

Abstract
Heparan sulfate (HS) plays essential roles in assisting herpes simplex virus infection and other biological processes. The biosynthesis of HS includes numerous specialized sulfotransferases that generate a variety of sulfated saccharide sequences, conferring the selectivity of biological functions of HS. We report a structural study of human HS 3-O-sulfotransferase isoform 3 (3-OST-3), a key sulfotransferase that transfers a sulfuryl group to a specific glucosamine in HS generating an entry receptor for herpes simplex virus 1. We have obtained the crystal structure of 3-OST-3 at 1.95 Å in a ternary complex with 3′-phosphoadenosine 5′-phosphate and a tetrasaccharide substrate. Mutational analyses were also performed on the residues involved in the binding of the substrate. Residues Gln255 and Lys368 are essential for the sulfotransferase activity and lie within hydrogen bonding distances to the carboxyl and sulfo groups of the uronic acid unit. These residues participate in the substrate recognition of 3-OST-3. This structure provides atomic level evidence for delineating the substrate recognition and catalytic mechanism for 3-OST-3.
Heparan sulfate (HS)1 is ubiquitous on the cell surface and in the extracellular matrix. It has diverse roles in regulating embryonic development and homeostasis as well as in assisting viral infections (1, 2). HS is a highly sulfated polysaccharide consisting of 1→4-linked sulfated glucosamine and sulfated glucuronic/iduronic acid units. HS is initially synthesized as a copolymer of glucuronic acid and N-acetylated glucosamine by D-glucuronyl and N-acetyl-D-glucosaminyltransferase, followed by various modifications in the Golgi apparatus (3). These modifications include N-deacetylation and N-sulfonation of glucosamine, C5 epimerization of glucuronic acid to form iduronic acid units, 2-O-sulfonation of iduronic and glucuronic acid units, and 6-O-sulfonation and 3-O-sulfonation of glucosamine units. The specific sulfated saccharide sequences of HS determine its biological function (4).
Infections caused by herpes simplex virus type 1 (HSV-1) are highly prevalent in human and result in localized mucocutaneous lesions and, in rare cases, encephalitis (5). HS plays critical roles in assisting HSV-1 infections in both viral attachment and entry steps (6). In the attachment step, HSV-1 binds to the cells through the interactions of envelope glycoproteins gC or/and gB with cell surface HS (7). Viral entry requires the interaction of a third viral glycoprotein, gD, with a specific cell surface entry receptor to induce fusion of the viral envelope with the cell membrane (in the presence of gB, gH, and gL) to establish an infection (8). The 3-O-sulfated HS, representing the culmination of a series of specific sulfated saccharide sequences, binds to gD and serves as an entry receptor of HSV-1 (9, 10). The 3-O-sulfated HS is synthesized by heparan sulfate 3-O-sulfotransferase isoform 3 (3-OST-3) and by 3-OST-5 (9, 10). A gD-binding HS octasaccharide was isolated and characterized, confirming that HSV-1 utilizes a unique HS sequence for its entry (11). Understanding the biosynthesis of 3-O-sulfated HS could potentially lead to a new strategy for developing therapeutic agents against HSV infection.
HS 3-O-sulfotransferase is present in at least six different isoforms with unique expression patterns in human tissues (10, 12). The amino acid sequences of the different isoforms have greater than 60% homology in the sulfotransferase domains (12). These different 3-OST isoforms transfer the sulfuryl group from 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to the 3-OH position of glucosamine units residing within the context of different saccharide sequences. As a result, the HS products generated by these different isoforms exhibit unique and distinctive biological activities (Fig. 1A). It is known that the HS modified by 3-OST-1 and 3-OST-5 displays anticoagulant activity. The HS modified by 3-OST-3 and 3-OST-5 serves as an entry receptor for HSV-1 (9, 10, 13). The molecular mechanism used by 3-OSTs to distinguish substrates with unique saccharide sequences remains largely unknown.
Fig. 1. Substrate specificities of 3-OST-1 and 3-OST-3 and the structure of the tetrasaccharide substrate.

A shows the enzymatic reactions catalyzed by 3-OST-1 and 3-OST-3. 3-OST-1 transfers sulfate to the 3-OH position of a glucosamine unit (GlcNS±6S) that is linked to a glucuronic acid unit (GlcUA), thereby forming the 3-O-sulfated HS containing GlcUA-GlcNS3S±6S (an anticoagulant HS). 3-OST-3 transfers sulfate to the 3-OH position of a glucosamine unit (GlcNH2±6S) that is linked to a 2-O-sulfo iduronic acid unit (IdoUA2S), thereby forming the 3-O-sulfated HS containing IdoUA2S-GlcNH 3S±6S (an entry receptor for HSV-1). R represents a proton (−H) or a sulfate group (−SOa− ). B shows the structure of the tetrasaccharide used in the study. The 3-O-sulfonation site is circled. For clarity, U4 represents δUA2S; G3 represents GlcNS6S linked to δUA2S; I2 represents IdoUA2S; and G1 represents GlcNS6S at the reducing end. The numbers indicate the positions of the saccharide units.
Previously, x-ray crystal structures have been obtained for the sulfotransferase domains of human N-deacetylase/N-sulfotransferase (NST-1) and mouse 3-OST-1 in the complex with 3′-phosphoadenosine 5′-phosphate (PAP) (14, 15). The structural information combined with the results of mutational analysis provided us with preliminary insight into the HS sulfotransferase mechanism (14, 16). Furthermore, potential roles of specific amino acid residues from NST-1 and 3-OST-1 for directing the transfer of the sulfuryl group were suggested (14). However, because these structures lacked acceptor substrate, they have provided limited knowledge to our understanding of what dictates HS binding, in particular, among the 3-OST isoforms.
In this study, we report the crystal structure of a ternary complex of human 3-OST-3, PAP, and a tetrasaccharide substrate with the structure of δUA2S-GlcNS6S-IdoUA2S-GlcNS6S (for structure see Fig. 1B). This ternary complex structure, combined with the mutagenesis data presented here, reveals the residues involved in catalysis and in substrate binding. The data provide structural evidence for which residues confer substrate specificity between the 3-OST isoforms. These results present new insight into the biosynthesis of the HS with various biological functions ranging from viral infection to blood coagulation.
