

Abstract
Chronic lung diseases are common causes of pulmonary hypertension. It ranks second after the left heart disease. Both obstructive and restrictive lung diseases are know to cause pulmonary hypertension.
The pathophysiology of the disease is complex, and includes factors affecting the blood vessels, airways, and lung parenchyma. Hypoxia and the inhalation of toxic materials are another contributing factors. Recent guidelines have further clarified the association between pulmonary hypertension and chronic lung disease and made general guidelines concerning the diagnosis and management.
In this article, we will provide a detailed revision about the new classification and give general recommendations about the management of pulmonary hypertension in chronic lung diseases.
Keywords: Hypoxia, lung diseases, pulmonary hypertension, chronic obstructive airways disease, pulmonary fibrosis, Saudi association for pulmonary hypertension guidelines
Pulmonary hypertension (PH) due to lung disease is a form of precapillary PH and is defined as mean pulmonary artery pressure (mPAP) ≥25 mmHg associated with pulmonary artery wedge pressure (PAWP) ≤15 mmHg and normal or reduced cardiac output (CO).
The 4th and 5th world symposium on pulmonary arterial hypertension (PAH) grouped these conditions under the heading “PH due to lung diseases and/or hypoxia.”[1] As the classification implies; the predominant cause of PH in this group of disorders is alveolar hypoxia as a result of either chronic lung disease, impaired control of breathing or residing at high altitude.
It is a common form of PH and ranks second after group II disease (PH due to left heart disease). In the author referral clinic, 25% of PH cases evaluated are patients with lung disease.
Pathophysiology
Chronic hypoxia as a contributing factor to the development of PH is very important. This made grouping of these disorders that involves a wide variety of clinical conditions possible.[2,3] Chronic hypoxia exerts its effect by causing vasoconstriction and shear stress on the vascular endothelium and smooth muscle cells to release vasoconstrictive, pro-proliferative, and mitogenic mediators. The pathologic features of hypoxic vasculopathy include concentric intimal thickening-secondary to the proliferation of endothelial cells, smooth muscle cells, and myofibroblasts and medial hypertrophy of the muscular pulmonary arteries, adventitial proliferation, and abnormal extracellular matrix deposition.[2]
Yet there is accumulating evidence that pathophysiologic mechanisms other than hypoxia can contribute to the development of pulmonary vasculopathy in these disorders such as cigarette smoke injury,[4] destruction of the pulmonary capillary bed,[5] and inflammation.[6,7]
Finally, as both PAH and PH due to lung disease are classified as precapillary PH, sometimes it is crucial to differentiate between the two condition, especially when the lung disease is present with the severe form of PH. Table 1 summaries the criteria that might help in differentiating between the two diseases.
Table 1.
Criteria distinguishing PAH from PH due to lung disease

Epidemiology
The exact prevalence of PH in chronic lung disease at large is not known and yet not accurately reported for individual diseases. The diagnostic methodology used in the studies is not always the same; some used echocardiography as a sole diagnostic tool, while in the studies used right heart catheterization (RHC) as the diagnostic tool, the cut-off pressure for the diagnosis was not standardized and the PAWP was not reported in all studies. These issues limit the ability to draw universal and firm conclusion about the prevalence of PH associated with lung diseases.[3,8,9,10,11]
Pulmonary hypertension due to chronic obstructive pulmonary disease (COPD)
Estimates of the prevalence of PH in patients with COPD vary widely based on the definition of PH, the physiologic characteristics of the underlying lung disease, and the methods used to determine pulmonary pressures. For example, in a study of 215 patients with severe COPD (forced expiratory volume in 1 s [FEV1] <24% predicted), who were candidates for lung volume reduction surgery or lung transplantation, PH, defined as mPAP > 25 mmHg, was present in 50%, although it was mostly mild (mPAP < 35 mmHg).[12] On the contrary, out of 998 patients with moderate to severe COPD evaluated for lung transplantation, only 27 had mPAP above 40 by RHC, 16 of which had other causes that can explain PH leaving only 1.1% of the study population with severe PH attributed to COPD.[13]
