

Abstract
The state-of-the-science in asymmetric free radical additions to imino compounds is presented, beginning with an overview of methods involving stereocontrol by various chiral auxiliary approaches. Chiral N-acylhydrazones are discussed with respect to their use as radical acceptors for Mn-mediated intermolecular additions, from design to scope surveys to applications to biologically active targets. A variety of aldehydes and ketones serve as viable precursors for the chiral hydrazones, and a variety of alkyl iodides may be employed as radical precursors, as discussed in a critical review of the functional group compatibility of the reaction. Applications to amino acid and alkaloid synthesis are presented to illustrate the synthetic potential of these versatile stereocontrolled carbon–carbon bond construction reactions. Asymmetric catalysis is discussed, from seminal work on the stereocontrol of radical addition to imino compounds by non-covalent interactions with stoichiometric amounts of catalysts, to more recent examples demonstrating catalyst turnover.
1. Background and Introduction
Chiral α-branched amine functionalities are present in a wide range of bioactive synthetic targets, both natural and unnatural. Accordingly, a variety of methods for asymmetric amine synthesis have been developed over recent years, many of which involve carbon–carbon bond constructions by addition to the C=N bond of carbonyl imino derivatives, represented in the retrosynthetic direction in Figure 1 [1].
Fig. 1.
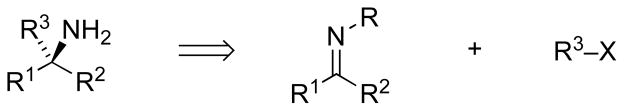
Disconnecting a C–C bond of a chiral amine suggests an imino compound (e.g., imine, oxime, hydrazone, etc.) and an organic halide as precursors.
As is typical in most of synthetic chemistry, methods in this area accrue higher impact if they offer configurational control under mild conditions compatible with a range of spectator functional groups. Unfortunately, many methods involving nucleophilic additions of carbanion-type reagents to C=N bonds have limited compatibility with electrophilic or acidic functionality, or may result in competing deprotonation at the imine α-carbon due to the basicity of the organometallic reagent.[2] These limitations have spurred the development of free radical additions [3] to imino compounds (Figure 2) as a C–C bond construction approach to chiral amines under mild conditions, offering a valuable complement to organometallic additions and expanding the scope of this α-C–C retrosynthetic disconnection.[4]
Fig. 2.
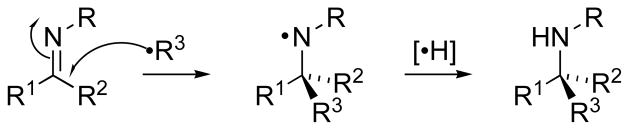
Radical Addition to Imino Compounds.
Seeking to probe the improved versatility which might be associated with the radical addition approach, we initiated a program to develop a variety of radical additions to imino compounds.[5] In the process, we introduced several new modes of stereocontrol using hydrazones as the C=N radical acceptor functionality. Imino compounds have been extensively used for cyclizations [6], and initially we built upon this foundation by introducing a temporary tethering approach [7,8] which has been effective in establishing relative configuration in a predictable diastereoselective fashion via reactions of α-hydroxyaldehyde hydrazones [9]. In this review, we will focus on the intermolecular variant of the reaction, for which we have introduced methodology to build chiral amines from achiral precursors using chiral auxiliaries or chiral catalysts for stereocontrol;[10] the design and experimental evaluation of these strategies will be described along with synthetic applications.
2. Intermolecular Radical Addition to Chiral N-Acylhydrazones
2.1. Use of Chiral Auxiliaries in Radical Additions to Imino Compounds
In many areas of synthetic methods development, the reaction methodology has progressed to a point where it is quite well-understood, and the exciting new advances are mostly focused on extension to asymmetric catalysis. In contrast, in the area of radical additions to C=N bonds, only rare examples of intermolecular coupling reactions were available prior to the initial work on asymmetric methods [11] Thus, the development of asymmetric induction for radical addition to imino compounds has been slow, pending important advances that are still needed in the fundamental reaction chemistry. As this chemistry emerges, the challenges of adapting the new chemistry to all types of stereocontrol modes, including auxiliary, reagent and catalyst, continue to attract active investigation. An overview of some of the main approaches to chiral auxiliary-mediated stereocontrol is provided below.
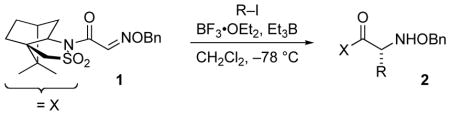
Naito and coworkers reported the first general method for achieving reductive addition of carbon-centered radicals to imino compounds; by using Et3B/oxygen initiation in the presence of BF3•OEt2, neutral radicals were added successfully to prochiral aldoximes.[12] Although unhindered formaldoximes and activated glyoxylic oxime ethers did not require Lewis acid activation, the beneficial effect of BF3•OEt2 allowed general application to various oxime ethers. This enabled the use of 1 bearing a chiral auxiliary for asymmetric induction (Table 1).[13] The proposed stereocontrol model invokes steric blocking by one of the sultam oxygens (Figure 3). Although difunctional alkyl iodides afforded lower yields, subsequent work has successfully employed more complex radicals.[14]
Table 1.
Radical Addition to Chiral Glyoxylic Oxime Ethers.
| |||
|---|---|---|---|
| Entry | R1 | Yielda | drb |
| 1 | Et | 80% | 95:5 |
| 2 | i-Bu | 83% | 97:3 |
| 3 | i-Pr | 80% | 96:4 |
| 4 | s-Bu | 69%c | >98:2 |
| 5 | c-C6H11 | 86% | 96:4 |
| 6 | t-Bu | 83% | >98:2 |
| 7 | AcO(CH2)4 | 41%d | >98:2 |
| 8 | Cl(CH2)4 | 15%d | >98:2 |
Isolated yield.
dr = diastereomer ratio.
Obtained as 1:1 mixture of diastereomers.
Major product was ethyl addition.
Fig. 3.
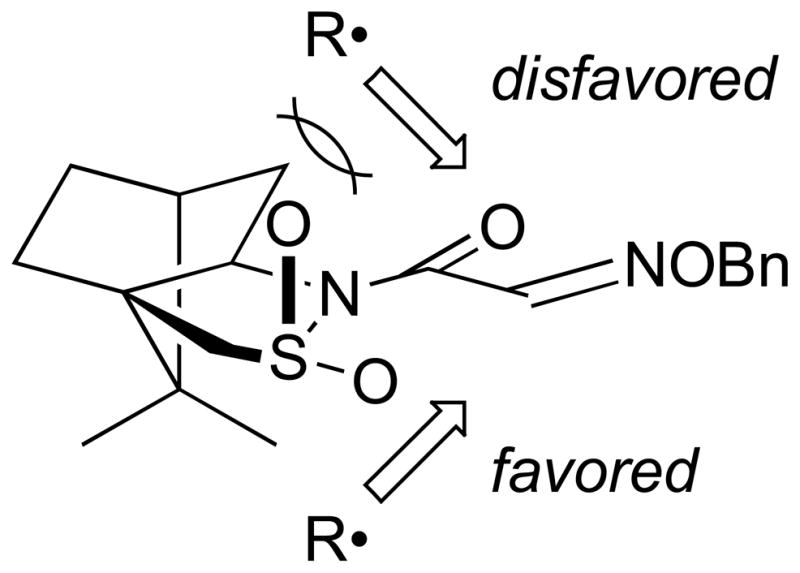
Naito stereocontrol model proposed for additions to camphorsultam-functionalized glyoxylic oxime ethers.
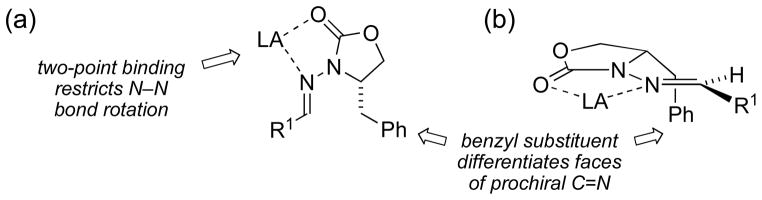
(a) Lewis acid chelation induces rigidity and electron-deficiency into the N-acylhydrazone radical acceptor (LA = Lewis acid). (b) Benzyl substituent of 4-benzyl-2-oxazolidinone provides facial diferentiation of the C=N bond.
Bertrand and coworkers reported the radical addition of various alkyl iodides to cyclic and acyclic chiral glyoxylate imines using either tin-mediated conditions initiated by AIBN, or triethylborane conditions initiated by oxygen.[15] Building on these initial studies, Bertrand discovered the combination of diethylzinc and air was useful to promote radical addition to chiral imines (Scheme 1).[16] Stereoselectivity was modest in additions to 1-phenylethylimines. However, two-point binding imines capable of chelating Zn(II), such as norephedrine-derived imine 3, led to improved diastereomer ratios. [17]
Scheme 1.

