

Abstract
VIP is highly expressed in the colon and regulates motility, vasodilatation, and sphincter relaxation. However, its role in the development and progress of colitis is still controversial. Our aim was to determine the participation of VIP on dextran sodium sulfate (DSS)-induced colonic mucosal inflammation using VIP−/− and WT mice treated with VIP antagonists. Colitis was induced in 32 adult VIP−/− and 14 age-matched WT litter-mates by giving 2.5 % DSS in the drinking water. DSS-treated WT mice were injected daily with VIP antagonists, VIPHyb (n=22), PG 97–269 (n=9), or vehicle (n=31). After euthanasia, colons were examined; colonic cytokines mRNA were quantified. VIP−/− mice were remarkably resistant to DSS-induced colitis compared to WT. Similarly, DSS-treated WT mice injected with VIPHyb (1 μM) or PG 97–269 (1 nM) had significantly reduced clinical signs of colitis. Furthermore, colonic expression of IL-1, TNF-α, and IL-6 was significantly lower in VIP−/− and VIPHyb or PG 97–269 compared to vehicle-treated WT. Genetic deletion of VIP or pharmacological inhibition of VIP receptors resulted in resistance to colitis. These data demonstrate a pro-inflammatory role for VIP in murine colitis and suggest that VIP antagonists may be an effective clinical treatment for human inflammatory bowel diseases.
Keywords: VIP, Colitis, VIP antagonist: IBD
Introduction
The enteric nervous system (ENS) modulates intestinal inflammation through neuropeptides acting on immune and central nervous systems (CNS) (Gross 2007). Vasoactive intestinal peptide (VIP), a 28-amino acid neuropeptide is widely distributed in central and peripheral neurons and is expressed in the colon with the highest concentration in the myenteric plexus (Harmar 2012). VIP exhibits broad physiological intestinal functions, regulating motility, secretory activity and vasodilatation, and inhibiting peristaltic reflex in the circular smooth muscle layer and sphincter relaxation (Harmar 2012). In the immune system, VIP triggers multiple complex effects through VPAC1 and VPAC2 receptors, which are expressed on T-cells and macrophages (Delgado 1996; Delgado et al. 2004a, b) and less consistently on dendritic cells, mast cells, and neutrophils (Delgado 2004a, b).
VIP is up-regulated in the peritoneal fluid during LPS-induced inflammation and inhibits LPS-induced TNF-α, IL-6, and IL-12 production (Delgado et al. 1999a, b). Inflammatory stimuli and cytokines can induce VIP expression in neurons and antigen-activated CD4 (Delgado 1999a, b, 2004a, b) cells. Similarly, endotoxic shock in humans elevated levels of VIP in plasma (Brandtzaeg 1989). Patients with multiple sclerosis have reportedly increased levels of VIP immunoreactivity in their cerebral spinal fluid (Andersen 1984). Furthermore, patients with Sjögren’s syndrome, rheumatoid arthritis, and Crohn’s disease have altered levels of VIP (Törnwall 1994; Belai 1997; Boyer 2007; Juarranz 2008). Administration of VIP following murine endotoxic shock was reported to lower inflammation (Delgado 2004a, b) while VIP and its analogs have been proposed as therapeutic agents in patients with chronic inflammatory and autoimmune diseases (Delgado 2004a, b).
The role of VIP in inflammatory bowel diseases (IBD) has been very controversial and not clearly defined. In murine TNBS-induced colitis some authors have shown that intraperitoneal (ip) VIP was protective against mucosal inflammation by inhibiting pro-inflammatory cytokines and downregulating Toll-like receptors 2 and 4 (Abad 2003). Others have demonstrated that prophylactic or therapeutic treatment with VIP by ip injection or continual infusion did not ameliorate colitis-induced weight loss, mortality, inflammatory cytokine response, and histological damage, even though it abrogated chemokine-induced chemotaxis (Newman 2005).
Recently, genetically engineered mouse models have allowed the characterization of the VIP pathway in inflammatory models. VIP−/− mice were resistant to experimental autoimmune encephalomyelitis (EAE) with reduced immune infiltrates in the brain parenchyma and spinal cord (Waschek 2013). VIP−/− mice were also resistant to LPS-induced shock (Waschek 2013) suggesting a functional deficit of myeloid cells, which are responsible for the elevated levels of TNF-a, IL-6, and IL-12. Furthermore, VPAC1-null mice were resistant to dextran sodium sulfate (DSS)-induced colitis, whereas VPAC2-null mice developed a more severe colitis (Yadav 2011). To study the pharmacological effects of VIP signaling, peptides with modified VIP sequences have been developed. VIPHyb, in which the first six C-terminal amino acids were replaced with the neurotensin sequence, is a broad spectrum VIP antagonist inhibiting human and mouse VPAC1, VPAC2, and PAC1 receptors (Moody 2002). VIPHyb has been shown to inhibit the growth of tumor cells of lung, breast, and pancreatic cancers (Moody et al. 2003; Zia 1996 ; Zia 2000). On T lymphocytes, VIPHyb causes a half-maximal inhibition of VIP binding at 5 mM, and maximal inhibition of VIP-induced cAMP generation at 10mM(Gozes 1991). Another VIP antagonist, PG 97–269, selectively inhibits only VPAC1 receptors (Banks 2005).
In the present study, we examined the importance of VIP deficiency and the therapeutic effects of VIP receptor antagonists in the DSS model of colitis. Consistent with the attenuation of inflammation in VIP−/− models of EAE and LPS-induced shock, VIP−/− mice with DSS-induced colitis exhibited reduced clinical signs and mucosa damage together with reduced or absent mRNA expression of proinflammatory cytokines in colonic tissue. To investigate whether pharmacological antagonists to VIP can play similar anti-inflammatory effects in the colonic mucosa, an ip treatment with VIPHyb or PG 97–269 was utilized in C57BL/6 WT mice using the same experimental models of DSS colitis.
Materials and Methods
Animals
Male VIP knockout C57BL/6J mice (VIP−/−) 6–12 weeks old, developed as described (Colwell 2003), and age-matched wild type (WT) were housed and fed ad libitum in a specific sterile animal facility at the Department of Veteran Affairs Greater Los Angeles Healthcare System (WLAVA) animal facility. All experimental protocols and procedures were approved by the WLAVA-IACUC and Research and Development Committee (11030-11).
Dextran Sodium Sulfate-Induced Colitis
To induce colitis, 2.5 % (wt/vol) DSS (MW, 40,000–50,000; USB Corp, Cleveland, OH) was given in drinking water as outlined in Fig. 1a (Melgar 2005). VIP−/− (n=32) and WT (n=14) mice were allowed free access to DSS-containing or normal water during the experiment. Body weight was recorded from day 0 to11. Control VIP−/− (n=25) and WT (n=16) mice were given normal water from day 0 to 11. The volumes of DSS containing- or normal water in the bottles were monitored. Stool appearance, physical activity, and general well-being were assessed daily from day 0 to 11.
Fig. 1.