EXPERIMENTAL PROCEDURES
Expression, Purification, and Crystallization of 3-OST-3
Preparation of 3-OST-3 Bacterial Expression Plasmid
The cDNA fragment encoding the catalytic domain of 3-OST-3 (Gly139–Gly406) was amplified from plasmid h3-OST-3A-pcDNA3 (17) with a 5′ overhang containing an EcoRI site and a 3′ overhang containing an NotI site. This construct was inserted into the pGEX4T3 vector (Amersham Biosciences) using the EcoRI and NotI restriction sites to produce a glutathione S-transferase fusion protein. The resultant plasmid was sequenced to confirm the reading frame and the lack of mutations within the coding region (University of North Carolina, DNA sequencing core facility). The plasmid was transformed into BL21(DE3)RIL cells (Stratagene).
Protein Expression and Purification
Cells containing the expression plasmid were grown in twelve 2.8-liter Fernbach flasks containing 1 liter of LB medium with 100 μg/ml of ampicillin at 37 °C. When the A600 reached 0.6–0.8, the temperature was lowered to 22 °C for 15 min. Isopropyl-β-D-thiogalactopyranoside was then added to a final concentration of 200 μM, and the cells were allowed to shake overnight. The cells were pelleted and resuspended in 120 ml of sonication buffer (25 mM sodium/potassium phosphate, pH 7.4, and 625 mM NaCl). The cells were sonicated, and the cell debris was pelleted by centrifugation. The soluble fraction was loaded onto 20 ml of glutathione-Sepharose 4B (Amersham Biosciences) resin in batch and washed repeatedly with sonication buffer. The catalytic domain of 3-OST-3 was cleaved from the fusion protein on the resin at 4 °C overnight using 400 units of thrombin in a total volume of 50 ml. PAP (Sigma) was added to the solution for a final concentration of 0.5 mM. The protein was concentrated to ~35 mg/ml. Protein was further purified by a Superdex 75 16/60 (Amersham Biosciences) column. The eluted fractions containing pure 3-OST-3 were pooled. The buffer was exchanged with the crystallization buffer (20 mM Tris, pH 7.5, 100 mM NaCl, and 1 mM PAP) by repeated concentration and dilution. The final concentration of the protein was 10 mg/ml, and additional PAP was added for a final concentration of 4 mM.
Protein Crystallization, Data Collection, and Structure Solution
Crystals of the catalytic domain of 3-OST-3 were obtained using the hanging drop method by mixing 2 μl of the above protein solution with 2 μl of the reservoir solution consisting of 12–13% polyethylene glycol 4000, 200 mM ammonium acetate, and 100 mM sodium citrate, pH 5.5. For data collection, the crystals were transferred in four steps into a cryo-protectant consisting of 15% polyethylene glycol 4000, 12.5% ethylene glycol, 4 mM PAP, 200 mM ammonium acetate, 100 mM NaCl, and 100 mM sodium citrate, pH 5.5. To obtain the ternary complex, the crystal was soaked overnight in cryo-protectant containing 20 mM of the tetrasaccharide. The crystals were frozen in liquid nitrogen and then placed in a stream of nitrogen gas cooled to −180 °C. The data were collected on a Rigaku RU3H generator fitted with Osmic mirrors and a Raxis IV area detector. The data were indexed, integrated, and scaled using HKL2000 (18). Coordinates from the 3-OST-1 isoform (Protein Data Bank code 1S6T) were used to find a solution for the molecular replacement problem using the program Molrep from CCP4 (19, 20). The model of 3-OST-3 was refined by iterative cycles of model building in O (21) and refinement in CNS (22). The starting model for the tetrasaccharide was obtained from the Protein Data Bank coordinates 1BFB (23).
Mutational Analysis of 3-OST-3
Preparation of 3-OST-3 Mutant Plasmids
The cDNA fragment encoding the catalytic domain of 3-OST-3 (Gly139–Gly406) was amplified from plasmid h3-OST-3A-pcDNA3 (17) with a 5′ overhang containing an NdeI site and a 3′ overhang containing an EcoRI site. This construct was inserted into the pET28a vector (Novagen) using the NdeI and EcoRI restriction sites to produce a His6-tagged protein. The mutants were prepared using Gene Tailor site-directed mutagenesis kit from Invitrogen as described elsewhere (14).
Expression and Characterization of 3-OST-3
The procedures for the expression and purification of 3-OST-3 were essentially identical to those for 3-OST-1 as described in a prior publication (14). To confirm that bacterial 3-OST-3 has similar activity to the protein expressed in SF9 cells, we conducted a kinetic analysis to determine the Km and Vmax toward PAPS and HS, respectively. The Km and Vmax toward PAPS of the 3-OST-3 expressed in Escherichia coli is 40 μM and 27 pmol of sulfate/min, respectively, which are close to the Km value (16 μM) and Vmax value (27 pmol of sulfate/min) of the 3-OST-3 expressed in SF9 cells. The Km and Vmax toward HS of the 3-OST-3 expressed in E. coli is 2.5 μM and 24 pmol of sulfate/min, respectively, which are close to the Km (1 μM) and Vmax (21 pmol of sulfate/min) of the 3-OST-3 expressed in SF9 cells.
Expression and Purification of 3-OST-3 Mutants
The expression plasmids for various 3-OST-3 mutants were transformed individually into BL21(DE3)RIL cells (Stratagene). Each mutant construct was grown in 500 ml of LB broth and induced by isopropyl-β-D-thiogalactopyranoside. The bacteria were harvested and sonicated. The lysate was subjected to a 400-μl nitrilotriacetic acid-agarose column (0.75 cm × 1 cm), followed by 5 ml of wash with 10 mM imidazole, 25 mM Tris, and 500 mM NaCl, pH 7.5. Mutant proteins were eluted with 1 ml of elution buffer containing 25 mM Tris, 500 mM NaCl, and 250 mM imidazole, pH 7.5. Approximately 25 μl of the eluent was subjected to the analysis on a 16.5% Tris-Tricine-PAGE gel (Bio-Rad), and the gel was stained by Coomassie Blue. The expression level of the mutant protein was estimated by determining the intensity of the Coomassie-stained protein band near 30 kDa. Wild type 3-OST-3 was expressed along with the mutants. The expression levels and the sulfotransferase activity of mutant proteins were normalized to those of wild type protein.
Determination of the Sulfotransferase Activity
Sulfotransferase activity was determined by incubating ~5 μl (10 –100 ng) of purified mutant or wild type 3-OST-3 proteins with 10 μg of HS (from bovine kidney; ICN), 5 × 106 cpm of [35S]PAPS (~10 μM) in 50 μl of buffer containing 50 mM MES, pH 7.0, 10 mM MnCl2, 5 mM MgCl2, and 1% Triton X-100. The reaction was incubated at 37 °C for 60 min and quenched by the addition of 6 M urea and 100 mM EDTA. The sample was then subjected to 200-μl DEAE-Sepharose chromatography to purify the [35S]HS.