Radical addition to glyoxylate imines.
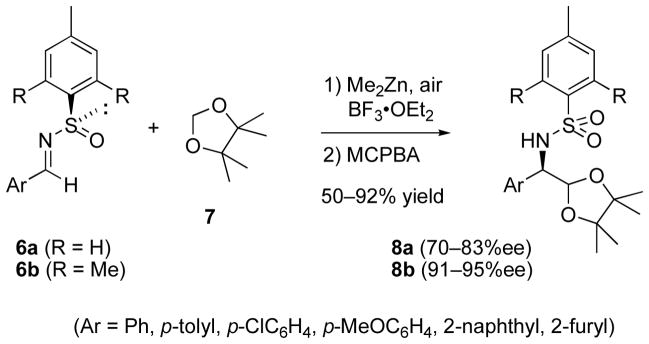
Chiral N-sulfinylimines, pioneered by Davis and Ellman for stereocontrolled additions of carbanion reagents,[18] have also been employed successfully in radical additions. Tomioka and coworkers have used the dimethylzinc/air method to generate radicals by H-atom abstraction from ethers and acetals which then add to the C=N bond of the N-sulfinylimines 6.[19] Although tetrahydrofuran and tert-butyl methyl ether gave modest stereocontrol, formaldehyde acetals were more promising, with the best results observed for addition of dioxolane 7 (Scheme 2) to substituted benzaldehyde imines (70–83% ee, measured after oxidation to sulfonamide). Subsequently, increasing the steric demand of the arenesulfinyl stereocontrol element led to improved enantioselectivities.[20] Recently the reaction has been extended to include triethylborane-mediated addition of an iodomethyl ester to N-alkoxycarbonylimines.[21]
Scheme 2.
Radical addition to N-sulfinylimines
2.2. Design of Chiral N-Acylhydrazones
In the late 1990s we began our work in developing hydrazones as promising radical acceptors for asymmetric tranformations. At that time, oxime ethers were the most commonly explored C=N radical acceptors.[22,23,24] We considered that linking an amino or amido substituent to the C=N nitrogen atom could afford the same reactivity enhancement seen in oxime ethers, and would also provide opportunities for superior rotamer control needed for stereoselectivity. Both activation and stereocontrol would be achieved through one removable N-substituent on the imine, and the substituents on the carbon of the C=N bond would then be freed from those roles, offering potential for broader scope.
Our new type of chiral hydrazone, tailored for use in free radical addition reactions, incorporates Lewis acid activation and restriction of rotamer populations as key design elements. We hypothesized that the restricted rotation achieved through two-point binding would fix a substituent relative to the plane of the C=N bond to differentiate the enantiotopic approach trajectories (Figure 3). The Lewis acid would also lower the LUMO energy of the C=N π bond, increasing its reactivity toward nucleophilic alkyl radicals.[25] This would ensure selective reactivity via the chelated structure, suppressing the non-selective background reaction. After radical addition, H-atom abstraction would occur to afford an N-acylhydrazine product; reductive cleavage of the N–N bond [26] would provide an enantiomerically pure amine and release the chiral auxiliary for reuse. For the first test of our design, N-acylhydrazones derived from N-aminooxazolidinones [27] were chosen to satisfy all these design criteria (Figure 3).
2.3. Preparation and Initial Reactivity Studies of Chiral N-Acylhydrazones
Although N-amino derivatives of oxazolidinones such as 9 (Scheme 3) were sporadically reported in the literature,[28] the installation of the N-amino group was not well-developed. Therefore we began with a study of electrophilic amination of commercial oxazolidinones. A variety of electrophilic amination reagents were employed for this purpose, and the preferred reagents were O-(p-nitrobenzoyl)hydroxylamine (NbzONH2), O-(diphenylphosphinyl)hydroxylamine (Ph2P(=O)ONH2) and NH2Cl.[29,30] The optimized procedure for N-amination with NbzONH2 entailed deprotonation with NaH (or KH) in hot dioxane, followed by introduction of NbzONH2 as a solid at ambient temperature.[31] With NH2Cl the same procedure may be followed, but the stoichiometry of base and chloramine should be carefully controlled to nearly one equivalent in order to obtain reproducible yields in the amination. A survey of representative condensations of N-aminooxazolidinone 9 with aldehydes and ketones (Scheme 3) shows that the sequence is robust and general, affording a wide range of chiral N-acylhydrazones 10.[32] The reaction is readily scaled to multigram quantities, and the carbonyl component of these N-acylhydrazones may be exchanged with other carbonyl compounds.[31]
Scheme 3.

Representative preparations of chiral N-acylhydrazones
Ketone-derived chiral N-acylhydrazones may also be prepared by direct condensation with N-aminooxazolidinones (Scheme 3).[30,33] Mixtures of E/Z isomers were usually obtained, although ketone N-acylhydrazones with highly branched tertiary butyl (t-Bu) substituents, were formed as single isomers. A pyruvate-derived hydrazone was formed with high selectivity, and the major isomer was readily separated from its minor (Z)-isomer by flash chromatography.[30] Others have recently used these amination and condensation procedures to prepare very similar chiral N-acylhydrazones from ketones with excellent results.[34]
2.3.1. Additions of Secondary and Tertiary Radicals
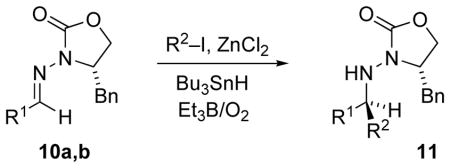
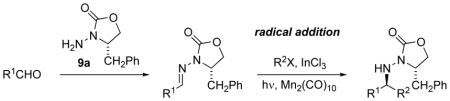
The first test of the chiral N-acylhydrazones was in tin-mediated radical addition of various secondary and tertiary iodides.[32] Using the tin hydride method with triethylborane initiation [35] (Bu3SnH, Et3B/O2), with ZnCl2 as a Lewis acid additive, addition of isopropyl iodide to N-acylhydrazone 10a afforded adduct 11a with a diastereomer ratio of 99:1 (Table 2). In contrast, 11a was produced with poor selectivity (dr 2:1) in the absence of Lewis acid, which validated the two-point binding stereocontrol model (Figure 3). Cyclopentyl, cyclohexyl, and tert-butyl iodides successfully added both to propionaldehyde N-acylhydrazone 10a and to benzaldehyde N-acylhydrazone 10b (Table 2). Ethyl radical generated from triethylborane can compete for the radical acceptor, and as a result, the ethyl radical adduct was observed (<10% yield) in all cases.
Table 2.
Tin-Mediated Radical Addition to Chiral N-Acylhydrazone 10a in the Presence of ZnCl2
| |||
|---|---|---|---|
| Entry | R1 | R2 | Product, Yieldb(dr) |
| 1 | Et (10a) | i-Pr | 11a, 60% (99:1) |
| 2 | Et (10a) | c-C5H9 | 11b, 59% (96:4) |
| 3 | Et (10a) | c-C6H11 | 11c, 28% (97:5) |
| 4 | Et (10a) | t-Bu | 11d, 54% (95:5) |
| 11 | Ph (10b) | i-Pr | 11e, 42% (99:1) |
| 12 | Ph (10b) | c-C5H9 | 11f, 59% (96:4) |
| 13 | Ph (10b) | c-C6H11 | 11g, 30% (99:1) |
| 14 | Ph (10b) | t-Bu | 11h, 83% (93:7) |
Reaction conditions: Bu3SnH (5 equiv) and O2 (7mL/mmol) by syringe pump, i-PrI (10 equiv), Et3B (10 equiv), and Lewis acid (2 equiv), 2:1 CH2Cl2/ether, −78°C → rt.
Recovered hydrazone, %.
Isolated yield, %.
Not determined.
In search of optimal stereocontrol, substituents on the oxazolidinone moiety were varied. Thus, isopropyl radical additions to several propionaldehyde N-acylhydrazones were compared for stereoselectivity (Scheme 4). High diastereoselectivities were observed in all adducts 12a–12e, although a rigorous measurement was not obtained on 12c (R = i-Pr). All of the auxiliaries impart stereocontrol suitable for practical synthetic application.[32b]
Scheme 4.

Role of oxazolidinone substituents on diastereoselectivity in isopropyl radical addition
2.3.2. Triethylborane-Mediated Radical Additions Without Tin
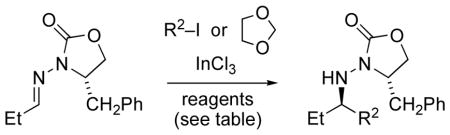
Triethylborane and diethylzinc may serve both as initiator and chain transfer agents in radical additions to C=N bonds.[17,22] This raised the question of whether similar additions to chiral N-acylhydrazones may occur in the absence of tin hydride. Accordingly, we attempted tin-free additions of various halides to the propionaldehyde hydrazone 10a in the presence of triethylborane, using InCl3 as the Lewis acid.[32b] These reactions were indeed successful with various secondary iodides (Table 3, Entries 2–4). Chloroiodomethane also gave successful addition, and the chloromethyl adduct bears functionality suitable for subsequent manipulations.
Table 3.
Tin-Free Radical Additions to 10a in the Presence of InCl3
The use of photolysis also enabled a tin-free method devised by Fernández and Alonso for addition of the 1,3-dioxolan-2-yl radical to these chiral N-acylhydrazones.[36] Irradiation in the presence of 1 equiv benzophenone led to H-atom abstraction from the solvent, 1,3-dioxolane, followed by intermolecular radical addition to chiral N-acylhydrazones (Scheme 5). The Lewis acid (InCl3) facilitated excellent diastereoselectivity in addition of this formyl radical equivalent. The preferred diastereomer was that suggested by the Lewis acid chelate model, consistent with the model supported by our own observations (see Figure 3). After N–N bond cleavage and oxidation at the formyl carbon, preparation of α-amino acids was achieved with high stereoselectivity. A one-pot protocol was also introduced for this reaction, preparing the N-acylhydrazone in situ for the radical addition; thus, for a series of aldehydes, the corresponding adducts 13a–13f were obtained with yields ranging from 75–99%.
Scheme 5.

