependent manner, with maximum effects at 300 mg/kg (n=4/group). For synaptic and non-synaptic mitochondria, only 300 mg/kg NACA dosage showed efficacy. Similar dosage (300mg/kg) also maintained mitochondrial GSH near normal levels. Other designated injured rats (n=21) received continuous NACA (150 or 300mg/kg/day) treatment starting at 15min post-injury for one week to assess long-term functional recovery over 6 weeks post-injury. Locomotor testing and novel gait analyses showed significantly improved hindlimb function with NACA that were associated with increased tissue sparing at the injury site. Overall, NACA treatment significantly maintained acute mitochondrial bioenergetics and normalized GSH levels following SCI, and prolonged delivery resulted in significant tissue sparing and improved recovery of hindlimb function.
Keywords: N-acetylcysteineamide (NACA), neuroprotective agent, gait analysis, therapeutics, contusion spinal cord injury
Introduction
Traumatic spinal cord injury (SCI) induces a cascade of secondary pathophysiological events that results in neuronal death at and around the site of injury, with accompanying loss of motor and sensory functions. Mitochondrial dysfunction is thought to be one of the major cause-effect factors for these secondary events (Benedict et al., 2012; Dumont et al., 2001; Huang et al., 2012; Patel et al., 2012; Patel et al., 2010; Patel et al., 2009; Sullivan et al., 2007; Teng et al., 2004), and thus can serve as a pivotal target for pharmacological strategies to foster neuroprotection after SCI. Mitochondria are known as the “powerhouse” of the cell which generates cellular energy in the form of ATP. On the other hand, they are also a major site for free radical production as well as a primary target for free radical attack, and we have documented the progressive nature of mitochondrial dysfunction and their oxidative damage over 24 hr post-SCI (Sullivan et al., 2007). Importantly, we have shown that administration of pharmacological compounds that maintain mitochondrial function confer neuroprotection and significantly improve long-term hindlimb function after injury (Patel et al., 2012; Patel et al., 2010).
In the current study we assessed the neuroprotective efficacy of a novel thiol-containing antioxidant, N-acetylcysteineamide (NACA). NACA is a modified form of its parent compound N-acetylcysteine (NAC) that is a precursor of the most abundant endogenous antioxidant, glutathione (GSH) (De Flora et al., 1991). Depletion of GSH has been associated conditions such as with aging, diabetes mellitus, inflammation, and neurodegenerative diseases as well as CNS injuries (Drake et al., 2002; Droge, 2002; Kamencic et al., 2001; Thomas and Mallis, 2001). Thus, thiol-containing compounds have received growing attention due to the profound role of intracellular GSH in cellular antioxidant defense systems. Accordingly, NAC supplementation has been shown to have beneficial effects on oxidative stress induced diseases and clinical disorders (Arent et al., 2012; Foltz et al., 2012; Holdiness, 1991; Kelly, 1998; Parcell, 2002). Experimental evidence suggests that NAC facilitates biosynthesis of intracellular GSH by reducing extracellular cystine to cysteine (Issels et al., 1988). NAC, itself, can serve as a potent free radical scavenger due to its nucleophilic interactions with reactive oxygen species (ROS) (Aruoma et al., 1989; Ozaras et al., 2003). However, the carboxyl group of NAC loses its proton at physiological pH, which makes the compound negatively charged and reduces its permeability through biological membranes. Thus, bioavailability of NAC is very low, efforts were made to synthesize a modified compound NACA (an amide form of NAC) that possesses a neutral charge at physiological pH (Grinberg et al., 2005; Offen et al., 2004). NACA is more membrane permeable than NAC and reacts with the oxidized form of glutathione (GSSG) to generate GSH, possibly by exchanging disulfide with GSSG (Grinberg et al., 2005). NACA has been shown to cross the blood-brain barrier, chelate copper, scavenge free radicals and attenuate experimental autoimmune encephalomyelitis induced by myelin oligodendrocyte glycoprotein inoculation (Offen et al., 2004). Moreover, NACA protects mammalian cells in vitro from oxidants such as HIV proteins, glutamate and beta amyloid toxicity (Bartov et al., 2006; Penugonda et al., 2005; Price et al., 2006).
Based on these antioxidant properties of NACA and our reports that maintenance of mitochondrial function following SCI is neuroprotective (Patel et al., 2012; Patel et al., 2010), we herein investigated the protective effects of NACA on the mitochondrial GSH pool, acute mitochondrial function, long-term tissue sparing, and hindlimb locomotor function following upper lumbar (L1/L2) contusion SCI in adult rats. Importantly, we employed refined gait analyses to assess the functional recovery in addition to a conventional locomotor rating scale. We also report a novel method for the isolation of synaptic (neuronal), non-synaptic (neuronal somata and glia) and mixed population of synaptic + non-synaptic mitochondria from a single 1.5 cm of thoracolumbar spinal cord segment to assess the effect of NACA treatment on their bioenergetics using the Seahorse Bioscience XF24 extracellular flux analyzer and measuring activities of mitochondrial enzyme complexes: NADH dehydrogenase (Complex I), cytochrome c oxidase (Complex IV) and pyruvate dehydrogenase (PDHC).
Materials and Methods
Spinal cord injury and treatments
Female Sprague-Dawley rats (n=141, see Table 1 for detail) (Harlan Labs, IN) weighing 225–250 g were housed in the animal facility, Biomedical & Biological Science Research Building, University of Kentucky and allowed ad libitum access to water and food. All animal procedures were approved by the Institutional Animal Care and Use Committee, University of Kentucky and according to NIH guidelines. Prior to surgeries all the animals were randomly assigned into different experimental groups such that on any given day the surgeon and person administering drug or vehicle were blinded to treatment. Rats were anesthetized with Ketamine (80 mg/kg, Fort Dodge Animal Health, Fort Dodge, IA) and Xylazine (10 mg/kg, Lloyd Laboratories, Shenandoah, IA). A dorsal laminectomy was performed at the 12th thoracic vertebra to expose the first and second lumbar (L1/L2) spinal cord level as described earlier (Patel et al., 2012), after which spinal cord contusions (250 kdyn) were performed with an Infinite Horizon impactor device (PSI, Lexington, KY). After injury the wounds were irrigated with saline, the muscles sutured together in layers with 3–0 Vicryl (Ethicon, Inc., Somerville, NJ), and the skin openings closed with wound clips (Stoelting Co., Wood Dale, IL). Hydrogen peroxide and betadine were used to clean the wound area and animals injected (s.c.) with pre–warmed lactated Ringer’s solution (10 ml split into 2 sites bilaterally) and Cefazolin (33.3 mg/kg) before the rats were returned to their cages with food and water ad libitum. As soon as rats regained consciousness, Buprenorphine-HCl (0.03 mg/kg; Reckitt Benckiser Pharmaceuticals Inc. Richmond, VA) was administered (s.c.) every 12 hr for either 24hr (mitochondrial experiments) or 72 hr (long-term behavioral experiments). Injured rats used for acute mitochondrial OCR (24 hr survival times) were administered (i.p.) either Vehicle (saline) or NACA (75, 150, 300 or 600 mg/kg bodyweight; NACA was a gift from Sentient Lifesciences, New York, NY 10021) 15 min post- injury followed by a corresponding booster at 6 hr. For assessment of activities of mitochondrial enzyme complexes, injured animals were injected (i.p.) with either Vehicle or NACA (75 or 300 mg/kg bodyweight) 15 min post-injury followed by a corresponding booster at 6 hr. For long- term behavioral assessments, injured rats were treated (i.p.) with either Vehicle, 150 or 300 mg/kg body weight NACA at 15 min post-injury followed by immediate implantation (s.c.) of primed osmotic mini-pumps (Model 2ML1; Alzet, Cupertino, CA) to continuously deliver 150 or 300 mg/kg/day for 7 days post-injury; after which rats were anesthetized under isoflurane for pump removal. NACA used for the current study was provided by David Pharmaceuticals, New York, NY.
Table 1.
Number of animals used for the study
| Outcome Measure | Beginning Number of Animals | Number of experimental Groups | Final Number per Group |
|---|---|---|---|
| Mitochondrial OCR (24hr) | 24 | 6 | 4 |
| Mitochondrial Enzyme Activity (24hr) | 72 | 4 | 6* |
| GSH − Bolus (24hr) | 12 | 4 | 3 |
| GSH − Bolus + continuous (24hr) | 12 | 3 | 4 |
| Long-Term Behavior Experiment 2 | 21 | 3 | 5–7** |
Mitochondria isolated from 3 spinal cord segments were pooled in order to get sufficient protein to measure activities of enzyme complexes.
Due to post-surgical complications, one rat from Vehicle-treated group died and data for 1 rat was excluded from data analysis due to >2 Standard Deviation difference from group mean in histological assessment using ANOVA and student newman-keuls post hoc analysis.
