

Abstract
Hereditary hemoglobin disorders affecting the globin chain synthesis namely thalassemia syndromes and sickle cell disease (SCD) are the most common genetic disorders in human. Around 7% of the world population carries genes for these disorders, mainly the Mediterranean Basin, Middle and Far East, and Sub-Saharan Africa. An estimated 30 million people worldwide are living with sickle cell disease, while 60-80 million carry beta thalassemia trait. About 400,000 children are born with severe hemoglobinopathies each year.
Cardiovascular complications of hemoglobinopathies include left and right ventricular (RV) dysfunction, arrhythmias, pericarditis, myocarditis, valvular heart disease, myocardial ischemia, and notably pulmonary hypertension (PH).
Because of a unique pathophysiology, pulmonary hypertension associated with hemolytic disorders was moved from WHO group I to group V PH diseases. Treatment strategies are also unique and include blood transfusion, iron chelation, hydroxyurea, and oxygen therapy. The role of PH-specific agents has not been established.
Keywords: Hemolysis, pulmonary hypertension, sickle cell anemia, thalassemia, Saudi association for pulmonary hypertension guidelines
Hereditary hemoglobin disorders affecting the globin chain synthesis namely thalassemia syndromes and sickle cell disease (SCD) are the most common genetic disorders in human. Around 7% of the world population carries genes for these disorders, mainly the Mediterranean Basin, Middle and Far East, and Sub-Saharan Africa. An estimated 30 million people worldwide are living with SCD, while 60-80 million carry beta thalassemia trait. About 400,000 children are born with severe hemoglobinopathies each year. Cardiovascular complications of hemoglobinopathies include left and right ventricular (RV) dysfunction, arrhythmias, pericarditis, myocarditis, valvular heart disease, myocardial ischemia, and notably pulmonary hypertension (PH). PH associated with hemoglobinopathies is the main cause of morbidity and mortality in this group of the population.[1,2,3,4,5,6,7,8,9,10] Several studies using tricuspid-valve regurgitant velocity (TRV) on Doppler echocardiography of at least 2.5 m/s to diagnose PH have put the prevalence of PH at 20-30% in SCD and 10-75% in thalassemia syndromes. In a series of 65 patients with SCD in a tertiary care Saudi hospital, echocardiographic evidence of PH was present in 38% of patients.[11] The prevalence of PH in other hemolytic disorders, such as hereditary spherocytosis and stomatocytosis, paroxysmal nocturnal hemoglobinuria, microangiopathic hemolytic anemia, and pyruvate kinase deficiency are not well-studied [Table 1]. It is believed that hemolytic disorders both acute and chronic are associated with PH.[10,12,13] In a longitudinal study, Ataga et al. followed patients with SCD whose initial screening had normal TRV over 3 years. A repeat echocardiography showed that 13% had developed echocardiographic evidence of PH suggesting incidence of about 4%/year.[14]
Table 1.
Hemolytic disorders associated with PH

Although studies using TRV on Doppler echocardiography for diagnosis of PH have shown a high prevalence rate in patient with hemoglobinopathies, there appears to be a significant false positive rate as shown by Parent et al., who studies 398 patients with SCD, of which 109 patients (27%) had PH based on TRV >2.5 m/s. Ninety-six of these patients underwent right heart catheterization (RHC). Only 6% of these patients were found to have PH based on hemodynamic criteria.[15] Furthermore, RHC is not only an important tool in diagnosing PH, it also distinguishes precapillary PH from postcapillary PH, which is caused by left heart disease and frequently seen in hemolytic anemia.[16]
Pulmonary hypertension associated with chronic hemolytic anemia was classified as Group 1 with a subcategory of associated pulmonary arterial hypertension in the 4th PH World Congress.[17] However, a new proposed NICE (5th World Congress) classification has moved this category of PH to Group 5 due to its complex nature, as shown in Table 2.[18]
Table 2.
WHO Group 5

Pathophysiology
The pathophysiology of PH in SCD and other chronic hemolytic disorders is complex. The common link for development of PH in all hemolytic disorders is probably the chronic hemolysis, as shown in Figure 1. Several studies have shown a strong association between the severity of hemolysis and development of PH.[19,20,21] Free hemoglobin released during hemolysis scavenges the intrinsic vasodilator nitric oxide (NO) and red cell breakdown releases arginase, which is an enzyme responsible for depletion of L-arginine, a substrate for NO synthesis. NO is one of the most potent vasodilators known and is an essential tool for vascular homeostasis. It plays an important role in the maintenance of vasomotor tone, limits platelet aggregation and ischemia-reperfusion injury, modulates endothelial proliferation, and has anti-inflammatory properties. Inactivation and reduced synthesis of NO leads to impaired NO dependent vasodilatation of pulmonary vasculature. Arginase is also responsible for altered metabolism of L-arginine to L-ornitine resulting in the synthesis of L-proline, which contributes to the smooth muscle proliferation and collagen synthesis leading to vascular remodeling and intimal thickening.[10,13,19,22,23,24,25]
Figure 1.