A one-pot condensation–radical addition of N-acylhydrazones
A limitation of the aforementioned methods is that they are unsuitable for the use of primary alkyl iodides. Under Et3B/O2 initiation conditions, the desired radical is generated by I-atom transfer from the alkyl iodide to ethyl radicals; this is not favorable in the case of primary iodides. Thus ethyl radical (from Et3B) competes with the desired addition of a primary radical in these circumstances. Also, generating radicals by H-atom transfer from ethers or acetals has limited applicability because the radical precursor is generally also a solvent. Because of the expanded synthetic potential of primary alkyl iodides as radical precursors, there is great import in finding alternatives to accomplish I-atom transfers of broader utility for radical additions.
2.4. Manganese-Mediated Radical Addition: Discovery and Method Development
Our attention was drawn to photolytic radical generation with hexamethylditin because it showed promise in Kim’s prior work with C=N radical acceptors, which included additions of primary radicals.[37] We noted, however, that manganese carbonyl [38] [Mn2(CO)10] (λmax 340 nm, σMn–Mn → σ*Mn–Mn) may be photolyzed without any sensitizer, leading to homolytic metal–metal bond cleavage. This produces two •Mn(CO)5 radicals, which are well-known to abstract halogen atoms from alkyl halides.[39] Despite its longtime familiarity in organometallic chemistry, the first studies of the synthetic scope of this reactivity mode appeared in a series of papers by Parsons.[40,41]
Armed with these precedents, we applied these manganese-mediated photolytic conditions to the addition of ethyl iodide to N-acylhydrazone 10a (Table 4).[42] Irradiation (300 nm) with Mn2(CO)10 using InCl3 as a Lewis acid furnished the ethyl adduct in 85% yield (entry 1), a dramatic improvement over use of triethylborane or hexamethylditin. Control experiments revealed a requirement for both irradiation and Mn2(CO)10. Without InCl3, the reaction proceeded sluggishly (21% yield, 2 d).
Table 4.
Mn-Mediated Radical Additions to N-Acylhydrazones.
| |||||
|---|---|---|---|---|---|
| entry | aldehyde (or acetal) | yield, hydrazonea | halide R2X | yield, radical additionb | dr |
| 1 | CH3CH2CHO | 81% | CH3CH2I | 85% | - |
| 2 | CH3I | 48%c,d (S) | 95:5e | ||
| 3 | n-PrI | 66% (R) | 94:6e | ||
| 4 | n-BuI | 78% (R) | 95:5e | ||
| 5 | n-C5H11I | 79% (R) | 96:4e | ||
| 6 | i-BuI | 54%c (R) | 95:5f | ||
| 7 | i-PrI | 75% (R) | 95:5f | ||
| 8 | ClCH2I | 63% (R) | 93:7e | ||
| 9 | Cl(CH2)3I | 52% (R) | 96:4f | ||
| 10 | Cl(CH2)4I | 55% (R) | 96:4e | ||
| 11 | Cl2CHBr | 38%c,d (R) | 98:2f | ||
| 12 | CH3CHO | 66% | CH3CH2I | 66% (R) | 95:5e |
| 13 | n-PrCHO | 87% | 63% (S) | 95:5e | |
| 14 | n-BuCHO | 89% | 72% (S) | 97:3e | |
| 15 | n-C5H11CHO | 88% | 77% (S) | 97:3e | |
| 16 | i-BuCHO | 85% | 65% (S) | 95:5f | |
| 17 | ClCH2CH(OMe)2 | 85% | 57% (S) | 93:7e | |
| 18 | Cl(CH2)3CHO | 95% | 60% (S) | 93:7f | |
| 19 | Cl(CH2)4CHO | 89% | 62% (S) | 97:3e | |
| 20 | Cl2CHCH(OEt)2 | 54% | 34%c (S) | 89:11f | |
Reaction conditions: (1) Aldehyde or acetal (5–10 equiv), 9a, p-toluenesulfonic acid, CH2Cl2, rt. (2) Hydrazone in deoxygenated CH2Cl2 (0.1 M), InCl3 (2.2 equiv), Mn2(CO)10 (1–2 equiv), R2X (10 equiv), hν (300 nm, pyrex), 1–2 d, ca. 35 °C.
Isolated yield.
Isolated yields of purified diastereomer mixtures. R or S denotes the configuration of the new stereogenic center. Addition of methyl iodide gives opposite configurations due to the lower priority of the methyl ligand.
20 equiv of R2X was used.
1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) was used in removal of Mn byproducts.
Ratio by HPLC (Chiralcel OD, 2-PrOH/hexane).
Ratio by 1H NMR.
A variety of other halides, including methyl iodide and dihaloalkanes, were also effective (Table 4, entries 2–11). An exception was a low-yielding addition of 2-chloroethyl iodide, which presumably was compromised by fragmentation of the 2-chloroethyl radical. Similar versatility was displayed with respect to the hydrazone component; ethyl radical addition to various N-acylhydrazones occurred in good yields (entries 12–20). These adducts are epimeric to those derived from hydrazone 10a with respect to the new stereogenic center, as a result of simply changing the roles of the aldehyde and iodide precursors. From the strategy standpoint, this combination of stereocontrol flexibility and functional group compatibility is advantageous for total synthesis applications.
2.5. Hybrid Radical–Ionic Annulation
2.5.1. Pyrrolidine Synthesis
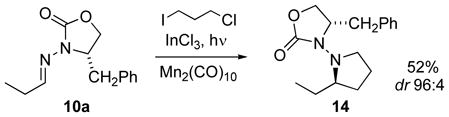
Applying radical addition reactions in the presence of electrophilic spectator functionalities was attractive, as such demonstrations of compatibility would illustrate the complementarity of radical conditions and strongly nucleophilic conditions. In this regard, it should be noted that dihalopropanes had been used as radical precursors (Table 4, entries 8–11), preserving halide functionality in most cases. However, in one case, addition of 3-chloro-1-iodopropane (eq. 1), characterization of the adduct indicated there was no chlorine present; this reaction had afforded pyrrolidine 14.[42] This outcome may be explained by sequential radical addition and SN2-type cyclization in situ. Support for such a process also was found upon addition of ethyl iodide to the 3-chlorobutyraldehyde hydrazone (Table 4, entry 18), which gave the epimeric pyrrolidine (epi-14). These radical–polar crossover reactions,[43] which also may be termed hybrid radical–ionic annulations, offer a novel way to achieve 1,2-bis-functionalization of the C=N bond.
 |
Equation 1 |
Control of the steps, interrupting the pyrrolidine annulation after the radical addition step, has been observed upon slight modifications to the conditions using hydrazone 15 (Scheme 6).[44] After a 15-hour reaction time, the initial radical adduct 16 was isolated in 45% yield, and on extending the reaction time to 65 hours, the radical adduct had mostly cyclized to the pyrrolidine 17. When the reaction was carried out in the absence of Lewis acid, the only observed product was pyrrolidine 17. Further studies of the scope of these pyrrolidine constructions are ongoing.
Scheme 6.

Control of stepwise annulation
2.5.2. Stepwise Annulation in Piperidine Synthesis
The simple piperidine alkaloid coniine[45] (Scheme 7) offered a preliminary test case for hybrid radical–ionic annulation in alkaloid synthesis. We envisioned a radical addition followed by cyclization, and two alternative bond constructions can be considered. The exocyclic propyl group may be introduced as part of the radical acceptor (path A), or may originate in the radical precursor (path B).
Scheme 7.

Retrosynthetic disconnection of coniine
In contrast to the pyrrolidine constructions, the addition of ethyl radical to a difunctional 5-chloropentanal hydrazone (Table 4, entry 19) did not result in cyclization in situ. Therefore, the chloride substituent was replaced with a tosylate to facilitate the polar cyclization. The Mn-mediated addition of 1-iodopropane to 5-tosyloxypentanal N-acylhydrazone 18 (Scheme 8) indeed provided the expected annulation product 19 in 59% yield. Unfortunately, unusually poor stereocontrol was observed (dr 3:1).[42b] The lack of high selectivity in the addition to hydrazone 18 is very unusual among all the examples of Mn-mediated radical addition observed to date. This, together with anomalously poor mass balance (no reactant hydrazone was recovered), provides evidence for a polar cyclization to form iminium ion 20 (Scheme 8) prior to radical addition; such a cyclization would be detrimental to stereoselectivity due to the loss of two-point binding of the Lewis acid.
Scheme 8.

Radical addition to 5-tosyloxypentanal hydrazone 18
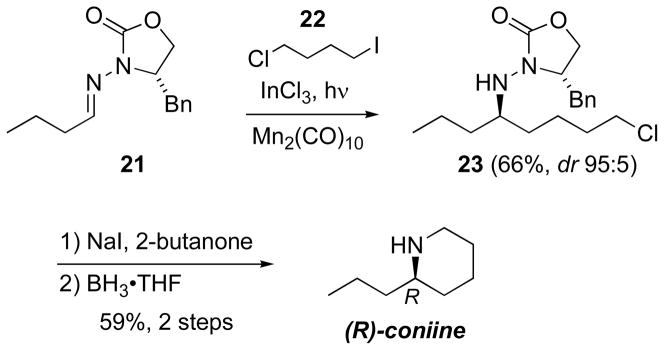
Because of the problems with the premature ionic cyclization noted above, a stepwise approach was adopted for piperidine construction. From butyraldehyde hydrazone 21 (Scheme 9) and 4-chloro-iodobutane (22), Mn-mediated photolysis afforded the acyclic adduct 23 in 66% yield (dr 95:5); the cyclization did not occur in situ.[42] Nevertheless, Finkelstein conditions afforded the piperidine, and reductive removal of the auxiliary afforded coniine in 34% overall yield for 4 steps. This reaction sequence offers a favorable comparison between radical- and carbanion-based syntheses using the same retrosynthetic disconnection.[45b,f]
Scheme 9.
Synthesis of (R)-coniine
2.5.3. Application to Formal Synthesis of Quinine
Our approach to the antimalarial alkaloid quinine focuses on strategic application of the Mn-mediated hybrid radical–ionic annulation.[46] Retrosynthetic disconnection of the azabicyclic ring system to a piperidine A (Scheme 10) suggests isoquinolizidine B as a potential precursor, where functionality of C6 and C7 would eventually be exploited for ring cleavage. The radical–ionic annulation is then applied via disconnection of either of two C–C bonds in structure B, involving either Path a or Path b. With enantiomeric chiral auxiliaries, these paths converge to structures B and B′, which differ only in the configuration of the removable chiral auxiliary. A pseudo-C2-symmetric precursor C would facilitate efficient access to the coupling components needed to test either of these strategies.
Scheme 10.