In another retrospective review of 4930 patients with COPD evaluable for lung transplantation, PH was present in 30.4% by RHC (mPAP > 25 mmHg and normal PAWP), with additional 17.2% of the patient having pulmonary venous hypertension with elevated PAWP.[14]
Finally, another study on 120 patients with severe emphysema (FEV1 <27% predicted) undergoing evaluation for lung volume reduction surgery found that the incidence of PH, defined as a mPAP > 20 mmHg, was 91%. Only, 5% of the patients showed a mPAP > 35 mmHg. The mPAP was closely related to PAWP, which was mildly elevated in the majority of these patients, suggesting the common presence of diastolic left ventricular dysfunction in advanced COPD.[15]
Pulmonary hypertension due to diffuse parenchymal lung diseases (DPLD)
Pulmonary hypertension is common in idiopathic pulmonary fibrosis (IPF). The prevalence of PH reported is between 32% and 46% depending on the time of the assessment relative to the disease stage and progression. This prevalence may rise to as high as 85% in advanced IPF patients before lung transplantation. In 2525 patients evaluated for lung transplantation for IPF between 1995 and 2004 who underwent RHC study, PH was found to affect 46.1% of the subjects; 9% had severe PH (mPAP > 40 mmHg).[16] In another study of 44 patients with IPF evaluated for lung transplantation and who had serial RHC at the time of referral and at the time of transplantation were reviewed. 38.6% (17/44) of the patients had PH at baseline. The majority of the non PH patients developed PH during the serial time interval with a subsequent incidence of 86.4% at the time of transplant. The mPAP at baseline and follow-up were 22.5 and 32.7 mmHg, respectively.[17]
Pulmonary hypertension due to combined pulmonary fibrosis and emphysema
Combined pulmonary fibrosis and emphysema syndrome is characterized by a mixed obstructive and restrictive pattern. It is now included in the updated clinical classification of PH due to lung disease.[1] In a study of 40 patients with combined emphysema and lung fibrosis who underwent RHC, 27 patients (68%) were found to have a mPAP > 35 mmHg (and >40 mmHg in 48% of patients).[18] The incidence of emphysema in IPF patient cohort is estimated to be 28%; these patients do have more advanced disease and more severe functional impairment. They were also found to carry a worse prognosis than patients with IPF without emphysema despite the preserved lung volumes on pulmonary function testing. This is partly related to the development of PH.[19]
Pulmonary hypertension due to other lung disease
Pulmonary hypertension in the setting of obstructive sleep apnea (OSA) generally appears to be mild to moderate; severe PH being less common.[20] The true prevalence of PH in OSA is unknown due to a lack of large, population-based studies. Prevalence data on resting, awake PH in OSA is based primarily on retrospective case series or prospective cohort studies with poorly defined entry criteria. The reported prevalence of PH (determined by RHC or echocardiography) in patients with OSA ranges from 17% to 41%. In the largest series published to date, 17% of a sample of 220 patients with OSA met diagnostic criteria for PH; the mPAP by RHC was >25 mmHg.[21] The mild nature of PH was observed in the majority of PH in OSA studies. Also, large number of patients had co-morbid elevation in the PAWP as an indicator of left heart disease or diastolic dysfunction, which is common in OSA patients.[21]
Obesity-hypoventilation syndrome (OHS) seems to carry a higher chance for development of PH due to the added effect of hypercapnia. In a study of 27 patients with OHS, defined as a body mass index >30 and PaCO2 > 45 mmHg, 59% of cases had mPAP of > 20 mmHg, the average PAP was 23 ± 10 mmHg.[22]
The PH associated with residing at high altitude and developmental abnormalities is not discussed in this manuscript.
Definitions of Pulmonary Hypertension Due to Chronic Lung Diseases
During the 5th Nice PH World Congress, it was suggested that the term “out of proportion” be abandoned in describing PH due to chronic lung disease and instead use the following definitions:
Chronic lung disease without PH when the mPAP is <25 mmHg,
Chronic lung disease with PH when the mPAP ≥ 25 mmHg,
Chronic lung disease with severe PH when the mPAP is ≥35 mmHg, or ≥25 but <35 mmHg with low cardiac index (<2.0 L/min/m2), as assessed by RHC.
This will standardize the definition of the disease in this group of patients for the sake of future research purposes.