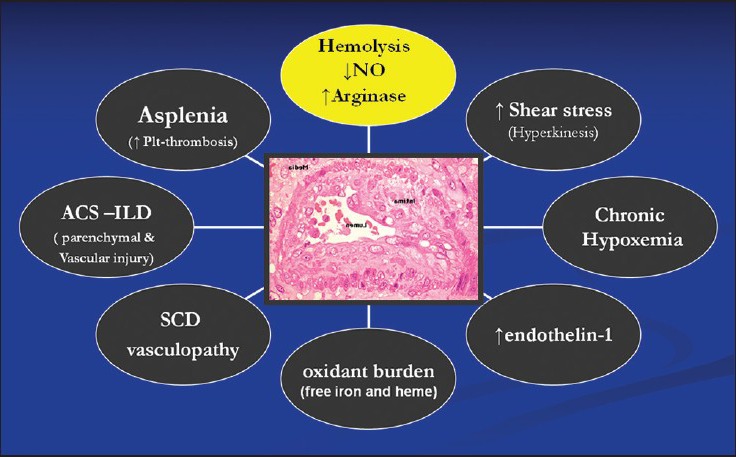
Pathophysiology of pulmonary hypertension in hemoglobinopathies
There is an increased risk of thrombosis as factors released during red cell destruction leads to platelet activation, thrombin generation, and tissue factors activation leading to obliterative pulmonary vasculopathy. Hypercoagulable state develops from a variety of causes in these patients, including red cell precoagulant surface, genetic coagulation defects, splenectomy, endothelial dysfunction, and vaculopathy. Thromboembolic complications including pulmonary emboli and in situ thrombi have been reported in patients with hemoglobinopathy.[25,26,27,28,29,30,31] Since many patients with hemoglobinopathies have asplenia either due to auto or surgical splenectomy, the role of splenectomy as a risk factor for development of PH is well-established. Spleen plays an important role in removal of senescent and damaged red cells, and hence, its absence leads to platelet activation promoting microthromosis of pulmonary circulation and red cell adhesion to the capillary endothelial lining leading to vascular obliteration.[32,33,34,35] There is a role of endothelin pathway in development of PH in hemolytic disorders as hemolysis induces increased endothelin-1 mediated responses leading to pulmonary vasoconstriction. It has been shown that plasma endothelin-1 levels are increased in patients with SCD both in steady state and in sickle cell crisis.[36] Red cells in patients with hemoglobinopathies have increased concentrations of reactive oxygen species, such as superoxide, which can disrupt NO hemostasis by scavenging NO in pulmonary vascular system due to oxidative stress. As the glutathione buffering system is overwhelmed by oxidative stress, the red cells in these patients are more prone to hemolysis. It has been shown that erythrocyte glutathione depletion is associated with severity of PH in SCD.[37,38,39,40] Pulmonary complications in hemoglobinopathies, especially SCD, are linked to dysregulated arginine metabolism. Compromised oxygenation leads to increased sickling and vice versa. Chronic lung injury leads to pulmonary fibrosis and chronic hypoxemia, which in turn can cause increased pulmonary vascular resistance (PVR) and PH. There is probably no strong association between the number of episodes of acute chest syndrome and development of PH, as it occurs with equal prevalence in patients with thalassemia who do not develop acute chest syndrome.[19,41] Pulmonary venous hypertension due to left heart dysfunction is not uncommon in patients with hemoglobinopathies. Even in well-treated patients with thalassemia major, 7% were found to have systolic dysfunction, while 38% had diastolic dysfunction. In addition, mitral valve disease is much more common in these patients than in the normal population. Left heart disease in hemoglobinopaties is due to multiple factors, including iron overload, high output cardiac state, myocarditis, and elastic tissue defect. Iron overload not only lead to left heart dysfunction, it also causes liver disease contributing further to the development of PH due to liver cirrhosis.[6,42,43,44] In short, the pathobiology of PH in hemolytic disorders is a rainbow of many colors. Mechanisms like NO depletion, dysregulated arginine metabolism, oxidative stress and hypercoagulable state result in pulmonary vasoconstriction, endothelial proliferation and hyperplasia, and in situ thrombi. However, the development of plexiform pulmonary arteriopathy in this group of patients has been recently challenged, as most reported plexiform lesions in old studies where indeed organized thrombi.
In reality, PH in hemolytic disorders is mainly a combination of precapillary and postcapillary PH and while a small proportion of patients have hypoxia-induced PH and thromboembolic PH, as shown in Figure 2.[19,45,46,47,48,49]
Figure 2.
