

Abstract
The explosive growth of medical literature on pulmonary hypertension (PH) has led to a steady increase in awareness of this disease within the medical community during the past decade. The recent revision of the classification of PH is presented in in the main guidelines.
Group 1 PH or pulmonary arterial hypertension (PAH) is a heterogeneous group and includes PH due to inheritable, drug-induced, and toxin-induced causes and to such underlying systemic causes as connective tissue diseases, human immunodeficiency viral infection, portal hypertension, congenital heart disease, and schistosomiasis.
Systemic sclerosis (SSc) is an autoimmune multisystem disorder, which affects over 240 persons per million in the United States.[1] Its manifestations are not confined to the skin but may also involve the lungs, kidneys, peripheral circulation, musculoskeletal system, gastrointestinal tract, and heart.
The outcome of PAH associated with SSc is worse when compared to other subtypes of PAH.
In this review, we summarize available information about the pulmonary vascular and cardiac manifestations of SSc with special emphasis on their prognostic implications as well as the peculiarity of their detection.
Keywords: Pulmonary arterial hypertension, connective tissue disease, systemic sclerosis, right ventricular failure, Saudi association for pulmonary hypertension guidelines
Pulmonary arterial hypertension (PAH) is caused by a remodeling of precapillary arterioles leading to a progressive increase in pulmonary vascular resistance and right ventricular (RV) failure, which is a cause of significant morbidity and eventually the cause of death in patients afflicted with this syndrome.[2,3,4,5] PAH is typically diagnosed by right heart catheterization (RHC) when the mean pulmonary arterial pressure is ≥25 mmHg in the absence of elevation of the pulmonary arterial wedge pressure (PAWP). PAH includes a heterogeneous group of clinical entities sharing similar pathological changes that have been subcategorized as idiopathic PAH (IPAH), heritable PAH, and PAH associated with other diseases such as connective tissue diseases (CTD), including systemic sclerosis (SSc), systemic lupus erythematosus (SLE), and mixed connective tissue disease (MCTD), portopulmonary hypertension, and PAH-related to HIV infection, drugs, and toxins.
The exact mechanisms involved in the pathogenesis of PAH remain vastly unknown but are likely to involve significant alterations in endothelial function,[6] an understanding of which has led over the past two decades to targeted therapy for this disease.[7] In addition to endothelial dysfunction, there are several lines of evidence implicating autoimmunity in the development of the pulmonary vascular changes, including the presence of circulating autoantibodies,[8] pro-inflammatory cytokines (e.g., IL-1 and IL-6),[9] and association of PAH with auto-immune diseases and CTD such as systemic sclerosis (SSc), systemic lupus erythematosus (SLE), and MCTD. The pathologic pulmonary vascular lesions SSc-associated PAH (SSc-APAH) are indistinguishable from those of IPAH (with the exception of a higher prevalence of veno-occlusive disease in SSc-APAH).[10] although it is now quite clear that the two diseases have quite divergent outcomes and response to therapy.[3] Survival is significantly worse in SSc-APAH compared to IPAH despite the use of modern medical therapy.[11,12,13] While there are serologic and pathologic features suggestive of inflammation in both IPAH and SSc-APAH, inflammatory pathways and autoimmunity are likely more pronounced in SSc-APAH, perhaps explaining clinical discrepancies between the two syndromes.[11,12] Other connective tissue diseases such as SLE, MCTD, and to a lesser extent rheumatoid arthritis (RA), dermatomyositis, and Sjogren's syndrome, can also be complicated by PAH and will be discussed separately in this review.
Scleroderma
SSc is a heterogeneous disorder characterized by dysfunction of the endothelium, dysregulation of fibroblasts resulting in excessive production of collagen, and abnormalities of the immune system.[14] Progressive fibrosis of the skin and internal organs is a pathologic hallmark of the disease resulting in major organ damage and failure explaining the high morbidity and early death. Genetic and environmental factors are thought to contribute to host susceptibility in the context of autoimmune dysregulation.[15] Whether presenting in the limited or diffuse form, SSc is a systemic disease with the propensity to involve multiple organ systems such as the gastrointestinal tract, the heart, kidneys and lungs.[16] Pulmonary manifestations include PAH, interstitial fibrosis, and increased susceptibility to lung neoplasms.
The use of a standard classification system for SSc has allowed more accurate estimates of incidence and prevalence of SSc, which vary according to geographic location, supporting a role for environmental factors in disease pathogenesis.[17] Prevalence of SSc ranges from 30-70 cases per million in Europe and Japan[18,19,20] to ~240 cases per million in the United States.[17] Incidence varies similarly by geographic area, with the highest rates found in the US (~19 persons per million per year).[21]
Scleroderma-associated PAH
The prevalence of PAH in SSc patients, when the diagnosis is based on rigorous RHC for assessment of filling pressures, is about 8-14%.[22,23]
Previous assessments based on echocardiographic measurements have overestimated the real prevalence of SSc-APAH[24,25,26,27] and should not be relied upon for establishing the diagnosis and initiating treatment given the inaccuracy of the Doppler signal in assessing true right ventricular systolic pressure[28] and the fact that, if pressure is indeed elevated, other diagnoses such as pulmonary venous hypertension cannot be excluded with certainty. However, it is likely that SSc-APAH remains overall under-diagnosed as suggested by the lower than expected prevalence of the disease in the few registries available.[29,30,31]
The vascular structural (intimal hyperplasia, fibrosis, and occlusion) and functional (endothelial activation, vasoreactivity and disorganized angiogenesis) abnormalities seen in SSc lead to widespread clinical manifestations such as Raynaud's phenomenon, digital ulcers, pulmonary-renal disease, renal crises, and PAH. The prevalence of these complications is variable with nearly 100% of SSc patients having evidence of Raynaud's phenomenon,[32] 5% developing acute renal crises,[33] and an estimated 7 to 13% developing PAH as diagnosed by right heart catheterization (RHC).[34]
It has become increasingly apparent that the disease course and responsiveness to therapeutic interventions in SSc-APAH are different from that observed in other forms of PAH, e.g. idiopathic pulmonary arterial hypertension or (IPAH). In retrospective cohort studies, the risk of death was higher in SSc-APAH patients when compared with those with IPAH.[35] In addition, unlike IPAH patients whose mortality rate has probably improved with modern therapies, the survival rate of SSc patients remains relatively poor.[36] In a retrospective cohort study, Girgis et al compared the long-term outcomes of first-line bosentan monotherapy in 19 patients with IPAH and 17 patients with SSc-APAH. Functional class improved in 58% of IPAH patients compared with only 25% of SSc-APAH patients and overall survival at 1 and 2 years was 100% and 100% versus 87% and 79% for IPAH and PAH-SSc patients, respectively.[37]
The reasons for the substantial prognostic differences between IPAH and SSc-APAH are poorly understood. They might be in part explained by comorbid scleroderma disease features (such as interstitial lung disease) and age-related factors secondary to the later onset of PAH in SSc,[38] although age alone was not thought to be a predisposing factor for increased mortality in one study.[32] It has also been suggested that esophageal dysmotility, gastroparesis and small bowel malabsorption that are common in SSc may contribute to altered absorption and metabolism of oral PAH-specific therapy, thereby leading to sub-therapeutic concentrations of these medications in the sera.
Pathophysiology
More details about the pathophysiology of the RV and pulmonary vascular disease in Scleroderma is discussed in the review (Pulmonary Vascular Disease in Scleroderma) in the current issue of the Journal.
There are early vascular changes in SSc, which include gaps between endothelial cells, cellular apoptosis, endothelial activation with expression of cell adhesion molecules, inflammatory cell recruitment, a procoagulant state, and remodeling of the small vessels with intimal proliferation and adventitial fibrosis leading to vessel obliteration.[39,40,41,42] The extent of these vascular lesions in vital organs such as the lungs, kidneys and heart defines the prognosis of patients with SSc.[43]
Specific endothelial injury is reflected by increased levels of soluble vascular cell adhesion molecule (sVCAM-1),[44] disturbances in angiogenesis reflected by increased levels of circulating vascular endothelial growth factor (VEGF),[45,46] and presence of angiostatic factors.[45,47] Increased VEGF, may be a consequence of increased angiogenesis or profound disturbances in signaling in SSc. Dysregulated angiogenesis in SSc-APAH, exemplified by the upregulation of VEGF, a glycoprotein with potent angiogenic and vascular permeability-enhancing properties, is a predominant pathological feature of the disease and is a logical candidate for therapeutic targeting.
Autoantibodies
Several antibodies are frequently found in SSc-APAH such as antifibrillarin antibodies (anti-U3-RNP)[48] and the poorly characterized anti-endothelial antibodies (aECA), which correlate with digital infarcts.[49] Antibodies to fibrin-bound tissue plasminogen activator in CREST patients[50] and in IPAH patients with HLA-DQ7 antigen,[51] and anti-topoisomerase II- alpha antibodies, particularly in association with HLA-B35 antigen,[52] have been reported in SSc-APAH. aECA antibodies which can activate endothelial cells, induce the expression of adhesion molecules, and trigger apoptosis, are thought to play a role in the pathogenesis of PAH.[53]
Fibroblasts are essential components of the pulmonary vascular wall remodeling in PAH and are found in the remodeled neointimal layer in both SSc-APAH and IPAH. In that regard, the detection of anti-fibroblast antibodies in the serum of SSc and IPAH patients[54,55] has significant pathogenic importance since these antibodies can activate fibroblasts and induce collagen synthesis, thus contributing potentially directly to the remodeling process. Antibodies from sera of patients with SSc induce a pro-adhesive and pro-inflammatory response in normal fibroblasts.[55] Antifibroblast antibodies from sera of IPAH and SSc-APAH patients have distinct reactivity profiles[56] and react with fibroblast proteins involved in regulation of cytoskeletal function, cell contraction, cell and oxidative stress, cell energy metabolism and in different key cellular pathway.[57]
Taken together, particularly in light of the positive response to immunosuppressive therapy for some patients with PAH associated with SLE and MCTD,[58] these studies suggest that inflammation and autoimmunity play a major role in the pathogenesis of PAH, perhaps more specifically in CTD-associated PAH.