Retrosynthetic analysis of quinine
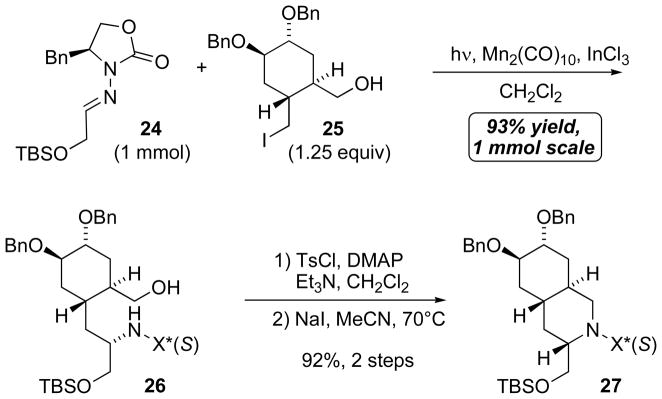
The ultimately successful radical–ionic annulation was carried out in the presence of protected 1,2-diol functionality at C6 and C7, specifically the coupling of 24 with 25 (Scheme 11). Stoichiometry was a concern, because iodide 25 had been prepared through several synthetic steps, making it impractical to rely on large excesses of radical precursor, as is commonly required for many intermolecular radical additions. Fortunately, the Mn-mediated coupling of 24 and 25 with only 1.25-fold excess of 25 proceeded in 93% yield in 1 mmol scale, giving 26 as a single diastereomer. Although completion of the hybrid radical–ionic annulation in situ during the Mn-mediated coupling has not yet been achieved, a stepwise process provided decahydroisoquinoline 27 in a quite satisfactory overall yield (85% for 3 steps).[46a] The low stoichiometric requirement in the coupling of the multifunctional iodide 25 to an imino compound is attractive and should enable broader applications of this Mn-mediated coupling process in complex target synthesis.
Scheme 11.
Mn-mediated radical addition en route to quinine
To complete a formal synthesis of quinine, quinolizidine 28 was converted in three steps to the piperidine 29 (Scheme 12). Unfortunately the cyclization to form an azabicyclic ring system was not regioselective; both hydroxyethyl groups cyclized, and the preferred product contained the azabicyclo[3.2.1]octane rather than the desired azabicyclo[2.2.2]octane. This necessitated a 6-step sequence to differentiate the hydroxyethyl groups of 13. This eventually furnished quincorine, which has previously been converted in two steps to quinine.[46b]
Scheme 12.

Conversion of quinolizidine 28 to quincorine
2.6. Applications in Amino Acid Synthesis
2.6.1. Synthesis of γ-amino acids
Although α- and β-amino acids have drawn more attention in synthetic chemistry, γ-amino acids such as 30 and 31 (Figure 4) are also important targets from the perspective of bioorganic and medicinal chemistry.[47,48,49,50] Disconnections of C–C bonds as shown calls for iodides and hydrazones bearing oxygen-containing functional groups, an important challenge to the synthetic versatility of the Mn-mediated coupling reactions. With this in mind, we employed Mn-mediated radical addition for a novel synthesis of γ-amino acids 30 and 31.[51]
Figure 4.

Representative γ-amino acids with strategic bond disconnections at the γ–δ and β–γ carbons.
Because disconnection of α-alkoxy-γ-amino acid 30 calls for a base-sensitive β-alkoxyhydrazone 32 (Scheme 13), there is a potential for β-elimination of the alkoxy group from the hydrazone precursor 32 which makes non-basic conditions critical. In fact, treatment of 32 with TBAF in THF led to just such a β-elimination.[52] However, the Mn-mediated radical addition of isopropyl iodide proceeded in 77% yield, without any evidence of β-elimination, to afford 33 as a single diastereomer. Reductive removal of the chiral auxiliary and oxidation to the carboxylic acid gave 30 in good overall yield.[51]
Scheme 13.
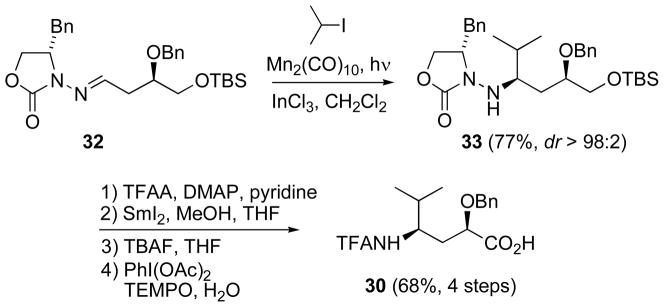
Addition to a β-alkoxyhydrazone without β-elimination
Phenylacetaldehyde N-acylhydrazone 34 served as the radical acceptor for assembly of γ-amino acid 31 (Scheme 14), employing difunctional iodide 35 in the Mn-mediated radical addition (56% yield, single diastereomer).[51] As with 33 (shown above), this radical adduct 36 was converted through the same 4-step sequence to γ-amino acid 31.
Scheme 14.

Addition of a 3-silyloxyalkyl iodide to an N-acylhydrazone
Although the aforementioned routes provided the desired γ-amino acids, it was desirable to develop a synthesis which incorporates the carboxylic acid oxidation state prior to coupling. We hypothesized that Mn-mediated radical addition would accomplish this objective, and therefore initiated a study of Mn-mediated coupling of alkyl iodides with γ-hydrazonoesters (Figure 5).[44] We had already shown that the Mn-mediated radical addition conditions offer excellent chemoselectivity, but it remained to be seen whether the stereocontrol model would be disrupted; would an additional Lewis basic ester function in the hydrazone interfere with the role of In(III) in two-point binding and rotamer control?
Fig. 5.
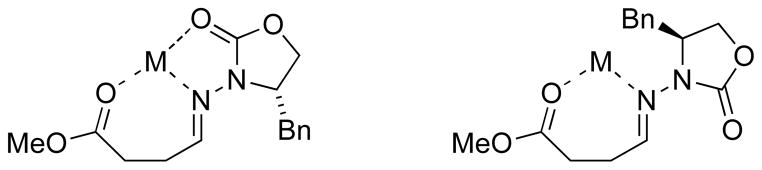
Some hypothetical multipoint binding of a Lewis acid by N-acylhydrazones bearing additional ester functionality
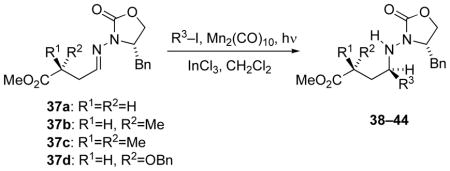
Prototypical radical additions were examined under Mn-mediated photolysis conditions with InCl3 as the Lewis acid, coupling isopropyl iodide with a variety of γ-hydrazonoesters 37a–37d (Table 5) bearing varied substitution at the position α to the ester. The α-methyl, αα, -dimethyl, and α-benzyloxy substituents appeared to have little effect on reaction efficiency and selectivity, as all provided the isopropyl adducts with consistently high diastereoselectivities and excellent yields (91–98%). Surprisingly, the selectivity was only slightly diminished in the absence of InCl3 (entries 5 and 6); the yield in the absence of Lewis acid activation was modest but synthetically useful.
Table 5.
Additions of Alkyl Iodides to γ-Hydrazonoesters
| ||||
|---|---|---|---|---|
| entry | hydrazone | iodide R3I | product, yield | dr |
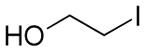
| 1 | 37a | i-PrI | 38a, >99% | 94:6 |
| 2 | 37b | 38b, 98% | 95:5 | |
| 3 | 37c | 38c, 96% | 99:1 | |
| 4 | 37d | 38d, 96% | 90:10 | |
| 5 | 37a |
|
39, 82% | -- |
| 6 |
|
40, 66% | 94:6 | |
| 7 |
|
41, 37% | 96:4 | |
| 8 |
|
42, 61% | 97:3 | |
| 9 |
|
43, 33% | 96:4 | |
| 10 |
|
44, 45% | 85:15 | |
For the γ-hydrazonoesters, NMR experiments substantiated the typical two-point chelation model. A mixture of 37a with InCl3 in CD2Cl2 exhibited the H–C=N absorbance of the γ-hydrazonoester at 7.74 ppm, 0.30 ppm upfield from 37a alone, consistent with precedent regarding Lewis acid coordination to imino compounds.[53] Also, the carbons of the oxazolidinone C=O and the hydrazone C=N were shifted downfield by 8 and 5 ppm on mixing with InCl3. In contrast, the C=O carbon of the ester showed minimal change (<1 ppm). This suggests that the InCl3 is chelated by the imino nitrogen and the oxazolidinone carbonyl in the usual way, without significant interference by the ester function.
A range of iodides were next examined in reactions with and without InCl3, starting with a comparison of secondary and primary iodides (Table 5). When secondary iodides were subjected to coupling with γ-hydrazonoester 37a the yields were excellent (entries 1 and 5), while primary iodides gave the desired adducts in moderate yields (33–66%, entries 6–10). All of these reactions occurred with good-to-excellent diastereoselectivities, and it was worth noting that both silyl ether and alcohol functionality were compatible with the coupling.
To document the synthetic applicability of these reactions, N–N bond cleavage was needed. After trifluoroacetylation of 38a (Scheme 15) under microwave irradiation,[54] exposure to SmI2 smoothly furnished known γ-aminoester 45 and offered proof of absolute configuration.[55]
Scheme 15.
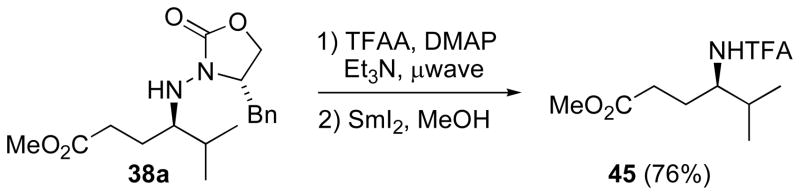
Conversion of radical adduct to N-trifluoroacetamide
2.6.2. Synthesis of α,α-Disubstituted α-Amino Acids
Although radical additions to aldimine-type acceptors have now become well-established, intermolecular additions to ketimine radical acceptors are rare by comparison.[56,57] We envisioned that the versatility of the Mn-mediated radical additions might offer potential access to a diverse range of tert-alkyl amines which are difficult to prepare by other means.[58] In planning a study of such reactions we were cognizant of the importance of ensuring the reaction takes place exclusively through a single C=N pi-bond geometry. Aldehyde N-acylhydrazone derivatives are exclusively obtained in E geometry, making this issue of little relevance, but ketone hydrazones are generally formed as mixtures of E and Z isomers. Therefore we sought a ketone hydrazone which could be obtained predominantly as one isomer.
The N-amino-2-oxazolidinone 9a was condensed with methyl pyruvate (46) to give hydrazone 47 (Scheme 16) as an E/Z mixture (dr 92:8), from which the minor (Z)-isomer was removed via flash chromatography to give pure (E)-47 in 75% yield. Addition of ethyl iodide under Mn-mediated photolysis conditions in the presence of InCl3 gave a moderate yield of 48a (66% yield, dr 70:30), while the corresponding isopropyl adduct 48b was very effectively produced (85% yield, dr 92:8). The N–N bond was cleaved upon conversion of isopropyl adduct 48b to the benzoyl derivative and treatment with SmI2/MeOH (Scheme 10) to afford known benzamide (S)-(+)-49[59] and confirm the assigned configuration.
Scheme 16.
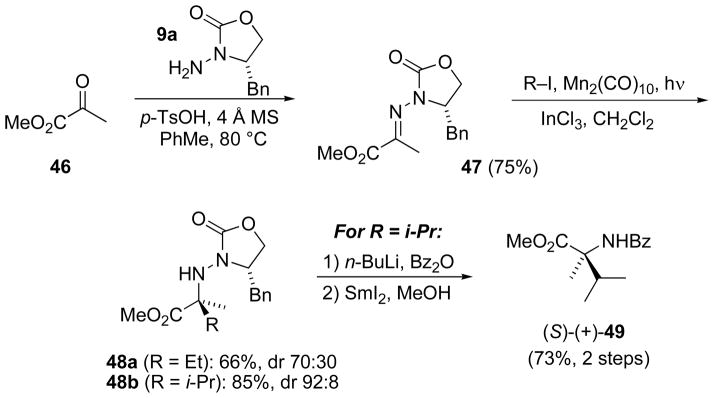
Radical addition to ketimine 47
In the additions to 47 it was noted, through variations of the stoichiometric loading of Lewis acid, that amounts of InCl3 less than 2 equiv resulted in lower diastereoselectivity. From this it may be inferred that the Lewis acid, aside from its usual chelation by the N-acylhydrazone, may interact with another Lewis basic site (e.g., the ester).
2.7. Considerations for Synthesis Design Using Mn-Mediated Radical Addition
2.7.1. Functional Group Compatibility
In the forgoing sections, there are numerous examples illustrating the use of Mn-mediated radical additions to couple compounds containing more than one functional group. Although there are still combinations left to be explored, the examples published to date already illustrate that various useful functionalities may be tolerated within either of the precursors.
In the radical precursor, the alkyl iodide may be accompanied by alkyl chloride, alcohol, benzylic ether, or silyl ether functionalities. The alkyl chlorides have certain limitations on the location relative to the radical; 2-chloroethyl radical may eliminate chloride prior to radical addition, and the adduct from 3-chloropropyl radical may cyclize after radical addition.
In the N-acylhydrazone radical acceptor, the functionalities tolerated include alkyl chloride, benzylic ether, silyl ether, and ester. An alkoxy leaving group may be accomodated at the β-carbon of an N-acylhydrazone without β-elimination, which complements the functional group tolerance of basic organometallic reagents. Depending on the chain length, a subsequent cyclization may occur in radical adducts containing alkyl chloride.
What types of functionalities are not compatible? Applications to total synthesis are rigorous proving grounds, often revealing that potentially useful methodologies are in fact limited to simple monofunctional precursors. Despite the tolerances for functional groups noted above, application of Mn-mediated radical additions to total synthesis objectives uncovered some cases of incompatibility which deserve further mention.
In the early stages of developing a synthetic route to quinine, the Mn2(CO)10-mediated coupling of iodide 50 and N-acylhydrazone 51 was attempted in 10:1 PhH/MeCN (eq. 2). Unfortunately no coupling product could be found, so each of the spectator functionalities had to be examined in control experiments in order to find the structural features which might be interfering with the reaction. Mn-mediated radical additions of iodides containing the silyl ether moiety had previously been successful in various contexts,[42,44,46,51] so interference by the silyl ether was ruled out. However, it was noted that a precipitate formed on mixing hydrazone 51 with InCl3 in 10:1 PhH/MeCN, and the precipitate remained insoluble even at higher ratios of CH3CN. This suggested closer examination of the N-acylhydrazone to determine whether complexation of the N-acylhydrazone with InCl3, normally required to facilitate radical addition, had been disrupted by the basic methoxyquinoline. A second concern was that the compatibility of an electron-rich methoxyaryl group with the Mn-mediated radical additions had not previously been established.
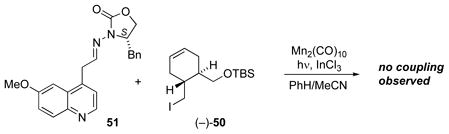 |
Equation 2 |
To address this question, a series of control experiments was carried out with aromatic hydrazones 52a, 52b, and 52c, using Mn-mediated radical addition of iodide 53. From hydrazones 52a and 52b, the expected products 54a and 54b were obtained in moderate yield (ca. 40%), confirming that the electron-rich methoxyaryl substituent was compatible with the coupling. Pyridine-containing hydrazone 52c, however, gave none of the desired coupling product 54c, indicating that heteroaromatic nitrogen may have interfered with the Mn-mediated coupling reaction.
When attempting the coupling of iodide 50 (eq. 2) with simplified hydrazones en route to quinine, another issue of compatibility arose. Although OH and OTBS groups were already known to be well-tolerated, the couplings of 50 were inefficient. This raised suspicion about the potential side reactions of an alkene moiety in the radical intermediate. Despite changing the identity of the spectator functional groups, or the roles of the two precursors, so that the alkene was in the radical acceptor (e.g., 55a–55d), the reaction remained inefficient. Yields of the desired product remained generally less than 30% despite extensive efforts toward improvement. Finally, a control experiment with saturated iodide 50c revealed that the alkene indeed was detrimental to the success of this coupling. It is unclear whether this incompatibility is a general one or a peculiarity of the examples shown in Scheme 18.
Scheme 18.