Disaccharide Analysis of the HS Modified by Wild Type 3-OST-3 and 3-OST-3 Mutants
3-OST-3-modified HS was degraded with nitrous acid at pH 1.5, followed by reduction with sodium borohydride (24). The resultant 35S-labeled disaccharides were resolved by a C18 reversed phase column (0.46 × 25 cm) (Vydac) under reversed phase ion pairing HPLC conditions. The identities of the disaccharides were determined by coeluting with appropriate 35S-labeled disaccharide standards (25).
Determination of 3-O-[35S]Sulfonation Site on the Tetrasaccharide
We utilized a combination of the digestions of heparin lyase and δ4,5-glycuronate-2-sulfatase to determine the 3-O-[35S]sulfonation site. The 3-O-[35S]sulfated tetrasaccharide was prepared by incubating 3-OST-3 enzyme, the tetrasaccharide, and [35S]PAPS. The resultant 3-O-[35S]sulfated tetrasaccharide was eluted from the anion exchange HPLC column at a salt concentration consistent with the introduction of one additional sulfo group. Heparin lyase cleaves the glycosidic bond between G3 and I2 of the 3-O-[35S]sulfated tetrasaccharide to yield two disaccharides, δUA2S-[3-35S]GlcNS3S6S and δUA2S-GlcNS6S (unlabeled) (illustrated in Fig. 5) (26). To this point, it is not possible to distinguish whether the 3-O-[35S]sulfo group was originally on the G3 or G1 unit of the original tetrasaccharide because the digestion would yield an identical 35S-labeled disaccharide for both cases. Pretreatment of 3-O-[35S]sulfated tetrasaccharide with δ4,5-glycuronate-2-sulfatase, however, removes the 2-O-sulfo group from the U4 unit (27) to yield δUA unit at the nonreducing end (U4 unit). Subsequent treatment with heparin lyase (a mixture of heparin lyase I, II, and IV were used because of the reported resistance of small oligosaccharides to heparin lyases (26)) afforded the δUA-[3-35S]GlcNS3S6S consistent with the presence of the 3-O-[35S]sulfo group on the G3 residue, the 35S-labeled disaccharide with a structure of δUA2S-[3-35S]GlcNS3S6S would be obtained if the 3-O-[35S]sulfo group is on the G1 residue. The resultant disaccharides were identified by determining the elution positions on an anion exchange HPLC using an on-line radioactive detector.
Fig. 5. Anion exchange HPLC chromatograms of enzymatic digested and undigested 3-O-[35S]sulfated tetrasaccharide.

A and B show the chromatogram of 3-O-[35S]sulfated tetrasaccharide without and with δ4,5-glycuronate-2-sulfatase digestion, respectively. C shows the chromatogram of heparin lyases digested 3-O-[35S]sulfated tetrasaccharide that was treated with δ4,5-glycuronate-2-sulfatase (solid line) and the chromatogram of the heparin lyases digested 3-O-[35S]sulfated tetrasaccharide without the treatment of δ4,5-glycuronate-2-sulfatase (dashed line). The enzymatic digestion reactions are illustrated besides the chromatograms. The anion exchange HPLC was performed on a silica based amino-bound anion exchange HPLC (0.46 × 24 cm; Waters) eluted with a linear gradient of KH2PO4 (300 –1000 mM in 60 min) followed by 1 M KH2PO4 for additional 20 min at a flow rate of 0.5 ml/min. Arrow 1, elution position of [35S]sulfate; arrow 2, elution position of δUA-[3-35S]GlcNS3S6S; arrow 3, elution position of δUA2S-[3-35S]GlcNS3S6S; arrow 4, elution position of δUA-[3-35S]GlcNS3S6S-IdoUA2S-GlcNS6S; arrow 5, elution position of δUA2S-[3-35S]GlcNS3S6S-IdoUA2S-GlcNS6S.
The conditions for δ4,5-glycuronate-2-sulfatase digestion are described elsewhere (11). Digestion with heparin lyase of the tetrasaccharide was carried out using a mixture of heparin lyase I, II, and IV. The digestion condition for the mixture of heparin lyases was described in a prior publication (11). The anion exchange HPLC was performed on a silical based amino-bound anion exchange HPLC (0.46 × 24 cm; Waters) eluted with a linear gradient of KH2PO4 (300–1000 mM in 60 min) followed by 1 m KH2PO4 for an additional 20 min at a flow rate of 0.5 ml/min. We noted that this 3-O-sulfated tetrasaccharide is resistant to nitrous acid (at pH 1.5) degradation.
RESULTS AND DISCUSSION
Crystal Analysis
Human 3-OST-3 crystallizes in space group C2221 and diffracts to 1.85 Å resolution. Two molecules were present in the asymmetric unit (molecules A and B), and each protein molecule contains one molecule of PAP. The crystals of 3-OST-3 are densely packed, generating a unit cell with a Matthews coefficient of 2.0 and an overall solvent content of 38.5% (28). Both molecules in the asymmetric unit are well ordered and have similar B-factors (Table I). The data were collected on a similar crystal soaked in the tetrasaccharide at 1.95 Å. One of the two molecules in the asymmetric unit has clear electron density for the tetrasaccharide that allows for precise positioning. The other molecule has some electron density in the binding pocket region, but it is not clear enough to model.
Table I.
Crystallographic data statistics
| Data set | 3-OST-3 and PAP | 3-OST-3 and tetrasaccharide and PAP |
|---|---|---|
| Unit cell | a = 80.34, b = 155.39, c = 92.32 | a = 80.81, b = 154.53, c = 91.93 |
| α = β = γ = 90° | α = β = γ = 90° | |
| Space group | C222(1) | C222(1) |
| Resolution (Å) | 50–1.85 | 50-1.95 |
| Number of observations | 183,877 | 160,766 |
| Unique reflections | 49,160 | 42,327 |
| Rsvm (%) (last shell)a | 11.0 (29.7) | 5.4 (39.3) |
| I/σ (I) (last shell) | 23.1 (3.5) | 18.2 ( 2.4) |
| Mosaicity | 0.7 | 0.7 |
| Completeness (%) (last shell) | 99.3 (99.1) | 99.9 (99.4) |
| Refinement statistics | ||
| Rcryst (%)b | 20.6 | 20.1 |
| Rfree (%)c | 23.6 | 22.5 |
| Number of waters | 422 | 333 |
| Average B values (Å) | ||
| Protein | 29.1 | 27.9 |
| PAP | 20.2 | 17.9 |
| Tetrasaccharide | 43.1 | |
| Root mean square deviation from ideal valuesd | ||
| Bond length (Å) | 0.006 | 0.006 |
| Bond angle (°) | 1.2 | 1.2 |
| Dihedral angle (°) | 21.2 | 21.0 |
| Improper angle (°) | 0.87 | 0.79 |
| Ramachandran statisticsd | ||
| Residues in favored (98%) regions (%) | 97.56 | 97.94 |
| Residues in allowed (>99.8%) regions (%) | 100 | 100 |
Rsym = Σ (Ii – ⟨I⟩/Σ(Ij) where Ij is the intensity of the ith observation and ⟨I⟩ is the mean intensity of the reflection.