Genetic factors
Polymorphisms involving the bone morphogenetic protein receptor-2 (BMPRII) are present in over 80% of heritable IPAH[59,60] and up to 25% of sporadic cases of IPAH.[61,62,63] Additional candidate genes have been proposed to influence the pathogenesis of PAH.[64] Polymorphisms of the activin-receptor-like kinase 1 (ALK1) gene, another member of the TGF β receptor superfamily, have been reported in patients with hereditary hemorrhagic telangiectasia (HHT) and PAH.[64] However, to date BMPR2 mutations have not been identified in two small cohorts of SSc-APAH patients.[65,66]
Candidate genes associated with SSc have been reported in different populations and include a variant in the promoter of monocyte chemotactic protein-1 (MCP-1);[67] two variants in CD19 (-499G>T and a GT repeat polymorphism in the 3’-UTR region);[68] a promoter and coding polymorphism in TNFA (TNFA-238A>G, TNFA 489A>G);[69] a variant in the promoter of the IL- 1 alpha gene (IL1A -889T);[70,71] and a 3-SNP haplotype in IL10.[72] A genome-wide association analysis provided evidence for association to multiple loci in a Native American population.[73]
Recently, an association between an endoglin gene (ENG) polymorphism and SSc-related PAH was identified. Wipf and colleagues demonstrated a significant lower frequency of the 6bINS allele in SSc-APAH patients as compared to controls or patients with SSc but no PAH.[74] Endoglin, a homodimeric membrane glycoprotein primarily present on human vascular endothelium, is part of the TGF-β receptor complex. The functional significance of the ENG polymorphism in SSc patients remains to be determined.
More detailed information about the genetic factors can be found in the review of “genetic of Pulmonary Hypertension” in this issue of the journal.
Thus, there is compelling data supporting a genetic basis for SSc. However, aside from the few examples cited above, the genes relevant to the pathogenesis and poor outcome associated with SSc-APAH have not been identified, and their definition will require robust, well-characterized large patient populations to provide adequate power for analysis.
Clinical Features
Risk factors for the development of PAH in SSc patients include late-onset disease,[75] an isolated reduction in DLCO, an FVC %/DLCO % ratio greater than 1.6,[76,77] or a combined decreased DLCO/alveolar volume with elevation of serum N-terminal pro-brain natriuretic peptide levels, (Figure).[78,79]
Typically, patients with SSc-APAH are predominantly women, have limited sclerosis with predominantly anticentromere antibodies, are older and have seemingly less severe hemodynamic impairment compared to IPAH patients.[12] Clinical symptoms are non specific, including dyspnea, functional limitation which may be more severe than in IPAH due not only to older age but also to frequent involvement of the musculoskeletal system in these patients. SSc-APAH patients also tend to have other organ involvement such as renal dysfunction and intrinsic heart disease. Indeed patients with SSc (even in the absence of PAH) tend to have depressed RV function as well as left ventricular systolic and diastolic dysfunction.[80,81,82] Like IPAH patients, SSc-APAH patients have severe RV dysfunction already at time of presentation but have more severely depressed RV contractility compared top IPAH patients.[83]
In addition, SSc- APAH patients tend to have more commonly LV diastolic dysfunction and a high prevalence of pericardial effusion (34% compared to 13% for IPAH).[12] In both groups, pericardial effusion portends a particularly poor prognosis.[12]
SSc-APAH patients also tend to have more severe hormonal and metabolic dysfunctions, such as high levels of N-terminal brain natriuretic peptide (N-TproBNP)[84] and hyponatremia.[85] Both N-TproBNP and hyponatremia have been shown, at baseline and with serial changes (for N- TproBNP),[84] to correlate with survival in PAH.[84,85]
Early Detection
While IPAH is usually diagnosed late (patients presenting in WHO functional status III and IV), a population at risk for PAH such as SSc theoretically allows establishing measures for early detection. An algorithm for early detection of PAH may be helpful if based on a combination of symptoms and screening echocardiography [Figure 1] as exemplified by a recent large French registry where patients with SSc with tricuspid regurgitation velocity (TRV) jet by transthoracic echocardiography greater than 3 m/sec or between 2.5 and 3 m/sec if accompanied by unexplained dyspnea, were systematically referred for RHC.[22] Investigators were able to detect incident cases of SSc-APAH with less severe disease (as judged on hemodynamic data) compared to patients with known disease. Therefore, unexplained dyspnea should prompt a search for PAH in these patients, in particular in the setting of a low single breath DLCO or declining DLCO over time,[77] echocardiographic findings suggestive of the disease (elevated TRV jet or dilated RV or right atrium), or elevated levels of N-TproBNP, which can reflect cardiac dysfunction and have been found to predict the presence of SSc-APAH.[84] Systematic screening should allow detection of early disease and prompt therapy which may theoretically be beneficial from a prognostic standpoint.[86]
Figure 1.
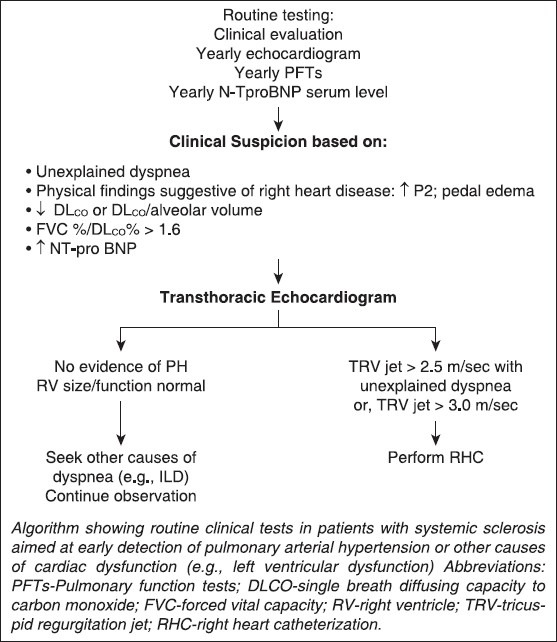
Algorithm for detection of PAH in patients with systemic sclerosis
Diagnostic Work up
ECG and chest X-ray together with clinical evaluation have a poor sensitivity and do not allow a prompt diagnosis at a pre-clinical stage, when therapeutics are supposed to be more effective. More elaborate techniques are becoming more available to accurately identify early perturbations of normal cardiac configuration and performance in patients with SSc.
Transthoracic echocardiography
Transthoracic echocardiography (TTE) is the most widely available semi-quantitative cardiac assessment modality. This non-invasive diagnostic tool gives estimates of elevated pulmonary artery pressures and of RV structural changes consistent with RV overload, such as ventricular dilatation and septal wall motion abnormalities. Overall, echocardiographic measures are useful in detecting RV and/or left ventricular (LV) dysfunction that usually occur in later disease stages, but are less helpful in identifying patients with early subtle cardiac involvement. Furthermore, unlike the LV, the non-invasive evaluation of the RV is hindered by its complex cavity geometry that does not lend itself to mathematical modeling. Additionally, there is poor endocardial definition and muscle fiber contraction occurs predominantly in the longitudinal direction and in a peristaltic fashion, which complicates estimations of RV ejection fraction and performance.[87] The appraisal of RV function is also difficult because of the interplay between intrinsic myocardial performance and right ventricular loading conditions.[88] Nevertheless, evaluation of overall RV performance has recently been proposed in PAH with TTE measures that are not reliant on geometric measurements of the structurally intricate RV. This has allowed an earlier detection of cardiac alterations.
The Tei-index (or myocardial performance index), a mathematical quotient of isovolumetric contraction and relaxation time divided by ejection time, has been utilized as such a measure.[89] Patients with pulmonary hypertension have increased isovolumetric contraction and relaxation times and a decreased ejection time. In combination with standard TTE measures of pulmonary artery pressure, a Tei-index > 0.36 was found to be highly predictive of SSc patients with PAH based upon RHC.[90] It is important to note that the value of these parameters as outcome measures in clinical practice remains unknown.
Tricuspid annular systolic excursion (TAPSE) is another TTE method for the detection of global RV compromise that is independent of RV geometry. TAPSE is measured by the total displacement of the tricuspid annulus from end-diastole to end- systole. While prior studies demonstrated a strong relationship with RV ejection fraction, more recent investigations have shown that TAPSE scores can be influenced by RV and LV dysfunctions.[91] In PAH, TAPSE measurements have been shown to correlate with RHC parameters and to predict survival.[92] Preliminary studies suggest that PAH-SSc patients exhibit worse TAPSE measurements and worse prognosis compared to other patients with PAH.
Data related to Tei-indices and TAPSE scores for detection of global RV dysfunction and to the diagnosis and prognosis of PAH-SSc continue to accumulate. However, the utilization of these imaging studies as outcome measures in response to pharmacologic interventions has yet to be adequately evaluated. The studies available so far do not provide sufficiently detailed information regarding the components of RV structure and function to account for the disparity in the observed survival of PAH-SSc patients compared to other PAH patients.
Tissue doppler and doppler strain imaging
Tissue Doppler imaging (TDI) allows echocardiographic measurement of myocardial velocities for determination of global as well as segmental systolic and diastolic ventricular dysfunction. Although originally developed for the examination of the LV, application of this technique to the RV is gaining interest. TDI has many advantages over conventional Doppler when deriving time-dependent myocardial performance indices. However, its major drawback is that nonfunctioning segments are often not observed due to interdependence with functioning segments.[93] In contrast, Doppler strain imaging (DSI) allows for examination of segmental deformation regardless of normal surrounding segments.[93] Therefore, the addition of TDI and Doppler strain and strain rate imaging to standard TTE provides additional information; TDI detects abnormalities not shown on TTE, and DSI finds discrepancies not found on TDI. Strain and strain rate imaging are thus promising new sensitive and accurate modalities to evaluate SSc patients for early cardiac involvement.