Assessment of Pulmonary Hypertension Due to Chronic Lung Diseases
The symptoms of lung disease and of PH can be similar, as dyspnea and fatigue are the cardinal features of both diseases. The presence of PH in patients with underlying lung disease should be suspected when the symptoms or degree of hypoxia and exercise limitation are “not in proportion” to the pulmonary function and ventilatory abnormalities, or decline in clinical status does not parallel the declines in lung function. Very low diffusion capacity is an important indicator of pulmonary vasculopathy.[23,24] Clinical features of right ventricular failure may also indicate the development of PH in patients with chronic lung disease.
Doppler echocardiography is the screening tool of choice for PH. Unfortunately, the accuracy of echocardiography seems to be worse in patients with lung disease than patients without lung disease in regards to the ability to detect tricuspid regurgitation jet and estimation of systolic PAP (sPAP). Arcasoy et al., showed that estimation of sPAP was possible only in 44% of patients who underwent Doppler echocardiography, out of these patients the estimated pressure was accurate (within 10 mmHg) in only 48% when compared to RHC.[8] Another study looked at right ventricular systolic pressure estimation by echocardiography in COPD patients found that echocardiography had a sensitivity of 60%, specificity of 74%, positive predictive value of 68% and a negative predictive value of 67%, compared with the invasive measurement.[9]
Brain natriuretic peptide (BNP) is a marker that is used for prognostication purpose in cases of PH. Its significance in patients with advanced lung disease was looked at in small studies that showed elevated levels in patients with lung disease may indicate the development of PH. The level of elevation correlates with the PAP and does carry prognostic significance. Although it lacks specificity, it may prove to be a useful screening test for PH development in chronic lung disease patients.[10,11]
The gold standard for diagnosing PH remains to be RHC. Patient with lung disease are likely to be older and might have co-morbid left heart disease and diastolic dysfunction, which will be diagnosed by RHC. Selecting patients for RHC is important and should be reserved for people in whom the diagnosis will make a difference in their clinical management, like patients who are candidates for lung transplantation, and patients who are suspected to have PAH independent to their primary lung disease severity (used to known as “out of proportion”). In these patients, inclusion into clinical trials is recommended and encouraged to better characterize their features and treatment options.
Impact of Pulmonary Hypertension on Chronic Lung Disease
The presence of PH in patients with chronic lung disease complicates the clinical presentation and is usually associated with more severe symptoms of dyspnea and fatigue. The deterioration of the patient clinical status is more accelerated in these patients than what is expected solely from their underlying lung disease. The functional status of the patient is usually worse with its negative impact on quality of life, exercise capacity and distance walked, and mortality.[19,22,25,26,27]
The presence of PH influences definitive treatment options for the patient underlying disease like transplantation as well as the survival outcome after transplantation.[28,29]
Treatment
The underlying lung disease should be optimally managed according to guidelines and clinical practice measures. Such as optimizing bronchodilator use, smoking cessation, pulmonary rehabilitation and weight-loss.
Surgical option including lung transplantation should be considered in advanced cases, and whenever that is suitable for the disease condition. Attention should be paid to co-morbidities that might contribute to PH in these patients and managing them adequately, such as fluid overload, left heart disease, OSA, and pulmonary thromboembolic diseases.
Aggressive management of hypoxia at rest, during exertion, and during sleep is crucial for all hypoxic patients. This recommendation is extrapolated from COPD literature, but certainly makes sense in any hypoxia-related pulmonary disease, especially when PH is also present.
In severe COPD patients, who require initiation of long-term O2, such therapy has shown to halt the progression of PH.[30]
In patients with OSA, OHS, and chronic respiratory failure who have associated PH, initiating positive airway pressure therapy to correct respiratory failure parameters during the daytime has shown to improve the hemodynamic measurements in these cases.[31,32]
Using pulmonary arterial hypertension specific therapy for pulmonary hypertension due to lung diseases and/or hypoxia
Patients with PH and chronic diseases may continue to be very symptomatic and significantly limited despite optimal medical therapy of the lung disease and other co-morbid conditions. In these situations, attempt to target the moderately severe PH (mPAP > 35-40 mmHg) might be logical in an era where effective and approved medications are available to treat PAH.[3]
However, due to unproven efficacy in this situation, the role of the PAH-specific drugs in PH secondary to lung disease and hypoxia is controversial and not well studied in large randomized controlled trials. Data available are from the small number of patients, short duration of treatment, open label and single center trials. Furthermore, the effect of these medications was studied mainly in COPD and interstitial lung diseases (ILD).