Functionality variations in Mn-mediated coupling attempts for quinine synthesis
In summary, there are some useful functional groups bearing either acidic protons (alcohol) or electrophilic centers (alkyl chloride, ester) which are tolerated as spectators in the Mn-mediated radical addition reaction. These compatibilities complement those of other chiral amine synthesis methods involving strongly nucleophilic (and basic) organometallic reagents, thereby offering some useful options for synthetic design. Ethers and silyl ethers are also compatible. Selected examples involving pyridines and alkenes as spectator groups revealed some complications, the scope of which remain unclear, so for synthetic design purposes, these types of moieties may need to be introduced later in the route.
2.7.2. Stereoconvergence for Flexibility in Synthetic Application
Considering the examples discussed above, it is clear that the Mn-mediated radical additions offer useful functional group compatibilities in both the radical precursor and N-acylhydrazone acceptor. And, the epimeric configuration can be selected by either (A) employing the enantiomeric auxiliary, or (B) interchanging the roles of R1 and R2 in the alkyl halide and aldehyde precursors of Table 4.[60] Thus, stereoconvergent construction of alternative C–C bonds at the chiral amine stereocenter (Scheme 19) can be readily conceived, so that the roles of these precursors can be chosen on the basis of synthetic strategy rather than rather than on the basis of functional group limitations of the methodology. Such strategic flexibility contributes to the synthetic potential of these radical addition reactions.
Scheme 19.

Stereoconvergent routes to chiral amines
3. Asymmetric Catalysis of Radical Addition
Although the forgoing sections have illustrated the viability of stereocontrolled radical addition to C=N bonds as a route to chiral amines, imparting the stereocontrol through asymmetric catalysis remains a challenge. To date, most efforts toward this goal have required very high catalyst loading (usually stoichiometric) or have limited scope. In this section, a summary of these studies illustrates some of the highlights and limitations.
Naito reported the first asymmetric radical additions to C=N bonds with a non-covalent mode of stereocontrol. Building upon earlier work exploiting chiral glyoxylate imines as radical acceptors in the presence of Lewis acids, Naito employed Lewis acids bearing a chiral bisoxazoline ligand for addition to achiral glyoxylate oxime ether 56 (Scheme 20). With MgBr2 and ligand 58, enantioselectivity up to 52% ee was obtained at stoichiometric loading.[61] Jorgensen published an alternative approach, attempting catalysis with a combination of Cu(I) and to lBINAP (59).[62] Unfortunately this gave very low enantioselectivity, with a yield lower than the catalyst loading.
Scheme 20.