Nuclear imaging
Nuclear imaging can be a good adjunctive method for evaluating cardiac complications even in the early stages of SSc. Earlier studies investigating RV abnormalities using radionuclide imaging with 99mTc ventriculography showed a significant decrease in RV ejection fraction in SSc patients (with no evidence of PAH) when compared to controls.[94] Steen and colleagues followed 48 patients who had undergone cardiac evaluation, and found that thallium perfusion defect scoring was the single most powerful predictor of mortality and subsequent development of clinical cardiac disease in patients with either limited or diffuses scleroderma.[95] However, the clinical implications of these defects remain controversial.
Cardiac MRI
Cardiac MRI is becoming the gold standard for non-invasive evaluation of RV structural changes; it allows accurate and reproducible measurements of ventricular dimensions, wall thickness, and myocardial mass without relying on geometric assumptions. It enables precise analysis of the different patterns of heart involvement in SSc by differentiating morphological, functional, perfusion and delayed contrast enhancement abnormalities. Compared to other imaging modalities, cardiac MRI detected significantly compromised RV function in a higher number of asymptomatic SSc patients.[96] The ventricular mass index, ratio of the right and left ventricular end diastolic mass, was found to correlate strongly with the mean pulmonary artery pressure in patients with SSc and may have a role in non-invasively excluding clinically significant PH in breathless SSc patients in whom echocardiographic screening has failed.[97]
Moreover, MRI provides imaging of the coronary arteries and assessment of the viability of the myocardium. It can also potentially be used as a guide for myocardial biopsy or ablation studies, when indicated, and as a mean for monitoring therapeutic effects. For instance, Wilkins et al were able to show significant improvement in cardiac MRI morphological alterations (i.e., decreased RV mass) with sildenafil as compared to bosentan therapy.[98] Taken together, these results suggest that MRI is a reliable and sensitive technique to diagnose heart involvement in SSc and identify potential mechanisms such as inflammation and fibrosis.
Right heart catheterization
RHC remains the gold standard to evaluate RV hemodynamic changes following pressure overload. In patients with SSc, RHC is essential for diagnosing pulmonary hypertension, monitoring disease progression, and assessing the response to therapeutic interventions.
Endomyocardial biopsy
The technique of endomyocardial biopsy is now recognized as an established and safe procedure that can significantly increase the frequency of detection of cardiac involvement in patients with SSc.
In addition, several investigators advocate the use of myocardial biopsies in this patients’ population as a tool to exclude specific cardiac pathologies such as hydroxychloroquine toxicity and cardiac amyloid and to distinguish between inflammatory and fibrotic forms of heart involvement in SSc patients, distinctions that might lead to specific therapies. However, this diagnostic method is somewhat limited by its invasive nature and high rate of sampling errors, as suggested by its relatively low diagnostic yield compared with autopsy results.
Prognosis
Patients with SSc-APAH have a worse prognosis compared to patients with other forms of PAH such as IPAH. Indeed, one-year survival rates for SSc-APAH patients range from 50-81%,[11,13,23,27,99] considerably lower than the estimated 88% one year survival for IPAH patients.[100] In all patients with SSc, PAH significantly worsens survival and is one of the leading causes of mortality in these patients.[17,99,101]
PAH Associated with Other Connective Tissue Diseases
PAH can complicate any connective tissue disease, most frequently SSc as discussed above, but also SLE, MCTD, RA, or other diseases such as Sjogren's syndrome and dermatomyositis.
Systemic lupus erythematosus
Pulmonary vascular involvement is common in SLE and, like in SSc, there is evidence of endothelial dysfunction with an imbalance between vasodilators and vasoconstrictors. Endothelin levels are high in patients with SLE, particularly in those patients with pulmonary hypertension. Other factors contributing to pulmonary vascular derangement in SLE include recurrent thromboembolic disease, particularly in patients with a hypercoagulable state from antiphospholipid antibodies, which are present in up to 10% of patients with SLE,[102] pulmonary vasculitis, and parenchymal disease (e.g., interstitial lung disease, and the shrinking lung syndrome from myositis of the diaphragm). Combined vasculitis and chronic hypoxia are frequent contributing offenders in these syndromes. Finally, pulmonary venous hypertension may be a consequence of left ventricular dysfunction, myocarditis, or Libman Scahs endocarditis.
The prevalence of PAH in SLE is unclear but is likely less than in SSc, affecting about 0.5- 14% patients with SLE in a large review of the literature encompassing over 100 patients.[103] The patients are predominantly female (90%), young (average age of 33 at time of diagnosis), and often suffer from Raynaud's phenomenon.
The pathological lesions are often indistinguishable from IPAH or SSc-APAH lesions, with intimal hyperplasia, smooth muscle cell hypertrophy, and medial thickening. Survival, which was quite poor (25-50% at 2 years) even compared to SSc-APAH in studies antedating specific PAH therapy, is now improved and estimated at 75%.[13]
Mixed connective tissue disease
Patients with MCTD have clinical features, which overlap between those of SSc, SLE, RA, and polymyositis. The exact prevalence of PAH in MCTD is unknown but is thought to be as high as 50%.[104] PAH in these patients may occasionally respond to immunosuppressive drugs.[58]
Rheumatoid arthritis
Both the prevalence and impact of PAH in patients with RA have not been well characterized but are thought to be relatively low compared to other CTD such as SSc, SLE and MCTD.
Primary Sjogren's syndrome
Although primary Sjogren's syndrome (pSS) is a relatively common autoimmune disease with glandular and extra-glandular manifestations, it is very rarely complicated by PAH. In a recent review by Launay et al of patients with pSS and PAH,[105] the mean age at diagnosis of PAH of these almost exclusively female patients was 50 years. Patients had severe functional class (FC III and IV) and hemodynamic impairment. Standard therapy (with endothelin receptor antagonists, phosphodiesterase inhibitors or prostanoids) was typically ineffective despite an initial improvement. Some patients were reported to respond to immunosuppressive treatment. However, any conclusion regarding treatment is limited by the small size of this case report. Survival rate was low (66% at 3 years).
Therapy for PAH Associated with CTD
More detailed information about specific drug therapy in the management of PAH can be about in the review (Specific Treatment For Pulmonary Arterial Hypertension) in this issue of the Journal. This section applies essentially to the treatment of patients with SSc-APAH, which represents the largest group of CTD-associated PAH patients in most clinical therapeutic trials.
Evidence of chronically impaired endothelial function,[106,107,108] affecting vascular tone and remodeling, is the basis for current PAH therapy. Vasodilator therapy using high dose calcium channel blockers is an effective long-term therapy for a minority of patients with IPAH who demonstrate acute and sustained vasodilation (e.g., to NO or adenosine) during hemodynamic testing.[109,110] However, since most patients with CTD-associated PAH patients fail to demonstrate a vasodilator response to acute testing,[29] high dose calcium channel therapy is usually not indicated for these patients except at low dosage for Raynaud's syndrome.
Anti-inflammatory drugs
Inflammation may play a significant role in PAH associated with CTD. In that respect, it is interesting to note that occasional patients with severe PAH associated with some forms of CTD (such as SLE, primary Sjögren's syndrome, and MCTD) have had dramatic improvement of their pulmonary vascular disease with corticosteroids and/or immunosuppressive therapy.[58] Unfortunately, this has not been the case for patients with SSc-APAH, whose PAH is usually notoriously refractory to immunosuppressive drugs.[58]
Prostaglandins
Prostacyclin (epoprostenol) has potent pulmonary vasodilator, anti-platelet aggregating and antiproliferative properties,[111] and has proven effective in improving exercise capacity, cardiopulmonary hemodynamics, modified NYHA functional class, symptoms, as well as survival in patients with PAH when given by continuous infusion.[112,113,114] In SSc-APAH, continuous intravenous epoprostenol improves exercise capacity and hemodynamics,[115] compared to conventional therapy, and may have long-term beneficial effects[116] although a clear effect on survival in these patients there has yet to be demonstrated.
Treprostinil, an analogue of epoprostenol suitable for continuous subcutaneous administration, has modest effects on symptoms and hemodynamics in PAH.[117] In a small study of 16 patients (among whom 6 had CTD related PAH), intravenous treprostinil improved 6 minute walk distance (6MWD), modified NYHA functional class, and hemodynamics after 12 weeks of therapy.[118]
Although the safety profile of this drug is similar to IV epoprostenol, required maintenance doses are usually twice as much as for epoprostenol. However, for patients with SSc-APAH and often severe and debilitating Raynaud's phenomenon, the lack of requirement of ice packing and less frequent mixing of the drug offer significant advantages.
Several reports of pulmonary edema in SSc-APAH patients treated with prostaglandin derivatives, both in acute and chronic settings, have raised the suspicion of increased prevalence of veno-occlusive disease in these patients,[119,120] and concern about usefulness of these drugs for this entity. Nevertheless, intravenous prostaglandin therapy remains an option for patients with CTD- APAH with modified NYHA class IV. Considering the frequent digital problems and disabilities that these patients often experience, this form of therapy can be quite challenging and may increase the already heavy burden of disease in these patients. In summary, both epoprostenol and treprostinil are FDA approved for PAH, but are cumbersome therapies requiring continuous parenteral administration with the attendant numerous adverse effects (e.g., infection and possibility of pump failure),[121] which make these drugs less than ideal.