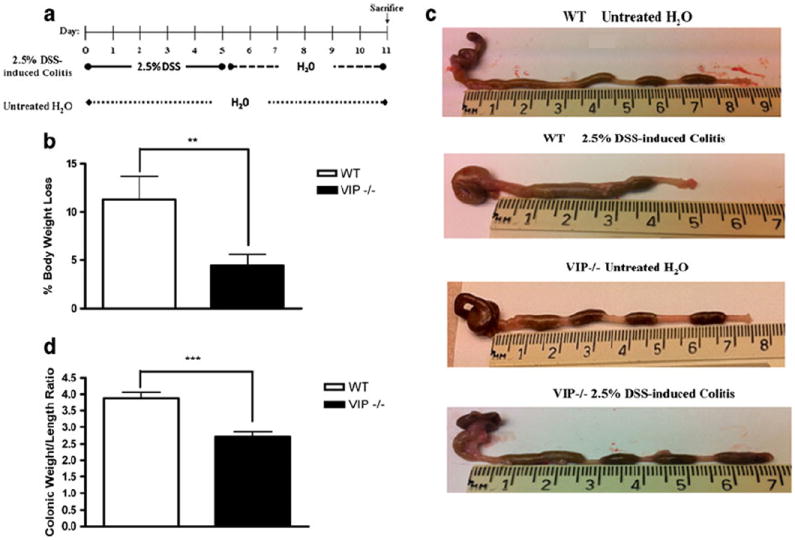
a Diagram of the experimental DSS-induced colitis study protocol: 2.5 % DSS in drinking water was administered from day 0 to 5, followed by drinking water from day 6 to 11. Mice were euthanized on day 11. b Body weight change percentage was calculated in DSS-treated WT (white) and VIP−/− (black) mice. Data expressed as mean±SEM. c Photographs of large intestine, oriented from cecum to rectum. Control WT (no DSS; top panel), DSS-treated WT (second panel from top), control VIP−/− (no DSS; third panel from top), DSS-treated VIP−/− (bottom panel). d Colonic weight/length ratio was measured in WT (white) and VIP−/− (black) mice. Data expressed as mean±SEM. Statistical analysis was performed using two-way ANOVA with Bonferroni post-test to calculate p values. Significant resistance to colitis, as established by percent of weight loss (p<0.01) and colonic weight/length ratio (p<0.001) were observed in DSS-treated VIP−/− as compared to WT mice. *p<0.05, **p<0.01, ***p<0.001
VIP Antagonists Treatment
VIP inhibitors, [Lys1, Pro 2, 5, Arg3, 4, Tyr6] VIP (VIPHyb) (American Peptides, Sunnyvale, CA), or [acetyl-] VIP(3–7)/His1, D-Phe2, Lys15, Arg16, Leu17GRF(8–27) (PG 97–269), VPAC1 receptor-specific antagonist (SM Biochemicals, Anaheim, CA) were injected daily from day 0 to 11, by ip in sterile saline in DSS-treated WT mice. Age- and weight-matched WT (6–12 weeks old) were administered VIPHyb at 100 nM (n=9) or 1 μM (n=22) or PG 97–269 at 1 nM (n=8), 10 nM (n=7), or 100 nM (n=9) in 200 μL of saline, as outlined in Fig. 2a or saline (n=31) from day 0 to 11.
Fig. 2.
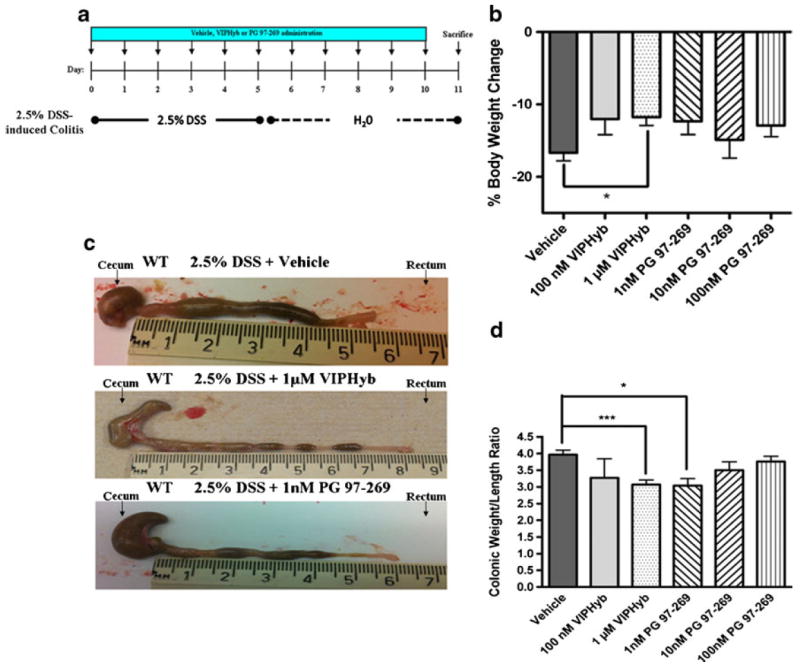
a Diagram of the experimental protocol followed to study the effects of VIP antagonists, VIPHyb or PG 97–269, or vehicle, in DSStreated WT mice. b Percent of body weight change in mice treated with DSS and daily ip injections with vehicle (saline), or 100 nM VIPHyb, or 1 μM VIPHyb, or 1 nM PG 97–269, or 10 nM PG 97–269, or 100 nM PG 97–269. Significant resistance to colitis was documented by assessing percentage of body weight loss in mice injected with 1 μM VIPHyb compared to vehicle (p<0.01). Data expressed as mean±SEM. c Photographs of large intestine oriented from cecum to rectum. WT mice treated with DSS and vehicle (top panel), WT treated with DSS and 1 μM VIPHyb (second from top panel), WT treated with DSS and 1 nM PG 97–269 (bottom panel). d Colonic weight/length ratio in DSS-treated WT mice injected with vehicle, or 100 nM VIPHyb, or 1 μM VIPHyb, or 1 nM PG 97–269, or 10 nM PG 97–269, or 100 nM PG 97–269. Significant resistance to colitis, assessing colonic weight/length ratio was observed in 1 μM VIPHyb- and 1 nM PG 97–269-treated WT (p<0.001). Data are expressed as mean±SEM. Statistical analysis was performed using one-way ANOVAwith Bonferroni post-test to calculate the p values. *p<0.05, **p<0.01, ***p<0.001
Assessment of Histologic Injury Scores
After euthanasia, colons were removed and colonic weight to length ratio, a parameter of mucosal inflammation (Okayasu 1990) was measured. Then, tissue segments were placed in 4 % paraformaldehyde or saved for RNA extraction.
Five micron paraffin sections, stained by H&E protocol, were analyzed in blinded fashion to assess colitis using the following criteria: edema, crypt shortening, ulceration, and immune-cellular infiltration. The proximal and distal colon of each animal was graded using a scale from 0 to 4, with 0 indicating no sign and 4 being most severe.