Asymmetric addition of isopropyl iodide to glyoxylate oxime ether
We sought to demonstrate the first example of catalytic asymmetric induction in radical addition to C=N bonds. Our effort toward this goal was built upon the two-point binding motif of N-acylhydrazones (Figure 6), which could potentially transmit stereocontrol from chiral ligands accompanying a Lewis acid.
Fig. 6.
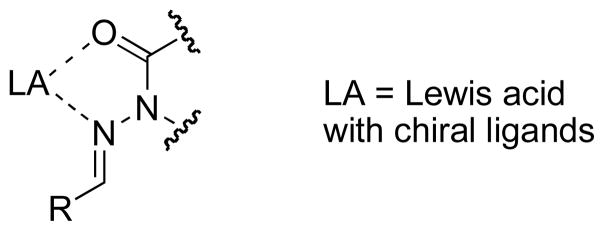
Two-point binding of a Lewis acid by a generalized N-acylhydrazone structure.
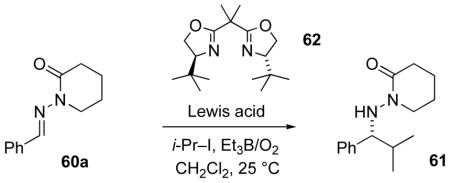
The first successes exploited additions of isopropyl iodide, with radical initiation by triethylborane and oxygen. Using valerolactam-derived achiral N-acylhydrazone acceptor 60a (Table 6), highly enantioselective isopropyl radical additions were promoted by one equivalent each of Lewis acid and bisoxazoline ligand 62. With InCl3, Mg(ClO4)2, and Cu(OTf)2 as the Lewis acids only modest yields of 61 were obtained (entries 1–3), but selectivities in the range of 57–66%ee set a new standard for selectivity in radical additions to C=N bonds.[63] Using benzene/CH2Cl2 as the solvent (entry 4), the selectivity increased further, to 95%ee (66% yield). The less polar solvent system presumably facilitated the assembly of a ternary complex from the ligand, Lewis acid, and substrate. The yield improved to 94% with larger amounts of 2-iodopropane and Et3B (entry 5), but this came at the expense of some selectivity.
Table 6.
Comparison of Lewis acids in isopropyl addition to 60a.a
| ||||
|---|---|---|---|---|
| entry | Lewis acid | solvent, additive | yield[b] | ee[c] |
| 1 | InCl3 | CH2Cl2 | 33% | 57% |
| 2 | Mg(ClO4)2 | CH2Cl2 | 41% | 66% |
| 3 | Cu(OTf)2 | CH2Cl2 | 41% | 59% |
| 4d,e | Cu(OTf)2 | PhH/CH2Cl2 (2:1), 4Å MS | 66% | 95% |
| 5d,e,f | Cu(OTf)2 | PhH/CH2Cl2 (2:1), 4Å MS | 94% | 86% |
Reaction conditions: Lewis acid (1 equiv), tert-butylbisoxazoline ligand (1 equiv), 2-iodopropane (6 equiv), Et3B/O2 (6 equiv).
Isolated yield, %.
Enantiomeric excess, determined by HPLC.
Et3N was added during workup.
Preformed aquo complex Cu(tBu-Box)(H2O)2(OTf)2 was used.
10 equiv of i-PrI and Et3B were used.
A series of radical precursors and acceptors were employed, showing some versatility accompanied by high enantioselectivity (Table 7).[63] Using stoichiometric amounts of the preformed aquo complex Cu(tBu-Box)(H2O)2(OTf)2, isopropyl additions to N-acylhydrazones prepared from benzaldehyde, p-chlorobenzaldehyde and p-methoxybenzaldehyde were highly enantioselective (entries 1 and 2). Additions of various radicals, including chloromethyl, to 60a were also successful (entries 3–6). Turnover of the chiral catalyst was examined by lowering the catalyst loading (Table 7, entries 8–10). The yield remained high, while enantioselection decreased. However, the yield and enantioselection (74% yield, 46%ee) at 10 mol% catalyst loading showed evidence of catalyst turnover — the first example of asymmetric catalysis in radical addition to C=N bonds.
Table 7.
Varying the radical precursors and acceptors in Cu(II) catalyzed addition to N-acylhydrazones.
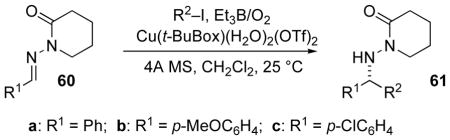
| |||||
|---|---|---|---|---|---|
| entry | halide | hydrazone | catalyst load | % yieldb | % eec |
| 1 | iPrI | 60a | 1 equiv | 66 | 95 |
| 2 | i-PrI | 60b | 1 equiv | 46 | 90 |
| 3 | i-PrI | 60c | 1 equiv | 53 | 81 |
| 4 | EtId | 60a | 1 equiv | 88 | 83 |
| 5 | c-C5H9Id | 1 equiv | 86 | 84 | |
| 6 | c-C6H11Id | 1 equiv | 84 | 89 | |
| 7 | ClCH2Id | 1 equiv | 44e | 95 | |
| 8 | iPrI | 0.5 equiv | 71 | 81 | |
| 9 | iPrI | 0.2 equiv | 83 | 58 | |
| 10 | iPrI | 0.1 equiv | 74 | 46 | |
Reaction conditions: see Table 6.
Isolated yield, %.
Enantiomeric excess, % (hexane:2-propanol, Chiralcel OD or AD).
10 equiv of alkyl halide was used.
56% recovery of unreacted hydrazone.
The glyoxylic oxime ether 62 was subjected by Jang et al. to radical addition of several simple alkyl iodides in with triethylborane/oxygen initiation in the presence of Cinchona alkaloid salts of hypophosphorous acid (Scheme 21).[64] Although an excess of the Cinchona alkaloid was required, transmission of stereochemical information was confirmed. A stereocontrol model was proposed with a combination of hydrogen bonding and pi stacking to activate the radical acceptor and constrain its structure.
Scheme 21.

Radical addition in the presence of cinchona alkaloid salts of hypophosphorous acid.
More recently, the Jang group has reported that N-benzoylhydrazones are also suitable substrates for stereocontrolled radical addition in the presence of Cinchona alkaloids.[65] With hydrazones prepared from a series of substituted benzaldehydes (Scheme 22), very high enantioselectivities (98–99%ee) were observed using the O-benzyl alkaloid derivative 68 (40 mol%). The high selectivity observed with aromatic hydrazones was not retained during addition to the N-benzoylhydrazone prepared from octanal (80%ee, not shown). Cyclohexyl, tert-butyl, and adamantyl radical additions were effective, but the use of primary radical (from n-octyl iodide) was detrimental to both efficiency and selectivity (55% yield, 81%ee).
Scheme 22.

Catalytic asymmetric radical addition to N-benzoylhydrazones in the presence of a cinchona alkaloid salt
Binaphthol-derived chiral Bronsted acids have been exploited to catalyze addition to N-aryl imines (Scheme 23).[66] In these reactions, the use of 30 mole % of catalyst 71 was sufficient to promote addition of isopropyl and tert-butyl iodides to imine 69 with enantiomeric excesses in the 73–84% range, although the isolated yields were moderate. As in most reactions using the triethylborane-oxygen initiation system, the products were accompanied by significant amounts of the ethyl adduct (76, R = Et, from triethylborane).
Scheme 23.