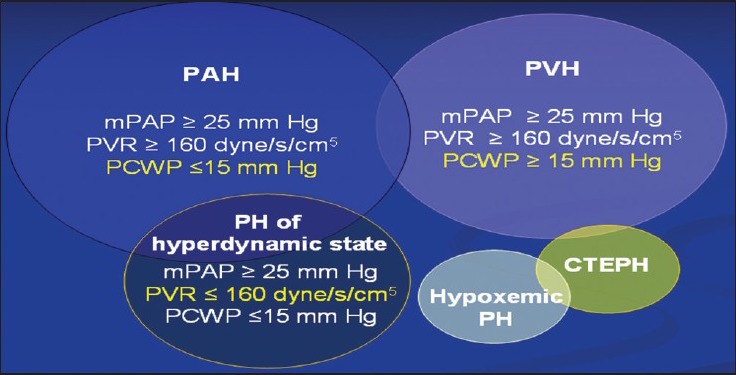
Different types of pulmonary hypertension in patients with hemolytic disorders
Clinical Features and Diagnosis
Dyspnea, which is a typical symptom associated with PH, is very common in patients with hemoglobinopathies due to anemia. It is very important to have an index of suspicion in these patients and perform screening echocardiography. Even a mild degree of PH in these patients is poorly tolerated due to chronic anemia, which results in very high cardiac output usually in the range of 10 L/min resulting in significant morbidity and possibly mortality.
Patients with SCD and PH (mean pulmonary artery pressure [mPAP] of 36 ± 1.5 mmHg) when compared with patients with normal PAP are found to have walked significantly lower distance on 6-min walk test (435 ± 31 vs. 320 ± 20 m; P = 0.002) and had lower maximum oxygen uptake (50 ± 3% vs. 41 ± 2% of normality; P = 0.02).[16] PVR in patients with SCD sharply rises with exercise, suggesting that pulmonary vascular disease contributes to functional impairment in this group of patients.[49] Patients with hemoglobinopathies are more symptomatic as compared to patients with idiopathic pulmonary artery hypertension (IPAH) despite lower mPAP and PVR. The workup of exercise intolerance should also include aggressive search for other conditions contributing to PH such as chronic liver disease, HIV, iron overload, sleep apnea, and thromboembolism.[50]
Doppler echocardiography is an excellent screening tool for cardiovascular complications in patients with hemoglobinopathies.[24] It may overestimate PAP resulting in false positive results especially in patients with hemoglobinopathies where several factors lead to high output state. RHC in patients with hemoglobinopathies is recommended in making a diagnosis of precapillary PH defined by mPAP ≥25 mmHg and pulmonary arterial wedge pressure (PAWP) <15 mmHg. In a multi-center study mentioned earlier, the prevalence of PH in SCD by RHC was 6%, while only about 2% patients had true precapillary PH with a PAWP of 15 mmHg or less. The positive predictive value of echocardiography for the detection of PH was only 25%.[15] A TRV of 2.5 m/s or higher on Doppler echocardiogram is a strong predictor of death in patients with SCD with about 40% mortality risk within 3 years of diagnosis.[51] Patients with SCD have a high-risk of death with mild elevation of pulmonary pressure as compared to patients with IPAH.[52] Several reports support the use of TRV of 2.5 m/s as a good threshold for intervention. About 10% of patients with SCD have TRV >3 m/s and a majority of these have mean PAP >25 mmHg on RHC.[13,16,19] Evidence of diastolic dysfunction on echocardiography is common in these patients, which is an independent mortality risk in patient with SCD. In a cohort of 141 patients with SCD, Sachdev et al. have reported a relative risk of death of 4.8 (95% confidence interval [CI], 1.9-12.1), whereas relative risk of death when both PH and diastolic dysfunction are present was 12.0 (95% CI, 3.8-38.1).[53] The N-terminal pro-brain natriuretic peptide (NT-pro-BNP) has been found to have a correlation with the severity of PAP and RV dysfunction in IPAH.[52] Levels of NT-pro BNP are also found to be higher in patients with sickle cell-induced PH and correlate directly with TRV.[54] An NT-pro-BNP level of 160 pg/mL or higher has a 78% positive predictive value for the diagnosis of PH and is an independent predictor of mortality with a risk ratio of 5.1 (95% CI, 2.1-12.5).[41]
Treatment
There are no specific treatment guidelines on the management of patients with hemoglobinopathy associated PH. It is essential that treatment of primary hemoglobinopathy should be maximized, hypoxia should be corrected by chronic oxygen use and associated complications like cardiopulmonary conditions should be treated appropriately. There are two aspects of management in these patients, mainly hemoglobinopathy specific treatment and PH-specific treatment. Hemoglobinopathy specific treatment includes blood transfusion, iron chelation and hydroxyurea, while PH-specific therapy comprises of anticoagulation, diuretics, digoxin, oxygen, and PAH specific vasodilator agents. Chronic blood transfusion in patients with SCD has been shown to reduce the synthesis of sickle cells and its associated complications including pulmonary events and central nervous system vasculopathy.[55,56] Aggressive transfusion therapy and iron chelation in patients with thalassemia major has been shown to completely prevent the development of PH in one study.[42] Although transfusion therapy lower plasma free hemoglobin, which is an important trigger in development of PH in these patients, there are no prospective studies evaluating its efficacy to decrease PH in this group of patients. There are few case reports of improvement of TRV with transfusion therapy;[21] and a retrospective study of SCD patients who were transfused has shown significantly lower TRV as compared to those who were not transfused.[57] The aim of transfusion therapy is to maintain Hb level of ≥8 g/dl and HbS level of <40%.
