

Abstract
Duplicated sequences are important sources of genetic instability and in the evolution of new gene function within species. Hominids have a preponderance of intrachromosomal duplications organized in an interspersed fashion, as opposed to tandem duplications, which are common in other mammalian genomes such as mouse, dog, and cow. Multiple lines of evidence, including sequence divergence, comparative primate genomes, and fluorescence in situ hybridization (FISH) analyses, point to an excess of segmental duplications in the common ancestor of humans and African great apes. We find that much of the interspersed human duplication architecture within chromosomes is focused around common sequence elements referred to as “core duplicons.” These cores correspond to the expansion of gene families, some of which show signatures of positive selection and lack orthologs present in other mammalian species. This genomic architecture predisposes apes and humans not only to extensive genetic diversity, but also to large-scale structural diversity mediated by nonallelic homologous recombination. In humans, many de novo large-scale genomic changes mediated by these duplications are associated with neuropsychiatric and neurodevelopmental disease. We propose that the disadvantage of a high rate of new mutations is offset by the selective advantage of newly minted genes within the cores.
Geneticists have long appreciated the dual nature of duplicated sequences as sources of evolutionary innovation and regions of genomic instability. Muller et al. (1936), Bridges (1936), and Sturtevant (1925) were among the first to recognize the role duplicated sequences have in both phenotype and genetic instability by their association of unequal crossing-over of the Bar locus in Drosophila and the eye-reduction phenotype. The frequency and phenotypic consequences of new mutations among tandem duplicates were noted by Bridges in 1936 when he commented, “The production of Bar-double and of Bar-reverted is seen to be the insertion of this extra section twice, or conversely, its total loss—both presumably by a process of unequal crossing-over. A remarkable peculiarity of the mutant is that occasionally the homozygous stock gives rise to a fly indistinguishable in appearance and genetic behavior from wild-type.” Ohno highlighted the importance of duplication in the “birth” of new genes during evolution. To Ohno, the process of duplication liberated genes from the constraint of ancestral function, allowing new mutations to give rise to modified or novel function. This was an extension of Muller’s dictum “all life from pre-existing life…and every gene from a pre-existing gene” (Muller et al. 1936). Ohno posited that the origin of vertebrate complexity lies in the large whole-genome duplications providing a burst of functional redundancy and subsequent specialization (Ohno et al. 1968).
It follows that if one is interested in areas of rapid evolutionary change and the discovery of genes important in the specification of the human condition, then the recently duplicated regions of our genome represent fertile areas of investigation (Eichler 2001). The study of these regions has revealed unexpected complexities in the evolution of our genome, led to the identification of novel human/great ape genes, and provided a road map for the discovery of new mutations associated with a wide range of pediatric and adult-onset disease. Although the sequencing of entire genomes has accelerated at a breakneck pace, sequencing of recently duplicated regions of the genome has proved more challenging and proceeded much more slowly. By dint of their high sequence identity and their large size (frequently >100 kbp in length) (She et al. 2004), sequence assemblies based strictly on short whole-genome shotgun sequences (<600 bp) have often failed to resolve these aspects of genome organization. Among mammals, only two genomes—mouse and human—have been sequenced to the level of rigor required to accurately infer the structure and organization from the assembled genome sequence.
HUMAN VERSUS MOUSE SEGMENTAL DUPLICATION PROPERTIES
The most recent comparisons of the mouse and human finished genomes (Collins et al. 2004; Church et al. 2009) show that the two species are comparable in terms of the number of base pairs mapping to high-identity (>90%) duplications. However, there are three notable differences. Almost all large segmental duplications (SDs) in the mouse lineage are tandemly organized, whereas >59% of the duplications in humans are interspersed—being separated from their nearest paralog by more than 1 Mbp or mapping to a nonhomologous chromosome (She et al. 2008). Experimental and computational analyses of other genomes, such as the dog, rat, and cow, suggest that the tandem configuration likely represents the mammalian archetype (Tuzun et al. 2004; Elsik et al. 2009; Nicholas et al. 2009). Second, human duplications tend to be significantly enriched in spliced transcripts when compared to mouse, which appear to be more deficient in transcripts and, possibly, genes (She et al. 2008). Third, within the human genome, there is a skew toward higher sequence identity duplications, which suggests a potential excess of evolutionarily young SDs (Fig. 1). The presence of large, high-identity duplications at more locations has sensitized more of the human genome to the dosage and potential position effects as a result of unequal crossing-over.