Binaphthol-derived Bronsted acid catalyst for asymmetric radical addition to N-arylimines
4. Summary
Radical addition to imino compounds has emerged as a general approach with broad versatility that complements non-radical methodology. Our work has discovered and elaborated this concept into synthetically useful methodology: First, highly stereoselective intermolecular additions of alkyl iodides to chiral hydrazones in the presence of Mn2(CO)10 accomodate a broad range of functionality, including esters and unprotected hydroxyl groups which may not be compatible with carbanion chemistry. The viability of this strategies for stereocontrol has now been established in applications to target-directed synthesis. Secondly, we have developed a means of catalytic asymmetric induction; excellent enantioselectivity is obtained at stoichiometric loading, and moderate enantioselectivity is observed in conditions which demonstrate catalyst turnover. Although these are promising results, a method for asymmetric catalysis in radical addition to C=N bonds which offers high turnover numbers and broad scope is still an unmet challenge.
Scheme 17.
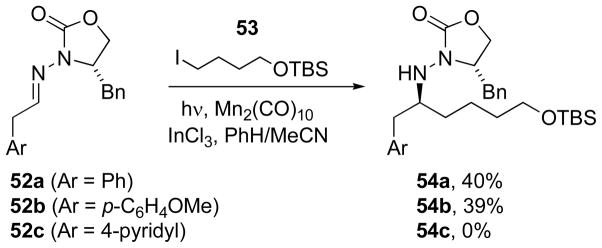
Control experiments with electron-rich aromatics
Acknowledgments
The generous support by NSF (CHE-0096803 and CHE-0749850) and NIH (R01-GM67187) of our radical addition chemistry method development and applications to natural product synthesis is acknowledged with gratitude. Portions of this chapter are excerpted from a previous Topics in Current Chemistry volume: Gregory K. Friestad “Asymmetric Radical Addition to Chiral Hydrazones.” In Topics In Current Chemistry: Radicals in Synthesis III; Gansauer A.; Heinrich, M., Eds.; Springer-Verlag: Berlin, 2012, vol. 320, pp. 61–92.
References and Notes
- 1.Recent reviews: Friestad GK. Addition of Carbanions to Azomethines. In: Enders D, Shaumann E, editors. Science of Synthesis Vol 40a: Compounds with One Saturated Carbon-Heteroatom Bond: Amines and Ammonium Salts. Thieme; Stuttgart: 2009. Yamada K-I, Tomioka K. Chem Rev. 2008;108:2874–2886. doi: 10.1021/cr078370u.Friestad GK, Mathies AK. Tetrahedron. 2007;63:2541–2569. doi: 10.1016/j.tet.2007.06.117.Ding H, Friestad GK. Synthesis. 2005:2815–2829.Alvaro G, Savoia D. Synlett. 2002:651–673.Kobayashi S, Ishitani H. Chem Rev. 1999;99:1069–1094. doi: 10.1021/cr980414z.Bloch R. Chem Rev. 1998;98:1407–1438. doi: 10.1021/cr940474e.Davis FA, Zhou P, Chen B-C. Chem Soc Rev. 1998;27:13–18.Enders D, Reinhold U. Tetrahedron Asymmetry. 1997;8:1895–1946.Denmark SE, Nicaise OJ-C. J Chem Soc Chem Commun. 1996:999–1004.
- 2.For examples of aza-enolization of imino compounds by organometallic reagents see: Stork G, Dowd SR. J Am Chem Soc. 1963;85:2178–2180.Wittig G, Frommeld HD, Suchanek P. Angew Chem Int Ed Engl. 1963;2:683.Guerrier L, Royer J, Grierson DS, Husson H-P. J Am Chem Soc. 1983;105:7754–7755.Enders D, Diez E, Fernandez R, Martin-Zamora E, Munoz JM, Pappalardo RR, Lassaletta JM. J Org Chem. 1999;64:6329–6336.
- 3.Reviews of free radical reactions in synthesis: Ischay MA, Yoon TP. Eur J Org Chem. 2012:3359–3372.Rowlands GJ. Tetrahedron. 2009;65:8603–8655.Renaud P, Sibi M, editors. Radicals in Organic Synthesis. Wiley-VCH; New York: 2001. Curran DP, Porter NA, Giese B. Stereochemistry of Radical Reactions: Concepts Guidelines and Synthetic Applications. VCH; New York: 1995. Jasperse CP, Curran DP, Fevig TL. Chem Rev. 1991;91:1237–1286.Giese B. Radicals in Organic Synthesis: Formation of Carbon-Carbon Bonds. Pergamon Press; New York: 1986. Hart DJ. Science. 1984;223:883–887. doi: 10.1126/science.223.4639.883.
- 4.Reviews of radical additions to imines and related acceptors: Friestad GK. In: Topics In Current Chemistry: Radicals in Synthesis III. Gansauer A, Heinrich M, editors. Vol. 320. Springer-Verlag; Berlin: 2012. pp. 61–92.Friestad GK. Chiral Amine Synthesis. In: Nugent T, editor. Methods, Developments and Applications. Wiley-VCH; Weinheim, Germany: 2010. pp. 51–74.Miyabe H, Yoshioka E, Kohtani S. Curr Org Chem. 2010;14:1254–1264.Yamada K, Tomioka K. Chem Rev. 2008;108:2874–2886. doi: 10.1021/cr078370u.Friestad GK. Tetrahedron. 2001;57:5461–5496.
- 5.Friestad GK. Eur J Org Chem. 2005:3157–3172. [Google Scholar]
- 6.Fallis AG, Brinza IM. Tetrahedron. 1997;53:17543–17594. [Google Scholar]
- 7.(a) Nishiyama H, Kitajima T, Matsumoto M, Itoh K. J Org Chem. 1984;49:2298–2300. [Google Scholar]; (b) Stork G, Kahn M. J Am Chem Soc. 1985;107:500–501. [Google Scholar]
- 8.Reviews of temporary tethers: Cusak A. Chem Eur J. 2012;18:5800–5824. doi: 10.1002/chem.201103186.Gauthier DR, Jr, Zandi KS, Shea KJ. Tetrahedron. 1998;54:2289–2338.Fensterbank L, Malacria M, Sieburth SM. Synthesis. 1997:813–854.Fleming I, Barbero A, Walter D. Chem Rev. 1997;97:2063–2192. doi: 10.1021/cr941074u.Bols M, Skrydstrup T. Chem Rev. 1995;95:1253–1277.
- 9.Friestad GK. Org Lett. 1999;1:1499–1501.Friestad GK, Massari SE. Org Lett. 2000;2:4237–4240. doi: 10.1021/ol0067991.Friestad GK, Jiang T, Fioroni GM. Tetrahedron: Asymmetry. 2003;14:2853–2856.Friestad GK, Massari SE. J Org Chem. 2004;69:863–875. doi: 10.1021/jo035405r.Friestad GK, Fioroni GM. Org Lett. 2005;7:2393–2396. doi: 10.1021/ol050663r.Friestad GK, Jiang T, Mathies AK. Tetrahedron. 2007;63:3964–3972. doi: 10.1016/j.tet.2007.06.117.Friestad GK, Mathies AK. Tetrahedron. 2007;63:9373–9381. doi: 10.1016/j.tet.2007.06.117.(Corrigendum: Friestad GK, Mathies AK. Tetrahedron. 2007;63:13039. doi: 10.1016/j.tet.2007.06.117.Friestad GK, Jiang T, Mathies AK. Org Lett. 2007;9:777–780. doi: 10.1021/ol063010z.Friestad GK, Jiang T, Fioroni GM. Tetrahedron. 2008;64:11549–11557.
- 10.Friestad GK. Eur J Org Chem. 2005:3157–3172. [Google Scholar]
- 11.(a) Hart DJ, Seely FL. J Am Chem Soc. 1988;110:1631–1633. [Google Scholar]; (b) Hart DJ, Krishnamurthy R, Pook LM, Seely FL. Tetrahedron Lett. 1993;34:7819–7822. [Google Scholar]; (c) Bhat B, Swayze EE, Wheeler P, Dimock S, Perbost M, Sanghvi YS. J Org Chem. 1996;61:8186–8199. doi: 10.1021/jo961549c. [DOI] [PubMed] [Google Scholar]; (d) Hanamoto T, Inanaga J. Tetrahedron Lett. 1991;32:3555–3556. [Google Scholar]; (e) Russell GA, Yao C-F, Rajaratnam R, Kim BH. J Am Chem Soc. 1991;113:373–375. [Google Scholar]; (f) Russell GA, Wang L, Rajaratnam R. J Org Chem. 1996;61:8988–8991. doi: 10.1021/jo961544f. [DOI] [PubMed] [Google Scholar]
- 12.(a) Miyabe H, Shibata R, Ushiro C, Naito T. Tetrahedron Lett. 1998;39:631–634. [Google Scholar]; (b) Miyabe H, Shibata R, Sangawa M, Ushiro C, Naito T. Tetrahedron. 1998;54:11431–11444. [Google Scholar]
- 13.(a) Miyabe H, Ushiro C, Naito T. J Chem Soc, Chem Commun. 1997:1789–1790. [Google Scholar]; (b) Miyabe H, Ushiro C, Ueda M, Yamakawa K, Naito T. J Org Chem. 2000;65:176–185. doi: 10.1021/jo991353n. [DOI] [PubMed] [Google Scholar]
- 14.McNabb SB, Ueda M, Naito T. Org Lett. 2004;6:1911–1914. doi: 10.1021/ol049671i. [DOI] [PubMed] [Google Scholar]
- 15.Bertrand MP, Feray L, Nouguier R, Stella L. Synlett. 1998:780–782. [Google Scholar]
- 16.Bertrand MP, Feray L, Nouguier R, Perfetti P. Synlett. 1999:1148–1150. [Google Scholar]; Bertrand MP, Coantic S, Feray L, Nouguier R, Perfetti P. Tetrahedron. 2000;56:3951–3961. [Google Scholar]
- 17.Review: Bertrand M, Feray L, Gastaldi S. Comptes Rend Acad Sci Paris Chimie. 2002;5:623–638.
- 18.(a) Zhou P, Chen B, Davis FA. Tetrahedron. 2004;60:8003–8030. [Google Scholar]; (b) Ellman JA, Owens TD, Tang TP. Acc Chem Res. 2002;35:984–995. doi: 10.1021/ar020066u. [DOI] [PubMed] [Google Scholar]
- 19.Akindele T, Yamamoto Y, Maekawa M, Umeki H, Yamada K, Tomioka K. Org Lett. 2006;8:5729–5732. doi: 10.1021/ol0621093. [DOI] [PubMed] [Google Scholar]
- 20.Akindele T, Yamada K, Sejima T, Maekawa M, Yamamoto Y, Nakano M, Tomioka K. Chem Pharm Bull. 2010;58:265–269. doi: 10.1248/cpb.58.265. [DOI] [PubMed] [Google Scholar]
- 21.Yamada K, Konishi T, Nakano M, Fujii S, Cadou R, Yamamoto Y, Tomioka K. J Org Chem. 2012;77:1547–1553. doi: 10.1021/jo2025042. [DOI] [PubMed] [Google Scholar]
- 22.Review: Miyabe H, Ueda M, Naito T. Synlett. 2004:1140–1157.
- 23.Booth SE, Jenkins PR, Swain CJ, Sweeney JB. J Chem Soc Perkin Trans. 1994;1:3499–3508. [Google Scholar]
- 24.Booth SE, Jenkins PR, Swain CJ. J Braz Chem Soc. 1998;9:389–395. [Chemical Abstracts 130:38274] [Google Scholar]
- 25.(a) Russell GA, Yao C-F, Rajaratnam R, Kim BH. J Am Chem Soc. 1991;113:373–375. [Google Scholar]; (b) Renaud P, Gerster M. Angew Chem Int Ed. 1998;37:2563–2579. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2562::AID-ANIE2562>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]; (c) Guerin B, Ogilvie WW, Guindon Y. In: Radicals in Organic Synthesis. Renaud P, Sibi M, editors. Wiley-VCH; New York: 2001. [Google Scholar]
- 26.(a) Burk MJ, Feaster JE. J Am Chem Soc. 1992;114:6266–6267. [Google Scholar]; (b) Sturino CF, Fallis AG. J Am Chem Soc. 1994;116:7447–7448. [Google Scholar]