Rcryst = Σ||Fo – Fc calculated from working data set.
Rfree was calculated from 5% of data randomly chosen not to be included in refinement.
The Ramachandran results were determined by MolProbity (35).
Overall Fold
The catalytic domain of human 3-OST-3 (Gly139–Gly406) is roughly spherical with a large open cleft running across the surface of the sphere (Fig. 2). At the core of the sphere is an α/β motif. This α/β motif is comprised of a five-stranded parallel β-sheet (β7-β6-β2-β5-β3), laterally flanked by α-helices. Central to this structural motif is the phosphosulfate-binding loop (K162-R166), contained within a strand-loop-helix comprised of β2 and α2 (29). The phosphosulfate-binding loop allows for several strong hydrogen bonding interactions with the 5′-phosphate of PAP (Fig. 3 and Table II). These interactions are believed to help position the donor substrate, PAPS, in the precise location for catalysis (30). This binding site is well conserved with the previously reported 3-OST-1 and NST-1 (14, 15). The 5′-phosphate of PAP is found at the bottom of the large open cleft that provides the binding pocket for HS substrate (Figs. 2 and 3). A conserved α-helix (α5) composed of residues Ala244–Lys259 is positioned in close proximity to the PAP-binding site and protrudes outward into the open cleft. The C-terminal region of the protein contains a small three-stranded anti-parallel β-sheet (β8-β9-β10). The second and third strands of this sheet are structurally reinforced by a disulfide bond between residues Cys351 and Cys363. Strand 10 of this sheet is connected to the C-terminal β-helix (β11) via a long coil (Lys366–Arg377) that lies along one side of the open cleft, partially obscuring the PAP-binding site.
Fig. 2. Ribbon diagram of the crystal structure of ternary complex 3-OST-3/PAP (blue)/tetrasaccharide (green) and sequence alignment of human 3-OST-3 and mouse 3-OST-1.

β-Strands and α-helices are colored purple and cyan, respectively. This figure was created using Molscript and Raster3d (33, 34). Structural alignment of 3-OST-3 with 3-OST-1 is shown besides the ribbon diagram. Cyan barrels represent residues in helices, and purple represents regions in β-strands. Residues that are structurally dissimilar are shaded with a yellow background. Residues that form interactions with the tetrasaccharide are colored green, and the cysteines that form a disulfide are colored orange. Red asterisks represent the three residues that may form a catalytic triad in the 3-OSTs.
Fig. 3. Stereo diagram of the PAPS binding site of 3-OST-3 with PAP bound (blue).
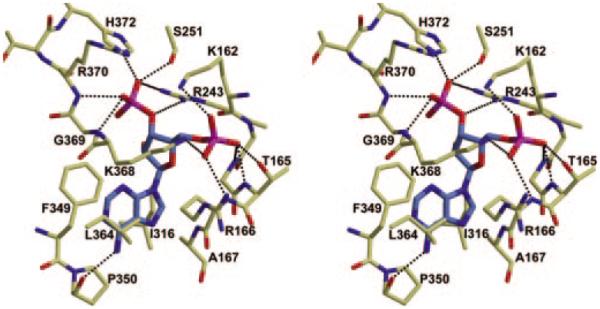
Residues that are within hydrogen bonding distance or that line the binding pocket are pictured. Possible hydrogen bonds between the protein and the PAP are displayed as black dashed lines. This figure was created using Molscript and Raster3d (33, 34).
Table II.
Distances of potential hydrogen bonds between PAP and 3-OST-3 protein
| PAP | Protein contact | Distance |
|---|---|---|
| Å | ||
| 5′P | ||
| O6P | Lys162 NZ | 2.8 |
| O6P | Lys368 NZ | 3.1 |
| O5P | Lys368 NZ | 2.7 |
| O5P | Arg166 N | 3.1 |
| O4P | Thr165 OG1 | 2.7 |
| O4P | Thr165 N | 2.8 |
| O4P | Gly164 N | 3.2 |
| O5′ | Gly164 N | 3.1 |
| 3′P | ||
| O3′ | Arg243 NH1 | 3.0 |
| O2P | His372 NE2 | 2.6 |
| O2P | Ser251 OG | 2.6 |
| O3P | Arg370 N | 2.8 |
| O3P | Gly369 N | 2.9 |
| Ade N6 | Pro350 O | 2.9 |
The structure of 3-OST-3 is remarkably similar to that of 3-OST-1, with a root mean square deviation of 1.1 Å for 246 equivalent Cα atoms as calculated by the software program O (14, 21), although some minor differences present within looped regions were notable (Fig. 2). In contrast to 3-OST-1, 3-OST-3 has an insertion of three amino acids (Ala355–Gly357) into the loop formed by the disulfide bond between Cys351 and Cys363 in strands 9 and 10 of the C-terminal β-sheet. These additional residues cause the loop to swing in the direction of the open cleft. Replacing the disulfide bond loop regions of 3-OST-1 (Cys260–Cys269) with the sequence of 3-OST-3 (Cys351–Cys363) has no effect on the substrate specificity of 3-OSTs (data not shown). This finding suggests that this loop structure does not directly contribute to the substrate specificity of 3-OST-1 and 3-OST-3. Another notable difference between 3-OST-1 and 3-OST-3 is the presence of a N-terminal α-helix, Pro136–Glu147 (residues Pro136–Ser138 are cloning artifacts from the expression vector), found in 3-OST-3. This helix packs along the side of the protein in a shallow groove formed by the C-terminal three-stranded β-sheet of a neighboring molecule in the crystal. However, it is external to the conserved sulfotransferase domain.