Figure 1.
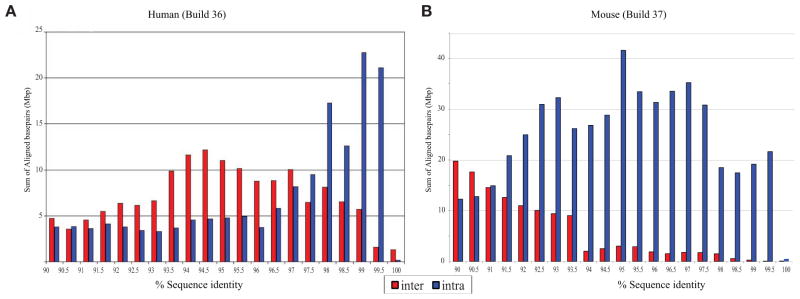
Percentage of identity distribution of mouse versus human SDs. Note the increase of interchromosomal duplications and the higher proportion of recent SDs in humans and the excess of intrachromosomal (tandem) duplications in mouse.
PRIMATE COMPARISONS
Despite the working draft nature of other nonhuman primate genome assemblies, the random nature of the underlying whole-genome shotgun (WGS) sequence data provides a means to detect duplications in the absence of an assembly. By mapping regions of excess WGS read-depth against the finished human reference sequence, we can predict the content of duplication in closely related primates such as chimpanzee, orangutan, and macaque. We can, then, parsimoniously infer the age of human duplications based on their shared or lineage-specific nature within the context of the generally accepted primate phylogeny. The analysis shows that the proportion of lineage-specific duplications in the chimpanzee and human lineages is approximately equal (Cheng et al. 2005; Marques-Bonet et al. 2009). We, however, predict a two to fourfold excess of new SDs in the common ancestor of humans and African great apes when compared to Asian apes (orangutan) and Old World monkey lineages (represented by macaque) (Fig. 2). The effect is most pronounced for intrachromosomal SDs. These findings are consistent with the excess of high-identity (>97%) pairwise alignments noted within the human genome assembly for intrachromosomal duplications (Fig. 1) and studies of gene duplication (Fortna et al. 2004; Dumas et al. 2007; Hahn et al. 2007) that suggest a burst of duplication activity during primate evolution. Notably, this duplication acceleration occurs at a period of time when most other mutational processes, including point mutation and retrotransposon activity, were slowing down (Wu and Li 1985; Li and Tanimura 1987; Waterston et al. 2002; Consortium 2005).
Figure 2.
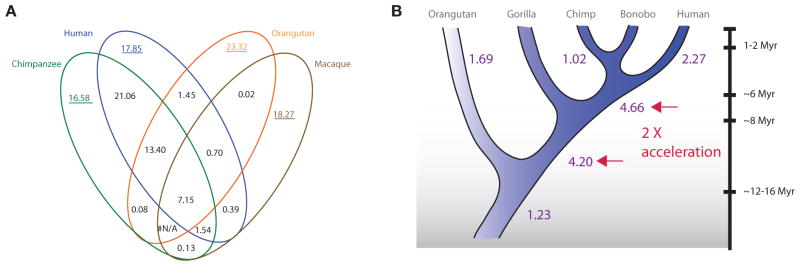
(A) Venn diagram showing shared and lineage-specific duplications among four primate genomes. Estimates were based on identifying regions of excess read-depth to the human assembly genome. Numbers underlined are copy-number corrected to avoid the bias of nonhuman-specific SDs. (B) Assignment of duplications and rate estimation of Mbp/Myr for each branch. Note the excess of duplication rate in the branch leading to the common ancestor of human and chimpanzee (Marques-Bonet et al. 2009).