Although studies have not shown a conclusive relationship between hydroxyurea use and reduction of TRV in patients with SCD,[19,41,58,59] hydroxyurea is a useful treatment tool. It helps to reduce hemolysis, increase hemoglobin, induces NO in endothelial cells, improves clinical symptoms, reduces the requirement for blood transfusion, and prevent acute episodes that exacerbate PH and potentially decrease overall mortality.[60,61] Two small case series have shown reduction in TRV with the use of hydroxyurea.[62,63] In a recent study of 584 patients with thalassemia intermedia, optimum therapy involving blood transfusion, chelation therapy and hydroxyurea has found that this strategy is protective against development of PH.[64]
Anticoagulation in IPAH has been shown to decrease mortality,[65,66,67] however, the potential benefit of warfarin in hemoglobinopathy associated PH has to be weighed against the risk of hemorrhagic complications. It has been recommended that patients with severe SCD associated PH may be considered for oral anticoagulation if there is no contraindication. However, the conclusive answer about this question will be answered once the ongoing randomized clinical trial is completed.
Due to the old belief that the pathobiological pathways of plexiform lesion similar to PAH also play a role in hemoglobinopathy associated PH, vasodilator therapies including prostanoids, endothelin receptor antagonists, and phosphodiesterase-5 inhibitors have been tried in these patients. As NO depletion is a critical factor in the development of PH in SCD patients, chronic NO administration would be an attractive method of therapy. However, chronic NO treatment is not practical for general use as the delivery system is very complicated and expensive.[47,68] L-arginine improves NO bioavailability and has been shown to decrease systolic PAP in patients with SCD.[69,70,71] Use of L-carnitine for 3 months in a case series of 32 patients with beta thalassemia major showed a significant reduction in systolic PAP.[72]
Targeted PH-specific therapy has certain setbacks in patients with hemoglobinopathies, especially SCD. In addition to their general adverse profile, there are specific side-effects that can interfere negatively with the pathophysiological aspects of SCD and other hemoglobinopathies [Table 3].
Table 3.
Adverse effects of PH-specific agents in SCD

Phosphodiesterase-5 inhibitors have been tried in few small case series with positive results. Derchi et al. have reported 7 patients, 4 with thalassemia intermedia, 2 with thalassemia major and 1 with sickle thalassemia. Treatment with sildenafil in these patients who had severe PH improved 6-min walk distance (6MWD) and the modified NYHA functional class and decreased TRV.[73] Another small study of sildenafil use in 12 patients with SCD associated PH has shown that sildenafil use for a mean of 6 months was resulted in improved 6MWD, decreased TRV, mPAP and NT-pro BNP levels.[74] Another case series of 14 patients in which sildenafil and L-arginine was used has shown a significant improvement in 6MWD and decreased TRV with sildenafil, but not L-arginine.[75] A multi-center randomized double-blind trial (walk-PHaSST trial) was terminated in July 7, 2009 because of the increased occurrence of painful crises in the sildenafil arm and the lack of benefit from this agent in patients who completed the study. Hence, it is recommended that phosphodiesterase-5 inhibitors should not be considered as first-line agents for the treatments of PH in patients with SCD in the light of walk-PHaSST study.
A clinical trial of use of endothelin receptor antagonists (bosentan or ambrisentan) as monotherapy or in combination with sildenafil in 14 patients with SCD-PH showed improvement in 6MWD and a reduction in TRV, mPAP, and NT-pro-BNP levels.[76]
A randomized, double-blind, multi-center trial to assess the efficacy, safety, and tolerability of bosentan in patients with symptomatic pulmonary arterial hypertension associated with SCD (ASSET-1 and ASSET-2) studies were prematurely terminated because of slow site activation and patient recruitment.