Tetrasaccharide-binding Site
The structure of the protein with bound substrate is virtually identical to that of the apo-protein with respect to the backbone positions. A small number of side chains were altered upon substrate binding, most of which are found in or near the HS-binding site. The tetrasaccharide (δUA2S-GlcNS6S-IdoUA2S-GlcNS6S; Fig. 1B) binds in an extended conformation along the open cleft. This position places the 3-OH group of the glucosamine unit (G3) in close proximity to the PAP-binding site within the open cleft. It is interesting to note that the tetrasaccharide appears to behave rather like two distinct disaccharide units (Fig. 4). The two units on the reducing end (I2-G1) have fewer potential hydrogen bonding partners than those (U4-G3) on the nonreducing end and thus are more disordered and consequently have higher B-factors (Table I). In addition, the sugar rings of U4-G3 lie in the same plane that is rotated almost 90° around the α(1→4) glycosidic bond, with respect to the plane of the sugar rings of I2-G1. A similar twist has been previously observed in the basic fibroblast growth factor/HS structure (23).
Fig. 4. Stereo diagrams of the binding of 3-OST-3 and tetrasaccharide.

a, stereo diagram of HS acceptor substrate binding cleft with tetrasaccharide (green) bound. Residues that line the cleft are pictured. Displayed in blue is the simulated annealing Fo – Fc omit electron density map contoured at 2 σ. b, stereo diagram displaying the interactions between 3-OST-3 and U4-G3 units. The O-3 oxygen of G3 unit is labeled. c, stereo diagram of the interactions between 3-OST-3 and the I2-G1 units. A metal ion modeled as a sodium ion is colored cyan, as are interactions between the metal and the coordinating atoms. Possible hydrogen bonds between the protein and tetrasaccharide are displayed as black dashed lines. These figures were created using Molscript and Raster3d (33, 34).
The saccharide that forms the most extensive interactions with the protein is the uronic acid (U4) (Fig. 4, a and b, Table IV). This observation suggests that this sugar unit contributes significantly to the substrate specificity for 3-OST-3. Residues Gln255, Lys259, and Arg370 are all within hydrogen bonding distance to the 2-O-sulfo group. Residues Arg166, Lys215, and Lys368 are all within hydrogen bonding distance to the carboxyl group (Table IV). In addition to interactions with the charged groups, the hydroxyl from residue Thr367 is within hydrogen bonding distance to the 3-OH of U4. It should be noted that δ4,5 unsaturated 2-O-sulfo uronic acid (δUA2S or U4 in this tetrasaccharide), a product of heparin lyase digestion, is a saturated saccharide unit (in the form of 2-O-sulfo iduronic acid, or IdoUA2S) in HS polysaccharide. The 2So conformation of 2-O-sulfo iduronic acid and the 2H1 (31) conformation of δUA2S (U4) unit allow similar positioning of the 2-O-sulfo, 3-OH, and 5-carboxyl groups. Thus, the δUA2S mimics IdoUA2S sufficiently well to be recognized by 3-OST-3. Furthermore, the solution conformation of this tetrasaccharide indicates that neither the I2 or U4 units substantially modify the conformations of the adjacent G1 and G3 residue (31). Indeed, this tetrasaccharide is a substrate for 3-OST-3 as described below.
Table IV.
Distances of potential hydrogen bonds between the tetrasaccharide and 3-OST-3 protein
| Saccharide | Protein contact | Distance |
|---|---|---|
| Å | ||
| U4 | ||
| 2 O1S | Lys259 NZ | 2.9 |
| 2 O1S | Gln255 NE2 | 3.0 |
| 2 O3S | Arg370 NH2 | 2.7 |
| 2 O3S | Gln255 OE1 | 3.1 |
| 6 O61 | Lys368 NZ | 2.7 |
| 6 O61 | Lys215 NZ | 2.7 |
| 6 O62 | Arg166 NE | 2.7 |
| 6 O62 | Arg166 NH2 | 2.5 |
| 3 OH | Thr367 OG1 | 2.6 |
| G3 | ||
| 2 N O2S | Ser218 N | 3.0 |
| 2 N O6S | Lys366 NZa | 2.5 |
| 6 O4S | Lys259 NZ | 2.9 |
| O6 | Lys259 NZ | 3.1 |
| 3 OH | Glu184 OE2 | 2.8 |
| 3 OH | H2O 158 | 3.0 |
| I2 | ||
| 3 OH | Arg190 NH2 | 2.6 |
| 6 O61 | Lys259 NZ | 2.6 |
| 6 O61 | Gln255 NE2 | 2.6 |
| 6 O62 | Lys161 NZ | 2.6 |
| G1 | ||
| 2 N | Thr256 OG1 | 2.9 |
| 2 N O1S | Ser284 OG | 2.9 |
| 2 N O1S | Trp283 NE1 | 2.9 |
| 2 N O3S | Asp252 OD1 | 2.8 |
This residue is from a crystallographic symmetry related protein molecule in the crystal lattice.
The G3 unit appears to be the acceptor site of sulfonation. This unit is present in a 4C1 conformation, and its 3-OH is positioned within hydrogen bonding distance to the glutamate residue (Glu184) (Table IV). It has been demonstrated that the equivalent residue in similar enzymes functions as the catalytic base (14, 16). A less ordered sulfo group at the C-6 position was observed, suggesting that it is not essential for substrate specificity. This observation is consistent with results from biochemical studies of 3-OST-3 (11, 25). The N-sulfo group is in a position to form a hydrogen bond with the backbone amide of Ser218. It has been determined that 3-OST-3 sulfonates an N-unsubstituted glucosamine unit at the polysaccharide level (Fig. 1A) (25). Both glucosamine units (G1 and G3) are N-sulfonated (Fig. 1B) in the tetrasaccharide used in this experiment. Furthermore, the N-sulfo group was well ordered in a confined pocket, although no positively charged amino acid residues are found nearby. The restricted location of the N-sulfo group is perhaps due to the fact that the acceptor 3-OH group is highly oriented, consequently reducing the flexibility of N-sulfo group. In light of these results, we are currently unable to conclusively determine whether this N-sulfo group is required for substrate recognition.
Numerous amino acid residues have contacts with the carboxyl group of the iduronic acid unit (I2). The amino acid residues include Lys161, Gln255, and Lys259 (Fig. 4c). In addition, the 3-OH position lies well within hydrogen bonding distance of Arg190. Interestingly, the 2-O-sulfo group is not positioned near any hydrogen bonding partners, yet it is reasonably well ordered. The I2 unit is present in the 2S0 skew boat conformation, which differs from the conformation of this saccharide bound to basic fibroblast growth factor (23). Recent work has demonstrated the importance of the iduronic acid skew boat conformation in exhibiting the anticoagulant activity (32).