DUPLICATION ORGANIZATION AND CORE DUPLICONS
Within the human genome, ancestral duplications (termed duplicons) of diverse interspersed origin juxtapose one another, forming complex mosaic duplication blocks that are hundreds of kilobase pairs in length (Rouquier et al. 1998; Johnson et al. 2006). This is in contrast to the organization in the mouse where most duplication blocks consist of tandemly organized SDs. Using a modified de Brujin graph theory approach along with comparative sequence data, we identified the ancestral origin of 4692 human duplication loci and deconvoluted the architecture of 437 duplication blocks in the human genome (Jiang et al. 2007). A complex pattern of duplication within duplications emerges, confirming the stepwise accretion of SDs on a genome-wide scale during hominid evolution (Eichler et al. 1997; Horvath et al. 2000; Courseaux et al. 2003; Stankiewicz et al. 2004; Johnson et al. 2006). Hierarchical clustering of these duplication blocks based on shared duplicon content organizes duplication blocks into 24 distinct groups (Fig. 3). Two distinct types of duplication blocks are distinguished: those in which the evolutionary flow of genetic information has occurred between nonhomologous chromosomes (n = 10) and those where the mosaic architectures have largely formed within a specific chromosome (n = 14). The former consists mainly of subtelomeric and pericentromeric duplications, and the latter corresponds almost exclusively to the intrachromosomal burst of SDs discussed above.
Figure 3.

Hierarchical clustering of human duplication blocks based on ancestral duplicon content. The termini of each branch represent one of 437 duplication blocks, which cluster into 24 distinct groups, 14 of which are restricted to a specific chromosome and 10 of which are mixed (M) among chromosomes mapping largely to subtelomeric (S) or pericentromeric (P) regions of the genome. An expanded view of chromosome 16 is shown (Fig. 5) (Jiang et al. 2007).
The hierarchical clustering suggests that the duplication blocks have been formed around a core or seed duplicon (defined as an ancestral duplicon that populates >67% of all duplication blocks within a group). These core sequences are among the most abundant and most ancient; they are particularly enriched for RefSeq genes and spliced expressed sequence tags (ESTs) when compared to flanking duplicons, and a few have been subjected to independent and recurrent duplications in different primate lineages (Johnson et al. 2006). Several of the corresponding genes and gene families encoded by these core duplicons lack orthologs in other mammalian species and have been highlighted as human–great ape gene family innovations (Johnson et al. 2001; Paulding et al. 2003; Ciccarelli et al. 2005). The TRE2 oncogene, for example, is a fusion of a USP32 protease and a TBC1D3 core duplicon. The resulting fusion gene is expressed solely in humans and African great apes (Paulding et al. 2003). The RANBP2, morpheus (NPIP), and NBPF11 (also known by its protein domain DUF1220) gene families show evidence of positive selection. Data from numerous copy-number variation studies (Sharp et al. 2005; Redon et al. 2006) suggest that these gene families are copy-number polymorphic in the human population. The functional significance of most of these genes is unknown. Functional characterization of the TBC1D3 core suggests that it may be important in modulating signaling of growth factors during development (Hodzic et al. 2006; Wainszelbaum et al. 2008). It is interesting that the copy-number polymorphism of one of these genes (NPBF23) has recently been implicated in pediatric neuroblastoma, with certain gene family members showing preferential expression in fetal brain and fetal sympathetic nervous tissue (Diskin et al. 2009).