ASSET-1 was for patients with precapillary PH while ASSET-2 for pulmonary venous hypertension. In a limited sample of 26 patients, bosentan use for 16 weeks was well tolerated and there was a nonsignificant increase in cardiac output and decrease in PVR. Because of the limited sample size, efficacy endpoints were not formally analyzed.[77] Acute administration of intravenous eposprostenol in patients with SCD-PH has shown to decrease PVR from 271 dyne/s/c5 to 170 dyne/s/c5 and to increase cardiac output from 7.1 l/min to 9.1 l/min. There are no studies on chronic use of prostanoid analogues in this group of patients.[52]
Conclusion
Cardiopulmonary complications associated with SCD and other hemoglobinopathies are major causes of morbidity and mortality. PH in these patients has a complex and multifactorial pathophysiology. Many disease-related mechanisms, especially hemolysis, play an important role in the development of PH. Treatment strategies include disease specific modalities, such as blood transfusion, iron chelation, hydroxyurea, and oxygen therapy that may prevent the development and progression of PH. The role of PH-specific agents is not established. Threshold of screening for PH in this group of patients should be low, so that the treatment can be optimized in order to improve the quality of life and prolong survival.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Alabdulaali MK. Sickle cell disease patients in eastern province of Saudi Arabia suffer less severe acute chest syndrome than patients with African haplotypes. Ann Thorac Med. 2007;2:158–62. doi: 10.4103/1817-1737.36550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: An increasing global health problem. Bull World Health Organ. 2001;79:704–12. [PMC free article] [PubMed] [Google Scholar]
- 3.Cavalli-Sforza LL, Menozzi P, Piazza A. Princeton, NJ: Princeton University Press; 1994. The History and Geography of Human Genes. [Google Scholar]
- 4.Weatherall DJ. The thalassaemias. BMJ. 1997;314:1675–8. doi: 10.1136/bmj.314.7095.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sutton LL, Castro O, Cross DJ, Spencer JE, Lewis JF. Pulmonary hypertension in sickle cell disease. Am J Cardiol. 1994;74:626–8. doi: 10.1016/0002-9149(94)90760-9. [DOI] [PubMed] [Google Scholar]
- 6.Aessopos A, Farmakis D, Karagiorga M, Voskaridou E, Loutradi A, Hatziliami A, et al. Cardiac involvement in thalassemia intermedia: A multicenter study. Blood. 2001;97:3411–6. doi: 10.1182/blood.v97.11.3411. [DOI] [PubMed] [Google Scholar]
- 7.Derchi G, Fonti A, Forni GL, Galliera EO, Cappellini MD, Turati F, et al. Pulmonary hypertension in patients with thalassemia major. Am Heart J. 1999;138:384. doi: 10.1016/s0002-8703(99)70129-8. [DOI] [PubMed] [Google Scholar]
- 8.Du ZD, Roguin N, Milgram E, Saab K, Koren A. Pulmonary hypertension in patients with thalassemia major. Am Heart J. 1997;134:532–7. doi: 10.1016/s0002-8703(97)70091-7. [DOI] [PubMed] [Google Scholar]
- 9.Grisaru D, Rachmilewitz EA, Mosseri M, Gotsman M, Lafair JS, Okon E, et al. Cardiopulmonary assessment in beta-thalassemia major. Chest. 1990;98:1138–42. doi: 10.1378/chest.98.5.1138. [DOI] [PubMed] [Google Scholar]
- 10.Barnett CF, Hsue PY, Machado RF. Pulmonary hypertension: An increasingly recognized complication of hereditary hemolytic anemias and HIV infection. JAMA. 2008;299:324–31. doi: 10.1001/jama.299.3.324. [DOI] [PubMed] [Google Scholar]
- 11.Aleem A, Jehangir A, Owais M, Al-Momen A, Al-Diab A, Abdulkarim H, et al. Echocardiographic abnormalities in adolescent and adult Saudi patients with sickle cell disease. Saudi Med J. 2007;28:1072–5. [PubMed] [Google Scholar]
- 12.Connor P, Veys P, Amrolia P, Haworth S, Ashworth M, Moledina S. Pulmonary hypertension in children with Evans syndrome. Pediatr Hematol Oncol. 2008;25:93–8. doi: 10.1080/08880010801888253. [DOI] [PubMed] [Google Scholar]
- 13.Ataga KI, Moore CG, Jones S, Olajide O, Strayhorn D, Hinderliter A, et al. Pulmonary hypertension in patients with sickle cell disease: A longitudinal study. Br J Haematol. 2006;134:109–15. doi: 10.1111/j.1365-2141.2006.06110.x. [DOI] [PubMed] [Google Scholar]
- 14.Farmakis D, Aessopos A. Pulmonary hypertension associated with hemoglobinopathies: Prevalent but overlooked. Circulation. 2011;123:1227–32. doi: 10.1161/CIRCULATIONAHA.110.988089. [DOI] [PubMed] [Google Scholar]
- 15.Parent F, Bachir D, Inamo J, Lionnet F, Driss F, Loko G, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med. 2011;365:44–53. doi: 10.1056/NEJMoa1005565. [DOI] [PubMed] [Google Scholar]