Endothelin receptor antagonists
In randomized, placebo-controlled trials, bosentan therapy was shown to have a beneficial effect on functional class, 6MWD, time to clinical worsening, and hemodynamics in PAH.[122,123] Roughly one fifth of these patients consisted of SSc- APAH patients while a large majority had a diagnosis of IPAH. In a subgroup analysis, there was a non-significant trend towards a positive treatment effect on 6MWD among the SSc-APAH patients treated with bosentan compared to placebo.[123] At most, bosentan therapy prevented deterioration in these patients. This less than optimal effect of therapy in patients with SSc-APAH is unclear but may be related to the severity of PAH at time of presentation, as well as other factors such as, hypothetically, more severe RV and pulmonary vascular dysfunction, as compared to patients with other forms of PAH (e.g., IPAH).
In a recent analysis of patients with CTD-APAH (e.g., patients with lupus, overlap syndrome, and other rheumatological disorders) included in randomized clinical trials of bosentan, there was a trend toward improvement in 6MWD and improved survival compared to historical cohorts.[124] Single center experience suggests that long-tem outcome of first-line bosentan monotherapy is inferior in SSc-APAH compared to IPAH patients, with no change in functional class and worse survival in the former group.[125] Since Endothelin-1 (ET-1) appears to play an important pathogenic role in the development of SSc-APAH, contributing to vascular damage and fibrosis, inhibiting ET-1 remains a rational therapeutic strategy in these patients.
As an example of mechanistic effect, in a small study of 35 patients with SSc (10 of whom had SSc-APAH), bosentan treatment appeared to reduce endothelial cell (as determined by endothelial soluble serum factors such as ICAM-1, VCAM-1, P-selection and PECAM-1) and T cell subset (assessed by expression of lymphocyte function- associated antigen-1, very late antigen-4, and L-selectin on CD3 Tcells) activation.[126] Aside from improving pulmonary hypertension, ET-1 receptor antagonists (specifically bosentan) cause significant reductions in the occurrence of new digital ulcerations without, however, healing preexisting ulcers.[127]
In an effort to target the vasoconstrictive effects of endothelin while preserving its vasodilatory action, selective endothelin-A receptor antagonists have been developed.
A large placebo- controlled, randomized trial of ambrisentan, the only currently FDA-approved selective endothelin receptor antagonist, improved 6MWD in PAH patients at week 12 of treatment, however, the effect was larger in patients with IPAH compared to patients with CTD-APAH (range of 50-60 meters versus 15-23 meters, respectively).[128] Ambrisentan is generally well tolerated although peripheral edema (in up to 20% of patients)[128] and congestive heart failure have been reported.
Phosphodiesterase inhibitors
Sildenafil, a phosphodiesterase type V inhibitor that reduces the catabolism of cGMP, thereby enhancing the cellular effects mediated by nitric oxide, has become a widely used and highly efficacious therapy for PAH. The pivotal SUPER trial demonstrated that sildenafil therapy causes an improvement in the 6MWD in patients with IPAH and CTD-APAH or pulmonary hypertension associated with repaired congenital heart disease (patients were predominantly FC II or III) at all three doses tested (20, 40, and 80 mg, given three times a day).[129] The current FDA- recommended dose is 20 mgs three times a day since there were no significant differences in clinical effects and time to clinical worsening at week 12 between all doses tested. In a post-hoc subgroup analysis of 84 patients with PAH related to CTD (forty-five percent of whom had SSc-APAH), data from the SUPER study suggest that sildenafil at a dose of 20 mgs improved exercise capacity (6MWD), hemodynamic measures, and functional class after 12 weeks of therapy.[130] However, there was no effect for the dose of 80 mgs three times a day on hemodynamics in this subgroup of patients with CTD-related PAH.[130] For this reason and because of the potential of increased side-effects (such as bleeding from arterio-venous malformations) at high doses, a sildenafil dosage of 20 mg three times a day is recommended for SSc-APAH patients (and perhaps patients with PAH associated with other forms of CTD) as standard therapy. Higher doses are occasionally attempted in case of limited response. The impact of long-term sildenafil therapy on survival in these patients remains to be determined.
Finally, tadalafil, another phosphodiesterase inhibitor, has now been shown to be effective for PAH,[131] although subgroup analysis has not been performed yet and thus its effects on CTD-APAH remain unclear. Tadalafil has the advantage over sildenafil of single daily dosage.
Combination therapy
It is now a common practice to add drugs when patients fail to improve on monotherapy. Adding inhaled iloprost to patients receiving bosentan has been shown to be beneficial in a small, randomized trial.[132] Combination therapy is mechanistically appealing and anecdotally efficacious as these drugs target separate, potentially synergistic pathways.[133] Several multicenter trials are now exploring the efficacy of various combinations of two oral drugs or one oral and one inhaled drug. The published results of the PACES trial demonstrate that adding sildenafil (at a dose of 80 mgs three times a day) to intravenous epoprostenol improves exercise capacity, hemodynamic measurements, time to clinical worsening, and quality of life.[134] About 21% of these patients had CTD, including 11% with SSc-APAH. Although no specific subgroup analysis is provided, improvement was apparently mainly in patients with IPAH.
In a smaller one center clinical trial, adding sildenafil to patients with IPAH or SSc-APAH after they failed initial monotherapy with bosentan, demonstrates that combination therapy improved the 6MWD and FC in IPAH patients. The outcome in patients with SSc-APAH was less favorable, although combination therapy may have halted clinical deterioration. In addition, there were more side effects reported in the SSc-APAH compared to the IPAH patients, including hepatotoxicity that developed after addition of sildenafil to bosentan monotherapy.[135]
Anticoagulation
Based essentially on retrospective data showing a survival advantage,[109,136] anticoagulation is routinely recommended in the treatment of IPAH patients. However, the role of anticoagulation in other forms of PAH, in particular in CTD-APAH is much less clear.
Theoretically, there is potential for increased bleeding in patients with CTD, particularly with SSc where intestinal telangiectasia may be common. In the author's experience, less than 50% of SSc-APAH patients started on anticoagulation remain on therapy long-term because of bleeding complications (e.g., often related to occult bleeding in the gastrointestinal tract), which are often difficult to diagnose.
Tyrosine kinase inhibitors
The finding that there is pathologically aberrant proliferation of endothelial and smooth muscle cells in PAH, as well increased expression of secreted growth factors such as VEGF and bFGF, has caused a shift in paradigm in treatment strategies for this disease as some investigators have likened this condition to a neoplastic process reminiscent of advanced solid tumors.[137] As a result, anti-neoplastic drugs have been tested in experimental models.[138,139] However, the results of a double-blinded, placebo-controlled, randomized phase III trial (IMPRES) have recently published and have created a major concern about the effectiveness of this drug, as despite the fact that patients treated with imatinib experienced better exercise tolerance (the primary endpoint) and significant improvement in hemodynamics, the time to clinical worsening and mortality were not different between the 2 groups. Furthermore, side effects profile, mainly cerebral hemorrhage, was another major concern in imatinib-treated patient.[140] Whether these new anti-neoplastic drugs with anti-tyrosine kinase activity will have a role in SSc (where there is evidence for both dysregulated proliferation and increased expression of growth factors such as VEGF)[141] or in IPAH remains to be determined. Interestingly, a single case report suggests significant improvement of right ventricular function with imatinib treatment in a SSc-APAH patient. Also of note is that imatinib is being investigated for SSc-related interstitial lung disease.[142]
Lung transplantation
Lung transplantation (LT) is typically offered as a last resort to patients with PAH who fail medical therapy. CTD is not an absolute contraindication to LT; however, patients with CTD-APAH frequently suffer from associated morbidity and organ dysfunction other than the lung, which places them at a significantly increased risk for LT. Motility disorder of the esophagus and gastroesophageal reflux in patients with SSc significantly enhance the post-operative potential of aspiration and damage to the recipient lung. For these reasons, patients with SSc-APAH are often denied LT consideration. However, if properly screened and approved for LT, patients with SSc experience similar rates of survival 2 years after the procedure compared with patients who receive LT for pulmonary fibrosis or IPAH.[133] In addition, a recent retrospective study suggests that the 1-year survival rate is similar for SSc patients (transplanted for respiratory failure related to PAH or interstitial lung disease) compared to patients with IPF although acute rejection appears to be more common for the former group.[144]
Conclusion
In summary, pulmonary hypertension is a common complication of CTD, particularly SSc where it carries a very poor prognosis. Despite modern therapy for PAH, survival of patients with CTD-APAH remains unacceptably low. Possible reasons include an increased prevalence of pulmonary veno-occlusive lung disease in SSc-APAH patients, or more severe vascular lesions affecting not only proximal and distal pulmonary vessels but also the heart (such as inflammatory myocarditis) in CTD. Thus, a better understanding of the underlying pathophysiology affecting the heart and pulmonary vessels in CTD is needed for better targeted therapy. Whether specific anti-inflammatory agents or drugs targeting tyrosine kinase activity will have any role in CTD-APAH is unclear at this time but needs to be explored.