PRIMATE SEQUENCE CHARACTERIZATION OF LCR16A
Detailed comparative primate sequencing of one of the core duplicons (LCR16a—seat of the NPIP/morpheus gene family expansion) is illustrative of the evolutionary dynamism that occurred during the human–great ape evolution. In the human genome reference sequence, there are 23 copies of the LCR16a sequence distributed among 17 complex duplication blocks ranging in size from ~40 to 609 kbp (Figs. 4 and 5). In addition to LCR16, 11 additional SDs of distinct evolutionary origin populate the duplication blocks on chromosome 16. Although the 20-kbp LCR16a occasionally occurs as a solitary duplicon (i.e., without flanking duplicons), almost all other LCR16 elements occur in association with the LCR16a core duplicon. Phylogenetic reconstruction indicates that the flanking duplicons duplicated more recently have accumulated at the periphery of LCR16a duplications, leading to the formation of the complicated duplication blocks now observed in the human genome. Comparative sequence analysis in macaque and baboon (Old World outgroup species) reveals that each of the SDs originated as a single-copy sequence on chromosome 16 (Fig. 4). Remarkably, bacterial artificial chromosome (BAC)-based sequencing of LCR16a elements in the orangutan shows that the LCR16a core duplicon has duplicated independently and to nonorthologous locations when compared to human and African great apes. Moreover, the LCR16a has colonized chromosome 13 in the orangutan and has accumulated its own set of orangutan-specific flanking SDs on the periphery. Most of these flanking duplicons are single copy in humans and African great ape genomes. These data suggest that the LCR16a core duplicon has an inherent proclivity to duplicate and has served to prime lineage-specific duplications contributing to the emergence of large duplication blocks in both lineages. Thus, two independent bursts of the LCR16a have occurred in the last 12 million years in two different ape lineages.
Figure 4.

Comparative schematic showing the distribution of LCR16 duplications. Color bars shows LCR16 duplicons. In human, the LCR16a core duplicon (red) is present within most duplication blocks on chromosome 16; all corresponding duplications are single copy in baboon, but in orangutan, LCR16a exists at nonorthologous locations and on different chromosomes (chromosome 13) in association with a new suite of orangutan-specific duplications at the periphery. Map locations are numbered according to the human reference with ancestral locations flagged by an asterisk.
Figure 5.

SDs and disease. Detailed duplicon composition of duplication blocks are shown along an ideogram of human chromosome 16. Duplications mediating recurrent deletions and duplications associated with disease are indicated (Ballif et al. 2007; Jiang et al. 2007; Ullmann et al. 2007; Kumar et al. 2008; Weiss et al. 2008; Hannes et al. 2009).
DISEASE CONSEQUENCES AND COPY-NUMBER VARIATION
Similar to Bridges and Muller’s Bar locus, the presence of these large, high-identity duplications predisposes to recurrent deletions and duplications as a result of unequal crossing-over events during meiosis and/or mitosis. Not surprisingly, SDs are significantly enriched for copy-number polymorphisms (Iafrate et al. 2004; Sharp et al. 2005; Redon et al. 2006) with most of the genic copy-number polymorphisms mapping to these regions of the genome (Cooper et al. 2007; Bailey et al. 2008). The fact that so many of these duplications are interspersed, however, is double jeopardy for humans and its most closely related ape species. An unequal crossover event between two directly oriented duplications separated by a unique gene-rich region of the genome means that both the duplicated sequence and the unique sequence are subjected to copy-number variation (Lupski 1998). Nearly 10% of human euchromatin maps to ~110 such hot-spot regions of the genome, which is now sensitized to recurrent copy-number changes due to the evolution of this genomic architecture. More than 30 of these regions have been associated with both syndromic and complex diseases (Stankiewicz et al. 2004; Lupski 2007; Mefford and Eichler 2009). Interestingly, the majority of the pathogenic rearrangements involve neurocognitive and neurobehavioral diseases including intellectual disability, developmental delay, autism, schizophrenia, and epilepsy. Ironically, the breakpoints of many of these disease-causing rearrangements map to the same duplication blocks carrying core duplicons that emerged specifically within the human–great ape lineage (Tables 1 and 2). Although most of these large-scale copy-number changes appear to be under strong negative selection (Itsara et al. 2009), there is also evidence that SD-mediated rearrangements, such as the inversion on 17q21.31, may be positively selected, resulting in increased fecundity in specific human populations (Stefansson et al. 2005).
Table 1.