- 16.Anthi A, Machado RF, Jison ML, Taveira-Dasilva AM, Rubin LJ, Hunter L, et al. Hemodynamic and functional assessment of patients with sickle cell disease and pulmonary hypertension. Am J Respir Crit Care Med. 2007;175:1272–9. doi: 10.1164/rccm.200610-1498OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S43–54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 18.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34–41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 19.Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350:886–95. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- 20.Kato GJ, McGowan V, Machado RF, Little JA, Taylor J, 6th, Morris CR, et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood. 2006;107:2279–85. doi: 10.1182/blood-2005-06-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kato GJ, Onyekwere OC, Gladwin MT. Pulmonary hypertension in sickle cell disease: Relevance to children. Pediatr Hematol Oncol. 2007;24:159–70. doi: 10.1080/08880010601185892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Castro LM, Jonassaint JC, Graham FL, Ashley-Koch A, Telen MJ. Pulmonary hypertension associated with sickle cell disease: Clinical and laboratory endpoints and disease outcomes. Am J Hematol. 2008;83:19–25. doi: 10.1002/ajh.21058. [DOI] [PubMed] [Google Scholar]
- 23.Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: Reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21:37–47. doi: 10.1016/j.blre.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: A novel mechanism of human disease. JAMA. 2005;293:1653–62. doi: 10.1001/jama.293.13.1653. [DOI] [PubMed] [Google Scholar]
- 25.Villagra J, Shiva S, Hunter LA, Machado RF, Gladwin MT, Kato GJ. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood. 2007;110:2166–72. doi: 10.1182/blood-2006-12-061697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagger D, Wolff S, Owen J, Samson D. Changes in coagulation and fibrinolysis in patients with sickle cell disease compared with healthy black controls. Blood Coagul Fibrinolysis. 1995;6:93–9. doi: 10.1097/00001721-199504000-00001. [DOI] [PubMed] [Google Scholar]
- 27.Ataga KI, Moore CG, Hillery CA, Jones S, Whinna HC, Strayhorn D, et al. Coagulation activation and inflammation in sickle cell disease-associated pulmonary hypertension. Haematologica. 2008;93:20–6. doi: 10.3324/haematol.11763. [DOI] [PubMed] [Google Scholar]
- 28.van Beers EJ, Spronk HM, Ten Cate H, Duits AJ, Brandjes DP, van Esser JW, et al. No association of the hypercoagulable state with sickle cell disease related pulmonary hypertension. Haematologica. 2008;93:e42–4. doi: 10.3324/haematol.12632. [DOI] [PubMed] [Google Scholar]
- 29.Westerman M, Pizzey A, Hirschman J, Cerino M, Weil-Weiner Y, Ramotar P, et al. Microvesicles in haemoglobinopathies offer insights into mechanisms of hypercoagulability, haemolysis and the effects of therapy. Br J Haematol. 2008;142:126–35. doi: 10.1111/j.1365-2141.2008.07155.x. [DOI] [PubMed] [Google Scholar]
- 30.Aessopos A, Kati M, Farmakis D. Heart disease in thalassemia intermedia: A review of the underlying pathophysiology. Haematologica. 2007;92:658–65. doi: 10.3324/haematol.10915. [DOI] [PubMed] [Google Scholar]
- 31.Sonakul D, Fucharoen S. Pulmonary thromboembolism in thalassemic patients. Southeast Asian J Trop Med Public Health. 1992;23(Suppl 2):25–8. [PubMed] [Google Scholar]
- 32.Atichartakarn V, Likittanasombat K, Chuncharunee S, Chandanamattha P, Worapongpaiboon S, Angchaisuksiri P, et al. Pulmonary arterial hypertension in previously splenectomized patients with beta-thalassemic disorders. Int J Hematol. 2003;78:139–45. doi: 10.1007/BF02983382. [DOI] [PubMed] [Google Scholar]
- 33.Chou R, DeLoughery TG. Recurrent thromboembolic disease following splenectomy for pyruvate kinase deficiency. Am J Hematol. 2001;67:197–9. doi: 10.1002/ajh.1107. [DOI] [PubMed] [Google Scholar]
- 34.Hayag-Barin JE, Smith RE, Tucker FC., Jr Hereditary spherocytosis, thrombocytosis, and chronic pulmonary emboli: A case report and review of the literature. Am J Hematol. 1998;57:82–4. doi: 10.1002/(sici)1096-8652(199801)57:1<82::aid-ajh15>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 35.Atichartakarn V, Angchaisuksiri P, Aryurachai K, Chuncharunee S, Thakkinstian A. In vivo platelet activation and hyperaggregation in hemoglobin E/beta-thalassemia: A consequence of splenectomy. Int J Hematol. 2003;77:299–303. doi: 10.1007/BF02983790. [DOI] [PubMed] [Google Scholar]
- 36.Rybicki AC, Benjamin LJ. Increased levels of endothelin-1 in plasma of sickle cell anemia patients. Blood. 1998;92:2594–6. [PubMed] [Google Scholar]
- 37.Ergul S, Brunson CY, Hutchinson J, Tawfik A, Kutlar A, Webb RC, et al. Vasoactive factors in sickle cell disease: In vitro evidence for endothelin-1-mediated vasoconstriction. Am J Hematol. 2004;76:245–51. doi: 10.1002/ajh.20107. [DOI] [PubMed] [Google Scholar]
- 38.Hebbel RP, Eaton JW, Balasingam M, Steinberg MH. Spontaneous oxygen radical generation by sickle erythrocytes. J Clin Invest. 1982;70:1253–9. doi: 10.1172/JCI110724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chakraborty D, Bhattacharyya M. Antioxidant defense status of red blood cells of patients with beta-thalassemia and Ebeta-thalassemia. Clin Chim Acta. 2001;305:123–9. doi: 10.1016/s0009-8981(00)00428-9. [DOI] [PubMed] [Google Scholar]