The G1 unit is also present in a 4C1 conformation (Fig. 4c). The N-sulfo group interacts with the side chain of Thr256 as well as with the side chains of Asp252, Trp283, and Ser284 (Table IV). The 6-O-sulfo group makes no hydrogen bonding contacts with the protein. Consequently, it is the most disordered group in the tetrasaccharide. Interestingly, a metal ion modeled as a sodium ion appears to bind to the I2 and G1 units. The backbone carbonyl and side chain of Asp252, as well as the side chain of Thr256, provide three of the ligands coordinated to the metal (Fig. 4c, Table III). The other three ligand interactions, which complete the octahedral coordination, are from the N-sulfo and 3-OH of the G1 unit as well as a carboxylate oxygen atom of the I2 unit.
Table III.
Distances of potential interactions with the metal ion
| Ion | Protein contact | Distance |
|---|---|---|
| Å | ||
| NA+1 | I2 O62 | 2.7 |
| NA+1 | G1 2N O3S | 2.4 |
| NA+1 | G1 3OH | 2.5 |
| NA+1 | Asp252 O | 2.5 |
| NA+1 | Thr256 OG1 | 2.3 |
| NA+1 | Asp252 OD1 | 2.4 |
Determination of 3-O-Sulfonation Site on the Tetrasaccharide
To confirm the conclusions from the structural data, we proved that 3-OST-3 transfers the [35S]sulfuryl group to the G3 unit (rather than to the G1 unit) when the tetrasaccharide is used as a substrate. We prepared the 3-O-[35S]sulfated tetrasaccharide by incubating the tetrasaccharide with purified 3-OST-3 enzyme and [35S]PAPS. We then identified the [35S]sulfonation site of the resultant tetrasaccharide. This was determined by comparing products of the heparin lyase digested 3-O-[35S]sulfated tetrasaccharide with and without pretreatment of δ4,5-glycuronate-2-sulfatase as described under “Experimental Procedures.” We observed that the product (δUA2S-[3-35S]GlcNS3S6S) of 3-O-[35S]sulfated tetrasaccharide digested by heparin lyase was eluted at 38 min on the anion exchange HPLC (Fig. 5C, dashed line), whereas the product (δUA-[3-35S]GlcNS3S6S) of the sulfatase-pretreated sample was eluted at 25 min (Fig. 5C, solid line). Therefore, our data demonstrated that the 3-O-[35S]sulfonation site is indeed on the G3 unit as predicted by the crystal structure.
Catalytic Mechanism
The ternary structure of 3-OST-3/PAP/tetrasaccharide provides atomic details about the reaction mechanism of HS sulfotransferases. When PAPS is superimposed onto PAP, the acceptor hydroxyl group of G3 is located 2.9 Å from the sulfur atom of PAPS and on the opposite of the sulfur from the leaving group PAP (Fig. 6). The sulfonate group is easily accommodated by the enzyme without rearrangement of side chain residues. The location of the sulfonate group is consistent with a SN2-like in-line displacement mechanism. Given that Glu184 is 2.8 Å from the 3-OH of the G3 unit (Fig. 4), it is reasonable to suppose that this residue likely functions as a catalytic base, deprotonating the 3-OH for nucleophilic attack on the sulfonate group. This glutamate residue is conserved among 3-OSTs and NSTs, thereby suggesting an essential role for the catalytic function of HS sulfotransferases (NSTs transfer the sulfuryl group to the amino position of a glucosamine unit within a HS polysaccharide.). Indeed, mutations of this glutamate residue result in a complete loss of sulfotransferase activities for NST-1 and 3-OST-1 (14, 16) and a 99.9% loss for 3-OST-3 (Table V).
Fig. 6. Active site of 3-OST-3 with PAPS superimposed.
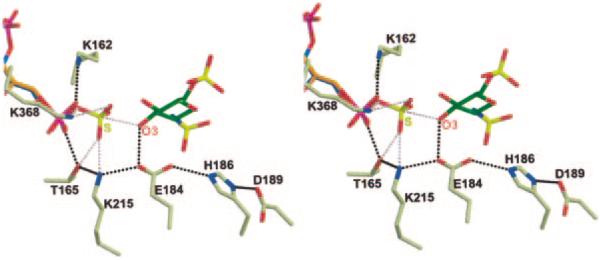
Stereo diagram of the superposition of the PAPS (orange) from the crystal structure of human estrogen sulfotransferase into the active site of 3-OST-3 (30). The superposition is based on the Cα atoms of the phosphosulfate-binding loop and selected atoms of the PAP and PAPS molecules (O-5′, C-5′, C-4′, C-3′, and the 5′phosphate). The PAP (blue) and the acceptor G3 (green) are pictured, as are the residues that may play a catalytic role. Possible hydrogen bonds between 3-OST-3 and its substrate are displayed as black dashed lines. Possible interactions between 3-OST-3 and the donor sulfuryl group based on the superposition are displayed as pink dashed lines. Also displayed as a pink dashed line is the line of attack of the O-3 oxygen of the sugar to the sulfur atom of the PAPS molecule. This figure was created using Molscript and Raster3d.
Table V.
Summary of the analysis of the sulfotransferase activity of 3-OST-3 mutants
| Name of the constructsa | Relative activityb | Expression levelc |
|---|---|---|
| % | ||
| 3-OST-3 wild type | 100 | + + |
| 3-OST-3 K161A | 0.4 | + |
| 3-OST-3 K162A | 0.4 | + |
| 3-OST-3 R166E | 0.2 | + + |
| 3-OST-3 E170Q | 83 | + + |
| 3-OST-3 R173S | 55.9 | + + + |
| 3-OST-3 G182A | 106 | + + |
| 3-OST-3 E184Q | 0.1 | + + |
| 3-OST-3 H186F | 0.0 | + |
| 3-OST-3 D189N | 0.9 | + |
| 3-OST-3 R190E | 68 | + + |
| 3-OST-3 K194A | 0.5 | + |
| 3-OST-3 K215A | 0.1 | + + + |
| 3-OST-3 S218A | 76.7 | + + |
| 3-OST-3 E224Q | 52.4 | + + |
| 3-OST-3 Q255A | 0.4 | + + |
| 3-OST-3 K259A | 51.7 | + + |
| 3-OST-3 I288A | 35 | + + |
| 3-OST-3 K293A | 66.4 | + + |
| 3-OST-3 H362A | 101.5 | + + |
| 3-OST-3 G365A | 57 | + + |
| 3-OST-3 K366A | 0.2 | + + |
| 3-OST-3 K368A | 0.1 | + + |
| 3-OST-3 R370E | 0.8 | + + |
The mutants were prepared using a site-directed mutagenesis kit from Invitrogen.