Core duplicons and disease-causing rearrangements
| Core | Locus | Phenotypea |
|---|---|---|
| NPIP | 16p11.2 | autism (1%), ID (0.6%) |
| NPIP | 16p13.1 | nonsyndromic ID (1%) |
| GLP/GOLGA -like protein | 15q11.2 | PW/AS, autism (1%) |
| GLP/GOLGA -like protein | 15q13.3 | epilepsy (1%), autism/ID (0.3%), schizophrenia (0.2%) |
| GLP/GOLGA -like protein | 15q24 | rare autism spectrum disorder |
| LRRC37 | 17q21.31 | 0.5% European ID syndrome |
| TBC1D3 | 17q12 | renal cyst and diabetes (RCAD) |
| TBC1D3 | 17p11.2 | Smith Magenis syndrome |
| NPBF | 1q21.1 | ID (0.5%), schizophrenia (0.3%), congenital heart defects |
ID indicates intellectual disability and developmental delay.
Table 2.
Examples of genes/gene families mapped to the most representative core duplication
| Duplicon clade | RefSeq gene | Gene name | Significant expression | Subcellular localization | Description | Possible function | Cancer association | Refs. |
|---|---|---|---|---|---|---|---|---|
| chr1 | NM_183372 | NBPF11 | soft tissue | cytoplasm | neuroblastoma breakpoint gene family, DUF1220 | unknown | neuroblastoma | 1,2 |
| chr2 | NM_005054 | RANBP2 | testis | nuclear pore | RANBP2-like and GRIP domain-containing 5 isoform | Ran GTPase binding | highly expressed in leukemia | 3 |
| chr7_2 | NM_174930 | PMS2L5 | ubiquitous | nuclear | postmeiotic segregation increased 2-like 5 | DNA mismatch repair | unknown | 4 |
| chr15 | NM_001012423 | GLP | exclusively | unknown in testis | golgin-like protein, golgi autoantigen, golgin subfamily a, 8E (GOLGA8E) | DNA binding | unknown | 5,6 |
| chr16 | NM_006985 | NPIP | ubiquitous | nuclear membrane | nuclear pore complex interacting protein, morpheus gene family | Nuclear-pore- associated | highly expressed | 7 |
| chr17_1 | NM_001006607 | LRRC37B | ubiquitous | unknown | leucine-rich repeat, c114 SLIT-like testicular protein | ATP-dependent peptidase activity | unknown | 8 |
| chr17_2 | NM_001001418 | TBC1/USP6 | testis | unknown | TBC1 domain family member 3C | GTPase activator activity | highly expressed in lymphoma | 9 |
CONCLUSIONS
Both experimental and computational data support an acceleration of SDs in the common ancestor of humans and African great apes. This apparent burst in mutational process occurred at a time when most other mutational processes such as single base pair substitutions experienced a slowdown. At a base per base level, SDs contribute to more genetic variation than single base pair changes. SDs have restructured great ape and human chromosomes, creating complex lineage-specific duplication blocks distributed throughout specific chromosomes where novel gene structures have been formed by shuffling and juxtaposition of different exon cassettes. Much of the intra-chromosomal duplication acceleration is centered around core duplicons that are also the seats of rapidly evolving genes that have expanded in the human and African great ape lineage. The concomitant large blocks of SDs are now predisposing to recurrent rearrangements that are associated with intellectual disability, autism, and schizophrenia. We hypothesize that the negative selection of disease-causing microdeletions and microduplications is balanced by positive selection of newly minted gene families embedded in cores and distributed to new locations. Elucidating the function of the genes embedded within the core duplicons remains an unmet challenge of human genetics and evolutionary biology.
Acknowledgments
We thank Lin Chen, Ze Cheng, Santhosh Girirajan, and Tonia Brown for valuable comments and help in the preparation of this manuscript. This work was supported, in part, by National Institutes of Health grants GM-058815 and HG002385 to E.E.E. T.M.-B. is supported by a Marie Curie fellowship. E.E.E. is an investigator of the Howard Hughes Medical Institute.