Administration of osmotic mini-pimps
For continuous delivery of NACA for glutathione and long- term behavioral assessments, injured rats were treated (i.p.) with either Vehicle, 150 or 300 mg/kg body weight NACA at 15 min post-injury followed by immediate sterile implantation (s.c.) of primed osmotic mini-pumps (Model 2ML1; Alzet, Cupertino, CA) to continuously deliver 150 or 300 mg/kg/day for 24 hr or 7 days post-injury. Briefly, a small subcutaneous pocket was created on the animals back by carefully separating the skin from the fascia layer and an osmotic pump was inserted with the delivery port facing rostral towards the injury site. The muscles layers and skin openings were closed as described above. After the 7-day delivery period, rats were re-anesthetized under isoflurane for pump removal.
Isolation of total, synaptic and non-synaptic mitochondria from a single spinal cord segment
All three mitochondrial populations were isolated from a single spinal cord segment by modifying our earlier described methods (Patel et al., 2009; Sullivan et al., 2007). At 24 hr post- injury, animals were decapitated and the thoraco-lumbar spinal cords from naïve as well as injured animals were rapidly dissected out and placed in an ice cold dissecting plate containing isolation buffer with 1mM EGTA (215mM mannitol, 75mM sucrose, 0.1% BSA, 20mM HEPES, 1mM EGTA, and pH adjusted to 7.2 with KOH). The following protocol is described for each sample and the steps are repeated for each animal. A 1.5 cm spinal cord segment centered on injury site was homogenized in 2 ml of ice cold isolation buffer (with EGTA). The homogenate was centrifuged at 1,400 × g for 3 min at 4°C. Supernatant was transferred to a new 2ml tube (S1, containing mitochondria and cytosol). The pellet was resuspended in 2ml isolation buffer and centrifuged at 1,400 × g for 3 min and this supernatant was transferred to a new tube (S2, containing mitochondria and cytosol) and the pellet was discarded. Both supernatant fractions (S1 & S2) were then topped off with isolation buffer and centrifuged at 13,000 × g for 10 min. The resultant two pellets (containing non-synaptic mitochondria and synaptosomes) were pooled and resuspended in 800 μl isolation buffer (with EGTA). This resuspension is the crude mitochondrial fraction that is used in later steps to isolate total, synaptic, and non-synaptic mitochondrial populations, as well as to measure GSH content.
Isolation of Total Mitochondria
Based on pilot experiments, 200 μl of the crude mitochondrial suspension (~25%) was burst in a nitrogen cell disruption bomb at 4°C for 10 min at 1200 psi (model 4639; Parr Instrument Co., Moline, IL) in order to release synaptic mitochondria from synaptosomes. This was used for isolation of total (mix population of synaptic and non-synaptic) mitochondria. The bombed crude total mitochondrial suspension was then placed on top of a discontinuous Ficoll gradient (7.5%, 10%), and centrifuged at 100,000 × g for 30 min using an ultracentrifuge (Beckman Coulter, Fullerton, CA). The pellet, consisting of purified total mitochondria, was resuspended in 600 ul of EGTA-free isolation buffer and centrifuged at 10,000 × g for 10 min at 4°C to wash off EGTA. The final pellet was then resuspended in EGTA-free isolation buffer (~20ul).
Isolation of Non-Synaptic and Synaptic Mitochondria
The remaining 600 μl of the crude mitochondrial suspension was placed on top of a discontinuous Ficoll gradient (7.5%, 10%) as described above and centrifuged at 100,000 × g for 30 min using an ultracentrifuge to isolate synaptosomes and non-synaptic mitochondria. Centrifugation separates non-synaptic mitochondria into the pellet and synaptosomes (containing synaptic mitochondria) into the interface between Ficoll layers. The synaptosomal fraction was carefully removed from the interface of Ficoll gradient and placed in a 2 mL centrifuge tube. This fraction was diluted 1:4 with isolation buffer and centrifuged at 13,000 × g for 10 min. The resultant pellet was resuspended in 350 μl of isolation buffer with EGTA and burst in a nitrogen cell disruption bomb at 4°C for 10 min at 1200 psi to release synaptic mitochondria from synaptosomes. The synaptosomal fractions were again run through Ficoll purification steps to get a purified synaptic mitochondria pellet. This pellet was resuspended in 600 ul of EGTA-free isolation buffer and centrifuged at 10,000 × g for 10 min at 4°C to washout EGTA. The resultant EGTA-free synaptic mitochondrial pellet was resuspended in EGTA-free isolation buffer (~20μl). The pellet obtained from the first Ficoll centrifugation of the crude mitochondrial suspension, consisting of non-synaptic mitochondria, was resuspended in 600 ul of EGTA-free isolation buffer and centrifuged at 10,000 × g for 10 min at 4°C. The resultant EGTA-free non- synaptic mitochondrial pellet was resuspended in EGTA-free isolation buffer (~20μl). All samples were kept on ice throughout the isolation process. The protein concentration was determined for each population of mitochondria with the BCA protein assay kit (Thermo Scientific, Rockford, IL) by measuring absorbance at 560 nm with a Biotek Synergy HT plate reader (Winooski, Vermont). Note: Isolation and measurements of OCRs were completed as quickly as possible after dissecting out the tissues (within 5–6 hrs).
In general, from a total of 800 μl crude mitochondrial fraction we obtain 1) ~60 μg of Total mitochondria from 200μl, 2) ~60μg of Synaptic and ~100 μg of Non-Synaptic mitochondria from the remaining 600 μl. Compared to the Total mitochondrial isolation procedure requiring only one centrifugation, there is about 10% mitochondrial protein loss during isolation of synaptic and non-synaptic mitochondria, which requires an additional Ficoll gradient centrifugation step. It is unclear if the loss of mitochondria is in equal proportion across synaptic and non-synaptic mitochondrial populations.
Measurements of mitochondrial function
Mitochondrial Respiration
Respiration of all three mitochondrial populations were measured in terms of mitochondrial oxygen consumption rate (OCR) using a Seahorse Bioscience XF24 extracellular flux analyzer as described previously with slight modifications (Sauerbeck et al., 2011). Briefly, 24 hr prior to each experiment, a 24 well dual-analyzer sensor cartridge (Seahorse Bioscience) was incubated with 1ml/well of XF Calibrant solution (Seahorse Bioscience) at 37°C in a CO2-free incubator (Seahorse Bioscience). On the day of the experiment, either A) pyruvate plus malate plus ADP, B) oligomycin, C) FCCP, or D) rotenone plus succinate were added in ports A–D, respectively, in the Seahorse Flux Pak cartridges to yield final concentrations of 5mM (pyruvate), 2.5mM (malate), 1mM (ADP), 1μg/ml (oligomycin), 3μM (FCCP), 100nM (rotenone), and 10mM (succinate) and placed into the Seahorse XF24 Flux Analyzer for automated calibration. During the sensor calibration, 50 μl volume of respiration buffer containing 5 μg of mitochondrial protein was added to each well of XF24 V7 cell culture microplates and centrifuged at 2000 rpm for 4 min at 4°C. This was followed by addition of 450 μl of respiration buffer (125 mM KCl, 2 mM MgCl2, 2.5 mM KH2PO4, 20 mM HEPES and 0.1% BSA, pH 7.2) at 37°C (gently added to each well) to make the final assay volume 500 μl per well. Plates were immediately placed into the calibrated seahorse XF24 flux analyzer for assessment of mitochondrial OCR in the presence of substrates and inhibitors in respective ports. The OCR data were analyzed using Excel software package (Microsoft), point by point rates were generated using the AKOS algorithm written by AKOS Gerencser for Seahorse Bioscience. For State III and State V–Complex I, the “middle point” (average) of OCR was generated from 4 time-dependent point-by-point rates for each well sample. Then, three independent middle points were averaged for each sample to generate n=1 per sample.
Mitochondrial Enzyme Assays
Activities of mitochondrial enzymes Complex I (NADH dehydrogenase) and Complex IV (cytochrome c oxidase) were measured using 96 well plate reader as described previously (Patel et al., 2009; Smith, 1955). Briefly, mitochondria were freeze-thawed for three cycles in 10 mM phosphate buffer (pH 7.4) and 8 μg of isolated mitochondrial protein was used. Complex I (NADH dehydrogenase) assay was performed in 25 mM KPO4 buffer (pH 7.2) containing mitochondrial protein (8 μg), 5 mM MgCl2, 1 mM KCN, 1 mg/ml BSA, and 150 μM NADH at 30°C. The reaction was initiated by addition of coenzyme Q-1 (50 μM). Decreased absorbance at 340 ηm was monitored. The assay was also performed in the presence of rotenone (10 μM) to determine the rotenone-insensitive and the rotenone-sensitive complex I enzyme activity. Complex IV (cytochrome c oxidase) activity was measured by monitoring a decrease in absorbance of reduced cytochrome c at 550 ηm. Pyruvate dehydrogenase complex (PDHC) assay was performed as described with modifications (Opii et al., 2007; Patel et al., 2010; Starkov et al., 2004). Briefly, PDHC activity was measured in a cocktail containing 8 μg mitochondria, 50mM KCl, 10mM HEPES, pH 7.4, 0.3 mM thiamine pyrophosphate (TPP), 10 μM CaCl2, 0.2 mM MgCl2, 5 mM pyruvate, 1 μM rotenone and 200 μM NAD+. The reaction was started with the addition of 0.14 mM CoASH. Decrease in NAD+ content was measured at 460 ηm emission after excitation at 346 ηm.