The activity of 3-OST-3 was assayed by incubating the purified mutant proteins with HS and [35S]PAPS, and the resultant [35S]HS was quantified by a DEAE chromatography, where 100% activity represents the transfer of 18 pmol of sulfate/μg of protein under the standard assay condition.
The expression level of the proteins was determined by the intensity of the Coomassie Blue-stained protein band migrated at 30 kDa on SDS-PAGE.
A hydrogen bonding network that involves Glu184, His186, and Asp189 is observed in the ternary complex (Fig. 6). The OE2 oxygen of Glu184 is 2.9 Å from the NE2 atom of His186. The ND1 atom of His186 is 2.8 Å from OD2 of Asp189. This hydrogen bonding pattern is reminiscent of a catalytic triad. The results of the mutational analysis demonstrated that His186 and Asp189 are essential for the sulfotransferase activity (Table V). This hydrogen bond network could either help properly position the glutamate for catalysis or serve as a charge relay system to regulate the pKa of the glutamate. A similar hydrogen bond network was also observed in 3-OST-1 but not in NST-1 (14, 15). Thus, the interactions within the “triad” appear to be specific for 3-OSTs.
Mutational analysis
Site-directed mutagenesis experiments were performed to decipher the roles of the amino acid residues in performing the catalytic functions and determining substrate specificity. The results are summarized in Table V. We conducted the disaccharide analysis of the HS modified by the mutants that maintain the sulfotransferase activity (catalytically active mutants). Our data indicate that wild type protein and all of the catalytically active mutants sulfonate identical disaccharide sequences (IdoUA2S-GlcNH2 and IdoUA2S-GlcNH26S; see Fig. 1A for structures), suggesting that these mutants maintain wild type 3-OST-3 substrate specificity (data not shown). This conclusion is further strengthened by our findings that none of the HS modified by the active mutants bind to antithrombin (data not shown). This is consistent with our understanding that 3-OST-1-modified HS binds to antithrombin, whereas 3-OST-3-modified HS does not (Fig. 1A).
Several mutations resulted in the loss of enzymatic activity (catalytically inactive mutants). Among these, K162A, E184Q, H186F, and D189N are probably due to defects in catalytic function. These conclusions are based on the crystal structural data as described above, as well as our previous results of the characterization of corresponding 3-OST-1 mutants (14). The roles of Lys215 and Arg370 are more complex. These two residues could be involved in both binding to PAPS and substrate. Like Glu184 and Lys162, Lys215 and Arg370 are conserved in the 3-OSTs and NST-1.
Two mutants, Q255A and K368A, are of special interest. Mutations at Gln255 and Lys368 nearly abolish sulfotransferase activity. However, mutations at the corresponding residues of 3-OST-1, Q166A and K274A, maintain 34.0 and 17.4% of the sulfotransferase activity, respectively (14). The data suggest that Gln255 and Lys368 contribute to the substrate specificity of 3-OST-3. Indeed, the structural data indicate that the side chains of Gln255 and Lys368 are within the hydrogen bonding distance to the carboxyl and 2-O-sulfo groups of the U4 unit and the carboxyl group of the I2 unit as described above. This is consistent with the fact that 3-OST-1 recognizes a disaccharide substrate (GlcUA-GlcNS±6S) that does not contain the 2-O-sulfo group and the orientation of the carboxyl group of the GlcUA unit is likely distinct from that of an iduronic acid unit (Fig. 1A).
Presently, we are unable to explain the mutation results for Lys194 and Lys366 because the side chains of both residues do not interact with the tetrasaccharide. Residue Lys194 is located over 14 Å from the tetrasaccharide. Residue Lys366 is located in a position where it could interact with the nonreducing end of the oligosaccharide if one more sugar (a glucosamine) were present, as is the case in the polymeric HS substrate used in the mutation studies. Interestingly, in the x-ray crystal structure, Lys366 forms a lattice contact with the 6-O-sufo group of G3 from a tetrasaccharide binding in a crystallographically related molecule. This interaction does not appear to be bio-chemically relevant because the 6-O-sulfo group is not required for 3-OST-3 activity (11), and the buried surface area between the crystallographic interface of the two protein molecules is less than 3% of the total surface area.
In summary, the structure of the 3-OST-3/PAP/tetrasaccharide has provided new insights into the requirements for the substrate recognition by 3-OSTs and a more complete description of the HS sulfotransferase mechanism. It appears that 3-OST-3 recognizes the carboxyl and sulfo groups of the iduronic acid (U4) at the nonreducing end of the glucosamine unit (G3) being sulfonated. The amino acid residues involved in binding to these structural motifs are essential for the activity of 3-OST-3. This conclusion is consistent with those from previous biochemical studies of 3-OSTs; 3-OST-1 sulfates a glucosamine linked to a glucuronic acid unit at the nonreducing end, whereas 3-OST-3 sulfates the glucosamine unit that is linked to a 2-O-sulfo iduronic acid unit at the nonreducing end (Fig. 1A) (9, 11, 17, 25). It is very important to note that the 3-OST-3 modification site is flanked by two iduronic acid units in a skew boat like conformation. In contrast, the conformation of the I2 unit is in the chair form in the fibroblast growth factor/tetrasaccharide complex (23). It is likely that the conformation of the flanking iduronic acid residues contribute to substrate recognition by 3-OST-3.
The 3-O-sulfonation of HS has been directly linked to the activities for regulating blood coagulation and for promoting infection by HSV-1. Consequently elucidation of the general mechanism and discovery of the atomic level requirements for specificity provide important insight for understanding the biosynthesis of biologically active forms of HS.
Acknowledgments
We thank Dr. Keiichi Yoshida (Seikagaku Corporation) for providing us purified δ4,5-glycuronate-2-sulfatase and heparin lyase IV and Drs. Jeffrey Vargason and Lee Pedersen for critical reading of the manuscript.