The pulmonary vasodilatation produced by these agents can be risky in patients with lung disease, as loss of the protective vasoconstriction mechanisms may consequently worsen ventilation perfusion mismatch and worsen the hypoxia. Furthermore, the reduction in the CO has been observed and subjected the patient to the risk of pulmonary edema.
Specific therapy in chronic obstructive pulmonary disease
Sildenafil was used in 20 patients with COPD and PH. Hemodynamics and gas exchange were assessed before and 1 h after sildenafil administration. Despite the initial improvement in pulmonary hemodynamics at rest and during exercise, there was an inhibition of the hypoxic vasoconstriction protective mechanism and impairment of arterial oxygenation at rest.[33]
Inhaled iloprost was also studied in 10 patients with COPD and PH. Assessment was performed before inhalation, 30 min after inhalation, and 2 h later. The overall results of this small study have showed that inhaled iloprost was safe in patients with COPD and PH, and was associated with improved V/Q matching and exercise tolerance.[34]
In a randomized controlled trial of bosentan in severe COPD, 30 patients enrolled with 2:1 randomization. Compared with placebo, patients treated with bosentan for 12 weeks showed no significant improvement in exercise capacity as measured by the 6-min walking distance (6MWD).
There was no change in lung function, PAP, maximal oxygen uptake, or regional pulmonary perfusion pattern. In contrast, arterial oxygen pressure dropped, the alveolar–arterial gradient increased, and quality of life deteriorated significantly in patients assigned to bosentan.[35]
Specific therapy in interstitial lung diseases
In ILD patients, a study on 15 ILD patients and severe PH who were treated with sildenafil for 6 months has found that the BNP levels were lower, 6MWD was higher, but no change in sPAP demonstrated.[36]
In IPF, bosentan failed to improve the predefined endpoint of time to the occurrence of lung fibrosis worsening in a Phase III trial.[37] Negative result was also reported for macitentan (MUSIC study) in IPF. In a recent randomized, double-blind, placebo-controlled, event-driven trial (ARTEMIS-IPF study) that was prematurely terminated for safety concern, ambrisentan has shown to be ineffective in treating IPF and was associated with an increased risk for disease progression and respiratory hospitalizations.[38] No results are yet available from a bosentan trial particularly focusing on IPF-associated PH (B-PHIT [Bosentan in PH in ILD] study.
A recent Phase II trial has demonstrated the efficacy of riociguat (soluble guanylate cyclase) in decreasing PVR and increasing CO and 6MWD in PH-DPLD.[39]
Due to this inhomogeneous data, the PAH specific therapy cannot be generally recommended to treat PH due to lung diseases or hypoxia at this stage. Such patients should be referred to the specialized center for evaluation and should be included in clinical trials in order to better evaluate their outcome.
Recommendations for Pulmonary Hypertension Due to Lung Disease and Hypoxia
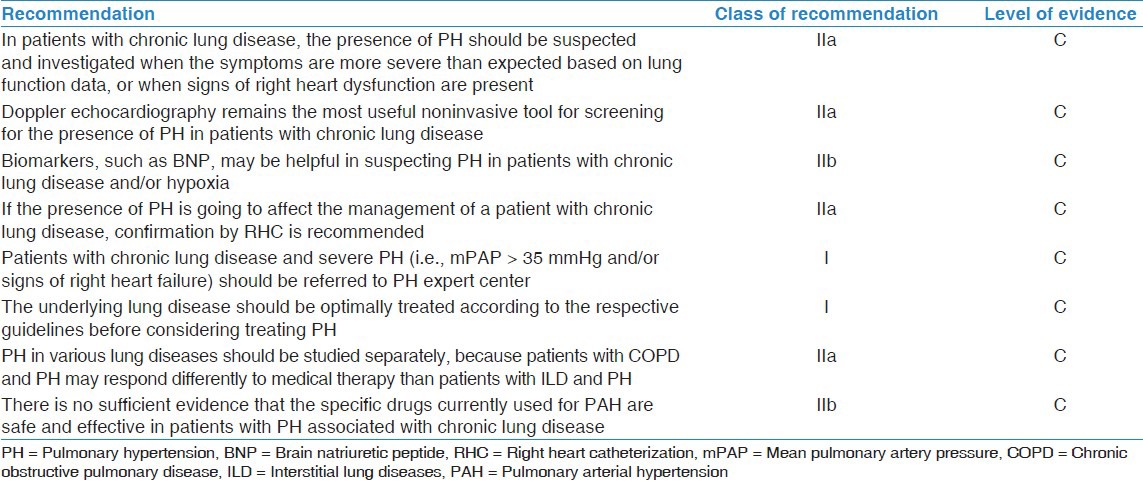
Most of the data available on PH due to chronic lung disease and hypoxia are insufficient to draw strong evidence based conclusions and recommendations.[40,41,42] The class of recommendation and level of evidence for different aspects of the management of PH due to lung diseases and/or hypoxia are shown in Table 2.