Quantification of Glutathione
Quantification of reduced form of glutathione (GSH), oxidized form of glutathione (GSSG) and total GSH (GSH + GSSG) was performed on the crude mitochondrial fraction (non-synaptic + synaptic mitochondria + synaptosomal membrane) using “Glutathione (total) detection kit” (Enzo Life Sciences, Farmingdale, NY, USA). Briefly, after nitrogen bombing at 1200 psi for 10 min at 4°C to break synaptosomes, 1 mg of crude mitochondrial fractions from each experimental group was added to chilled 400 μl 5% (w/v) m-phosphoric acid to precipitate proteins followed by rigorous vortex and sonication 3 times for 15 sec every 10 min before storing on ice between sonications. All the samples were then centrifuged at 15,000 × g for 15 min at 4°C. Supernatant (GSH extract) was collected and kept at 4°C for immediate assay or stored at −80°C for later use. For total GSH and GSSG estimation, 25 μl of GSH extracts were used and the reduced form of GSH was calculated as: Reduced GSH = Total GSH − GSSG. All the values are expressed as ηmols/mg protein.
Behavioral assessments
The Basso, Beattie, Bresnahan Locomotor Rating Scale (BBB LRS) was used to test the hindlimb function of injured rats after L1/L2 SCI (Basso et al., 1996; Patel et al., 2012). Testing began 2 days after SCI and continued weekly for up to 6 weeks post-injury. Assessments were made by individuals blinded to treatment groups.
Gait recording and analysis
Following the final BBB assessments all the rats were subjected to gait analysis (Kuerzi et al., 2010). Using a single high speed camera (Basler scA640), ventral-view videos were taken of each animal while passing across a customized transparent plexi-glass walkway. These videos, which allowed for viewing of forelimb and hindlimb paw placement during locomotion, were recorded at 60Hz and analyzed using MaxTraq and MaxMate software (Innovision Systems, MI). Six step cycles were digitized for each animal, specifically by marking when each foot was in contact with the plexiglass in a frame-by-frame manner. In addition, hindlimb dorsal steps were marked when observed.
From this data, it is possible to derive several refined measures of over ground locomotion, including the Regularity Index (RI) (Koopmans et al., 2005), Coordinated Pattern Index (CPI), Plantar Stepping Index (PSI), and the percentage of hindlimb steps that are dorsal. Briefly, the RI was developed as an objective measure of coordination which improves upon the more traditional BBB LRS which assesses forelimb-hindlimb coordination. It is calculated as: , where NCPP is the number of correct plantar placement patterns, and TNPP is the total number of plantar placement patterns. Any one of four typical stepping patterns observed in naïve animals would be considered a correct pattern. Thus, a fully coordinated animal will achieve 100% RI. Notably, the RI excludes dorsal steps in the analysis and therefore is not ideal for comprehensively assessing the coordination of stepping pattern in cases where plantar placement is not achieved.
CPI, a second measure of coordination which we developed, includes dorsal steps while also assessing the stepping pattern in a rolling fashion rather than by blocking every four foot-falls. Thus, the CPI will detect double stepping that may not be detected with the RI. Furthermore, the inclusion of dorsal steps provides a more comprehensive measurement of coordination as well as plantar vs. dorsal stepping. Like the RI, the CPI is expressed as a percentage where a fully coordinated animal will achieve a measure of 100%. Importantly, the CPI does not provide a direct indication of an animal’s ability to achieve plantar stepping. The percentage of dorsal steps is calculated as: . This provides insight into how any differences between RI and CPI can be attributed to dorsal stepping.
A third index of locomotor function we developed, the PSI, was calculated as: . This ratiometric parameter provides additional insight into the coordination between forelimb and hindlimb steps. A naïve animal would be expected to have a PSI of 100%, meaning equal frequencies of hindlimb and forelimb plantar steps. Notably, this ratio cannot exceed a value of 1.0 due to the fact that L1/L2 injured animals are unlikely to achieve more hindlimb plantar steps than forelimb plantar steps. A naïve animal would be expected to have a PSI of 100%, meaning equal frequencies of hindlimb and forelimb plantar steps. Notably, this ratio cannot exceed a value of 1.0 due to the fact that L1/L2 injured animals are unlikely to achieve more hindlimb plantar steps than forelimb plantar steps. Additionally, an animal with perfect coordination of forelimbs and hindlimbs would achieve 100% PSI.
Magnetic Resonance Imaging (MRI)
A 7T Bruker/Siemens Clinscan MRI scanner equipped with a 4-channel rat brain coil was used to capture magnetic resonance images (MRI). At 42 DPI, under continuous isoflurane anesthesia, animals were placed onto the coil in the supine position with the coil centered at the injury epicenter. After inserting the animal and coil into the scanner body, localizers in the three orthogonal planes were used to focus and homogenize the magnetic field around the thoracolumbar spine of the rat. Following localization, T2-weighted images were acquired in the sagittal plane yielding 11 subsequent slices with a field-of-view (FOV) of 30 mm × 22.5 mm, interpolated to a pixel resolution of 256 × 192. The imaging parameters were: Excitation Time (TE) = 50 ms, Relaxation Time (TR)= 2500 ms, Field of View (FOV) = 30 mm × 22.5mm, with the phase encoding direction set to Anterior-Posterior, and a total acquisition time of 14 minutes. MRI images were generated using Syngo Medical Imaging software (Siemens AG, Germany).
Spinal cord tissue processing
At 6 weeks post-injury, after terminal BBB LRS testing, rats were perfused and spinal cords dissected as described (Patel et al., 2012). Briefly, rats were overdosed with 0.2 ml Fatal- Plus solution containing sodium pentobarbital (390 mg/ml stock) (Vortech Pharma Ltd., Dearborn, MI, USA) and transcardially perfused with 0.1 M phosphate-buffered saline (PBS, pH 7.4) followed by 4% paraformaldehyde (PFA) in 0.1 M PBS. Each spinal cord from the L1/L2 injury cohorts was transected at the rostral T11 spinal root and a 30-mm segment of spinal cord extending caudally to L6/S1 was immediately dissected. All the dissected cords were post fixed in 4% PFA/PBS for 1–4 h at 4°C depending on quality of fixation. This was followed by overnight washing with 0.1 M PB at 4 C to remove excess 4% PFA before cryoprotecting in 20% sucrose/PBS at 4°C prior to embedding tissues as described (Rabchevsky et al., 2001). Frozen spinal cords were then serially cryosectioned at 20 μm in the coronal plane and processed for histological assessment of tissue sparing.
Histological Assessment
Transaxial sections of injured spinal cord from all the experimental groups were stained for Eriochrome Cyanine-Cresyl Violet (EC-CV) using our protocol that differentiates both white matter and neuropil (gray matter) as described earlier (Rabchevsky et al., 2002; Rabchevsky et al., 2007). Based on positive staining for myelin or normal cytoarchitecture of gray matter (GM), all the EC-CV-stained sections were assessed for spared tissue using Nikon microscope (Nikon Corporation, Tokyo, Japan) and Scion Imaging analysis software (Scion Corporation, Frederick, MD, USA). From a series of evenly spaced sections (1 mm apart) centered on the injury site, the volumes of injured/spared gray and white matter tissues were calculated using the Cavalieri method (Michel and Cruz-Orive, 1988; Rabchevsky et al., 2002). The % tissue sparing at the injury epicenter was calculated by dividing the circumferential area of spared tissue of the spinal cord section minus the area of injured tissue by the total circumferential area of the spinal cord, multiplied by 100 (Rabchevsky et al., 1999; Rabchevsky et al., 2000). All histological analyses were assessed blindly with respect to treatment.
Statistical Analysis
Data from mitochondrial preparations as well as histological and gait assessments were analyzed by an analysis of variance among the groups (ANOVA) using Graph Pad Prism 5. When appropriate, post hoc comparisons were made using Student-Newman-Keuls. Long-term behavioral (BBB LRS) analyses were carried out using a repeated measures ANOVA over the experimental period and and Student-Newman-Keuls post hoc test. For all data analysis, significance was set at p < 0.05.
Results
Effects of acute NACA treatment on mitochondrial bioenergetics
In the current study, assessments of mitochondrial function were carried out using 1) mitochondrial respiration, 2) activities of mitochondrial enzyme complexes, and 3) quantification of glutathione content as outcome measures. Mitochondrial respiration following SCI with or without NACA treatment was carried out by measuring mitochondrial oxygen consumption rate using the Seahorse Biosciences XF24 Flux Analyzer. Moreover, we modified our previous protocols (Sullivan et al., 2007, Patel et al., 2009) to isolate all three populations of mitochondria (total, synaptic and non-synaptic) from a single 1.5 cm spinal cord and assessed their respiration 24 hr after injury. After extensive standardization, the mitochondrial protein yield obtained from a single 1.5 cm peace of naïve or injured spinal cord was ~60ug for total and synaptic mitochondria each and ~100μg for non-synaptic mitochondria; this was sufficient to run the oxygen consumption assay in triplicate using the Seahorse Biosciences XF24 Flux Analyzer.