Cytokine Analysis by Quantitative RT-PCR
Cytokine expression was measured in homogenized colonic tissue biopsies immediately following necropsy. Total RNA was isolated using RNeasy kit (Qiagen, Valencia, CA) according to manufacturer’s instructions. RNA yield and purity were assessed by spectrometric measurements. RNA was applied for reverse transcriptase (RT) reaction using Superscript first-strand synthesis kit (Invitrogen, Carlsbad, CA). One microgram of RT product was then added in a reaction with SYBR premix Taq (Takara Bio, Shiga, Japan) for qPCR, which was performed on the DNA Engine Opticon 2 system (MJ Research, Waltham, MA). The cycle of threshold, C(T), was determined as the fluorescent signal of 1 standard deviation over background. The efficiency of each primer pair was measured by amplification of the known amount of cDNA starting material (10 pg–10 ng). IL-6, tumor necrosis factor α (TNF-α), IL-1β, IL-2, IL-23A, and T-bet target primer pair amplifications were compared with the reference primer pair (α-actin) amplification in the same experiment for each RT product tested. All reactions were carried out in duplicate. The relative expression ratio of the target gene, compared to the reference gene, was calculated according to the Pfaffl’s formula (Schmittgen 2008). Melting curves established the purity of the amplified band. PCR products were sequenced to confirm identity. Primer pair sequences and melting temperatures are listed in Supplemental Table 1.
Statistical Analysis
Data are expressed as mean±SEM. The statistical significance of differences between VIP−/−, WT, VIP antagonists, and vehicle-treated groups were determined by factorial ANOVA. Statistical analyses were performed using the SPSS software version-20 (IBM, Armonk, NY).
Results
Clinical and Macroscopic Evidence of VIP−/− Mice Resistance to Colitis
Colitis was induced in C57BL/6J WTor VIP−/− mice as outlined (Fig. 1a). In control groups, no change in physical activity or stool appearance was recorded. DSS-treated WT mice developed colitis with bloody diarrhea, altered physical behavior, and significant body weight loss percentage (−11.33±3.39, p<0.01). Conversely, DSS-treated VIP−/− presented no clinical alterations and a modest body weight loss percentage (−4.47±1.15; Fig. 1b).
Upon necropsy, severe colonic inflammation was observed only in DSS-treated WT (Fig. 1c). Conversely, DSS-treated and control VIP−/− showed no macroscopic sign of colitis (Fig. 1c). Colonic weight/length ratio, correlating with the clinical observations, was higher in DSS-treated WT than VIP−/− mice (3.88±0.17 vs. 2.71±0.14, p<0.0001), thus indicating greater tissue edema and immune-cellular infiltrates in WT than VIP−/−. Therefore, VIP absence resulted in a significantly reduced colitis severity. No difference was found in the colonic weight/length ratio of WT and VIP−/− controls.
VIP Antagonists, VIPHyb, and PG 97–269 Attenuate DSS-Induced Colitis
DSS-treated WT mice were administered daily ip injections of either increasing doses of VIP antagonists or vehicle (Fig. 2a). WT mice injected with vehicle only, manifested clinical signs of severe colitis with bloody diarrhea, reduced physical activity, and a percentage of body weight loss of (−16.69±1.13). One hundred nanomoles VIPHyb reduced weight loss (−12.05±2.16, p>0.05); however 1 μM VIPHyb treatment led to a significant reduction of weight loss percentage (−11.78±1.14, p<0.05) as well as bloody diarrhea, compared to vehicle-treated mice (Fig. 2b). To confirm that VIP antagonism ameliorates colitis, we next treated another group of DSS-induced WT mice with daily ip injections of VPAC1 antagonist, PG 97–269, at a dose of 1, 10, or 100 nM from day 1 to 11. PG 97–269 at 1 nM led to a weight loss improvement (−12.35±1.80) with absence of bloody diarrhea compared to the vehicle-treated group (Fig. 2b). PG 97–269 at 100 and 10 nM resulted in a lower weight loss percentage (−12.99±1.53 and −14.89±2.53 respectively) that was not statistically significant.
The degree of colonic inflammation in DSS-treated WT mice injected with VIPHyb or PG 97–269 was evaluated according to the colonic weight/length ratio. DSS-treated WT mice, vehicle-injected daily, presented significant intestinal inflammation with a colonic weight/length ratio of 3.97±0.13 (Fig. 2c, d). Treatment with VIPHyb at 1 μM gave a significantly lower colonic weight/length ratio (3.07±0.14, p<0.001; Fig. 2c, d). One hundred nanomoles VIPHyb resulted in lower inflammatory clinical signs with no significant reduction of colonic weight/length ratio compared to vehicle. One nanomole PG 97–269 was the most effective dose against colonic inflammation, producing a significantly reduced colonic weight/length ratio (3.04±0.21, p<0.05; Fig. 2c, d). PG 97–269 at 10 and 100 nM reduced clinical signs of colonic inflammation but not the weight/length ratio. Therefore, in our model of DSS-induced colitis in WT, treatment with VIPHyb at 1 μM or with PG 97–269 at 1 nM induced a significant resistance to colonic inflammation as seen in VIP−/− mice.
Histology of the Colon in DSS-Treated VIP−/− and VIP Antagonists-Treated WT Mice
Proximal colon sections from DSS-treated WT indicated the presence of significant inflammation, edema, and ulceration, with shortening of the normal crypt architecture compared to control WT mice (Fig. 3). However, in DSS-treated VIP−/− mice no microscopic damage was observed (Fig. 3). In the proximal colon, the histological score mirrored the clinical signs, with VIP−/− mice scoring significantly lower in edema (1.00±0.29 vs. 3.0±0.58, p<0.05), villi shortening (0±0 vs. 2.33±0.33, p<0.01), ulceration (0±0 vs. 2.33±0.33, p<0.01), and presence of immune-cellular infiltrates (0.78±0.22 vs. 2.67 ±0.33, p<0.05; Table 1). Similarly, in DSS-treated WT mice injected with 1 μM VIPHyb or 1 nM PG 97–269, lower colonic mucosal damage was observed compared to vehicle-injected mice (Fig. 3). Treatment with 1 μM VIPHyb resulted in significantly lower scoring compared to the vehicle-group considering edema (1.33±0.21 vs. 2.80±0.39, p<0.05), villi shortening (0.33±0.21 vs. 2.20±0.53, p<0.01), ulceration (0.33±0.21 vs. 2.30±0.54, p<0.001), and immune-cellular infiltrates (1.50 ±0.22 vs. 2.90±0.38, p<0.05; Table 1). Similarly, treatment with PG 97–269 at 1 nM, the most effective dose, significantly lowered inflammatory scores compared to vehicle when villi shortening (0.83±0.40, p<0.05), ulceration (0.83±0.40, p<0.05) and immune-cellular infiltrates (1.67±0.33, p<0.05; Table 1) were evaluated. Higher doses of PG 97–269 were excluded for lack of efficacy. Thus, VIP−/− demonstrated resistance to colitis as shown by their relative absence of tissue damage compared to WT mice.
Fig. 3.