References
- Bailey JA, Kidd JM, Eichler EE. Human copy number polymorphic genes. Cytogenet Genome Res. 2008;123:234–243. doi: 10.1159/000184713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballif BC, Hornor SA, Jenkins E, Madan-Khetarpal S, Surti U, Jackson KE, Asamoah A, Brock PL, Gowans GC, Conway RL, et al. Discovery of a previously unrecognized microdeletion syndrome of 16p11.2–p12.2. Nat Genet. 2007;39:1071–1073. doi: 10.1038/ng2107. [DOI] [PubMed] [Google Scholar]
- Bridges CB. The Bar “gene”—A duplication. Science. 1936;83:210–211. doi: 10.1126/science.83.2148.210. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Ventura M, She X, Khaitovich P, Graves T, Osoegawa K, Church D, DeJong P, Wilson RK, Paabo S, et al. A genome-wide comparison of recent chimpanzee and human segmental duplications. Nature. 2005;437:88–93. doi: 10.1038/nature04000. [DOI] [PubMed] [Google Scholar]
- Church DM, Goodstadt L, Hillier LW, Zody MC, Goldstein S, She X, Bult CJ, Agarwala R, Cherry JL, DiCuccio M, et al. Lineage-specific biology revealed by a finished genome assembly of the mouse. PLoS Biol. 2009;7:e1000112. doi: 10.1371/journal.pbio.1000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli FD, von Mering C, Suyama M, Harrington ED, Izaurralde E, Bork P. Complex genomic rearrangements lead to novel primate gene function. Genome Res. 2005;15:343–351. doi: 10.1101/gr.3266405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Lander ES, Rogers J, Waterston RH, et al. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- Consortium (Chimpanzee Sequencing and Analysis Consortium) Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- Cooper GM, Nickerson DA, Eichler EE. Mutational and selective effects on copy-number variants in the human genome. Nat Genet. 2007;39:S22–S29. doi: 10.1038/ng2054. [DOI] [PubMed] [Google Scholar]
- Courseaux A, Richard F, Grosgeorge J, Ortola C, Viale A, Turc-Carel C, Dutrillaux B, Gaudray P, Nahon JL. Segmental duplications in euchromatic regions of human chromosome 5: A source of evolutionary instability and transcriptional innovation. Genome Res. 2003;13:369–381. doi: 10.1101/gr.490303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diskin SJ, Hou CP, Glessner JT, Attiyeh EF, Laudenslager M, Bosse K, Cole K, Mosse YP, Wood A, Lynch JE, et al. Copy number variation at 1q21.1 associated with neuroblastoma. Nature. 2009;459:987–991. doi: 10.1038/nature08035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas L, Kim YH, Karimpour-Fard A, Cox M, Hopkins J, Pollack JR, Sikela JM. Gene copy number variation spanning 60 million years of human and primate evolution. Genome Res. 2007;17:1266–1277. doi: 10.1101/gr.6557307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler EE. Segmental duplications: What’s missing, mis-assigned, and misassembled—and should we care? Genome Res. 2001;11:653–656. doi: 10.1101/gr.188901. [DOI] [PubMed] [Google Scholar]
- Eichler EE, Budarf ML, Rocchi M, Deaven LL, Doggett NA, Baldini A, Nelson DL, Mohrenweiser HW. Inter chromosomal duplications of the adrenoleukodystrophy locus: A phenomenon of pericentromeric plasticity. Hum Mol Genet. 1997;6:991–1002. doi: 10.1093/hmg/6.7.991. [DOI] [PubMed] [Google Scholar]
- Elsik CG, Tellam RL, Worley KC, Gibbs RA, Muzny DM, Weinstock GM, Adelson DL, Eichler EE, Elnitski L, Guigo R, et al. The genome sequence of taurine cattle: A window to ruminant biology and evolution. Science. 2009;324:522–528. doi: 10.1126/science.1169588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortna A, Kim Y, MacLaren E, Marshall K, Hahn G, Meltesen L, Brenton M, Hink R, Burgers S, Hernandez-Boussard T, et al. Lineage-specific gene duplication and loss in human and great ape evolution. PLoS Biol. 2004;2:E207. doi: 10.1371/journal.pbio.0020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles F, Goy A, Remache Y, Manova K, Zelenetz AD. Cloning and characterization of a Golgin-related gene from the large-scale polymorphism linked to the PML gene. Genomics. 2000;70:364–374. doi: 10.1006/geno.2000.6388. [DOI] [PubMed] [Google Scholar]