Effects of NACA treatment on total, synaptic and non-synaptic mitochondrial respiration
Contusion SCI was found to significantly reduce mitochondrial respiration for all mitochondrial populations at 24 hr after injury compared to Naïve rats. A significant difference between groups was noted in ADP phosphorylation rate (State III) for total mitochondria [F(5,18) = 12.763, p<0.0001], synaptic mitochondria [F(5,18) = 4.419, p<0.01] and non-synaptic mitochondria [F(5,18) = 7.067, p<0.001] (Fig. 1A–C), as well as for complex I-driven maximum electron transport (State V-complex I) for total mitochondria [F(5,18) = 15.745, p<0.0001], synaptic mitochondria [F(5,18) = 6.699, p<0.001] and non-synaptic mitochondria [F(5,18) = 7.493, p<0.001] at 24 hr post-SCI for all the injury groups compared to Naïve. Post-hoc analysis revealed that for total mitochondria, treatment with NACA at 75, 150 or 300 mg/kg i.p. significantly (p<0.05) maintained mitochondrial respiration in a dose-dependent manner with maximum effects at 300 mg/kg dosage; NACA at 600 mg/kg was not effective in maintaining respiration (Fig. 1A). Notably, for synaptic and non-synaptic mitochondria, only 300 mg/kg NACA showed a significant (p<0.05) increase in mitochondrial respiration rates compared to Vehicle treatment (Figs. 1B and C).
Fig. 1.

Effects of NACA treatment on mitochondrial Oxygen Consumption Rate (OCR) following contusion SCI. Comparisons of the OCR in terms of nmol oxygen/min for (A) total (mixed population of synaptic and non-synaptic, (B) synaptic and (C) non-synaptic mitochondria that were assessed 24 hrs post-injury. 5μg mitochondrial protein was used for each sample. SCI significantly decreases ADP phosphorylation rate (State III) and State V-complex I (complex I- driven maximum electron transport) in all three population of mitochondria compared to Naïve (Panels A–C). Compared to Vehicle, treatment with NACA at 75, 150 or 300 mg/kg i.p. significantly maintained respiration for total mitochondria, in a dose-dependent manner (Panel A). For synaptic and non-synaptic mitochondria only 300mg/kg NACA significantly increase mitochondrial OCR compared to Vehicle (Panels B & C, respectively). Bars are group means ± SEM, n=4/group. *p<0.05 compared to Naïve; #p<0.05 compared to Vehicle.
Effects of NACA treatment on total, synaptic and non-synaptic mitochondrial enzyme activities
To complement the mitochondrial respiration results, we also assessed the effects of NACA treatment following SCI on activities of individual enzyme complexes that are involved in mitochondrial energy production. Mitochondria isolated from 3 spinal cord segments were pooled to generate one sample in order to get sufficient mitochondrial protein. Compared to the Naïve group, SCI significantly (p<0.05) decreased activity of NADH dehydrogenase (Complex I), cytochrome oxidase (Complex IV), and pyruvate dehydrogenase (PDHC) complexes in mitochondria of all three populations (Figs. 2A–C). One way ANOVA showed a significant difference between the experimental groups for 1) Complex I activity for total mitochondria [F(3,20) = 19.126, p<0.0001], synaptic mitochondria [F(3,20) = 14.551, p<0.0001] and non- synaptic mitochondria [F(3,20) = 12.629, p<0.0001], 2) Complex IV activity for total mitochondria [F(3,20) = 16.56, p<0.0001], synaptic mitochondria [F(3,20) = 8.551, p<0.001] and non-synaptic mitochondria [F(3,20) = 12.967, p<0.0001] and 3) PDHC activity for total mitochondria [F(3,20) = 23.528, p<0.0001], synaptic mitochondria [F(3,20) = 8.95, p<0.001] and non-synaptic mitochondria [F(3,20) = 13.707, p<0.0001]. Post-hoc analysis showed that treatment with 75 and 300 mg/kg NACA significantly (p<0.05) maintained these enzymatic activities to normal levels for total mitochondria (Fig. 2A). Similar to total mitochondria, activities of all enzyme complexes significantly (p<0.05) decreased in synaptic as well as in non- synaptic mitochondria after SCI. For synaptic mitochondria, treatment with NACA significantly improved complex I activity at both dosages, whereas complex IV and PDHC activities increased only with 300 mg/kg dosage (Fig. 2B). For non-synaptic mitochondria, NACA at 75 mg/kg significantly (p<0.05) improved only Complex IV and PDHC activities, whereas 300 mg/kg NACA completely restored activities of all three enzyme complexes to naïve levels (Fig. 2C).
Fig. 2.

Effects of NACA treatment on the activities of mitochondrial enzyme complexes following contusion SCI. Comparison among the activities of mitochondrial enzyme complexes in terms of nmols/min/mg protein for (A) total, (B) synaptic and (C) non-synaptic mitochondria. Compared to the Naïve group, SCI significantly decreased activity of NADH dehydrogenase (Complex I), cytochrome oxidase (Complex IV), and pyruvate dehydrogenase (PDHC) complexes in total mitochondria. Treatment with 75 and 300 mg/kg NACA significantly maintained these activities near normal levels in a dose dependent manner (A). Similar to total mitochondria, activities of all enzyme complexes significantly decreased in synaptic mitochondria after SCI. Treatment with 75mg NACA significantly increased only complex I activity, whereas, 300mg/kg NACA significantly improved activities of all three enzymes (B). For non-synaptic mitochondria, treatment with 75mg/kg NACA significantly improved Complex IV and PDHC activities whereas 300mg/kg NACA significantly improved activities of all three enzymes (C). Bars are group mean ± SEM, n=3/group. *p<0.05 compared to Naive and # p<0.05 compared to Vehicle-treated injured group.
Effects of NACA treatment on mitochondrial glutathione content
To assess whether NACA acts as a precursor of GSH, we measured reduced form (GSH), oxidized form (GSSG) and total form (GSH + GSSG) levels in total mitochondria isolated from different experimental groups. However, GSH content was not detectable in these samples due to low mitochondrial protein. Thus, we assessed GSH contents in crude mitochondria isolated 24 hr after injury using 300 mg/kg NACA dosage (15 min post-injury followed by a booster at 6 hr post injury). The results showed that reduced forms of GSH were significantly (p<0.05) reduced after injury compared to naïve, whereas the decrease in total form of GSH as well as the increase in GSSG were not statistically significant (Fig. 3). Compared to Vehicle treatment, 300 mg/kg NACA marginally improved total and reduced GSH compared to Vehicle (p>0.05). However, these values remained lower than naïve. Although values for GSSG content remained higher in all the injured groups compared to naïve, no significant differences were noted in GSSG content among groups. Therefore, in the next set of experiments injured rats were administered (i.p.) either Vehicle (saline) or 300 mg/kg NACA 15 min post-injury followed by immediate subcutaneous implantation of osmotic mini-pumps to continuously deliver Vehicle or NACA at 300 mg/kg/day. SCI significantly reduced total and reduced GSH levels after 24 hr (Fig. 4). In the NACA-treated group, total and reduced forms of GSH were significantly (p<0.05) higher compared to Vehicle and were comparable to naïve levels. No significant differences were noted for GSSG content among the experimental groups. Results from these experiments indicate that continuous delivery of NACA is necessary to maintain GSH levels to normality.
Fig. 3.

Effects of NACA treatment on the mitochondrial glutathione (GHS) content following acute contusion SCI. Comparison of total GSH (Total), reduced form of GSH and oxidized form of glutathione (GSSG) expressed as ηmols/mg protein in the crude mitochondrial fractions. Injured rats received 75 or 300 mg/kg NACA at 15 min post-injury followed by a booster at 6 hr post- injury. At 24 hr post-injury, total and reduced forms of GSH were decreased compared to naïve; only reduced GSH was significantly less (*p<0.05). Treatment with NACA marginally increased both total and reduced GSH levels to normality; however values remained lower than naïve (p>0.05). No significant difference was seen for oxidized form of glutathione (GSSG). Bars are group mean ± SEM, n=3/group.
Fig. 4.