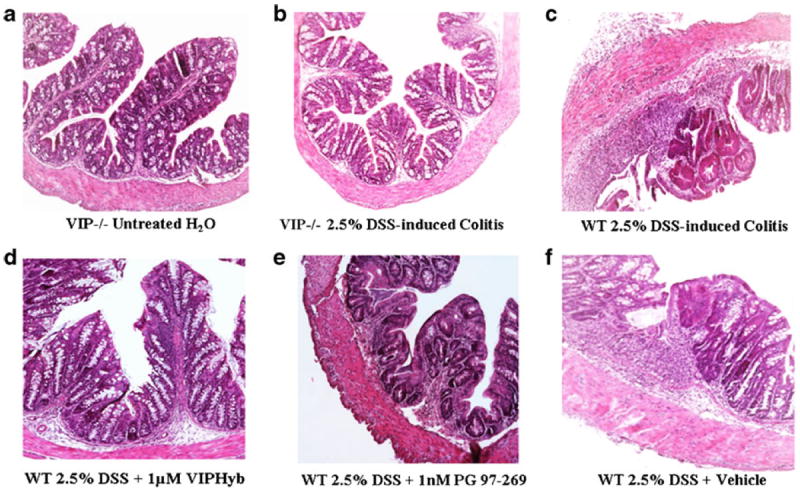
Proximal colon histopathology. a DSS-untreated VIP−/−. b 2.5 % DSS-treated mice VIP−/−; c 2.5 % DSS-treated WT mice. d WT mice treated with 2.5 % DSS and 1 μM VIPHyb. e WT mice treated with 2.5 % DSS and 1 nM PG 97–269. f WT mice treated with 2.5 % DSS and vehicle (saline)
Table 1.
Summary data showing proximal colon histopathological score in mice treated with DSS
| Proximal colon | Edema | Villi shortening | Ulceration | Immune cell infiltration |
|---|---|---|---|---|
| WT | 3.00±0.58 | 2.33±0.33 | 2.33±0.33 | 2.67±0.33 |
| VIP−/− | 1.00±0.29* | 0±0** | 0±0** | 0.78±0.22* |
| WT+vehicle | 2.80±0.39 | 2.20±0.53 | 2.30±0.54 | 2.90±0.38 |
| WT+1 μM VIPHyb | 1.33±0.21* | 0.33±0.21** | 0.33±0.21*** | 1.50±0.22* |
| WT+1 nM PG 97-269 | 1.67±0.33 | 0.83±0.40* | 0.83±0.40* | 1.67±0.33* |
p<0.05,
p<0.01,
p<0.001
In the distal colon DSS-treated WT showed significant mucosal inflammation with edema, ulceration, and shortening of the crypts compared to controls, whereas VIP−/− mice had significantly lower or absent mucosa damage than controls (Fig. 4). Scores of edema (1.20±0.20 vs. 2.00±0.41, p<0.05), villi shortening (1.60±0.40 vs. 3.25±0.48, p<0.05), ulceration (1.60±0.40 vs. 3.50±0.29, p<0.05), and immune-cellular infiltration (1.40±0.24 vs. 3.50±0.29, p<0.05) were significantly lower in VIP−/− than WT (Table 2). Additionally, DSS-treated WT mice, given either 1 μM VIPHyb or 1 nM PG 97– 269 developed lesser mucosal damage than vehicle group (Fig. 4). Treatment with 1 μM VIPHyb resulted in lower scores of villi shortening (1.63±0.50 vs. 2.33±0.48, p<0.05) and ulceration (1.38±0.53 vs. 2.25±0.41, p<0.05; Table 2). PG 97–269 at 1 nM also decreased villi shortening (0.83±0.40, p<0.01), ulceration (0.83±0.40, p<0.01), and immune cell infiltration (1.67±0.33, p<0.05). In summary, VIPHyb at 1 μM and PG 97–269 at 1 nM administered to DSS-treated WT, induced resistance to colitis in both proximal and distal colon, similar to that observed in VIP−/− mice.
Fig. 4.
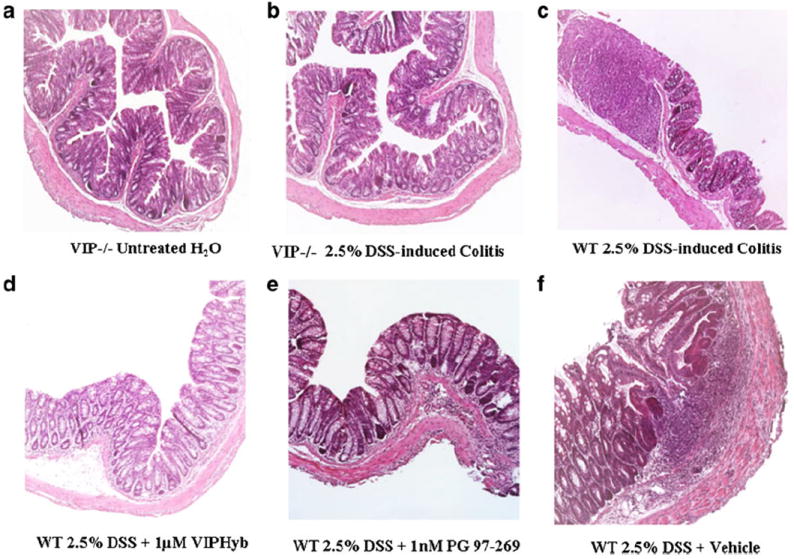
Distal colon histopathology. a DSS-untreated VIP−/−; b 2.5 % DSS-treated mice VIP−/−; c 2.5 % DSS-treated WT mice. d WT mice treated with 2.5 % DSS and 1 μM VIPHyb. e WT mice treated with 2.5 % DSS and 1 nM PG 97–269. f WT mice treated with 2.5 % DSS and vehicle (saline)
Table 2.
Summary data showing distal colon histopathological score in mice treated with DSS
| Distal colon | Edema | Villi shortening | Ulceration | Immune cell infiltrates |
|---|---|---|---|---|
| WT | 2.00±0.41 | 3.35±0.48 | 3.50±0.29 | 3.50±0.29 |
| VIP−/− | 2.20±0.20* | 1.60±0.40* | 1.60±0.40* | 1.40±0.24* |
| WT+vehicle | 2.67±0.26 | 2.33±0.43 | 2.25±0.41 | 2.83±0.34 |
| WT+1 μM VIPHyb | 2.25±0.25* | 1.63±0.50* | 1.38±0.53* | 2.75±0.41 |
| WT+1 nM PG 97–269 | 1.50±0.34 | 0.83±0.40** | 0.83±0.40** | 1.67±0.33* |
Pro-inflammatory Cytokines in DSS-Induced Colitis VIP−/− and VIP Antagonist-Treated WT Mice Colons
Consistent with the reduced mucosal inflammation in both proximal and distal colon, VIP−/− mice showed no change in mRNA expression for TNF-α (0.76-fold), IL-6 (0.34-fold), IL- 1β (1.16-fold), IL-2 (1.08-fold), IL-23A (0.73-fold), and T-bet (0.16-fold) compared to controls (Fig. 5). In contrast, DSS-treated WT had significantly up-regulated TNF-α (8.63-fold, p<0.01), IL-6 (13.93-fold, p<0.001), IL-1β (24.35-fold, p<0.01), IL-2 (18.76-fold, p<0.001), IL-23A (5.13-fold, p<0.05), and T-bet (14.47-fold, p<0.01) compared to WT and VIP−/− controls. In DSS-treated WT 1 μM VIPHyb or 1 nMPG 97–269 reduced significantly TNF-α (0.24-fold, p<0.05), IL-6 (0.24-fold, p<0.05), and IL-1β (0.68-fold, p<0.05) mRNA compared to vehicle-treated WT, Fig. 5. Similarly, PG 97–269 reduced significantly TNF-α (0.24-fold, p<0.05), IL-6 (0.36- fold, p<0.05), and IL-1β (0.70-fold, p<0.05) mRNA levels compared to vehicle-treated WT. However, IL-2, IL-23A, and T-bet levels were unchanged by the VIP antagonists’ treatment. In conclusion, DSS-treated VIP−/− mice showed suppressed colonic cytokine expression compared to WT. Similarly, 1 μM VIPHyb or 1 nM PG 97–269 antagonist treatment suppressed colonic cytokines in DSS-treated WT mice.