Heart involvement is a serious complication of SSc. It is often clinically occult and its detection depends on the sensitivity of the diagnostic method used. The etiology of cardiac involvement seems to be multifactorial, since different degrees of inflammation, fibrosis and vascular abnormalities have all been observed. Another important contributing factor is the presence of systemic and/or pulmonary hypertension that may overwhelm the ventricular compensatory mechanisms. In recent years, it has become evident that early diagnosis of scleroderma heart disease is fundamental for appropriate management of this patient population. For this reason, significant progress needs to be made in the development of simple, yet reliable, markers of cardiac function. However, major challenges remain in validating current assessment strategies in large and well-designed clinical studies, and determining longitudinally the effect of early therapeutic interventions. Furthermore, a better understanding of the fundamental cellular and molecular mechanisms that govern the adaptation of the right heart to its increased load may provide an explanation for the accelerated RV failure and less robust response to drug therapy observed in SSc patients. This will also allow exploration of novel treatment paradigms with potential positive impact on the quality of life and life expectancy of this patient population.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Mayes MD, Lacey JV, Jr, Beebe-Dimmerr J, Gillespie BW, Cooper B, Laing TJ, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003;48:2246–55. doi: 10.1002/art.11073. [DOI] [PubMed] [Google Scholar]
- 2.D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115:343–9. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- 3.Gaine SP, Rubin LJ. Primary pulmonary hypertension. Lancet. 1998;352:719–25. doi: 10.1016/S0140-6736(98)02111-4. [DOI] [PubMed] [Google Scholar]
- 4.Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107:216–23. doi: 10.7326/0003-4819-107-2-216. [DOI] [PubMed] [Google Scholar]
- 5.Idrees M. Pulmonary hypertension: Another light in the dark tunnel. Learning the lesson from cancer. Ann Thorac Med. 2013;8:69–70. doi: 10.4103/1817-1737.109813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109:159–65. doi: 10.1161/01.CIR.0000102381.57477.50. [DOI] [PubMed] [Google Scholar]
- 7.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425–36. doi: 10.1056/NEJMra040291. [DOI] [PubMed] [Google Scholar]
- 8.Isern RA, Yaneva M, Weiner E, Parke A, Rothfield N, Dantzker D, et al. Autoantibodies in patients with primary pulmonary hypertension: Association with anti-Ku. Am J Med. 1992;93:307–12. doi: 10.1016/0002-9343(92)90238-7. [DOI] [PubMed] [Google Scholar]
- 9.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot- Keros L, et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151:1628–31. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 10.Dorfmüller P, Humbert M, Perros F, Sanchez O, Simonneau G, Muller KM, et al. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum Pathol. 2007;38:893–902. doi: 10.1016/j.humpath.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 11.Kawut SM, Taichman DB, Archer-Chicko CL, Palevsky HI, Kimmel SE. Hemodynamics and survival in patients with pulmonary arterial hypertension related to systemic sclerosis. Chest. 2003;123:344–50. doi: 10.1378/chest.123.2.344. [DOI] [PubMed] [Google Scholar]
- 12.Fisher MR, Mathai SC, Champion HC, Girgis RE, Housten- Harris T, Hummers L, et al. Clinical differences between idiopathic and scleroderma-related pulmonary hypertension. Arthritis Rheum. 2006;54:3043–50. doi: 10.1002/art.22069. [DOI] [PubMed] [Google Scholar]
- 13.Condliffe R, Kiely DG, Peacock AJ, Corris PA, Gibbs JS, Vrapi F, et al. Connective tissue disease-associated pulmonary arterial hypertension in the modern treatment era. Am J Respir Crit Care Med. 2009;179:151–7. doi: 10.1164/rccm.200806-953OC. [DOI] [PubMed] [Google Scholar]
- 14.Jimenez SA, Derk CT. Following the molecular pathways toward an understanding of the pathogenesis of systemic sclerosis. Ann Intern Med. 2004;140:37–50. [PubMed] [Google Scholar]
- 15.Tan FK. Systemic sclerosis: The susceptible host (genetics and environment) Rheum Dis Clin North Am. 2003;29:211–37. doi: 10.1016/s0889-857x(03)00015-2. [DOI] [PubMed] [Google Scholar]
- 16.LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA, Jr, et al. Scleroderma (systemic sclerosis): Classification, subsets and pathogenesis. J Rheumatol. 1988;15:202–5. [PubMed] [Google Scholar]
- 17.Mayes MD. Scleroderma epidemiology. Rheum Dis Clin North Am. 2003;29:239–54. doi: 10.1016/s0889-857x(03)00022-x. [DOI] [PubMed] [Google Scholar]
- 18.Silman A, Jannini S, Symmons D, Bacon P. An epidemiological study of scleroderma in the West Midlands. Br J Rheumatol. 1988;27:286–90. doi: 10.1093/rheumatology/27.4.286. [DOI] [PubMed] [Google Scholar]
- 19.Tamaki T, Mori S, Takehara K. Epidemiological study of patients with systemic sclerosis in Tokyo. Arch Dermatol Res. 1991;283:366–71. doi: 10.1007/BF00371817. [DOI] [PubMed] [Google Scholar]
- 20.Allcock RJ, Forrest I, Corris PA, Crook PR, Griffiths ID. A study of the prevalence of systemic sclerosis in Northeast England. Rheumatology (Oxford) 2004;43:596–602. doi: 10.1093/rheumatology/keh124. [DOI] [PubMed] [Google Scholar]
- 21.Mayes MD, Lacey JV, Jr, Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003;48:2246–55. doi: 10.1002/art.11073. [DOI] [PubMed] [Google Scholar]
- 22.Hachulla E, Gressin V, Guillevin L, Carpentier P, Diot E, Sibilia J, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: A French nationwide prospective multicenter study. Arthritis Rheum. 2005;52:3792–800. doi: 10.1002/art.21433. [DOI] [PubMed] [Google Scholar]
- 23.Mukerjee D, St George D, Coleiro B, Knight C, Denton CP, Davar J, et al. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: Application of a registry approach. Ann Rheum Dis. 2003;62:1088–93. doi: 10.1136/ard.62.11.1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Battle RW, Davitt MA, Cooper SM, Buckley LM, Leib ES, Beglin PA, et al. Prevalence of pulmonary hypertension in limited and diffuse scleroderma. Chest. 1996;110:1515–9. doi: 10.1378/chest.110.6.1515. [DOI] [PubMed] [Google Scholar]
- 25.Stupi AM, Steen VD, Owens GR, Barnes EL, Rodnan GP, Medsger TA., Jr Pulmonary hypertension in the CREST syndrome variant of systemic sclerosis. Arthritis Rheum. 1986;29:515–24. doi: 10.1002/art.1780290409. [DOI] [PubMed] [Google Scholar]
- 26.Sacks DG, Okano Y, Steen VD, Curtiss E, Shapiro LS, Medsger TA., Jr Isolated pulmonary hypertension in systemic sclerosis with diffuse cutaneous involvement: Association with serum anti-U3RNP antibody. J Rheumatol. 1996;23:639–42. [PubMed] [Google Scholar]
- 27.MacGregor AJ, Canavan R, Knight C, Denton CP, Davar J, Coghlan J, et al. Pulmonary hypertension in systemic sclerosis: Risk factors for progression and consequences for survival. Rheumatology (Oxford) 2001;40:453–9. doi: 10.1093/rheumatology/40.4.453. [DOI] [PubMed] [Google Scholar]
- 28.Fisher MR, Forfia PR, Chamera E, Housten-Harris T, Champion HC, Girgis RE, et al. Accuracy of Doppler echocardiography in the hemodynamic assessment of pulmonary hypertension. Am J Respir Crit Care Med. 2009;179:615–21. doi: 10.1164/rccm.200811-1691OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France: Results from a national registry. Am J Respir Crit Care Med. 2006;173:1023–30. doi: 10.1164/rccm.200510-1668OC. [DOI] [PubMed] [Google Scholar]
- 30.Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. 2007;30:104–9. doi: 10.1183/09031936.00092306. [DOI] [PubMed] [Google Scholar]
- 31.Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, et al. Pulmonary arterial hypertension: Baseline characteristics from the REVEAL Registry. Chest. 2010;137:376–87. doi: 10.1378/chest.09-1140. [DOI] [PubMed] [Google Scholar]
- 32.Guiducci S, Giacomelli R, Cerinic MM. Vascular complications of scleroderma. Autoimmun Rev. 2007;6:520–3. doi: 10.1016/j.autrev.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 33.Teixeira L, Mouthon L, Mahr A, Berezné A, Agard C, Mehrenberger M, et al. Mortality and risk factors of scleroderma renal crisis: A French retrospective study of 50 patients. Ann Rheum Dis. 2008;67:110–6. doi: 10.1136/ard.2006.066985. [DOI] [PubMed] [Google Scholar]
- 34.Wigley FM, Lima JA, Mayes M, McLain D, Chapin JL, Ward- Able C. The prevalence of undiagnosed pulmonary arterial hypertension in subjects with connective tissue disease at the secondary health care level of community-based rheumatologists (the UNCOVER study) Arthritis Rheum. 2005;52:2125–32. doi: 10.1002/art.21131. [DOI] [PubMed] [Google Scholar]
- 35.Fisher MR, Mathai SC, Champion HC, Girgis RE, Housten-Harris T, Hummers L, et al. Clinical differences between idiopathic and scleroderma-related pulmonary hypertension. Arthritis Rheum. 2006;54:3043–50. doi: 10.1002/art.22069. [DOI] [PubMed] [Google Scholar]