- Hahn MW, Demuth JP, Han SG. Accelerated rate of gene gain and loss in primates. Genetics. 2007;177:1941–1949. doi: 10.1534/genetics.107.080077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannes FD, Sharp AJ, Mefford HC, de Ravel T, Ruivenkamp CA, Breuning MH, Fryns JP, Devriendt K, Van Buggenhout G, Vogels A, et al. Recurrent reciprocal deletions and duplications of 16p13.11: The deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J Med Genet. 2009;46:223–232. doi: 10.1136/jmg.2007.055202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodzic D, Kong C, Wainszelbaum MJ, Charron AJ, Su XO, Stahl PD. TBC1D3, a hominoid oncoprotein, is encoded by a cluster of paralogues located on chromosome 17q12. Genomics. 2006;88:731–736. doi: 10.1016/j.ygeno.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Horii A, Han HJ, Sasaki S, Shimada M, Nakamura Y. Cloning, characterization and chromosomal assignment of the human genes homologous to yeast PMS1, a member of mismatch repair genes. Biochem Biophys Res Commun. 1994;204:1257–1264. doi: 10.1006/bbrc.1994.2598. [DOI] [PubMed] [Google Scholar]
- Horvath JE, Schwartz S, Eichler EE. The mosaic structure of human pericentromeric DNA: A strategy for characterizing complex regions of the human genome. Genome Res. 2000;10:839–852. doi: 10.1101/gr.10.6.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, Krauss RM, Myers RM, Ridker PM, Chasman DI, et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Tang H, Ventura M, Cardone MF, Marques-Bonet T, She X, Pevzner PA, Eichler EE. Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nat Genet. 2007;39:1361–1368. doi: 10.1038/ng.2007.9. [DOI] [PubMed] [Google Scholar]
- Johnson ME, Viggiano L, Bailey JA, Abdul-Rauf M, Goodwin G, Rocchi M, Eichler EE. Positive selection of a gene family during the emergence of humans and African apes. Nature. 2001;413:514–519. doi: 10.1038/35097067. [DOI] [PubMed] [Google Scholar]
- Johnson ME, Cheng Z, Morrison VA, Scherer S, Ventura M, Gibbs RA, Green ED, Eichler EE. Recurrent duplication-driven transposition of DNA during hominoid evolution. Proc Natl Acad Sci. 2006;103:17626–17631. doi: 10.1073/pnas.0605426103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH, Dobyns WB, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- Laureys G, Speleman F, Opdenakker G, Benoit Y, Leroy J. Constitutional translocation t(1;17)(P36;Q12–21) in a patient with neuroblastoma. Genes Chromosome Cancer. 1990;2:252–254. doi: 10.1002/gcc.2870020315. [DOI] [PubMed] [Google Scholar]
- Li WH, Tanimura M. The molecular clock runs more slowly in man than in apes and monkeys. Nature. 1987;326:93–96. doi: 10.1038/326093a0. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Genomic rearrangements and sporadic disease. Nat Genet. 2007;39:S43–S47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- Marques-Bonet T, Kidd JM, Ventura M, Graves TA, Cheng Z, Hillier LW, Jiang ZS, Baker C, Malfavon-Borja R, Fulton LA, et al. A burst of segmental duplications in the genome of the African great ape ancestor. Nature. 2009;457:877–881. doi: 10.1038/nature07744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Eichler EE. Duplication hotspots, rare genomic disorders, and common disease. Curr Opin Genet Dev. 2009;19:196–204. doi: 10.1016/j.gde.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ, Prokofjeva-Belgovskaja AA, Kossikov KV. Unequal crossing-over in the bar mutant as a result of duplication of a minute chromosome section. C R Acad Sci USSR. 1936;2:87–88. [Google Scholar]