Fig. 5.
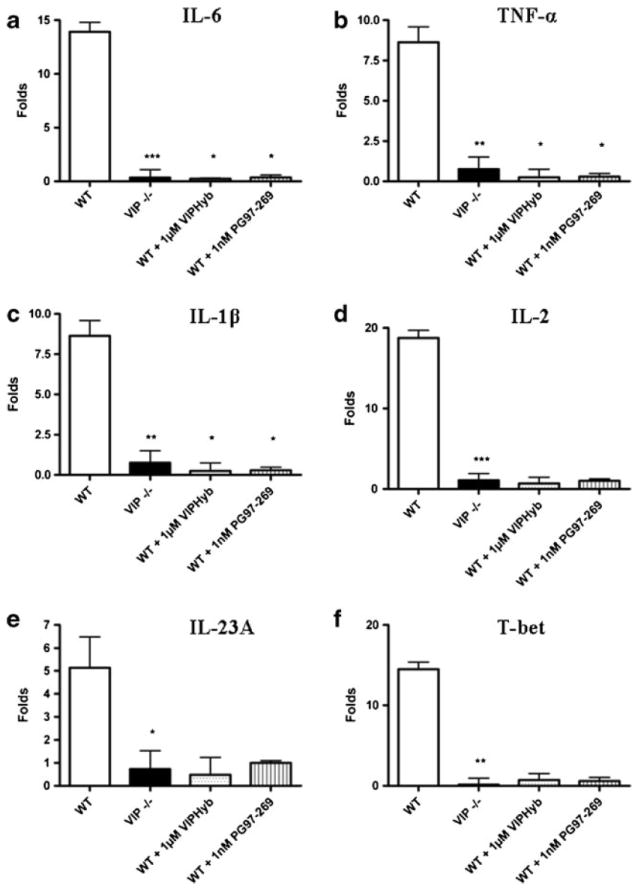
Cytokine profile, expressed as the ratio between the cytokine level in DSS-treated and untreated mice. Results are expressed as fold up- or down-regulation from controls. WT were compared to VIP−/− mice. Effect of attenuation of cytokine stimulation was evaluated in WT treated with either 1 μM VIPHyb, 1 nM PG 97–269, or vehicle. Cytokine levels of a IL-6, b TNF- α, c IL-1β, d IL-2, e IL-23A, and f T-bet were evaluated. Statistical analysis was performed using two-way ANOVA with Bonferroni post-test to calculate the p values. *p<0.05, **p<0.01, ***p<0.001
Discussion
In this study we report that VIP−/− mice are remarkably resistant to DSS-induced colitis, showing little to no clinical and histopathological damage. The data further show that treatment with VIPHyb or PG 97–269, in WT mice significantly improved the analyzed parameters. This study highlights the complex role of VIP signaling in colitis and suggests the potential VIP antagonists use as therapeutic modality in IBD.
VIP-Gene Deletion Confers Resistance to DSS-Colitis in C57Bl6 Mice
The development of a mouse model with VIP gene deletion (Colwell 2003) allowed us to study endogenous VIP role during colitis, and to clearly elucidate VIP effects in the colonic immune-inflammatory processes. VIP−/− mice showed a very significant resistance to colitis with absent clinical signs, immune-inflammatory infiltrates, and proinflammatory cytokines. Our results are consistent with the resistance to inflammation observed in the same VIP−/− mice by other investigators (Waschek 2013) using EAE and LPS inflammatory models, where immune-inflammatory response inhibition and impairment of NF-κB pathway was documented. Furthermore, in a LPS-induced endotoxic shock model, VIP−/− mice had suppressed levels of TNF-α and IL-6 mRNA as our DSS-treated VIP−/− (Waschek 2013).
Pharmacological Inhibition of VIP Attenuates DSS-Colitis in C57Bl/6 Mice
The role of VIP in colitis is still controversial. Several studies described an anti-inflammatory role for VIP in vitro (Delgado et al. 2004a, b). VIP analogues have been proposed as potential therapeutic agents against immune inflammation (Abad 2003, 2006; Delgado 2004a, b) in mouse models of rheumatoid arthritis, EAE, and endotoxic shock (Delgado et al. 2004a, b; Gonzalez-Rey 2006). However, in vivo data on the effects of VIP during inflammatory states are not consistent. In a model of EAE (Abad 2010), genetic deletion of VIP resulted in resistance to inflammation with mild or absent clinical signs and lack of CD4 cells infiltration into the brain parenchyma. In another study, VIP−/− mice exhibited resistance to LPS administration with decreased mortality, tissue damage, and pro-inflammatory cytokines (Abad 2012). However, Abad et al. (2003) in a Crohn’s disease murine model showed that TNBS-treated WT Balb/c mice injected daily with 1 or 5 nmol VIP showed reduced clinical and histopathologic severity of colitis as well as Th1 cytokine levels, whereas a higher 10 nmol VIP dose worsened the course of colitis. Conversely in the same experimental model, Newman et al. (2005) reported that VIP by constant infusion with mini-osmotic pumps or 1 or 5 nmol ip injections for 7 days enhanced the severity of colitis (Newman 2005).
In our study, VIPHyb treatment resulted in a significant clinical and histological improvement. VIPHyb at 1 μM significantly reduced weight loss and mucosal damage, inhibiting TNF-α, IL-6, and IL-1β in the colon. VIP, acting on macrophages, can stimulate or inhibit inflammatory response (Delgado 2004a, b). Furthermore, VIP through VPAC1 can suppress the function of chemokine receptors in vivo on monocytes and CD4+ T-cells, impairing their chemotaxis in both delayed type hypersensitivity (Grimm 2003) and TNBS-treated Balb/c mouse models but only at high doses as 5× 10−4 M (Newman 2005). Ottaway et al. (1987) previously showed that VIP plays a very important role in T-cells homing to mesenteric lymph nodes and Peyer’s patches. Others have observed that VIP plays a chemoattractant role, as potent as chemokines, on T-cells (Wang et al. 1993). Johnston et al. (1994) demonstrated in vitro that VIP at both 10−8 and 10−9 M induce chemotaxis in unstimulated CD4 and CD8 but not in neutrophils or antigen-stimulated T-cells, in which VPAC1 expression is suppressed. In this way, antigen-activated T-cells lacking VPAC1 become unable to infiltrate VIP expressing sites. Consequently, genetic or pharmacological inhibition of VIP and VPAC1 could promote intestinal mucosa homeostasis, preventing lymphocytes and neutrophils migration to damaged mucosa and consequently inflammatory immune-cellular infiltrates and cytokine release.