Table 2.
Class of recommendations and level of evidence for treatment of PH due to lung diseases or hypoxia
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: Cellular and molecular mechanisms. Circ Res. 2006;99:675–91. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 3.Alhamad EH, Cal JG, Alfaleh HF, Alshamiri MQ, Alboukai AA, Alhomida SA. Pulmonary hypertension in Saudi Arabia: A single center experience. Ann Thorac Med. 2013;8:78–85. doi: 10.4103/1817-1737.109816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santos S, Peinado VI, Ramírez J, Melgosa T, Roca J, Rodriguez-Roisin R, et al. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J. 2002;19:632–8. doi: 10.1183/09031936.02.00245902. [DOI] [PubMed] [Google Scholar]
- 5.Colombat M, Mal H, Groussard O, Capron F, Thabut G, Jebrak G, et al. Pulmonary vascular lesions in end-stage idiopathic pulmonary fibrosis: Histopathologic study on lung explant specimens and correlations with pulmonary hemodynamics. Hum Pathol. 2007;38:60–5. doi: 10.1016/j.humpath.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 6.Kwon YS, Chi SY, Shin HJ, Kim EY, Yoon BK, Ban HJ, et al. Plasma C-reactive protein and endothelin-1 level in patients with chronic obstructive pulmonary disease and pulmonary hypertension. J Korean Med Sci. 2010;25:1487–91. doi: 10.3346/jkms.2010.25.10.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaouat A, Savale L, Chouaid C, Tu L, Sztrymf B, Canuet M, et al. Role for interleukin-6 in COPD-related pulmonary hypertension. Chest. 2009;136:678–87. doi: 10.1378/chest.08-2420. [DOI] [PubMed] [Google Scholar]
- 8.Arcasoy SM, Christie JD, Ferrari VA, Sutton MS, Zisman DA, Blumenthal NP, et al. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med. 2003;167:735–40. doi: 10.1164/rccm.200210-1130OC. [DOI] [PubMed] [Google Scholar]
- 9.Fisher MR, Criner GJ, Fishman AP, Hassoun PM, Minai OA, Scharf SM, et al. Estimating pulmonary artery pressures by echocardiography in patients with emphysema. Eur Respir J. 2007;30:914–21. doi: 10.1183/09031936.00033007. [DOI] [PubMed] [Google Scholar]
- 10.Leuchte HH, Neurohr C, Baumgartner R, Holzapfel M, Giehrl W, Vogeser M, et al. Brain natriuretic peptide and exercise capacity in lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. 2004;170:360–5. doi: 10.1164/rccm.200308-1142OC. [DOI] [PubMed] [Google Scholar]
- 11.Leuchte HH, Baumgartner RA, Nounou ME, Vogeser M, Neurohr C, Trautnitz M, et al. Brain natriuretic peptide is a prognostic parameter in chronic lung disease. Am J Respir Crit Care Med. 2006;173:744–50. doi: 10.1164/rccm.200510-1545OC. [DOI] [PubMed] [Google Scholar]
- 12.Thabut G, Dauriat G, Stern JB, Logeart D, Lévy A, Marrash-Chahla R, et al. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest. 2005;127:1531–6. doi: 10.1378/chest.127.5.1531. [DOI] [PubMed] [Google Scholar]
- 13.Chaouat A, Bugnet AS, Kadaoui N, Schott R, Enache I, Ducoloné A, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172:189–94. doi: 10.1164/rccm.200401-006OC. [DOI] [PubMed] [Google Scholar]
- 14.Cuttica MJ, Kalhan R, Shlobin OA, Ahmad S, Gladwin M, Machado RF, et al. Categorization and impact of pulmonary hypertension in patients with advanced COPD. Respir Med. 2010;104:1877–82. doi: 10.1016/j.rmed.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 15.Scharf SM, Iqbal M, Keller C, Criner G, Lee S, Fessler HE, et al. Hemodynamic characterization of patients with severe emphysema. Am J Respir Crit Care Med. 2002;166:314–22. doi: 10.1164/rccm.2107027. [DOI] [PubMed] [Google Scholar]