- 27.Evans DA, Kim AS. In: Encyclopedia of Reagents for Organic Synthesis. Paquette LA, editor. Vol. 1. Wiley; New York: 1995. [Google Scholar]
- 28.(a) Kim M, White JD. J Am Chem Soc. 1977;99:1172–1180. [Google Scholar]; (b) Ciufolini MA, Shimizu T, Swaminathan S, Xi N. Tetrahedron Lett. 1997;38:4947–4950. [Google Scholar]; (c) Evans DA, Johnson DS. Org Lett. 1999;1:595–598. doi: 10.1021/ol990113r. [DOI] [PubMed] [Google Scholar]
- 29.Hynes J, Jr, Doubleday WW, Dyckman AJ, Godfrey JD, Jr, Grosso JA, Kiau S, Leftheris K. J Org Chem. 2004;69:1368–1371. doi: 10.1021/jo035587p. [DOI] [PubMed] [Google Scholar]
- 30.Friestad GK, Ji A. Org Lett. 2008;10:2311–2313. doi: 10.1021/ol800733b. [DOI] [PubMed] [Google Scholar]
- 31.Shen Y, Friestad GK. J Org Chem. 2002;67:6236–6239. doi: 10.1021/jo0259663. [DOI] [PubMed] [Google Scholar]
- 32.(a) Friestad GK, Qin J. J Am Chem Soc. 2000;122:8329–8330. [Google Scholar]; (b) Friestad GK, Draghici C, Soukri M, Qin J. J Org Chem. 2005;70:6330–6338. doi: 10.1021/jo050756m. [DOI] [PubMed] [Google Scholar]
- 33.Qin J, Friestad GK. Tetrahedron. 2003;59:6393–6402. [Google Scholar]
- 34.Lim D, Coltart DM. Angew Chem Int Ed. 2008;47:5207–5210. doi: 10.1002/anie.200800848. [DOI] [PubMed] [Google Scholar]
- 35.Nozaki K, Oshima K, Utimoto K. Bull Chem Soc Jpn. 1991;64:403–409. [Google Scholar]; Brown HC, Midland MM. Angew Chem Int Ed Engl. 1972;11:692–700. [Google Scholar]
- 36.Fernández M, Alonso R. Org Lett. 2003;5:2461–2464. doi: 10.1021/ol034696n. [DOI] [PubMed] [Google Scholar]
- 37.(a) Kim S, Lee IY, Yoon J-Y, Oh DH. J Am Chem Soc. 1996;118:5138–5139. [Google Scholar]; (b) Kim S, Yoon J-Y. J Am Chem Soc. 1997;119:5982–5983. [Google Scholar]; (c) Ryu I, Kuriyama H, Minakata S, Komatsu M, Yoon J-Y, Kim S. J Am Chem Soc. 1999;121:12190–12191. [Google Scholar]; (d) Jeon G-H, Yoon J-Y, Kim S, Kim SS. Synlett. 2000:128–130. [Google Scholar]; (e) Kim S, Kim N, Yoon J-Y, Oh DH. Synlett. 2000:1148–1150. [Google Scholar]; (f) Kim S, Song H-J, Choi T-L, Yoon J-Y. Angew Chem Int Ed. 2001;40:2524–2526. [PubMed] [Google Scholar]; (g) Kim S, Kavali R. Tetrahedron Lett. 2002;43:7189–7191. [Google Scholar]
- 38.(a) Pauson PL. In: Encyclopedia of Reagents for Organic Synthesis. Paquette LA, editor. Vol. 2. Wiley; New York: 1995. [Google Scholar]; (b) Meyer TJ, Caspar JV. Chem Rev. 1985;85:187. [Google Scholar]; (c) Gilbert BC, Parsons AF. J Chem Soc Perkin Trans. 2002;2:367–387. [Google Scholar]
- 39.Herrick RS, Herrinton TR, Walker HW, Brown TL. Organometallics. 1985;4:42–45. [Google Scholar]
- 40.For seminal examples see: Giese B, Thoma G. Helv Chim Acta. 1991;74:1135–1142.
- 41.(a) Gilbert BC, Kalz W, Lindsay CI, McGrail PT, Parsons AF, Whittaker DTE. Tetrahedron Lett. 1999;40:6095–6098. [Google Scholar]; (b) Gilbert BC, Lindsay CI, McGrail PT, Parsons AF, Whittaker DTE. Synth Commun. 1999;29:2711–2718. [Google Scholar]; (c) Gilbert BC, Kalz W, Lindsay CI, McGrail PT, Parsons AF, Whittaker DTE. J Chem Soc Perkin Trans. 2000;1:1187–1194. [Google Scholar]
- 42.(a) Friestad GK, Qin J. J Am Chem Soc. 2001;123:9922–9923. doi: 10.1021/ja011312k. [DOI] [PubMed] [Google Scholar]; (b) Friestad GK, Qin J, Suh Y, Marié J-C. J Org Chem. 2006;71:7016–7027. doi: 10.1021/jo061158q. [DOI] [PubMed] [Google Scholar]
- 43.For selected recent examples of radical-polar crossover reactions see: Callaghan O, Lampard C, Kennedy AR, Murphy JA. J Chem Soc Perkin Trans. 1999;1:995–1001.Jahn U, Muller M, Aussieker S. J Am Chem Soc. 2000;122:5212–5213.Harrowven DC, Lucas MC, Howes PD. Tetrahedron. 2001;57:791–804.Rivkin A, Nagashima T, Curran DP. Org Lett. 2003;5:419–422. doi: 10.1021/ol0272491.Denes F, Chemla F, Normant JF. Angew Chem Int Ed. 2003;42:4043–4046. doi: 10.1002/anie.200250474.Bazin S, Feray L, Vanthuyne N, Bertrand MP. Tetrahedron. 2005;61:4261–4274.Ueda M, Miyabe H, Sugino H, Miyata O, Naito T. Angew Chem Int Ed. 2005;44:6190–6193. doi: 10.1002/anie.200502263.Denes F, Cutri S, Perez-Luna A, Chemla F. Chem Eur J. 2006;12:6506–6513. doi: 10.1002/chem.200600334.Maruyama T, Mizuno Y, Shimizu I, Suga S. J Am Chem Soc. 2007;129:1902–1903. doi: 10.1021/ja068589a.Miyata O, Takahashi S, Tamura A, Ueda M, Naito T. Tetrahedron. 2008;64:1270–1284.
- 44.Friestad GK, Banerjee K. Org Lett. 2009;11:1095–1098. doi: 10.1021/ol802932v. [DOI] [PubMed] [Google Scholar]
- 45.For selected asymmetric syntheses of coniine see: Guerrier L, Royer J, Grierson DS, Husson H-P. J Am Chem Soc. 1983;105:7754–7755.Enders D, Tiebes J. Liebigs Ann Chem. 1993:173–177.Yamazaki N, Kibayashi C. Tetrahedron Lett. 1997;38:4623–4626.Reding MT, Buchwald SL. J Org Chem. 1998;63:6344–6347. doi: 10.1021/jo980808q.Wilkinson TJ, Stehle NW, Beak P. Org Lett. 2000;2:155–158. doi: 10.1021/ol9912534.Kim YH, Choi JY. Tetrahedron Lett. 1996;37:5543–5546.
- 46.(a) Korapala CS, Qin J, Friestad GK. Org Lett. 2007;9:4246–4249. doi: 10.1021/ol7017938. [DOI] [PubMed] [Google Scholar]; (b) Friestad GK, Ji A, Baltrusaitis J, Korapala CS, Qin J. Org Chem. 2012J;77:3159–3180. doi: 10.1021/jo2026349. [DOI] [PubMed] [Google Scholar]
- 47.Reviews: Ordonez M, Cativiela C. Tetrahedron: Asymmetry. 2007;18:3–99. doi: 10.1016/j.tetasy.2009.01.002.Trabocchi A, Guarna F, Guarna A. Curr Org Chem. 2005;9:1127–1153.
- 48.Examples: Matthew S, Schupp PJ, Leusch H. J Nat Prod. 2008;71:1113–1116. doi: 10.1021/np700717s.Kunze B, Bohlendorf B, Reichenbach H, Hofle G. J Antibiot. 2008;61:18–26. doi: 10.1038/ja.2008.104.Oh D-C, Strangman WK, Kauffman CA, Jensen PR, Fenical W. Org Lett. 2007;9:1525–1528. doi: 10.1021/ol070294u.Milanowski DJ, Gustafson KR, Rashid MA, Pannell LK, McMahon JB, Boyd MR. J Org Chem. 2004;69:3036–3042. doi: 10.1021/jo0303113.Williams PG, Luesch H, Yoshida WY, Moore RE, Paul VJ. J Nat Prod. 2003;66:595–598. doi: 10.1021/np030011g.Horgen FD, Kazmierski EB, Westenburg HE, Yoshida WY, Scheuer PJ. J Nat Prod. 2002;65:487–491. doi: 10.1021/np010560r.
- 49.(a) Dado GP, Gellman SH. J Am Chem Soc. 1994;116:1054–1062. [Google Scholar]; (b) Hanessian S, Luo X, Schaum R, Michnick S. J Am Chem Soc. 1998;120:8569–8570. [Google Scholar]; (c) Sanjayan GJ, Stewart A, Hachisu S, Gonzalez R, Watterson MP, Fleet GWJ. Tetrahedron Lett. 2003;44:5847–5851. [Google Scholar]; (d) Seebach D, Schaeffer L, Brenner M, Hoyer D. Angew Chem Int Ed. 2003;42:776–778. doi: 10.1002/anie.200390205. [DOI] [PubMed] [Google Scholar]; (e) Farrera-Sinfreu J, Zaccaro L, Vidal D, Salvatella X, Giralt E, Pons M, Albericio F, Royo M. J Am Chem Soc. 2004;126:6048–6057. doi: 10.1021/ja0398621. [DOI] [PubMed] [Google Scholar]; (f) Vasudev PG, Ananda K, Chatterjee S, Aravinda S, Shamala N, Balaram P. J Am Chem Soc. 2007;129:4039–4048. doi: 10.1021/ja068910p. [DOI] [PubMed] [Google Scholar]
- 50.Sasse F, Steinmetz H, Heil J, Höfle G, Reichenbach H. J Antibiot. 2000;53:879–885. doi: 10.7164/antibiotics.53.879. [DOI] [PubMed] [Google Scholar]; Höfle G, Glaser N, Leibold T, Karama U, Sasse F, Steinmetz H. Pure Appl Chem. 2003;75:167–178. [Google Scholar]
- 51.Friestad GK, Deveau AM, Marié J-C. Org Lett. 2004;6:3249–3252. doi: 10.1021/ol048986v. [DOI] [PubMed] [Google Scholar]
- 52.Marié J-C. University of Iowa; unpublished results. [Google Scholar]
- 53.Lin CH. Synth React Inorg Met-Org Chem. 1993;23:1097–1106. [Google Scholar]
- 54.(a) Salazar J, Lopez SE, Rebollo O. J Fluorine Chem. 2003;124:111–113. [Google Scholar]; (b) Iranpoor N, Zeynizadeh B. J Chem Res (S) 1999:124–125. [Google Scholar]; (c) Prashad M, Hu B, Har D, Repic O, Blacklock TJ. Tetrahedron Lett. 2000;41:9957–9961. [Google Scholar]
- 55.Allylation of the α-carbon of 15 established correlation with a known derivative Schaum R. PhD Thesis. Université de Montreal; Montreal Canada: 1998.
- 56.Torrente S, Alonso R. Org Lett. 2001;3:1985–1987. doi: 10.1021/ol015930h. [DOI] [PubMed] [Google Scholar]
- 57.Miyabe H, Yamaoka Y, Takemoto Y. J Org Chem. 2005;70:3324–3327. doi: 10.1021/jo050135t. [DOI] [PubMed] [Google Scholar]
- 58.Review: Ramon DJ, Yus M. Curr Org Chem. 2004;8:149–183.
- 59.Obrecht D, Bohdal U, Broger C, Bur D, Lehmann C, Ruffieux R, Schönholzer P, Spiegler C, Müller K. Helv Chim Acta. 1995;78:563–580. [Google Scholar]
- 60.For other applications of this strategy see: Enders D. In: Asymmetric Synthesis. Morrison JD, editor. Academic Press; New York: 1984. Husson H-P, Royer J. Chem Soc Rev. 1999;28:383–394.
- 61.Miyabe H, Ushiro C, Ueda M, Yamakawa K, Naito T. J Org Chem. 2000;65:176–185. doi: 10.1021/jo991353n. [DOI] [PubMed] [Google Scholar]
- 62.Halland N, Jørgensen KA. J Chem Soc, Perkin Trans. 2001;1:1290–1295. [Google Scholar]
- 63.Friestad GK, Shen Y, Ruggles EL. Angew Chem Int Ed. 2003;42:5061–5063. doi: 10.1002/anie.200352104. [DOI] [PubMed] [Google Scholar]
- 64.Cho DH, Jang DO. Chem Commun. 2006:5045–5046. doi: 10.1039/b613045c. [DOI] [PubMed] [Google Scholar]
- 65.Jang DO, Kim SY. J Am Chem Soc. 2008;130:16152–16153. doi: 10.1021/ja807685r. [DOI] [PubMed] [Google Scholar]
- 66.Lee S, Kim S. Tetrahedron Lett. 2009;50:3345–3348. [Google Scholar]