- 36.McLaughlin VV. Survival in patients with pulmonary arterial hypertension treated with first-line bosentan. Eur J Clin Invest. 2006;36(Suppl 3):10–5. doi: 10.1111/j.1365-2362.2006.01688.x. [DOI] [PubMed] [Google Scholar]
- 37.Girgis RE, Mathai SC, Krishnan JA, Wigley FM, Hassoun PM. Long-term outcome of bosentan treatment in idiopathic pulmonary arterial hypertension and pulmonary arterial hypertension associated with the scleroderma spectrum of diseases. J Heart Lung Transplant. 2005;24:1626–31. doi: 10.1016/j.healun.2004.12.113. [DOI] [PubMed] [Google Scholar]
- 38.Schachna L, Wigley FM, Chang B, White B, Wise RA, Gelber AC. Age and risk of pulmonary arterial hypertension in scleroderma. Chest. 2003;124:2098–104. doi: 10.1378/chest.124.6.2098. [DOI] [PubMed] [Google Scholar]
- 39.LeRoy EC. Systemic sclerosis. A vascular perspective. Rheum Dis Clin North Am. 1996;22:675–94. doi: 10.1016/s0889-857x(05)70295-7. [DOI] [PubMed] [Google Scholar]
- 40.Fleischmajer R, Perlish JS. Capillary alterations in scleroderma. J Am Acad Dermatol. 1980;2:161–70. doi: 10.1016/s0190-9622(80)80396-3. [DOI] [PubMed] [Google Scholar]
- 41.Sgonc R, Gruschwitz MS, Boeck G, Sepp N, Gruber J, Wick G. Endothelial cell apoptosis in systemic sclerosis is induced by antibody-dependent cell-mediated cytotoxicity via CD95. Arthritis Rheum. 2000;43:2550–62. doi: 10.1002/1529-0131(200011)43:11<2550::AID-ANR24>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 42.Cerinic MM, Valentini G, Sorano GG, D’Angelo S, Cuomo G, Fenu L, et al. Blood coagulation, fibrinolysis, and markers of endothelial dysfunction in systemic sclerosis. Semin Arthritis Rheum. 2003;32:285–95. doi: 10.1053/sarh.2002.50011. [DOI] [PubMed] [Google Scholar]
- 43.Altman RD, Medsger TA, Jr, Bloch DA, Michel BA. Predictors of survival in systemic sclerosis (scleroderma) Arthritis Rheum. 1991;34:403–13. doi: 10.1002/art.1780340405. [DOI] [PubMed] [Google Scholar]
- 44.Denton CP, Bickerstaff MC, Shiwen X, Carulli MT, Haskard DO, Dubois RM, et al. Serial circulating adhesion molecule levels reflect disease severity in systemic sclerosis. Br J Rheumatol. 1995;34:1048–54. doi: 10.1093/rheumatology/34.11.1048. [DOI] [PubMed] [Google Scholar]
- 45.Distler O, Del Rosso A, Giacomelli R, Cipriani P, Conforti ML, Guiducci S, et al. Angiogenic and angiostatic factors in systemic sclerosis: Increased levels of vascular endothelial growth factor are a feature of the earliest disease stages and are associated with the absence of fingertip ulcers. Arthritis Res. 2002;4:R11. doi: 10.1186/ar596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi JJ, Min DJ, Cho ML, Min SY, Kim SJ, Lee SS, et al. Elevated vascular endothelial growth factor in systemic sclerosis. J Rheumatol. 2003;30:1529–33. [PubMed] [Google Scholar]
- 47.Hebbar M, Peyrat JP, Hornez L, Hatron PY, Hachulla E, Devulder B. Increased concentrations of the circulating angiogenesis inhibitor endostatin in patients with systemic sclerosis. Arthritis Rheum. 2000;43:889–93. doi: 10.1002/1529-0131(200004)43:4<889::AID-ANR21>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 48.Okano Y, Steen VD, Medsger TA., Jr Autoantibody to U3 nucleolar ribonucleoprotein (fibrillarin) in patients with systemic sclerosis. Arthritis Rheum. 1992;35:95–100. doi: 10.1002/art.1780350114. [DOI] [PubMed] [Google Scholar]
- 49.Negi VS, Tripathy NK, Misra R, Nityanand S. Antiendothelial cell antibodies in scleroderma correlate with severe digital ischemia and pulmonary arterial hypertension. J Rheumatol. 1998;25:462–6. [PubMed] [Google Scholar]
- 50.Fritzler MJ, Hart DA, Wilson D, García-De La Torre I, Salazar- Páramo M, Vázquez-Del Mercado M, et al. Antibodies to fibrin bound tissue type plasminogen activator in systemic sclerosis. J Rheumatol. 1995;22:1688–93. [PubMed] [Google Scholar]
- 51.Morse JH, Barst RJ, Fotino M, Zhang Y, Flaster E, Gharavi AE, et al. Primary pulmonary hypertension, tissue plasminogen activator antibodies, and HLA-DQ7. Am J Respir Crit Care Med. 1997;155:274–8. doi: 10.1164/ajrccm.155.1.9001324. [DOI] [PubMed] [Google Scholar]
- 52.Grigolo B, Mazzetti I, Meliconi R, Bazzi S, Scorza R, Candela M, et al. Anti-topoisomerase II alpha autoantibodies in systemic sclerosis-association with pulmonary hypertension and HLA-B35. Clin Exp Immunol. 2000;121:539–43. doi: 10.1046/j.1365-2249.2000.01320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nicolls MR, Taraseviciene-Stewart L, Rai PR, Badesch DB, Voelkel NF. Autoimmunity and pulmonary hypertension: A perspective. Eur Respir J. 2005;26:1110–8. doi: 10.1183/09031936.05.00045705. [DOI] [PubMed] [Google Scholar]
- 54.Tamby MC, Chanseaud Y, Humbert M, Fermanian J, Guilpain P, Garcia-de-la-Peña-Lefebvre P, et al. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. Thorax. 2005;60:765–72. doi: 10.1136/thx.2004.029082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chizzolini C, Raschi E, Rezzonico R, Testoni C, Mallone R, Gabrielli A, et al. Autoantibodies to fibroblasts induce a proadhesive and proinflammatory fibroblast phenotype in patients with systemic sclerosis. Arthritis Rheum. 2002;46:1602–13. doi: 10.1002/art.10361. [DOI] [PubMed] [Google Scholar]
- 56.Tamby MC, Humbert M, Guilpain P, Servettaz A, Dupin N, Christner JJ, et al. Antibodies to fibroblasts in idiopathic and scleroderma-associated pulmonary hypertension. Eur Respir J. 2006;28:799–807. doi: 10.1183/09031936.06.00152705. [DOI] [PubMed] [Google Scholar]
- 57.Terrier B, Tamby MC, Camoin L, Guilpain P, Broussard C, Bussone G, et al. Identification of target antigens of antifibroblast antibodies in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;177:1128–34. doi: 10.1164/rccm.200707-1015OC. [DOI] [PubMed] [Google Scholar]
- 58.Sanchez O, Sitbon O, Jaïs X, Simonneau G, Humbert M. Immunosuppressive therapy in connective tissue diseases associated pulmonary arterial hypertension. Chest. 2006;130:182–9. doi: 10.1378/chest.130.1.182. [DOI] [PubMed] [Google Scholar]
- 59.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737–44. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, et al. International PPH Consortium. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–4. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 61.Newman JH, Trembath RC, Morse JA, Grunig E, Loyd JE, Adnot S, et al. Genetic basis of pulmonary arterial hypertension: Current understanding and future directions. J Am Coll Cardiol. 2004;43:33S–9S. doi: 10.1016/j.jacc.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 62.Koehler R, Grünig E, Pauciulo MW, Hoeper MM, Olschewski H, Wilkens H, et al. Low frequency of BMPR2 mutations in a German cohort of patients with sporadic idiopathic pulmonary arterial hypertension. J Med Genet. 2004;41:e127. doi: 10.1136/jmg.2004.023101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morse JH, Deng Z, Knowles JA. Genetic aspects of pulmonary arterial hypertension. Ann Med. 2001;33:596–603. doi: 10.3109/07853890109002105. [DOI] [PubMed] [Google Scholar]
- 64.Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2001;345:325–34. doi: 10.1056/NEJM200108023450503. [DOI] [PubMed] [Google Scholar]
- 65.Morse J, Barst R, Horn E, Cuervo N, Deng Z, Knowles J. Pulmonary hypertension in scleroderma spectrum of disease: Lack of bone morphogenetic protein receptor 2 mutations. J Rheumatol. 2002;29:2379–81. [PubMed] [Google Scholar]
- 66.Tew MB, Arnett FC, Reveille JD, Tan FK. Mutations of bone morphogenetic protein receptor type II are not found in patients with pulmonary hypertension and underlying connective tissue diseases. Arthritis Rheum. 2002;46:2829–30. doi: 10.1002/art.10487. [DOI] [PubMed] [Google Scholar]
- 67.Karrer S, Bosserhoff AK, Weiderer P, Distler O, Landthaler M, Szeimies RM, et al. The -2518 promotor polymorphism in the MCP-1 gene is associated with systemic sclerosis. J Invest Dermatol. 2005;124:92–8. doi: 10.1111/j.0022-202X.2004.23512.x. [DOI] [PubMed] [Google Scholar]
- 68.Tsuchiya N, Kuroki K, Fujimoto M, Murakami Y, Tedder TF, Tokunaga K, et al. Association of a functional CD19 polymorphism with susceptibility to systemic sclerosis. Arthritis Rheum. 2004;50:4002–7. doi: 10.1002/art.20674. [DOI] [PubMed] [Google Scholar]
- 69.Tolusso B, Fabris M, Caporali R, Cuomo G, Isola M, Soldano F, et al. −238 and +489 TNF-alpha along with TNF-RII gene polymorphisms associate with the diffuse phenotype in patients with Systemic Sclerosis. Immunol Lett. 2005;96:103–8. doi: 10.1016/j.imlet.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 70.Kawaguchi Y, Tochimoto A, Ichikawa N, Harigai M, Hara M, Kotake S, et al. Association of IL1A gene polymorphisms with susceptibility to and severity of systemic sclerosis in the Japanese population. Arthritis Rheum. 2003;48:186–92. doi: 10.1002/art.10736. [DOI] [PubMed] [Google Scholar]
- 71.Hutyrová B, Lukác J, Bosák V, Buc M, du Bois R, Petrek M. Interleukin 1alpha single-nucleotide polymorphism associated with systemic sclerosis. J Rheumatol. 2004;31:81–4. [PubMed] [Google Scholar]