- 16.Shorr AF, Wainright JL, Cors CS, Lettieri CJ, Nathan SD. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J. 2007;30:715–21. doi: 10.1183/09031936.00107206. [DOI] [PubMed] [Google Scholar]
- 17.Nathan SD, Shlobin OA, Ahmad S, Koch J, Barnett SD, Ad N, et al. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration. 2008;76:288–94. doi: 10.1159/000114246. [DOI] [PubMed] [Google Scholar]
- 18.Cottin V, Le Pavec J, Prévot G, Mal H, Humbert M, Simonneau G, et al. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur Respir J. 2010;35:105–11. doi: 10.1183/09031936.00038709. [DOI] [PubMed] [Google Scholar]
- 19.Mejía M, Carrillo G, Rojas-Serrano J, Estrada A, Suárez T, Alonso D, et al. Idiopathic pulmonary fibrosis and emphysema: Decreased survival associated with severe pulmonary arterial hypertension. Chest. 2009;136:10–5. doi: 10.1378/chest.08-2306. [DOI] [PubMed] [Google Scholar]
- 20.Minai OA, Ricaurte B, Kaw R, Hammel J, Mansour M, McCarthy K, et al. Frequency and impact of pulmonary hypertension in patients with obstructive sleep apnea syndrome. Am J Cardiol. 2009;104:1300–6. doi: 10.1016/j.amjcard.2009.06.048. [DOI] [PubMed] [Google Scholar]
- 21.Chaouat A, Weitzenblum E, Krieger J, Oswald M, Kessler R. Pulmonary hemodynamics in the obstructive sleep apnea syndrome. Results in 220 consecutive patients. Chest. 1996;109:380–6. doi: 10.1378/chest.109.2.380. [DOI] [PubMed] [Google Scholar]
- 22.Kessler R, Chaouat A, Schinkewitch P, Faller M, Casel S, Krieger J, et al. The obesity-hypoventilation syndrome revisited: A prospective study of 34 consecutive cases. Chest. 2001;120:369–76. doi: 10.1378/chest.120.2.369. [DOI] [PubMed] [Google Scholar]
- 23.Ghanem MK, Makhlouf HA, Agmy GR, Imam HM, Fouad DA. Evaluation of recently validated non- invasive formula using basic lung functions as new screening tool for pulmonary hypertension in idiopathic pulmonary fibrosis patients. Ann Thorac Med. 2009;4:187–96. doi: 10.4103/1817-1737.56013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Idrees MM, Al-Hajjaj M, Khan J, Al-Hazmi M, Alanezi M, Saleemi S, et al. Saudi guidelines on diagnosis and treatment of pulmonary arterial hypertension. Ann Thorac Med. 2008;3:1–57. [Google Scholar]
- 25.Sims MW, Margolis DJ, Localio AR, Panettieri RA, Kawut SM, Christie JD. Impact of pulmonary artery pressure on exercise function in severe COPD. Chest. 2009;136:412–9. doi: 10.1378/chest.08-2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lettieri CJ, Nathan SD, Barnett SD, Ahmad S, Shorr AF. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2006;129:746–52. doi: 10.1378/chest.129.3.746. [DOI] [PubMed] [Google Scholar]
- 27.Alhamad EH, Idrees MM, Alanezi MO, Alboukai AA, Shaik SA. Sarcoidosis-associated pulmonary hypertension: Clinical features and outcomes in Arab patients. Ann Thorac Med. 2010;5:86–91. doi: 10.4103/1817-1737.62471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lederer DJ, Caplan-Shaw CE, O’Shea MK, Wilt JS, Basner RC, Bartels MN, et al. Racial and ethnic disparities in survival in lung transplant candidates with idiopathic pulmonary fibrosis. Am J Transplant. 2006;6:398–403. doi: 10.1111/j.1600-6143.2005.01205.x. [DOI] [PubMed] [Google Scholar]