We showed also that treatment with PG 97–269, a VPAC1 antagonist, at 1 nM significantly reduced colonic damage. PG 97–269 at 10 and 100 nM did not significantly reduce the colonic weight to length ratio; this effect could perhaps be attributed to a potential VPAC2 inhibition or molecular aggregation at those high doses. VIPHyb at 1 μM and PG 97–269 1 nM showed similar efficacies in reducing mucosal inflammation as observed in VIP−/− mice. However, in the distal colon PG 97–269 treatment was more efficacious in reducing the amount of immune-cellular infiltrates, ulceration, and villi shortening compared to VIPHyb, consistent with the observation that DSS-induced colitis affects mainly the middle and distal third of the large bowel (Perše 2012). Even though VIPHyb and PG 97–269 were both efficient in targeting inflammation in the distal colon and provided a significant decrease in the severity of DSS-induced colitis, VIPHyb required higher doses than PG 97–269, which is VPAC1 specific. Conversely, PG 97–269 showed its highest efficacy in reducing colonic inflammation at 1 nM, but not at 10 and 100 nM suggesting a potential activation of VPAC2 receptor or a phenomenon of molecular aggregation and precipitation at high doses. Considering that VPAC2 receptors are mainly expressed on antigen-activated T lymphocytes while VPAC1 receptors are down-regulated on activated T-cells; the high capability of PG97–269 to significantly reduce colonic inflammation suggest that VIP inhibition plays an anti-inflammatory role by blocking cell chemotaxis, providing an explanation for the absence of immune-inflammatory infiltrates and cytokines.
VPAC1 Mediates the Pro-inflammatory Effect of VIP in DSS Colitis
Similarly, WT mice injected with either VIPHyb at 1 μM or PG 97–269 at 1 nM, presented lower immune cellular-infiltrates. IL-1β and IL-6 are mainly produced by innate immune system cells, as macrophages, mast cells, and monocytes, which can indirectly recruit neutrophils to the colon. Both cytokines were significantly lower in VIP−/− as well as in VIPHyb- and PG 97-269-treated mice. Our data suggest that the VIP pro-inflammatory effects are mediated through VPAC1 receptors. This is in agreement with Yadav et al. (2011), where VPAC1-deficient mice were resistant to DSS-induced colitis as our VIP−/−. These observations on the effects of VIP or VPAC1 inhibition, point to the crucial role of VIP and VPACs in regulating colonic inflammation. In contrast, DSS-treated VPAC2-null mice developed a worse colitis (Yadav 2011), while over-expression of VPAC2 on T-cells in VPAC1-null mice did not alter their resistance to DSS-induced colitis (Yadav 2011). These results suggest that in the colon, VIP signals predominantly through VPAC1 receptors, which are critical in modulating mucosal inflammation. Furthermore, blockade of VPAC1 signaling through PKA inhibitors in VPAC2-deficient mice inhibited clinical disease and reduced IL-1β and IL-6 to levels as those that we found in our DSS-treated VIP−/− mice (Yadav 2011). Therefore, targeting the VIP signaling axis primarily through VPAC1 is critical in maintaining intestinal mucosa immune homeostasis and reducing inflammation.
VPAC1 are expressed on immune cells in the gastrointestinal tract, especially on the lamina propria CD68+ cells during both ulcerative colitis and Crohn’s disease (Yukawa 2007). The entry of immune cells into mesenteric lymph nodes and Peyer’s patches is regulated by VPAC1 signaling (Von der Weid 2012). Furthermore, in a model of EAE in VIP−/− mice, T-cells and macrophages accumulated in the subarachnoid space but failed to enter the CNS parenchyma (Abad 2010); the levels of chemokine expression in the CNS were significantly lower in EAE-induced VIP−/− mice. Pharmacological inhibition of VIP in our model was effective in inducing resistance to colitis as that observed in VIP−/− mice.
The apparent discrepancy between the different studies on the role of VIP in colitis may be due to the complex VPAC1 and VPAC2 signaling, that may play differential roles. For instance, VPAC1 and VPAC2 are highly localized on immune cells (Delgado 2004a, b), but upon T-cell activation VPAC2 is upregulated, whereas VPAC1 is down-regulated (Delgado 1999a, b). VIP inflammatory effects during colitis appear to be mediated through the VPAC1 pathway. The current report using VPACs antagonists seems to be in contrast with those previous studies that showed that lower doses of VIP reduced intestinal inflammation (Abad 2003), whereas it can explain the proinflammatory effects induced by higher doses of VIP in the studies by Abad et al. (2003) and Newman et al. (2005). Our model along with Yadav et al. uses DSS-induced colitis in C57Bl/6 mice, whereas Abad et al. (2003) and Newman et al. (2005) used a TNBS model in Balb/c mice. DSS-induced colitis, considered a model of ulcerative colitis, directly injures host intestinal epithelia allowing for the uptake of bacterial endotoxin and peptidoglycan polysaccharide polymers by immune cells (Dieleman 1998). While TNBS-induced colitis is considered a model of Crohn’s disease, involves a T-cellmediated immune responses and induces a Th1 cytokine profile (Alex 2009). It should also be considered that C57Bl/6 mice have higher sensitivity to colitis compared to Balb/c, and develop a severe colitis with only one cycle of DSS-treatment (Melgar 2005).
Studies using different therapeutic agents for IBD have shown that DSS-induced colitis is a relevant model for the translation of mice data to human IBD studies (Melgar 2008). VIPHyb and PG 97–269 have been both previously administered in vivo without adverse effects and have very low neurotoxicity compared to other VIP antagonists (Banks 2005). While VIPHyb is pharmacologically less potent than PG 97–269, VIPHyb was selected for its ability to inhibit cAMP concentration and lack of toxicity in mice (Banks 2005). The selective antagonism of VPAC1 may represent an efficacious clinical target to elicit anti-inflammatory effects without the undesired side effects of a simultaneous VPAC2 antagonism, such as vasodilatation, hypotension, and diarrhea. Our data suggests that partial or transient blockade of VPAC1 receptors may be sufficient in inhibiting the pathophysiologic mechanisms of colitis.
Conclusion
Our findings reveal that absence of VIP expression induces resistance to colitis in our DSS-induced murine model, as well as VIP antagonism that inhibits VIP/VPAC1 signaling, thus leading to a suppression of colonic mucosa inflammation and damage. Currently, treatment options for human patients are associated with significant adverse effects (Truelove 1960; Reshef 1992; Ballinger; 2008). Anti-TNF-α targeting agents have been effective; however, up to half of the patients have developed no response, loss of response, or intolerance along with significant side effects (Perrier 2012). A major advance in IBD therapy would be the ability to target the site of inflammation and minimize systemic side effects. In humans there is a predominant VPAC1 expression in the colonic mucosa, and on inflammatory immune cells (Margolis 2009). VIPHyb and PG 97–269 represent potential novel candidates for the treatment of colitis. These studies support the potential use of VIP antagonists for a clinical treatment of patients with IBD.