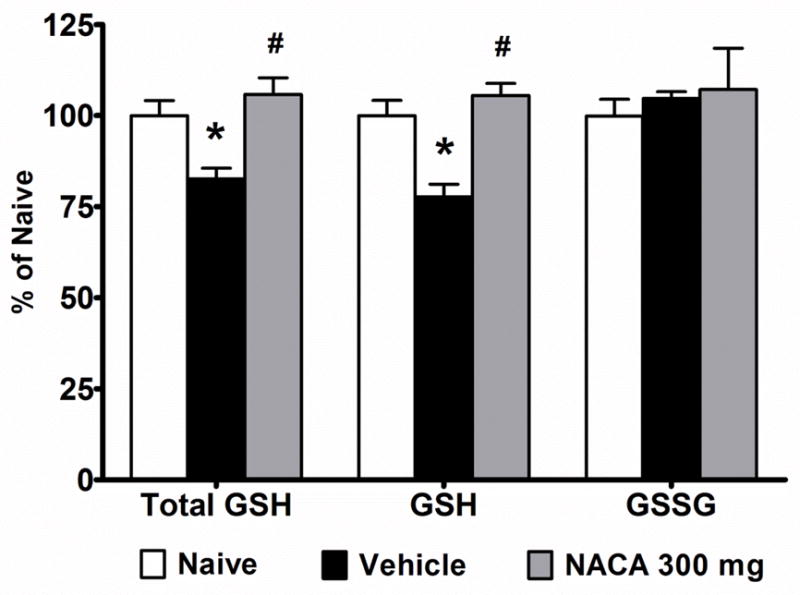
Effects of continuous NACA treatment on the mitochondrial glutathione (GHS) content following contusion SCI. Comparison of total glutathione (Total), reduced form of glutathione (GSH) and oxidized form of glutathione (GSSG) expressed as ηmols/mg protein in crude mitochondrial fractions. Injured rats received 300 mg/kg NACA at 15 min post-injury and osmotic mini-pumps containing Vehicle or NACA for continuous delivery (300 mg/kg/day). At 24 hr post-injury, crude mitochondria were isolated and subjected to glutathione estimation. SCI resulted in depletion of total and reduced forms of GSH. Continuous NACA treatment significantly maintained GSH levels compared to Vehicle treatment. The values for total and reduced forms of GSH were comparable to naïve. No significant difference was seen for oxidized form of glutathione (GSSG). Bars are group mean ± SEM, n=4/group. *p<0.05 compared to Naive and # p<0.05 compared to Vehicle-treated injured groups.
Effects of NACA treatment on behavioral recovery
Based on the results from biochemical assessment of mitochondrial function, we chose to treat injured rats with two of the maximum effective dosages of NACA (150 and 300 mg/kg/day) up to 7 DPI as described in “Materials and Method” section. Injury parameters were not significantly different (p>0.05) between the groups in terms of actual force (Vehicle: 255.00 ± 1.76, NACA 150mg/kg: 258.14 ± 1.37 and NACA 300mg/kg: 256.86 ± 3.04), displacement (Vehicle: 1587.0 ± 21.02, NACA 150mg/kg: 1493.86 ± 29.23 and NACA 300mg/kg: 1506.43 ± 49.68) and velocity (Vehicle: 122.60 ± 2.21, NACA 150mg/kg: 121.14 ± 1.37 and NACA 300mg/kg: 122.14 ± 1.91). Due to post-surgical complications, one rat from Vehicle-treated group died and another Vehicle-treated rat was excluded from data analysis due to >2x standard deviation from group means in histological assessments. At day 2 post-injury, all injured animals demonstrated complete hindlimb paralysis (Fig. 5). Over the course of 6 weeks, both dosages of NACA showed significantly [F(2,16) = 7.921, p<0.004] improved hindlimb locomotor recovery compared to Vehicle. Beginning at 21 DPI BBB scores for both NACA treatment dosages were significantly (p<0.05) higher compared to Vehicle (Fig. 5). Critically, at the terminal time point, animals in both the NACA-treated groups were able to walk with frequent to consistent weight supported plantar steps versus Vehicle-treated rats that demonstrated primarily dorsal and only occasional planter stepping.
Fig. 5.

Effects of prolonged continuous NACA treatment on hindlimb functional recovery following contusion SCI. Graphic representation of the mean behavioral (BBB) scores for rats treated with Vehicle, 150mg/kg NACA or 300mg/kg NACA following L1/L2 spinal cord injury. Compared to Vehicle-treated animals, 7 days of continuous NACA treatments resulted in improved hindlimb locomotor function after 42 days post-injury (DPI), when, both NACA- treated cohorts showed plantar stepping with weight-support with frequent to consistent coordination. Conversely, Vehicle-treated animals demonstrated primarily dorsal and occasional planter stepping. Symbols represent group means ± SEM, n=5–7/group. *p<0.05 Vehicle compared to 150mg/kg NACA and #p<0.05 Vehicle compared to 300mg/kg NACA.
As a more refined measure of locomotor function, we carried out terminal gait assessments and calculated the Coordinated Pattern Index (CPI; FL-HL coordination), the Plantar Stepping Index (PSI; fidelity of plantar stepping), the Regularity Index (RI; mirrors CPI, but includes dorsal steps) and the % of dorsal steps after NACA treatments following SCI, as described in the “Materials and Methods” section. Results showed that the PSI, CPI, and RI were all significantly higher (p<0.05) for the 150mg NACA-treated group compared to the Vehicle group (Fig. 6). However, the PSI, RI and % Dorsal steps for the 300mg/kg NACA dosage group were not statistically different from Vehicle. Importantly, however, the data for PSI, RI and % dorsal steps show that these refined gait analyses could detect differences between the 150 and 300mg/Kg NACA-treated groups even when the differences in their final BBB scores were not significant. Overall, these refined measurements validate our findings of improved hindlimb functional recovery following NACA treatment, in conjunction with standardized BBB testing.
Fig. 6.

Effects of prolonged continuous NACA treatment on hindlimb functional recovery (gait analysis) following contusion SCI. Comparison of the terminal gait analysis using the Coordinated Pattern Index (CPI), Plantar Stepping Index (PSI), Regularity Index (RI) and % Dorsal Steps. Compared to Vehicle, both NACA treatments resulted in significantly improved CPI. However, only 150mg/kg NACA showed significant increases in PSI and RI along with a decrease in % Dorsal Steps compared to Vehicle. Bars are group mean ± SEM, n=5–7/group. *p<0.05 compared to Vehicle-treated injured group.
Effects of NACA treatment on Tissue Sparing
Immediately following gait assessments, rats were subjected to in vivo MRI scanning. Qualitative T2-weighted sagittal MRI images revealed reduced lesion volume in both NACA treated groups compared to Vehicle (Figs. 7A–C). Histological assessment of spinal cord sections revealed increased tissue sparing at injury epicenter with NACA treatments compared to Vehicle (Figs. 7D–F). Subsequent quantitative morphometric assessment showed that NACA treatments following injury significantly (p<0.05) reduced the volume of injured spinal cord tissue (3 mm rostral and 3 mm caudal to the epicenter, total 7 mm) compared to vehicle treatment (Fig. 7G). While volumetric white and gray matter sparing in the NACA-treated groups were marginally increased (p>0.05) versus Vehicle (Fig. 7H), both NACA treatments showed significant (p<0.05) tissue sparing at the injury epicenter (Figs. 7I&J). Overall, the significant epicenter tissue sparing with both NACA dosages was, in fact, inversely correlated with their effects on reducing lesion volume.
Fig. 7.

Effects of prolonged continuous NACA treatment on histopathological outcome measures following contusion SCI. (A–C) T2-weighted sagittal MRI images showing reduced lesion volume with NACA treatment compared to Vehicle (white arrows in MRI images indicate injured tissue and yellow arrows pointed at dorsal root ganglion). D–F Photomicrographs of EC- CV-stained injured spinal cord sections from same animals in MRI images, illustrating that the NACA-treated injured groups showed increased sparing of spinal cord tissue at the injury epicenter compared to Vehicle. (G–J) Morphometric quantification of histopathology at 6 weeks post-injury. (G) Both NACA treatment groups showed less volume of injured tissue compared with the Vehicle-treated group. (H) Compared to Vehicle, both gray matter (GM) and white matter (WM) tissues sparing marginally increased with either NACA-treated groups. However, (I) and (J) demonstrate that both NACA treatments significantly increased tissue sparing at the injury epicenter. Bars are group mean ± SEM, n=5–7/group. * p<0.05 compared with vehicle- treated injured group. Scale bar = 1 mm for all histological photos.
Discussion
Collectively, the results of this study demonstrate that after severe contusion SCI, treatment with NACA significantly improved function of all three populations of mitochondria as well as maintained levels of antioxidant GSH that were associated with greater tissue sparing at the injury site. Such pronounced histological preservation ultimately resulted in the significant recovery of weight-supported hindlimb stepping.