- 29.Oswald-Mammosser M, Weitzenblum E, Quoix E, Moser G, Chaouat A, Charpentier C, et al. Prognostic factors in COPD patients receiving long-term oxygen therapy. Importance of pulmonary artery pressure. Chest. 1995;107:1193–8. doi: 10.1378/chest.107.5.1193. [DOI] [PubMed] [Google Scholar]
- 30.Cooper CB, Waterhouse J, Howard P. Twelve year clinical study of patients with hypoxic cor pulmonale given long term domiciliary oxygen therapy. Thorax. 1987;42:105–10. doi: 10.1136/thx.42.2.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schönhofer B, Barchfeld T, Wenzel M, Köhler D. Long term effects of non-invasive mechanical ventilation on pulmonary haemodynamics in patients with chronic respiratory failure. Thorax. 2001;56:524–8. doi: 10.1136/thorax.56.7.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olson AL, Zwillich C. The obesity hypoventilation syndrome. Am J Med. 2005;118:948–56. doi: 10.1016/j.amjmed.2005.03.042. [DOI] [PubMed] [Google Scholar]
- 33.Blanco I, Gimeno E, Munoz PA, Pizarro S, Gistau C, Rodriguez-Roisin R, et al. Hemodynamic and gas exchange effects of sildenafil in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Am J Respir Crit Care Med. 2010;181:270–8. doi: 10.1164/rccm.200907-0988OC. [DOI] [PubMed] [Google Scholar]
- 34.Dernaika TA, Beavin M, Kinasewitz GT. Iloprost improves gas exchange and exercise tolerance in patients with pulmonary hypertension and chronic obstructive pulmonary disease. Respiration. 2010;79:377–82. doi: 10.1159/000242498. [DOI] [PubMed] [Google Scholar]
- 35.Stolz D, Rasch H, Linka A, Di Valentino M, Meyer A, Brutsche M, et al. A randomised, controlled trial of bosentan in severe COPD. Eur Respir J. 2008;32:619–28. doi: 10.1183/09031936.00011308. [DOI] [PubMed] [Google Scholar]
- 36.Corte TJ, Gatzoulis MA, Parfitt L, Harries C, Wells AU, Wort SJ. The use of sildenafil to treat pulmonary hypertension associated with interstitial lung disease. Respirology. 2010;15:1226–32. doi: 10.1111/j.1440-1843.2010.01860.x. [DOI] [PubMed] [Google Scholar]
- 37.King TE, Jr, Brown KK, Raghu G, du Bois RM, Lynch DA, Martinez F, et al. BUILD-3: A randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184:92–9. doi: 10.1164/rccm.201011-1874OC. [DOI] [PubMed] [Google Scholar]
- 38.Raghu G, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: A parallel, randomized trial. Ann Intern Med. 2013;158:641–9. doi: 10.7326/0003-4819-158-9-201305070-00003. [DOI] [PubMed] [Google Scholar]
- 39.Hoeper MM, Halank M, Wilkens H, Günther A, Weimann G, Gebert I, et al. Riociguat for interstitial lung disease and pulmonary hypertension: A pilot trial. Eur Respir J. 2013;41:853–60. doi: 10.1183/09031936.00213911. [DOI] [PubMed] [Google Scholar]
- 40.Galiè N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT) Eur Heart J. 2009;30:2493–537. doi: 10.1093/eurheartj/ehp297. [DOI] [PubMed] [Google Scholar]
- 41.Hoeper MM, Barberà JA, Channick RN, Hassoun PM, Lang IM, Manes A, et al. Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension pulmonary hypertension. J Am Coll Cardiol. 2009;54:S85–96. doi: 10.1016/j.jacc.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 42.Barst RJ, Gibbs JS, Ghofrani HA, Hoeper MM, McLaughlin VV, Rubin LJ, et al. Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S78–84. doi: 10.1016/j.jacc.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]