Supplementary Material
{kind=link}
Acknowledgments
Grant Support Department of Veterans Affairs Merit Review (PG, JP) NIH DK-41301 & RO1 DK-078676 (MM)
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s12031-013-0205-3) contains supplementary material, which is available to authorized users.
Conflict of Interest None to declare.
Contributor Information
John P. Vu, CURE/Digestive Diseases Research Center, Department of Medicine, David Geffen School of Medicine, University of California at Los Angeles and VA Greater Los Angeles Health Care System, Los Angeles, CA, USA
Mulugeta Million, CURE/Digestive Diseases Research Center, Department of Medicine, David Geffen School of Medicine, University of California at Los Angeles and VA Greater Los Angeles Health Care System, Los Angeles, CA, USA.
Muriel Larauche, CURE/Digestive Diseases Research Center, Department of Medicine, David Geffen School of Medicine, University of California at Los Angeles and VA Greater Los Angeles Health Care System, Los Angeles, CA, USA.
Leon Luong, CURE/Digestive Diseases Research Center, Department of Medicine, David Geffen School of Medicine, University of California at Los Angeles and VA Greater Los Angeles Health Care System, Los Angeles, CA, USA.
Joshua Norris, CURE/Digestive Diseases Research Center, Department of Medicine, David Geffen School of Medicine, University of California at Los Angeles and VA Greater Los Angeles Health Care System, Los Angeles, CA, USA.
James A. Waschek, CURE/Digestive Diseases Research Center, Department of Medicine, David Geffen School of Medicine, University of California at Los Angeles and VA Greater Los Angeles Health Care System, Los Angeles, CA, USA Department of Psychiatry, David Geffen School of Medicine, University of California at Los Angeles, Los Angeles, CA, USA.
Charalabos Pothoulakis, CURE/Digestive Diseases Research Center, Department of Medicine, David Geffen School of Medicine, University of California at Los Angeles and VA Greater Los Angeles Health Care System, Los Angeles, CA, USA; Inflammatory Bowel Disease Center, Division of Digestive Diseases, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
Joseph R. Pisegna, CURE/Digestive Diseases Research Center, Department of Medicine, David Geffen School of Medicine, University of California at Los Angeles and VA Greater Los Angeles Health Care System, Los Angeles, CA, USA Division of Gastroenterology and Hepatology, Department of Medicine, VA Greater Los Angeles Healthcare System, Los Angeles, CA, USA.
Patrizia M. Germano, Email: pgermano@ucla.edu, CURE/Digestive Diseases Research Center, Department of Medicine, David Geffen School of Medicine, University of California at Los Angeles and VA Greater Los Angeles Health Care System, Los Angeles, CA, USA; Division of Pulmonary and Critical Care, Department of Medicine, VA Greater Los Angeles Healthcare System, Los Angeles, CA, USA; CURE/UCLA West Los Angeles VA Healthcare System, Bldg. 115 Rm 313, Los Angeles, CA, USA.
References
- Abad C, Martinez C, Juarranz MG, et al. Therapeutic effects of vasoactive intestinal peptide in the trinitrobenzene sulfonic acid mice model of Crohn’s disease. Gastroenterology. 2003;124:961–971. doi: 10.1053/gast.2003.50141. [DOI] [PubMed] [Google Scholar]
- Abad C, Gomariz RP, Waschek JA. Neuropeptide mimetics and antagonists in the treatment of inflammatory disease: focus on VIP and PACAP. Curr Top Med Chem. 2006;6:151–163. doi: 10.2174/156802606775270288. [DOI] [PubMed] [Google Scholar]
- Abad C, Tan YV, Lopez R, et al. Vasoactive intestinal peptide loss leads to impaired CNS parenchymal T-cell infiltration and resistance to experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010;107:19555–19560. doi: 10.1073/pnas.1007622107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abad C, Tan YV, Cheung-Lau G, Nobuta H, Waschek JA. VIP deficient mice exhibit resistance to lipopolysaccharide induced endotoxemia with an intrinsic defect in proinflammatory cellular responses. PLoS One. 2012;7:e36922. doi: 10.1371/journal.pone.0036922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alex P, Zachos NC, Nguyen T, et al. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBSinduced colitis. Inflamm Bowel Dis. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen O, Fahrenkrug J, Wikkelsø C, Johansson BB. VIP in cerebrospinal fluid of patients with multiple sclerosis. Peptides. 1984;5:435–437. doi: 10.1016/0196-9781(84)90249-3. [DOI] [PubMed] [Google Scholar]
- Ballinger A. Adverse effects of nonsteroidal anti-inflammatory drugs on the colon. Curr Gastroenterol Rep. 2008;10:485–489. doi: 10.1007/s11894-008-0089-5. [DOI] [PubMed] [Google Scholar]
- Banks MR, Farthing MJ, Robberecht P, Burleigh DE. Antisecretory actions of a novel vasoactive intestinal polypeptide (VIP) antagonist in human and rat small intestine. Br J Pharmacol. 2005;144:994–1001. doi: 10.1038/sj.bjp.0706128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belai A, Boulos PB, Robson T, Burnstock G. Neurochemical coding in the small intestine of patients with Crohn’s disease. Gut. 1997;40:767–774. doi: 10.1136/gut.40.6.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer L, Sidpra D, Jevon G, Buchan AM, Jacobson K. Differential responses of VIPergic and nitrergic neurons in paediatric patients with Crohn’s disease. Auton Neurosci. 2007;134:106–114. doi: 10.1016/j.autneu.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Brandtzaeg P, Oktedalen O, Kierulf P, Opstad PK. Elevated VIP and endotoxin plasma levels in human gram-negative septic shock. Regul Pept. 1989;24:37–44. doi: 10.1016/0167-0115(89)90209-7. [DOI] [PubMed] [Google Scholar]
- Colwell CS, Michel S, Itri J, et al. Disrupted circadian rhythms in VIP- and PHI-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2003;285:R939–R949. doi: 10.1152/ajpregu.00200.2003. [DOI] [PubMed] [Google Scholar]
- Delgado M, Martinez C, Johnson MC, Gomariz RP, Ganea D. Differential expression of vasoactive intestinal peptide receptors 1 and 2 (VIP-R1 and VIP-R2) mRNA in murine lymphocytes. J Neuroimmunol. 1996;68:27–38. doi: 10.1016/0165-5728(96)00063-x. [DOI] [PubMed] [Google Scholar]
- Delgado M, Pozo D, Martinez C, et al. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit endotoxin-induced TNF-alpha production by macrophages: in vitro and in vivo studies. J Immunol. 1999a;162:2358–2367. [PubMed] [Google Scholar]
- Delgado M, Martinez C, Leceta J, Gomariz RP. Vasoactive intestinal peptide in thymus: synthesis, receptors and biological actions. Neuroimmunomodulation. 1999b;6:97–107. doi: 10.1159/000026369. [DOI] [PubMed] [Google Scholar]