- 72.Crilly A, Hamilton J, Clark CJ, Jardine A, Madhok R. Analysis of the 5’ flanking region of the interleukin 10 gene in patients with systemic sclerosis. Rheumatology (Oxford) 2003;42:1295–8. doi: 10.1093/rheumatology/keg420. [DOI] [PubMed] [Google Scholar]
- 73.Zhou X, Tan FK, Wang N, Xiong M, Maghidman S, Reveille JD, et al. Genome-wide association study for regions of systemic sclerosis susceptibility in a Choctaw Indian population with high disease prevalence. Arthritis Rheum. 2003;48:2585–92. doi: 10.1002/art.11220. [DOI] [PubMed] [Google Scholar]
- 74.Wipff J, Kahan A, Hachulla E, Sibilia J, Cabane J, Meyer O, et al. Association between an endoglin gene polymorphism and systemic sclerosis-related pulmonary arterial hypertension. Rheumatology (Oxford) 2007;46:622–5. doi: 10.1093/rheumatology/kel378. [DOI] [PubMed] [Google Scholar]
- 75.Schachna L, Wigley FM, Chang B, White B, Wise RA, Gelber AC. Age and risk of pulmonary arterial hypertension in scleroderma. Chest. 2003;124:2098–104. doi: 10.1378/chest.124.6.2098. [DOI] [PubMed] [Google Scholar]
- 76.Chang B, Schachna L, White B, Wigley FM, Wise RA. Natural history of mild-moderate pulmonary hypertension and the risk factors for severe pulmonary hypertension in scleroderma. J Rheumatol. 2006;33:269–74. [PubMed] [Google Scholar]
- 77.Steen V, Medsger TA., Jr Predictors of isolated pulmonary hypertension in patients with systemic sclerosis and limited cutaneous involvement. Arthritis Rheum. 2003;48:516–22. doi: 10.1002/art.10775. [DOI] [PubMed] [Google Scholar]
- 78.Allanore Y, Borderie D, Avouac J, Zerkak D, Meune C, Hachulla E, et al. High N-terminal pro-brain natriuretic peptide levels and low diffusing capacity for carbon monoxide as independent predictors of the occurrence of precapillary pulmonary arterial hypertension in patients with systemic sclerosis. Arthritis Rheum. 2008;58:284–91. doi: 10.1002/art.23187. [DOI] [PubMed] [Google Scholar]
- 79.Ghanem MK, Makhlouf HA, Agmy GR, Imam HM, Fouad DA. Evaluation of recently validated non- invasive formula using basic lung functions as new screening tool for pulmonary hypertension in idiopathic pulmonary fibrosis patients. Ann Thorac Med. 2009;4:187–96. doi: 10.4103/1817-1737.56013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hsiao SH, Lee CY, Chang SM, Lin SK, Liu CP. Right heart function in scleroderma: Insights from myocardial Doppler tissue imaging. J Am Soc Echocardiogr. 2006;19:507–14. doi: 10.1016/j.echo.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 81.Lee CY, Chang SM, Hsiao SH, Tseng JC, Lin SK, Liu CP. Right heart function and scleroderma: Insights from tricuspid annular plane systolic excursion. Echocardiography. 2007;24:118–25. doi: 10.1111/j.1540-8175.2007.00365.x. [DOI] [PubMed] [Google Scholar]
- 82.Meune C, Avouac J, Wahbi K, Cabanes L, Wipff J, Mouthon L, et al. Cardiac involvement in systemic sclerosis assessed by tissuedoppler echocardiography during routine care: A controlled study of 100 consecutive patients. Arthritis Rheum. 2008;58:1803–9. doi: 10.1002/art.23463. [DOI] [PubMed] [Google Scholar]
- 83.Overbeek MJ, Lankhaar JW, Westerhof N, Voskuyl AE, Boonstra A, Bronzwaer JG, et al. Right ventricular contractility in systemic sclerosis-associated and idiopathic pulmonary arterial hypertension. Eur Respir J. 2008;31:1160–6. doi: 10.1183/09031936.00135407. [DOI] [PubMed] [Google Scholar]
- 84.Williams MH, Handler CE, Akram R, Smith CJ, Das C, Smee J, et al. Role of N-terminal brain natriuretic peptide (N-TproBNP) in scleroderma-associated pulmonary arterial hypertension. Eur Heart J. 2006;27:1485–94. doi: 10.1093/eurheartj/ehi891. [DOI] [PubMed] [Google Scholar]
- 85.Forfia PR, Mathai SC, Fisher MR, Housten-Harris T, Hemnes AR, Champion HC, et al. Hyponatremia predicts right heart failure and poor survival in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;177:1364–9. doi: 10.1164/rccm.200712-1876OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Williams MH, Das C, Handler CE, Akram MR, Davar J, Denton CP, et al. Systemic sclerosis associated pulmonary hypertension: Improved survival in the current era. Heart. 2006;92:926–32. doi: 10.1136/hrt.2005.069484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chin KM, Kim NH, Rubin LJ. The right ventricle in pulmonary hypertension. Coron Artery Dis. 2005;16:13–8. doi: 10.1097/00019501-200502000-00003. [DOI] [PubMed] [Google Scholar]
- 88.Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, et al. Right ventricular function and failure: Report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation. 2006;114:1883–91. doi: 10.1161/CIRCULATIONAHA.106.632208. [DOI] [PubMed] [Google Scholar]
- 89.Tei C, Nishimura RA, Seward JB, Tajik AJ. Noninvasive Doppler-derived myocardial performance index: Correlation with simultaneous measurements of cardiac catheterization measurements. J Am Soc Echocardiogr. 1997;10:169–78. doi: 10.1016/s0894-7317(97)70090-7. [DOI] [PubMed] [Google Scholar]
- 90.Vonk MC, Sander MH, van den Hoogen FH, van Riel PL, Verheugt FW, van Dijk AP. Right ventricle Tei-index: A tool to increase the accuracy of non-invasive detection of pulmonary arterial hypertension in connective tissue diseases. Eur J Echocardiogr. 2007;8:317–21. doi: 10.1016/j.euje.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 91.Gupta S, Khan F, Shapiro M, Weeks SG, Litwin SE, Michaels AD. The associations between tricuspid annular plane systolic excursion (TAPSE), ventricular dyssynchrony, and ventricular interaction in heart failure patients. Eur J Echocardiogr. 2008;9:766–71. doi: 10.1093/ejechocard/jen147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Forfia PR, Fisher MR, Mathai SC, Housten-Harris T, Hemnes AR, Borlaug BA, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med. 2006;174:1034–41. doi: 10.1164/rccm.200604-547OC. [DOI] [PubMed] [Google Scholar]
- 93.Gondi S, Dokainish H. Right ventricular tissue Doppler and strain imaging: Ready for clinical use? Echocardiography. 2007;24:522–32. doi: 10.1111/j.1540-8175.2007.00430.x. [DOI] [PubMed] [Google Scholar]
- 94.Meune C, Allanore Y, Devaux JY, Dessault O, Duboc D, Weber S, et al. High prevalence of right ventricular systolic dysfunction in early systemic sclerosis. J Rheumatol. 2004;31:1941–5. [PubMed] [Google Scholar]
- 95.Steen VD, Follansbee WP, Conte CG, Medsger TA., Jr Thallium perfusion defects predict subsequent cardiac dysfunction in patients with systemic sclerosis. Arthritis Rheum. 1996;39:677–81. doi: 10.1002/art.1780390421. [DOI] [PubMed] [Google Scholar]
- 96.Bezante GP, Rollando D, Sessarego M, Panico N, Setti M, Filaci G, et al. Cardiac magnetic resonance imaging detects subclinical right ventricular impairment in systemic sclerosis. J Rheumatol. 2007;34:2431–7. [PubMed] [Google Scholar]
- 97.Hagger D, Condliffe R, Woodhouse N, Elliot CA, Armstrong IJ, Davies C, et al. Ventricular mass index correlates with pulmonary artery pressure and predicts survival in suspected systemic sclerosis-associated pulmonary arterial hypertension. Rheumatology (Oxford) 2009;48:1137–42. doi: 10.1093/rheumatology/kep187. [DOI] [PubMed] [Google Scholar]
- 98.Wilkins MR, Paul GA, Strange JW, Tunariu N, Gin-Sing W, Banya WA, et al. Sildenafil versus endothelin receptor antagonist for pulmonary hypertension (SERAPH) study. Am J Respir Crit Care Med. 2005;171:1292–7. doi: 10.1164/rccm.200410-1411OC. [DOI] [PubMed] [Google Scholar]
- 99.Koh ET, Lee P, Gladman DD, Abu-Shakra M. Pulmonary hypertension in systemic sclerosis: An analysis of 17 patients. Br J Rheumatol. 1996;35:989–93. doi: 10.1093/rheumatology/35.10.989. [DOI] [PubMed] [Google Scholar]
- 100.McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: The impact of epoprostenol therapy. Circulation. 2002;106:1477–82. doi: 10.1161/01.cir.0000029100.82385.58. [DOI] [PubMed] [Google Scholar]
- 101.Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972-2002. Ann Rheum Dis. 2007;66:940–4. doi: 10.1136/ard.2006.066068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pope J. An update in pulmonary hypertension in systemic lupus erythematosus — do we need to know about it? Lupus. 2008;17:274–7. doi: 10.1177/0961203307087188. [DOI] [PubMed] [Google Scholar]
- 103.Haas C. Pulmonary hypertension associated with systemic lupus erythematosus. Bull Acad Natl Med. 2004;188:985–97. 997. [PubMed] [Google Scholar]
- 104.Sullivan WD, Hurst DJ, Harmon CE, Esther JH, Agia GA, Maltby JD, et al. A prospective evaluation emphasizing pulmonary involvement in patients with mixed connective tissue disease. Medicine (Baltimore) 1984;63:92–107. doi: 10.1097/00005792-198403000-00003. [DOI] [PubMed] [Google Scholar]
- 105.Launay D, Hachulla E, Hatron PY, Jais X, Simonneau G, Humbert M. Pulmonary arterial hypertension: A rare complication of primary Sjögren syndrome: Report of 9 new cases and review of the literature. Medicine (Baltimore) 2007;86:299–315. doi: 10.1097/MD.0b013e3181579781. [DOI] [PubMed] [Google Scholar]