Our findings from mitochondrial studies showed that acute (24 hr) SCI resulted in compromised mitochondrial bioenergetics in terms of ADP phosphorylation rate (State III) and complex I – driven maximum electron transport (State V – Complex I), as well as activities of key mitochondrial enzyme complexes: complex I, IV and PDHC in all three populations of mitochondria. These results further validate our previous findings showing compromised mitochondrial bioenergetics in total (Patel et al., 2012; Patel et al., 2010; Sullivan et al., 2007) as well as synaptic and non-synaptic (Patel et al., 2009) mitochondrial populations. In the present study we showed for the first time that we can isolate all three populations of mitochondria (total, synaptic and non-synaptic) from a single 1.5 cm rat spinal cord sufficiently enough to assess their bioenergetics using Seahorse Bioscience XF24 extracellular flux analyzer. This novel method will be instrumental in studying the effects of injury/treatment on synaptic (purely neuronal) and non- synaptic (neuronal somata and glia) mitochondria separately as well as combined from single spinal cord tissue samples. It is important to note that in our previous studies, total as well as synaptic and non-synaptic mitochondria were isolated from different spinal cord samples whereas all three populations of mitochondria were isolated from a single spinal cord segment from each rat in different experimental groups in the current study. Notably, for naïve rats State III respiration (complex I driven ADP phosphorylation rate) for synaptic mitochondria are ~20% lower than non-synaptic mitochondria (Figs. 1B and C); a similar pattern was found in our previous study using a miniature Clark-type electrode (Hansatech Instruments, Norfolk, England) (Patel et al., 2009). Moreover, differential responses to SCI as well as therapeutic interventions are noted among these different populations (Figure 1) (Patel et al., 2009). Such differences were also evident in the enzymatic activities measured for complex I of the ETS in the non-synaptic mitochondrial fraction (Figs. 2B and C), which is suggestive of a higher complex I driven-ETS activity and capacity of non- synaptic mitochondria compared with synaptic mitochondria under basal conditions. Although the precise mechanism underlying these differences is not clear, these findings indicate the importance of comparative studies of various populations of mitochondria.
The current study demonstrates the protective effects of acute NACA treatment on bioenergetics of total, synaptic and non-synaptic mitochondria, as well as prolonged continuous treatment on neuroprotection and improved functional recovery following contusion SCI. For total mitochondria, all the dosages except 600 mg showed protective effects on mitochondrial respiration in a dose-dependent manner with maximum effects with 300 mg/kg dosage. For synaptic as well as non-synaptic mitochondria only 300 mg/kg dosage was effective in maintaining mitochondrial respiration near normal levels. Interestingly, in synaptic mitochondria the 150 mg/kg dosage increased mitochondrial bioenergetics to a level that was comparable to naïve samples, as was the case for total mitochondria. It is possible that the synaptic population was the driving force behind the response demonstrated in the total mitochondrial pool since the 150 mg/kg dosage did not result in a significant difference in synaptic mitochondria compared to vehicle. However, the extent of recovery of respiration rates as well as complex I activities following NACA treatment was higher in non-synaptic mitochondria than synaptic mitochondria, which indicates the higher susceptibility of neuronal mitochondria to oxidative stress following SCI. This suggests the need for more refined future dose-response studies. Regardless, these results show that treatment with NACA significantly maintains bioenergetics of all three populations of mitochondria, albeit differentially with respect to effective dosages. Notably, this could be due to further loss of mitochondria during isolation of synaptic and non-synaptic mitochondria as a result of an additional purification step required compared to Total mitochondria as described in Materials and Methods section.
The functional deficits following contusion SCI are contributed to both direct mechanical injury and secondary pathophysiology associated with spinal cord trauma due to oxidative stress, mainly lipid peroxidation cascades, and inflammation (Anderson and Hall, 1993; Brown and Hall, 1992; Springer et al., 1997). After SCI, improved mitochondrial function is associated with reduced oxidative stress and damage (Patel et al., 2009). It should also decrease post-traumatic inflammation since oxidative stress and cellular damage drive activation of pro-inflammatory cascades (Christman et al., 2000).
GSH plays an important role in attenuation of cellular oxidative stress and is shown to be depleted following SCI (Kamencic et al., 2001). Therefore, we reasoned that maintaining normal GSH levels using its precursor NACA would be an effective way to reduce post-traumatic oxidative stresses and improve mitochondrial function acutely, thereby promoting sparing of spinal cord tissue and improving functional recovery at later stages after spinal cord trauma. GSH is synthesized in the body from the amino acids L-cysteine, L-glutamic acid and glycine. Bio-availability of cysteine is the rate-limiting step in glutathione synthesis (Meister, 1989) and NAC has been used clinically as well as experimentally as a nutritional supplement to replenishment intracellular cysteine. However, it does not readily cross the blood–brain barrier (McLellan et al., 1995) whereas NACA does (Grinberg et al., 2005; Offen et al., 2004). In addition, NACA has been shown to cross cell membranes more efficiently than NAC due to its neutral carboxy group. In turn, this increased bioavailability gives NACA a higher capacity for replenishing GSH levels and scavenging free radicals (Grinberg et al., 2005). Likewise, it has been showed that NACA attenuates airway inflammation and hyper -responsiveness by regulating activation of NF-κB and HIF-1α as well as reducing ROS generation in allergic airway disease (Lee et al., 2007). NACA reversed the increased lactate dehydrogenase levels following diesel exhaust particles (DEPs)-induced toxicity in the lungs of C57BL/6 mice (Banerjee et al., 2009). Moreover, recent studies have demonstrated beneficial effects of NACA in animal models for neurological disorders including Parkinson’s disease, Multiple sclerosis and Tardive dyskinesia (Bahat-Stroomza et al., 2005; Offen et al., 2004; Sadan et al., 2005).
It is reported that depletion of GSH due to oxidative stress could not be recovered as oxidized glutathione (GSSG), as it forms glutathiyl-protein adducts during oxidative stress which cannot be reduced by glutathione reductase to GSH (Bellomo et al., 1987; Schuppe et al., 1992; Shivakumar et al., 1995). This could jeopardize the de novo synthesis of GSH following SCI, thus resulting in GSH depletion (Figure 3&4). Based on our results, administration of NACA facilitates de novo synthesis of GSH following SCI. Moreover, SCI induced oxidative stress may lead to continuous consumption of GSH due to antioxidant activities; therefore, replenishment of GSH after NACA treatment might be expected to be limited. Accordingly, when we administered NACA 15 min post-injury and 6 hr post-injury we found a marginal replenishment of GSH at 24 hr post-injury (Fig. 3). Alternatively, continuous delivery of NACA completely restored GSH levels (Fig. 4). This indicates that due to its short half-life, NACA should be administered continuously to maintain GSH levels. It will be interesting to study the effects of continuous delivery of NACA on mitochondrial bioenergetics following SCI. Also, it will be important to correlate reported improved mitochondrial outcome measures with oxidative stress markers such as protein oxidation (protein carbonyls) and lipid peroxidation (4-HNE) or activities of antioxidant enzymes such as glutathione reductase.
For the two maximum effective dosages of acute NACA treatment that improved mitochondrial bioenergetics, we also found that NACA significantly improved hindlimb weight-bearing and functional locomotion after SCI. Importantly, the indices of coordination employed were able to detect group differences between the 150 and 300mg/kg NACA-treated animals despite the similar terminal BBB scores. These differences were due mainly to the higher proportion of plantar steps exhibited by the 300mg/kg NACA group compared to the lower (150mg/kg) dose. These refined gait and coordination measurements validate the improvements in hindlimb functional recovery following NACA treatment, in conjunction with standardized BBB testing. Histological assessments revealed decreased injury volume and marginal sparing of gray and white matter volume following NACA treatment. Notably, our upper lumbar injury paradigm induces hindlimb locomotor deficits accounted for by damaging of critical locomotor circuitry (central pattern generator circuitry, CPG). The CPG extends from the low thoracic (T12/T13) to mid- lumbar (L3/L4) segments in the rat spinal cord (Bertrand and Cazalets, 2002; Kremer and Lev-Tov, 1997; Magnuson et al., 1998). We believe that even with marginal sparing of white matter at the injury epicenter and gray matter over the length of the injury, the NACA-treated groups had improved forelimb-hindlimb (FL-HL) coordination while the vehicle-treated animals were stepping with FL-HL de-coupling. This argues strongly for one of two potential mechanisms: spared white matter at the epicenter allowing inter-enlargement communication and/or spared cell bodies of inter-enlargement interneurons that mediate FL-HL coordination (Bareyre et al., 2004; Conta Steencken and Stelzner, 2010; Reed et al., 2006). Importantly, the NACA-treated groups showed improved functional recovery beginning the first week of administration, which supports earlier reports that maximal propriospinal axonal loss occurs by 2 weeks post-contusion SCI (Conta Steencken and Stelzner, 2010). Accordingly, our findings indicate that even a small amount of gray matter sparing over the injury length accompanied by significantly increased white matter sparing at the injury epicenter after NACA treatment was associated with improved functional recovery assessed by over ground locomotion and gait assessments. Moreover, the differences observed in the effects of NACA among different outcome measures (biochemical vs BBB vs gait analysis) may be due to acute versus chronic treatment delivery methods using different routes of administration (i.p. versus s.c.), as well as duration of delivery (24 hr versus 7 DPI). Based on the histological and BBB assessments, the 300 mg/kg dosage did not have detrimental effects. In fact, despite significant differences in refined gait analyses between NACA dosage groups, there were no differences in BBB scores or tissue sparing assessments. While dosages lower than 150mg/kg may have resulted in further enhanced functional recovery and tissue sparing, we did not pursue this based on our biochemical data. Future studies will consider delivery of lower NACA dosages, perhaps over more prolonged periods of time post-injury.