- 40.Morris CR, Suh JH, Hagar W, Larkin S, Bland DA, Steinberg MH, et al. Erythrocyte glutamine depletion, altered redox environment, and pulmonary hypertension in sickle cell disease. Blood. 2008;111:402–10. doi: 10.1182/blood-2007-04-081703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Machado RF, Anthi A, Steinberg MH, Bonds D, Sachdev V, Kato GJ, et al. N-terminal pro-brain natriuretic peptide levels and risk of death in sickle cell disease. JAMA. 2006;296:310–8. doi: 10.1001/jama.296.3.310. [DOI] [PubMed] [Google Scholar]
- 42.Aessopos A, Farmakis D, Hatziliami A, Fragodimitri C, Karabatsos F, Joussef J, et al. Cardiac status in well-treated patients with thalassemia major. Eur J Haematol. 2004;73:359–66. doi: 10.1111/j.1600-0609.2004.00304.x. [DOI] [PubMed] [Google Scholar]
- 43.Aessopos A, Farmakis D, Deftereos S, Tsironi M, Tassiopoulos S, Moyssakis I, et al. Thalassemia heart disease: A comparative evaluation of thalassemia major and thalassemia intermedia. Chest. 2005;127:1523–30. doi: 10.1378/chest.127.5.1523. [DOI] [PubMed] [Google Scholar]
- 44.Aessopos A, Farmakis D, Trompoukis C, Tsironi M, Moyssakis I, Tsaftarides P, et al. Cardiac involvement in sickle beta-thalassemia. Ann Hematol. 2009;88:557–64. doi: 10.1007/s00277-008-0661-y. [DOI] [PubMed] [Google Scholar]
- 45.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. Task force on expert consensus documents on pulmonary hypertension. J Am Coll Cardiol. 2009;53:1573–619. doi: 10.1016/j.jacc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 46.Morris CR. Mechanisms of vasculopathy in sickle cell disease and thalassemia. Hematology Am Soc Hematol Educ Program. 2008:177–85. doi: 10.1182/asheducation-2008.1.177. doi: 10.1182/asheducation-2008.1.177. [DOI] [PubMed] [Google Scholar]
- 47.Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO, 3rd, Schechter AN, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8:1383–9. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 48.Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Machado RF, Mack AK, Martyr S, Barnett C, Macarthur P, Sachdev V, et al. Severity of pulmonary hypertension during vaso-occlusive pain crisis and exercise in patients with sickle cell disease. Br J Haematol. 2007;136:319–25. doi: 10.1111/j.1365-2141.2006.06417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McGoon M, Gutterman D, Steen V, Barst R, McCrory DC, Fortin TA, et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126:14S–3. doi: 10.1378/chest.126.1_suppl.14S. [DOI] [PubMed] [Google Scholar]
- 51.Morris CR. Vascular risk assessment in patients with sickle cell disease. Haematologica. 2011;96:1–5. doi: 10.3324/haematol.2010.035097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Castro O, Hoque M, Brown BD. Pulmonary hypertension in sickle cell disease: Cardiac catheterization results and survival. Blood. 2003;101:1257–61. doi: 10.1182/blood-2002-03-0948. [DOI] [PubMed] [Google Scholar]
- 53.Sachdev V, Machado RF, Shizukuda Y, Rao YN, Sidenko S, Ernst I, et al. Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. J Am Coll Cardiol. 2007;49:472–9. doi: 10.1016/j.jacc.2006.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Voskaridou E, Tsetsos G, Tsoutsias A, Spyropoulou E, Christoulas D, Terpos E. Pulmonary hypertension in patients with sickle cell/beta thalassemia: Incidence and correlation with serum N-terminal pro-brain natriuretic peptide concentrations. Haematologica. 2007;92:738–43. doi: 10.3324/haematol.11136. [DOI] [PubMed] [Google Scholar]
- 55.Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339:5–11. doi: 10.1056/NEJM199807023390102. [DOI] [PubMed] [Google Scholar]
- 56.Pegelow CH, Adams RJ, McKie V, Abboud M, Berman B, Miller ST, et al. Risk of recurrent stroke in patients with sickle cell disease treated with erythrocyte transfusions. J Pediatr. 1995;126:896–9. doi: 10.1016/s0022-3476(95)70204-0. [DOI] [PubMed] [Google Scholar]
- 57.Joyce K, Sable C, Martin B, et al. Pulmonary artery hypertension in children with sickle cell disease: Is chronic transfusion protective? Blood. 2006;108:356a. [Google Scholar]
- 58.Gordeuk VR, Campbell A, Rana S, Nouraie M, Niu X, Minniti CP, et al. Relationship of erythropoietin, fetal hemoglobin, and hydroxyurea treatment to tricuspid regurgitation velocity in children with sickle cell disease. Blood. 2009;114:4639–44. doi: 10.1182/blood-2009-04-218040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Minniti CP, Sable C, Campbell A, Rana S, Ensing G, Dham N, et al. Elevated tricuspid regurgitant jet velocity in children and adolescents with sickle cell disease: Association with hemolysis and hemoglobin oxygen desaturation. Haematologica. 2009;94:340–7. doi: 10.3324/haematol.13812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: Risks and benefits up to 9 years of treatment. JAMA. 2003;289:1645–51. doi: 10.1001/jama.289.13.1645. [DOI] [PubMed] [Google Scholar]