Footnotes
This work is supported in part by National Institutes of Health Grants AI50050 (to J. L.) and HL52622 (to R. J. L.) and Grant-in-Aid 0355800U (to J. L.) from the American Heart Association MidAtlantic Affiliate. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The atomic coordinates and structure factors (codes 1T8U and 1T8T) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The abbreviations used are: HS, heparan sulfate; HSV-1, herpes simplex virus type 1; 3-OST-3, 3-O-sulfotransferase isoform 3; PAPS, 3′-phosphoadenosine 5′-phosphosulfate; NST, N-sulfotransferase; PAP, 3′-phosphoadenosine 5′-phosphate; Tricine, N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine; MES, 4-morpholineethanesulfonic acid; HPLC, high pressure liquid chromatography; GlcUA, glucuronic acid.
REFERENCES
- 1.Esko JD, Selleck SB. Annu. Rev. Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 2.Liu J, Thorp SC. Med. Res. Rev. 2002;22:1–25. doi: 10.1002/med.1026. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl U, Kusche-Gullberg M, Kjellen L. J. Biol. Chem. 1998;273:24979–24982. doi: 10.1074/jbc.273.39.24979. [DOI] [PubMed] [Google Scholar]
- 4.Sasisekharan R, Shriver Z, Venkataraman G, Narayanasami U. Nat. Rev. Cancer. 2002;2:521–528. doi: 10.1038/nrc842. [DOI] [PubMed] [Google Scholar]
- 5.Corey L, Spear PG. N. Engl. J. Med. 1986;314:686–691. doi: 10.1056/NEJM198603133141105. and 749–757. [DOI] [PubMed] [Google Scholar]
- 6.Spear PG, Eisenberg RJ, and Cohen GH. Virology. 2000;275:1–8. doi: 10.1006/viro.2000.0529. [DOI] [PubMed] [Google Scholar]
- 7.Shukla D, Spear PG. J. Clin. Invest. 2001;108:503–510. doi: 10.1172/JCI13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spear PG, Longnecker R. J. Virol. 2003;77:10179–10185. doi: 10.1128/JVI.77.19.10179-10185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. Cell. 1999;99:13–22. doi: 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 10.Xia G, Chen J, Tiwari V, Ju W, Li J-P, Malmstroöm A, Shukla D, Liu J. J. Biol. Chem. 2002;277:37912–37919. doi: 10.1074/jbc.M204209200. [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Shriver Z, Pope RM, Thorp SC, Duncan MB, Copeland RJ, Raska CS, Yoshida K, Eisenberg RJ, Cohen G, Linhardt RJ, Sasisekharan R. J. Biol. Chem. 2002;277:33456–33467. doi: 10.1074/jbc.M202034200. [DOI] [PubMed] [Google Scholar]
- 12.Shworak NW, Liu J, Petros LM, Zhang L, Kobayashi M, Copeland NG, Jenkins NA, Rosenberg RD. J. Biol. Chem. 1999;274:5170–5184. doi: 10.1074/jbc.274.8.5170. [DOI] [PubMed] [Google Scholar]
- 13.Liu J, Shworak NW, Fritze LMS, Edelberg JM, Rosenberg RD. J. Biol. Chem. 1996;271:27072–27082. doi: 10.1074/jbc.271.43.27072. [DOI] [PubMed] [Google Scholar]
- 14.Edavettal SC, Lee KA, Negishi M, Linhardt RJ, Liu J, Pedersen LC. J. Biol. Chem. 2004;279:25789–25797. doi: 10.1074/jbc.M401089200. [DOI] [PubMed] [Google Scholar]
- 15.Kakuta Y, Sueyoshi T, Negishi M, Pedersen LC. J. Biol. Chem. 1999;274:10673–10676. doi: 10.1074/jbc.274.16.10673. [DOI] [PubMed] [Google Scholar]
- 16.Kakuta Y, Li L, Pedersen LC, Pedersen LG, Negishi M. Biochem. Soc. Trans. 2003;31:331–334. doi: 10.1042/bst0310331. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Shworak NW, Sinayÿ P, Schwartz JJ, Zhang L, Fritze LMS, Rosenberg RD. J. Biol. Chem. 1999;274:5185–5192. doi: 10.1074/jbc.274.8.5185. [DOI] [PubMed] [Google Scholar]
- 18.Otwinowski Z, Minor V. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 19.Vagin A, Teplyakov A. J. Appl. Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 20.Bailey S. Acta Crystallogr. Sect. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 21.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Crystallogr. Sect. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 22.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 23.Faham S, Hileman RE, Fromm JR, Linhardt RJ, Rees DC. Science. 1996;271:1116–1120. doi: 10.1126/science.271.5252.1116. [DOI] [PubMed] [Google Scholar]
- 24.Shively JE, Conrad HE. Biochemistry. 1976;15:3932–3942. doi: 10.1021/bi00663a005. [DOI] [PubMed] [Google Scholar]
- 25.Liu J, Shriver Z, Blaiklock P, Yoshida K, Sasisekharan R, Rosenberg RD. J. Biol. Chem. 1999;274:38155–38162. doi: 10.1074/jbc.274.53.38155. [DOI] [PubMed] [Google Scholar]
- 26.Rice KG, Linhardt RJ. Carbohydr. Res. 1989;190:219–233. doi: 10.1016/0008-6215(89)84127-8. [DOI] [PubMed] [Google Scholar]
- 27.McLean MW, Bruce JS, Long WF, Williamson FB. Eur. J. Biochem. 1984;145:607–615. doi: 10.1111/j.1432-1033.1984.tb08600.x. [DOI] [PubMed] [Google Scholar]
- 28.Matthews BW. J. Mol. Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 29.Negishi M, Pedersen LG, Petrotchenko E, Shevtsov S, Gorokhov A, Kakuta Y, Pedersen LC. Arch. Biochem. Biophys. 2001;390:149–157. doi: 10.1006/abbi.2001.2368. [DOI] [PubMed] [Google Scholar]
- 30.Pedersen LC, Petrotchenko E, Shevtsov S, Negishi M. J. Biol. Chem. 2002;277:17928–17932. doi: 10.1074/jbc.M111651200. [DOI] [PubMed] [Google Scholar]
- 31.Mikhailov D, Mayo KH, Pervin A, Linhardt RJ. Biochem. J. 1996;315:447–454. doi: 10.1042/bj3150447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das SK, Mallet JM, Esnault J, Driguez PA, Duchaussoy P, Sizun P, Herault JP, Herbert JM, Petitou M, Sinay P. Chemistry. 2001;7:4821–4834. doi: 10.1002/1521-3765(20011119)7:22<4821::aid-chem4821>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 33.Kraulis PJ. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- 34.Merritt EA, Bacon DJ. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 35.Lovell SC, Davis IW, Arendall W. B. r., de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]