Conclusions
Most traditional therapeutic strategies for experimental SCI have either been technically challenging or limited to blocking downstream pathophysiological cascades after the initial insult, perhaps reflected in historically limited translational success. Therefore, our interest is to target upstream (e.g., mitochondria) of the end product measures (e.g., oxidative damage, neuronal cell death or a particular cascade) as this approach may hold more promise to take therapeutic interventions toward the clinical arena for traumatic SCI. Additionally, we have observed that NACA is therapeutic following traumatic brain injury (TBI) where it improves mitochondrial bioenergetics, behavioral outcome and offers neuroprotection (Pandya et al, this issue). We herein document that administration of NACA can effectively replenish GSH and improve mitochondrial bioenergetics at early stages following experimental SCI and, moreover, that prolonged continuous treatment significantly improves recovery of long-term hindlimb function and reduces lesion volume.
Highlights.
N-acetylcysteine amide (NACA) is a novel antioxidant therapy for spinal trauma
NACA acutely preserved synaptic, non-synaptic and total mitochondrial bioenergetics
Continuous NACA delivery over 24 h preserved mitochondrial glutathione levels
Prolonged NACA treatment increased long-term tissue sparing and functional recovery
Acknowledgments
Special thanks to Dr. David Powell for technical expertise in MRI analysis and Mr. Taylor Smith for helping with MRI scanning. This study was supported by NIH/NINDS R01NS069633 (A.G.R. and P.G.S.), The Craig H. Neilsen Foundation #190115 (AGR), NIH/NINDS P30 NS051220 (UK) and NIH/NIGMS P30 GM103507 (UofL).
Footnotes
Conflict of Interest Statement: Glenn A. Goldstein is president and founder of Sentient Lifesciences, Inc. which holds certain patent rights regarding NACA and supplied NACA for these studies. No other author conflict of interests exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson DK, Hall ED. Pathophysiology of spinal cord trauma. Annals of emergency medicine. 1993;22:987–992. doi: 10.1016/s0196-0644(05)82739-8. [DOI] [PubMed] [Google Scholar]
- Arent CO, Reus GZ, Abelaira HM, Ribeiro KF, Steckert AV, Mina F, Dal-Pizzol F, Quevedo J. Synergist effects of n-acetylcysteine and deferoxamine treatment on behavioral and oxidative parameters induced by chronic mild stress in rats. Neurochemistry international. 2012 doi: 10.1016/j.neuint.2012.07.024. [DOI] [PubMed] [Google Scholar]
- Aruoma OI, Halliwell B, Hoey BM, Butler J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free radical biology & medicine. 1989;6:593–597. doi: 10.1016/0891-5849(89)90066-x. [DOI] [PubMed] [Google Scholar]
- Bahat-Stroomza M, Gilgun-Sherki Y, Offen D, Panet H, Saada A, Krool-Galron N, Barzilai A, Atlas D, Melamed E. A novel thiol antioxidant that crosses the blood brain barrier protects dopaminergic neurons in experimental models of Parkinson’s disease. Eur J Neurosci. 2005;21:637–646. doi: 10.1111/j.1460-9568.2005.03889.x. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Trueblood MB, Zhang X, Manda KR, Lobo P, Whitefield PD, Hagen DE, Ercal N. N-acetylcysteineamide (NACA) prevents inflammation and oxidative stress in animals exposed to diesel engine exhaust. Toxicology letters. 2009;187:187–193. doi: 10.1016/j.toxlet.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Kerschensteiner M, Raineteau O, Mettenleiter TC, Weinmann O, Schwab ME. The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat Neurosci. 2004;7:269–277. doi: 10.1038/nn1195. [DOI] [PubMed] [Google Scholar]
- Bartov O, Sultana R, Butterfield DA, Atlas D. Low molecular weight thiol amides attenuate MAPK activity and protect primary neurons from Abeta(1–42) toxicity. Brain research. 2006;1069:198–206. doi: 10.1016/j.brainres.2005.10.079. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight-drop device versus transection. Experimental neurology. 1996;139:244–256. doi: 10.1006/exnr.1996.0098. [DOI] [PubMed] [Google Scholar]
- Bellomo G, Mirabelli F, DiMonte D, Richelmi P, Thor H, Orrenius C, Orrenius S. Formation and reduction of glutathione-protein mixed disulfides during oxidative stress. A study with isolated hepatocytes and menadione (2-methyl-1,4-naphthoquinone) Biochemical pharmacology. 1987;36:1313–1320. doi: 10.1016/0006-2952(87)90087-6. [DOI] [PubMed] [Google Scholar]
- Benedict AL, Mountney A, Hurtado A, Bryan KE, Schnaar R, Dinkova-Kostova AT, Talalay P. Neuroprotective Effects of Sulforaphane after Contusive Spinal Cord Injury. Journal of neurotrauma. 2012 doi: 10.1089/neu.2012.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand S, Cazalets JR. The respective contribution of lumbar segments to the generation of locomotion in the isolated spinal cord of newborn rat. Eur J Neurosci. 2002;16:1741–1750. doi: 10.1046/j.1460-9568.2002.02233.x. [DOI] [PubMed] [Google Scholar]
- Brown SA, Hall ED. Role of oxygen-derived free radicals in the pathogenesis of shock and trauma, with focus on central nervous system injuries. Journal of the American Veterinary Medical Association. 1992;200:1849–1859. [PubMed] [Google Scholar]
- Christman JW, Blackwell TS, Juurlink BH. Redox regulation of nuclear factor kappa B: therapeutic potential for attenuating inflammatory responses. Brain Pathol. 2000;10:153–162. doi: 10.1111/j.1750-3639.2000.tb00252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conta Steencken AC, Stelzner DJ. Loss of propriospinal neurons after spinal contusion injury as assessed by retrograde labeling. Neuroscience. 2010;170:971–980. doi: 10.1016/j.neuroscience.2010.06.064. [DOI] [PubMed] [Google Scholar]
- De Flora S, Izzotti A, D’Agostini F, Cesarone CF. Antioxidant activity and other mechanisms of thiols involved in chemoprevention of mutation and cancer. The American journal of medicine. 1991;91:122S–130S. doi: 10.1016/0002-9343(91)90295-9. [DOI] [PubMed] [Google Scholar]
- Drake J, Kanski J, Varadarajan S, Tsoras M, Butterfield DA. Elevation of brain glutathione by gamma-glutamylcysteine ethyl ester protects against peroxynitrite-induced oxidative stress. Journal of neuroscience research. 2002;68:776–784. doi: 10.1002/jnr.10266. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiological reviews. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Dumont RJ, Okonkwo DO, Verma S, Hurlbert RJ, Boulos PT, Ellegala DB, Dumont AS. Acute spinal cord injury, part I: pathophysiologic mechanisms. Clinical neuropharmacology. 2001;24:254–264. doi: 10.1097/00002826-200109000-00002. [DOI] [PubMed] [Google Scholar]
- Foltz WU, Wagner M, Rudakova E, Volk T. N-acetylcysteine prevents electrical remodeling and attenuates cellular hypertrophy in epicardial myocytes of rats with ascending aortic stenosis. Basic research in cardiology. 2012;107:290. doi: 10.1007/s00395-012-0290-4. [DOI] [PubMed] [Google Scholar]
- Grinberg L, Fibach E, Amer J, Atlas D. N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free radical biology & medicine. 2005;38:136–145. doi: 10.1016/j.freeradbiomed.2004.09.025. [DOI] [PubMed] [Google Scholar]
- Holdiness MR. Clinical pharmacokinetics of N-acetylcysteine. Clinical pharmacokinetics. 1991;20:123–134. doi: 10.2165/00003088-199120020-00004. [DOI] [PubMed] [Google Scholar]
- Huang SF, Tsai YA, Wu SB, Wei YH, Tsai PY, Chuang TY. Effects of Intravascular Laser Irradiation of Blood in Mitochondria Dysfunction and Oxidative Stress in Adults with Chronic Spinal Cord Injury. Photomedicine and laser surgery. 2012 doi: 10.1089/pho.2012.3228. [DOI] [PubMed] [Google Scholar]
- Issels RD, Nagele A, Eckert KG, Wilmanns W. Promotion of cystine uptake and its utilization for glutathione biosynthesis induced by cysteamine and N-acetylcysteine. Biochemical pharmacology. 1988;37:881–888. doi: 10.1016/0006-2952(88)90176-1. [DOI] [PubMed] [Google Scholar]
- Kamencic H, Griebel RW, Lyon AW, Paterson PG, Juurlink BH. Promoting glutathione synthesis after spinal cord trauma decreases secondary damage and promotes retention of function. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2001;15:243–250. doi: 10.1096/fj.00-0228com. [DOI] [PubMed] [Google Scholar]
- Kelly GS. Clinical applications of N-acetylcysteine. Alternative medicine review : a journal of clinical therapeutic. 1998;3:114–127. [PubMed] [Google Scholar]