- 61.Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332:1317–22. doi: 10.1056/NEJM199505183322001. [DOI] [PubMed] [Google Scholar]
- 62.Olnes M, Chi A, Haney C, May R, Minniti C, Taylor J, 6th, et al. Improvement in hemolysis and pulmonary arterial systolic pressure in adult patients with sickle cell disease during treatment with hydroxyurea. Am J Hematol. 2009;84:530–32. doi: 10.1002/ajh.21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pashankar FD, Carbonella J, Bazzy-Asaad A, Friedman A. Longitudinal follow up of elevated pulmonary artery pressures in children with sickle cell disease. Br J Haematol. 2009;144:736–41. doi: 10.1111/j.1365-2141.2008.07501.x. [DOI] [PubMed] [Google Scholar]
- 64.Taher AT, Musallam KM, Karimi M, El-Beshlawy A, Belhoul K, Daar S, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: The OPTIMAL CARE study. Blood. 2010;115:1886–92. doi: 10.1182/blood-2009-09-243154. [DOI] [PubMed] [Google Scholar]
- 65.Frank H, Mlczoch J, Huber K, Schuster E, Gurtner HP, Kneussl M. The effect of anticoagulant therapy in primary and anorectic drug-induced pulmonary hypertension. Chest. 1997;112:714–21. doi: 10.1378/chest.112.3.714. [DOI] [PubMed] [Google Scholar]
- 66.Fuster V, Steele PM, Edwards WD, Gersh BJ, McGoon MD, Frye RL. Primary pulmonary hypertension: Natural history and the importance of thrombosis. Circulation. 1984;70:580–7. doi: 10.1161/01.cir.70.4.580. [DOI] [PubMed] [Google Scholar]
- 67.Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327:76–81. doi: 10.1056/NEJM199207093270203. [DOI] [PubMed] [Google Scholar]
- 68.Channick RN, Newhart JW, Johnson FW, Williams PJ, Auger WR, Fedullo PF, et al. Pulsed delivery of inhaled nitric oxide to patients with primary pulmonary hypertension: An ambulatory delivery system and initial clinical tests. Chest. 1996;109:1545–9. doi: 10.1378/chest.109.6.1545. [DOI] [PubMed] [Google Scholar]
- 69.Morris CR, Morris SM, Jr, Hagar W, Van Warmerdam J, Claster S, Kepka-Lenhart D, et al. Arginine therapy: A new treatment for pulmonary hypertension in sickle cell disease? Am J Respir Crit Care Med. 2003;168:63–9. doi: 10.1164/rccm.200208-967OC. [DOI] [PubMed] [Google Scholar]
- 70.Morris CR, Vichinsky EP, van Warmerdam J, Machado L, Kepka-Lenhart D, Morris SM, Jr, et al. Hydroxyurea and arginine therapy: Impact on nitric oxide production in sickle cell disease. J Pediatr Hematol Oncol. 2003;25:629–34. doi: 10.1097/00043426-200308000-00008. [DOI] [PubMed] [Google Scholar]
- 71.Morris CR, Kuypers FA, Larkin S, Sweeters N, Simon J, Vichinsky EP, et al. Arginine therapy: A novel strategy to induce nitric oxide production in sickle cell disease. Br J Haematol. 2000;111:498–500. doi: 10.1046/j.1365-2141.2000.02403.x. [DOI] [PubMed] [Google Scholar]
- 72.El-Beshlawy A, Youssry I, El-Saidi S, El Accaoui R, Mansi Y, Makhlouf A, et al. Pulmonary hypertension in beta-thalassemia major and the role of L-carnitine therapy. Pediatr Hematol Oncol. 2008;25:734–43. doi: 10.1080/08880010802244035. [DOI] [PubMed] [Google Scholar]
- 73.Derchi G, Forni GL, Formisano F, Cappellini MD, Galanello R, D’Ascola G, et al. Efficacy and safety of sildenafil in the treatment of severe pulmonary hypertension in patients with hemoglobinopathies. Haematologica. 2005;90:452–8. [PubMed] [Google Scholar]
- 74.Machado RF, Martyr S, Kato GJ, Barst RJ, Anthi A, Robinson MR, et al. Sildenafil therapy in patients with sickle cell disease and pulmonary hypertension. Br J Haematol. 2005;130:445–53. doi: 10.1111/j.1365-2141.2005.05625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Little JA, Hauser KP, Martyr SE, Harris A, Maric I, Morris CR, et al. Hematologic, biochemical, and cardiopulmonary effects of L-arginine supplementation or phosphodiesterase 5 inhibition in patients with sickle cell disease who are on hydroxyurea therapy. Eur J Haematol. 2009;82:315–21. doi: 10.1111/j.1600-0609.2009.01210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Minniti CP, Machado RF, Coles WA, Sachdev V, Gladwin MT, Kato GJ. Endothelin receptor antagonists for pulmonary hypertension in adult patients with sickle cell disease. Br J Haematol. 2009;147:737–43. doi: 10.1111/j.1365-2141.2009.07906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barst RJ, Mubarak KK, Machado RF, Ataga KI, Benza RL, Castro O, et al. Exercise capacity and haemodynamics in patients with sickle cell disease with pulmonary hypertension treated with bosentan: Results of the ASSET studies. Br J Haematol. 2010;149:426–3. doi: 10.1111/j.1365-2141.2010.08097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]