- Delgado M, Pozo D, Ganea D. The significance of vasoactive intestinal peptide in immunomodulation. Pharmacol Rev. 2004a;56:249–290. doi: 10.1124/pr.56.2.7. [DOI] [PubMed] [Google Scholar]
- Delgado M, Gonzalez-Rey E, Ganea D. VIP/PACAP preferentially attract Th2 effectors through differential regulation of chemokine production by dendritic cells. FASEB J. 2004b;18:1453–1455. doi: 10.1096/fj.04-1548fje. [DOI] [PubMed] [Google Scholar]
- Dieleman LA, Palmen MJ, Akol H, et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol. 1998;114:385–391. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Rey E, Fernandez-Martin A, Chorny A, et al. Therapeutic effect of vasoactive intestinal peptide on experimental autoimmune encephalomyelitis: down-regulation of inflammatory and autoimmune responses. Am J Pathol. 2006;168:1179–1188. doi: 10.2353/ajpath.2006.051081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozes Y, Brenneman DE, Fridkin M, Asofsky R, Gozes I. AVIP antagonist distinguishes VIP receptors on spinal cord cells and lymphocytes. Brain Res. 1991;540:319–321. doi: 10.1016/0006-8993(91)90528-4. [DOI] [PubMed] [Google Scholar]
- Grimm MC, Newman R, Hassim Z, et al. Cutting edge: vasoactive intestinal peptide acts as a potent suppressor of inflammation in vivo by trans-deactivating chemokine receptors. J Immunol. 2003;17:4990–4994. doi: 10.4049/jimmunol.171.10.4990. [DOI] [PubMed] [Google Scholar]
- Gross KJ, Pothoulakis C. Role of neuropeptides in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:918–932. doi: 10.1002/ibd.20129. [DOI] [PubMed] [Google Scholar]
- Harmar AJ, Fahrenkrug J, Gozes I, et al. Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: IUPHAR review 1. Br J Pharmacol. 2012;166:4–17. doi: 10.1111/j.1476-5381.2012.01871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JA, Taubb DD, Lloyd AR, Conlon K, Oppenheim JJ, Kevlin DJ. Human T lymphocyte chemotaxis and adhesion induced by VIP. J Immunol. 1994;153:1762–1768. [PubMed] [Google Scholar]
- Juarranz Y, Gutiérrez-Cañas I, Santiago B, Carrión M, Pablos JL, Gomariz RP. Differential expression of vasoactive intestinal peptide and its functional receptors in human osteoarthritic and rheumatoid synovial fibroblasts. Arthritis Rheum. 2008;58:1086–1095. doi: 10.1002/art.23403. [DOI] [PubMed] [Google Scholar]
- Margolis KG, Gershon MD. Neuropeptides and inflammatory bowel disease. Curr Opin Gastroenterol. 2009;25:503–511. doi: 10.1097/MOG.0b013e328331b69e. [DOI] [PubMed] [Google Scholar]
- Melgar S, Karlsson A, Michaëlsson E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: correlation between symptoms and inflammation. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1328–G1338. doi: 10.1152/ajpgi.00467.2004. [DOI] [PubMed] [Google Scholar]
- Melgar S, Karlsson L, Rehnström E, et al. Validation of murine dextran sulfate sodium-induced colitis using four therapeutic agents for human inflammatory bowel disease. Int Immunopharmacol. 2008;8:836–844. doi: 10.1016/j.intimp.2008.01.036. [DOI] [PubMed] [Google Scholar]
- Moody TW, Jensen RT, Fridkin M, Gozes I. (N-stearyl, norleucine17)VIPhybrid is a broad spectrum vasoactive intestinal peptide receptor antagonist. J Mol Neurosci. 2002;18:29–35. doi: 10.1385/JMN:18:1-2:29. [DOI] [PubMed] [Google Scholar]
- Moody TW, Zia F, Draoui M, et al. Avasoactive intestinal peptide antagonist inhibits non-small cell lung cancer growth. Proc Natl Acad Sci U S A. 2003;90:4345–4349. doi: 10.1073/pnas.90.10.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman R, Cuan N, Hampartzoumian T, Connor SJ, Lloyd AR, Grimm MC. Vasoactive intestinal peptide impairs leucocyte migration but fails to modify experimental murine colitis. Clin Exp Immunol. 2005;139:411–420. doi: 10.1111/j.1365-2249.2005.02673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- Ottaway CA, Lewis DL, Asa SL. Vasoactive intestinal peptidecontaining nerves in Peyer’s patches. Brain Behav Immun. 1987;1:148–158. doi: 10.1016/0889-1591(87)90017-1. [DOI] [PubMed] [Google Scholar]
- Perrier C, Rutgeerts P. New drug therapies on the horizon for IBD. Dig Dis. 2012;30(Suppl 1):100–105. doi: 10.1159/000341133. [DOI] [PubMed] [Google Scholar]
- Perše M, Cerar A. Dextran sodium sulphate colitis mouse model: traps and tricks. J Biomed Biotechnol. 2012;2012:718617. doi: 10.1155/2012/718617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reshef R, Varkel J, Shiller M, Loberant N. Systemic effects of rectally administered corticosteroids. Isr J Med Sci. 1992;28:98–100. [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Törnwall J, Uusitalo H, Hukkanen M, Sorsa T, Konttinen YT. Distribution of vasoactive intestinal peptide (VIP) and its binding sites in labial salivary glands in Sjögren’s syndrome and in normal controls. Clin Exp Rheumatol. 1994;12:287–292. [PubMed] [Google Scholar]
- Truelove SC. Systemic and local corticosteroid therapy in ulcerative colitis. Br Med J. 1960;1:464–467. doi: 10.1136/bmj.1.5171.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von der Weid PY, Rehal S, Dyrda P, et al. Mechanisms of VIP-induced inhibition of the lymphatic vessel pump. J Physiol. 2012;590:2677–2691. doi: 10.1113/jphysiol.2012.230599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JM, McVicar DW, Oppenheim JJ, Kelvin DJ. Identification of RANTES receptors on human monocytic cells: competition for binding and desensitization by homologous chemotactic cytokines. J Exp Med. 1993;177:699–705. doi: 10.1084/jem.177.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waschek J. VIP and PACAP: neuropeptide modulators of CNS inflammation, injury and repair. Br J Pharmacol. 2013;169:512–523. doi: 10.1111/bph.12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M, Huang MC, Goetzl EJ. VPAC1 (vasoactive intestinal peptide (VIP) receptor type 1) G protein-coupled receptor mediation of VIP enhancement of murine experimental colitis. Cell Immunol. 2011;267:124–132. doi: 10.1016/j.cellimm.2011.01.001. [DOI] [PubMed] [Google Scholar]
- Yukawa T, Oshitani N, Yamagami H, Watanabe K, Higuchi K, Arakawa T. Differential expression of vasoactive intestinal peptide receptor 1 expression in inflammatory bowel disease. Int J Mol Med. 2007;20:161–167. [PubMed] [Google Scholar]
- Zia H, Hida T, Jakowlew S, et al. Breast cancer growth is inhibited by vasoactive intestinal peptide (VIP) hybrid, a synthetic VIP receptor antagonist. Cancer Res. 1996;56:3486–3489. [PubMed] [Google Scholar]
- Zia H, Leyton J, Casibang M, et al. (N-stearyl, norleucine17) VIP hybrid inhibits the growth of pancreatic cancer cell lines. Life Sci. 2000;66:379–387. doi: 10.1016/s0024-3205(99)00604-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.