- Koopmans GC, Deumens R, Honig WM, Hamers FP, Steinbusch HW, Joosten EA. The assessment of locomotor function in spinal cord injured rats: the importance of objective analysis of coordination. Journal of neurotrauma. 2005;22:214–225. doi: 10.1089/neu.2005.22.214. [DOI] [PubMed] [Google Scholar]
- Kremer E, Lev-Tov A. Localization of the spinal network associated with generation of hindlimb locomotion in the neonatal rat and organization of its transverse coupling system. Journal of neurophysiology. 1997;77:1155–1170. doi: 10.1152/jn.1997.77.3.1155. [DOI] [PubMed] [Google Scholar]
- Kuerzi J, Brown EH, Shum-Siu A, Siu A, Burke D, Morehouse J, Smith RR, Magnuson DS. Task-specificity vs. ceiling effect: step-training in shallow water after spinal cord injury. Experimental neurology. 2010;224:178–187. doi: 10.1016/j.expneurol.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Kim SR, Park HS, Park SJ, Min KH, Lee KY, Choe YH, Hong SH, Han HJ, Lee YR, Kim JS, Atlas D, Lee YC. A novel thiol compound, N-acetylcysteine amide, attenuates allergic airway disease by regulating activation of NF-kappaB and hypoxia-inducible factor-1alpha. Experimental & molecular medicine. 2007;39:756–768. doi: 10.1038/emm.2007.82. [DOI] [PubMed] [Google Scholar]
- Magnuson DS, Green DM, Sengoku T. Lumbar spinoreticular neurons in the rat: part of the central pattern generator for locomotion? Ann N Y Acad Sci. 1998;860:436–440. doi: 10.1111/j.1749-6632.1998.tb09069.x. [DOI] [PubMed] [Google Scholar]
- Meister A. Mechanism and regulation of the glutamine-dependent carbamyl phosphate synthetase of Escherichia coli. Advances in enzymology and related areas of molecular biology. 1989;62:315–374. doi: 10.1002/9780470123089.ch7. [DOI] [PubMed] [Google Scholar]
- Michel RP, Cruz-Orive LM. Application of the Cavalieri principle and vertical sections method to lung: estimation of volume and pleural surface area. Journal of microscopy. 1988;150:117–136. doi: 10.1111/j.1365-2818.1988.tb04603.x. [DOI] [PubMed] [Google Scholar]
- Offen D, Gilgun-Sherki Y, Barhum Y, Benhar M, Grinberg L, Reich R, Melamed E, Atlas D. A low molecular weight copper chelator crosses the blood-brain barrier and attenuates experimental autoimmune encephalomyelitis. Journal of neurochemistry. 2004;89:1241–1251. doi: 10.1111/j.1471-4159.2004.02428.x. [DOI] [PubMed] [Google Scholar]
- Opii WO, Nukala VN, Sultana R, Pandya JD, Day KM, Merchant ML, Klein JB, Sullivan PG, Butterfield DA. Proteomic identification of oxidized mitochondrial proteins following experimental traumatic brain injury. Journal of neurotrauma. 2007;24:772–789. doi: 10.1089/neu.2006.0229. [DOI] [PubMed] [Google Scholar]
- Ozaras R, Tahan V, Aydin S, Uzun H, Kaya S, Senturk H. N-acetylcysteine attenuates alcohol-induced oxidative stress in the rat. World journal of gastroenterology : WJG. 2003;9:125–128. doi: 10.3748/wjg.v9.i1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parcell S. Sulfur in human nutrition and applications in medicine. Alternative medicine review : a journal of clinical therapeutic. 2002;7:22–44. [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Lyttle TS, Magnuson DS, Rabchevsky AG. Acetyl-L-carnitine treatment following spinal cord injury improves mitochondrial function correlated with remarkable tissue sparing and functional recovery. Neuroscience. 2012;210:296–307. doi: 10.1016/j.neuroscience.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Lyttle TS, Rabchevsky AG. Acetyl-L-carnitine ameliorates mitochondrial dysfunction following contusion spinal cord injury. Journal of neurochemistry. 2010;114:291–301. doi: 10.1111/j.1471-4159.2010.06764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Pandya JD, Rabchevsky AG. Differential effects of the mitochondrial uncoupling agent, 2,4-dinitrophenol, or the nitroxide antioxidant, Tempol, on synaptic or nonsynaptic mitochondria after spinal cord injury. Journal of neuroscience research. 2009;87:130–140. doi: 10.1002/jnr.21814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penugonda S, Mare S, Goldstein G, Banks WA, Ercal N. Effects of N-acetylcysteine amide (NACA), a novel thiol antioxidant against glutamate-induced cytotoxicity in neuronal cell line PC12. Brain research. 2005;1056:132–138. doi: 10.1016/j.brainres.2005.07.032. [DOI] [PubMed] [Google Scholar]
- Price TO, Uras F, Banks WA, Ercal N. A novel antioxidant N-acetylcysteine amide prevents gp120- and Tat-induced oxidative stress in brain endothelial cells. Experimental neurology. 2006;201:193–202. doi: 10.1016/j.expneurol.2006.03.030. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Fugaccia I, Fletcher-Turner A, Blades DA, Mattson MP, Scheff SW. Basic fibroblast growth factor (bFGF) enhances tissue sparing and functional recovery following moderate spinal cord injury. Journal of neurotrauma. 1999;16:817–830. doi: 10.1089/neu.1999.16.817. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Fugaccia I, Sullivan PG, Blades DA, Scheff SW. Efficacy of methylprednisolone therapy for the injured rat spinal cord. Journal of neuroscience research. 2002;68:7–18. doi: 10.1002/jnr.10187. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Fugaccia I, Sullivan PG, Scheff SW. Cyclosporin A treatment following spinal cord injury to the rat: behavioral effects and stereological assessment of tissue sparing. Journal of neurotrauma. 2001;18:513–522. doi: 10.1089/089771501300227314. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Fugaccia I, Turner AF, Blades DA, Mattson MP, Scheff SW. Basic fibroblast growth factor (bFGF) enhances functional recovery following severe spinal cord injury to the rat. Experimental neurology. 2000;164:280–291. doi: 10.1006/exnr.2000.7399. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Sullivan PG, Scheff SW. Temporal-spatial dynamics in oligodendrocyte and glial progenitor cell numbers throughout ventrolateral white matter following contusion spinal cord injury. Glia. 2007;55:831–843. doi: 10.1002/glia.20508. [DOI] [PubMed] [Google Scholar]
- Reed WR, Shum-Siu A, Onifer SM, Magnuson DS. Inter-enlargement pathways in the ventrolateral funiculus of the adult rat spinal cord. Neuroscience. 2006;142:1195–1207. doi: 10.1016/j.neuroscience.2006.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadan O, Bahat-Stromza M, Gilgun-Sherki Y, Atlas D, Melamed E, Offen D. A novel brain-targeted antioxidant (AD4) attenuates haloperidol-induced abnormal movement in rats: implications for tardive dyskinesia. Clinical neuropharmacology. 2005;28:285–288. doi: 10.1097/01.wnf.0000191331.54649.e3. [DOI] [PubMed] [Google Scholar]
- Sauerbeck A, Pandya J, Singh I, Bittman K, Readnower R, Bing G, Sullivan P. Analysis of regional brain mitochondrial bioenergetics and susceptibility to mitochondrial inhibition utilizing a microplate based system. Journal of neuroscience methods. 2011;198:36–43. doi: 10.1016/j.jneumeth.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuppe I, Moldeus P, Cotgreave IA. Protein-specific S-thiolation in human endothelial cells during oxidative stress. Biochemical pharmacology. 1992;44:1757–1764. doi: 10.1016/0006-2952(92)90069-u. [DOI] [PubMed] [Google Scholar]
- Shivakumar BR, Kolluri SV, Ravindranath V. Glutathione and protein thiol homeostasis in brain during reperfusion after cerebral ischemia. The Journal of pharmacology and experimental therapeutics. 1995;274:1167–1173. [PubMed] [Google Scholar]
- Smith L. Spectrophotometric assay of cytochrome c oxidase. Methods of biochemical analysis. 1955;2:427–434. doi: 10.1002/9780470110188.ch13. [DOI] [PubMed] [Google Scholar]
- Springer JE, Azbill RD, Mark RJ, Begley JG, Waeg G, Mattson MP. 4-hydroxynonenal, a lipid peroxidation product, rapidly accumulates following traumatic spinal cord injury and inhibits glutamate uptake. Journal of neurochemistry. 1997;68:2469–2476. doi: 10.1046/j.1471-4159.1997.68062469.x. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Krishnamurthy S, Patel SP, Pandya JD, Rabchevsky AG. Temporal characterization of mitochondrial bioenergetics after spinal cord injury. Journal of neurotrauma. 2007;24:991–999. doi: 10.1089/neu.2006.0242. [DOI] [PubMed] [Google Scholar]
- Teng YD, Choi H, Onario RC, Zhu S, Desilets FC, Lan S, Woodard EJ, Snyder EY, Eichler ME, Friedlander RM. Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:3071–3076. doi: 10.1073/pnas.0306239101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JA, Mallis RJ. Aging and oxidation of reactive protein sulfhydryls. Experimental gerontology. 2001;36:1519–1526. doi: 10.1016/s0531-5565(01)00137-1. [DOI] [PubMed] [Google Scholar]