- 106.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333:214–21. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- 107.Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159:1925–32. doi: 10.1164/ajrccm.159.6.9804054. [DOI] [PubMed] [Google Scholar]
- 108.Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328:1732–9. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- 109.Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327:76–81. doi: 10.1056/NEJM199207093270203. [DOI] [PubMed] [Google Scholar]
- 110.Sitbon O, Humbert M, Jaïs X, Ioos V, Hamid AM, Provencher S, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111:3105–11. doi: 10.1161/CIRCULATIONAHA.104.488486. [DOI] [PubMed] [Google Scholar]
- 111.Vane JR, Anggård EE, Botting RM. Regulatory functions of the vascular endothelium. N Engl J Med. 1990;323:27–36. doi: 10.1056/NEJM199007053230106. [DOI] [PubMed] [Google Scholar]
- 112.Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334:296–301. doi: 10.1056/NEJM199602013340504. [DOI] [PubMed] [Google Scholar]
- 113.McLaughlin VV, Genthner DE, Panella MM, Rich S. Reduction in pulmonary vascular resistance with long-term epoprostenol (prostacyclin) therapy in primary pulmonary hypertension. N Engl J Med. 1998;338:273–7. doi: 10.1056/NEJM199801293380501. [DOI] [PubMed] [Google Scholar]
- 114.Rubin LJ, Mendoza J, Hood M, McGoon M, Barst R, Williams WB, et al. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann Intern Med. 1990;112:485–91. doi: 10.7326/0003-4819-112-7-485. [DOI] [PubMed] [Google Scholar]
- 115.Badesch DB, Tapson VF, McGoon MD, Brundage BH, Rubin LJ, Wigley FM, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med. 2000;132:425–34. doi: 10.7326/0003-4819-132-6-200003210-00002. [DOI] [PubMed] [Google Scholar]
- 116.Badesch DB, McGoon MD, Barst RJ, Tapson VF, Rubin LJ, Wigley FM, et al. Longterm survival among patients with scleroderma-associated pulmonary arterial hypertension treated with intravenous epoprostenol. J Rheumatol. 2009;36:2244–9. doi: 10.3899/jrheum.081277. [DOI] [PubMed] [Google Scholar]
- 117.Simonneau G, Barst RJ, Galie N, Naeije R, Rich S, Bourge RC, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: A double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165:800–4. doi: 10.1164/ajrccm.165.6.2106079. [DOI] [PubMed] [Google Scholar]
- 118.Tapson VF, Gomberg-Maitland M, McLaughlin VV, Benza RL, Widlitz AC, Krichman A, et al. Safety and efficacy of IV treprostinil for pulmonary arterial hypertension: A prospective, multicenter, open-label, 12-week trial. Chest. 2006;129:683–8. doi: 10.1378/chest.129.3.683. [DOI] [PubMed] [Google Scholar]
- 119.Farber HW, Graven KK, Kokolski G, Korn JH. Pulmonary edema during acute infusion of epoprostenol in a patient with pulmonary hypertension and limited scleroderma. J Rheumatol. 1999;26:1195–6. [PubMed] [Google Scholar]
- 120.Palmer SM, Robinson LJ, Wang A, Gossage JR, Bashore T, Tapson VF. Massive pulmonary edema and death after prostacyclin infusion in a patient with pulmonary veno-occlusive disease. Chest. 1998;113:237–40. doi: 10.1378/chest.113.1.237. [DOI] [PubMed] [Google Scholar]
- 121.Galiè N, Manes A, Branzi A. Emerging medical therapies for pulmonary arterial hypertension. Prog Cardiovasc Dis. 2002;45:213–24. doi: 10.1053/pcad.2002.130160. [DOI] [PubMed] [Google Scholar]
- 122.Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson VF, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: A randomized placebo-controlled study. Lancet. 2001;358:1119–23. doi: 10.1016/S0140-6736(01)06250-X. [DOI] [PubMed] [Google Scholar]
- 123.Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903. doi: 10.1056/NEJMoa012212. [DOI] [PubMed] [Google Scholar]
- 124.Denton CP, Humbert M, Rubin L, Black CM. Bosentan treatment for pulmonary arterial hypertension related to connective tissue disease: A subgroup analysis of the pivotal clinical trials and their open-label extensions. Ann Rheum Dis. 2006;65:1336–40. doi: 10.1136/ard.2005.048967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Girgis RE, Mathai SC, Krishnan JA, Wigley FM, Hassoun PM. Long-term outcome of bosentan treatment in idiopathic pulmonary arterial hypertension and pulmonary arterial hypertension associated with the scleroderma spectrum of diseases. J Heart Lung Transplant. 2005;24:1626–31. doi: 10.1016/j.healun.2004.12.113. [DOI] [PubMed] [Google Scholar]
- 126.Iannone F, Riccardi MT, Guiducci S, Bizzoca R, Cinelli M, Matucci- Cerinic M, et al. Bosentan regulates the expression of adhesion molecules on circulating T cells and serum soluble adhesion molecules in systemic sclerosis-associated pulmonary arterial hypertension. Ann Rheum Dis. 2008;67:1121–6. doi: 10.1136/ard.2007.080424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jain M, Varga J. Bosentan for the treatment of systemic sclerosis-associated pulmonary arterial hypertension, pulmonary fibrosis and digital ulcers. Expert Opin Pharmacother. 2006;7:1487–501. doi: 10.1517/14656566.7.11.1487. [DOI] [PubMed] [Google Scholar]
- 128.Galiè N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: Results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117:3010–9. doi: 10.1161/CIRCULATIONAHA.107.742510. [DOI] [PubMed] [Google Scholar]
- 129.Galiè N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–57. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 130.Badesch DB, Hill NS, Burgess G, Rubin LJ, Barst RJ, Galiè N, et al. Sildenafil for pulmonary arterial hypertension associated with connective tissue disease. J Rheumatol. 2007;34:2417–22. [PubMed] [Google Scholar]
- 131.Galiè N, Brundage BH, Ghofrani HA, Oudiz RJ, Simonneau G, Safdar Z, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119:2894–903. doi: 10.1161/CIRCULATIONAHA.108.839274. [DOI] [PubMed] [Google Scholar]
- 132.McLaughlin VV, Oudiz RJ, Frost A, Tapson VF, Murali S, Channick RN, et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174:1257–63. doi: 10.1164/rccm.200603-358OC. [DOI] [PubMed] [Google Scholar]
- 133.Hoeper MM, Faulenbach C, Golpon H, Winkler J, Welte T, Niedermeyer J. Combination therapy with bosentan and sildenafil in idiopathic pulmonary arterial hypertension. Eur Respir J. 2004;24:1007–10. doi: 10.1183/09031936.04.00051104. [DOI] [PubMed] [Google Scholar]
- 134.Simonneau G, Rubin LJ, Galiè N, Barst RJ, Fleming TR, Frost AE, et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: A randomized trial. Ann Intern Med. 2008;149:521–30. doi: 10.7326/0003-4819-149-8-200810210-00004. [DOI] [PubMed] [Google Scholar]
- 135.Mathai SC, Girgis RE, Fisher MR, Champion HC, Housten-Harris T, Zaiman A, et al. Addition of sildenafil to bosentan monotherapy in pulmonary arterial hypertension. Eur Respir J. 2007;29:469–75. doi: 10.1183/09031936.00081706. [DOI] [PubMed] [Google Scholar]
- 136.Fuster V, Steele PM, Edwards WD, Gersh BJ, McGoon MD, Frye RL. Primary pulmonary hypertension: Natural history and the importance of thrombosis. Circulation. 1984;70:580–7. doi: 10.1161/01.cir.70.4.580. [DOI] [PubMed] [Google Scholar]
- 137.Adnot S. Lessons learned from cancer may help in the treatment of pulmonary hypertension. J Clin Invest. 2005;115:1461–3. doi: 10.1172/JCI25399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115:2811–21. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Moreno-Vinasco L, Gomberg-Maitland M, Maitland ML, Desai AA, Singleton PA, Sammani S, et al. Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol Genomics. 2008;33:278–91. doi: 10.1152/physiolgenomics.00169.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galié N, et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: Results of the randomized IMPRES study. Circulation. 2013;127:1128–38. doi: 10.1161/CIRCULATIONAHA.112.000765. [DOI] [PubMed] [Google Scholar]
- 141.Grigoryev DN, Mathai SC, Fisher MR, Girgis RE, Zaiman AL, Housten-Harris T, et al. Identification of candidate genes in scleroderma-related pulmonary arterial hypertension. Transl Res. 2008;151:197–207. doi: 10.1016/j.trsl.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ten Freyhaus H, Dumitrescu D, Bovenschulte H, Erdmann E, Rosenkranz S. Significant improvement of right ventricular function by imatinib mesylate in scleroderma-associated pulmonary arterial hypertension. Clin Res Cardiol. 2009;98:265–7. doi: 10.1007/s00392-009-0752-3. [DOI] [PubMed] [Google Scholar]
- 143.Schachna L, Medsger TA, Jr, Dauber JH, Wigley FM, Braunstein NA, White B, et al. Lung transplantation in scleroderma compared with idiopathic pulmonary fibrosis and idiopathic pulmonary arterial hypertension. Arthritis Rheum. 2006;54:3954–61. doi: 10.1002/art.22264. [DOI] [PubMed] [Google Scholar]
- 144.Saggar R, Khanna D, Furst DE, Belperio JA, Park GS, Weigt SS, et al. Systemic sclerosis and bilateral lung transplantation: A single centre experience. Eur Respir J. 2010;36:893–900. doi: 10.1183/09031936.00139809. [DOI] [PMC free article] [PubMed] [Google Scholar]