- Nicholas TJ, Cheng Z, Ventura M, Mealey K, Eichler EE, Akey JM. The genomic architecture of segmental duplications and associated copy number variants in dogs. Genome Res. 2009;19:491–499. doi: 10.1101/gr.084715.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S, Wolf U, Atkin NB. Evolution from fish to mammals by gene duplication. Hereditas. 1968;59:169–187. doi: 10.1111/j.1601-5223.1968.tb02169.x. [DOI] [PubMed] [Google Scholar]
- Ota T, Suzuki Y, Nishikawa T, Otsuki T, Sugiyama T, Irie R, Wakamatsu A, Hayashi K, Sato H, Nagai K, et al. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat Genet. 2004;36:40–45. doi: 10.1038/ng1285. [DOI] [PubMed] [Google Scholar]
- Paulding CA, Ruvolo M, Haber DA. The Tre2 (USP6) oncogene is a hominoid-specific gene. Proc Natl Acad Sci. 2003;100:2507–2511. doi: 10.1073/pnas.0437015100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popesco MC, Maclaren EJ, Hopkins J, Dumas L, Cox M, Meltesen L, McGavran L, Wyckoff GJ, Sikela JM. Human lineage-specific amplification, selection, and neuronal expression of DUF1220 domains. Science. 2006;313:1304–1307. doi: 10.1126/science.1127980. [DOI] [PubMed] [Google Scholar]
- Pujana MA, Nadal M, Guitart M, Armengol L, Gratacos M, Estivill X. Human chromosome 15q11-q14 regions of rearrangements contain clusters of LCR15 duplicons. Eur J Hum Genet. 2002;10:26–35. doi: 10.1038/sj.ejhg.5200760. [DOI] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouquier S, Taviaux S, Trask BJ, Brand-Arpon V, van den Engh G, Demaille J, Giorgi D. Distribution of olfactory receptor genes in the human genome. Nat Genet. 1998;18:243–250. doi: 10.1038/ng0398-243. [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She XW, Jiang ZX, Clark RL, Liu G, Cheng Z, Tuzun E, Church DM, Sutton G, Halpern AL, Eichler EE. Shotgun sequence assembly and recent segmental duplications within the human genome. Nature. 2004;431:927–930. doi: 10.1038/nature03062. [DOI] [PubMed] [Google Scholar]
- She X, Cheng Z, Zöllner S, Church DM, Eichler EE. Mouse segmental duplication and copy number variation. Nat Genet. 2008;40:909–914. doi: 10.1038/ng.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Shaw CJ, Withers M, Inoue K, Lupski JR. Serial segmental duplications during primate evolution result in complex human genome architecture. Genome Res. 2004;14:2209–2220. doi: 10.1101/gr.2746604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J, Baker A, Jonasdottir A, Ingason A, Gudnadottir VG, et al. A common inversion under selection in Europeans. Nat Genet. 2005;37:129–137. doi: 10.1038/ng1508. [DOI] [PubMed] [Google Scholar]
- Sturtevant AH. The effects of unequal crossing over at the bar locus in Drosophila. Genetics. 1925;10:117–147. doi: 10.1093/genetics/10.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzun E, Bailey JA, Eichler EE. Recent segmental duplications in the working draft assembly of the brown Norway rat. Genome Res. 2004;14:493–506. doi: 10.1101/gr.1907504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullmann R, Turner G, Kirchhoff M, Chen W, Tonge B, Rosenberg C, Field M, Vianna-Morgante AM, Christie L, Krepischi-Santos AC, et al. Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum Mutat. 2007;28:674–682. doi: 10.1002/humu.20546. [DOI] [PubMed] [Google Scholar]
- Wainszelbaum MJ, Charron AJ, Kong C, Kirkpatrick DS, Srikanth P, Barbieri MA, Gygi SP, Stahl PD. The hominoid-specific oncogene TBC1D3 activates ras and modulates epidermal growth factor receptor signaling and trafficking. J Biol Chem. 2008;283:13233–13242. doi: 10.1074/jbc.M800234200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Weiss LA, Shen YP, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MAR, Green T, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Wu CI, Li WH. Evidence for higher rates of nucleotide substitution in rodents than in man. Proc Natl Acad Sci. 1985;82:1741–1745. doi: 10.1073/pnas.82.6.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]