

Abstract
Prior to the availability of the pulmonary arterial hypertension (PAH)-specific therapy, PAH was a dreadful disease with a very poor prognosis. Better understanding of the complex pathobiology of PAH has led to a major therapeutic evolution. International regulatory agencies have approved many specific drugs with different pharmacologic pathways and routes of administration. In the year 2013, two new drugs with great potentials in managing PAH have been added to the treatment options, macitentan and riociguat. Additional drugs are expected to come in the near future.
A substantial body of evidence has confirmed the effectiveness of pulmonary arterial hypertension (PAH)-specific therapies in improving the patients’ symptomatic status and slowing down the rate of clinical deterioration.
Although the newer modern medications have significantly improved the survival of patients with PAH, it remains a non-curable and fatal disease. Lung transplantation (LT) remains the only therapeutic option for selected patients with advanced disease who continue to deteriorate despite optimal therapy.
Keywords: Specific therapy, target therapy, pulmonary arterial hypertension, lung transplant, Saudi association for pulmonary hypertension guidelines
During the last decade, there have been enormous improvements in our understanding of the complex pathobiology of PAH, which subsequently led to tremendous advances in the treatment options and improved the disease outcome. A meta-analysis performed on 23 randomized clinical trials (RCTs) in PAH patients has reported a 43% reduction in mortality and a 61% decline in hospitalizations in patients treated with an average treatment period of 14.3 weeks of specific drug therapies.[1]
Despite these considerable improvements, PAH remains an incurable condition with poor outcome.[2] In addition, the complex treatments for advanced cases are still invasive and prone to significant side effects; hence, an extensive knowledge and experience of the multidiscipline treatment team are critical to optimize the use of available resources and to improve the outcomes.
Treatment Strategy for PAH Patients
The optimal choice of initial specific therapy depends upon a number of factors including the patient's functional status (see under Assessment of Disease Severity in the main guidelines), the vasoreactive response, and access to specific treatment. An evidence-based treatment algorithm is described in the Treatment section in the main guidelines.
The following discussion intends to give a detailed perspective regarding all of the specific-drug therapies available in Saudi Arabia.
Calcium-channel blockers (CCBs)
Class of Recommendation for vasoreactive patients: I & Level of Evidence: B
In the late 1980s, CCBs gained attention as a simple, yet effective, treatment for a subgroup of IPAH patients. Vasodilator therapy was based on evidence for the presence of smooth muscle medial hypertrophy and muscular contraction with a resultant elevation in the PVR.
Favorable outcomes in vasoreactive PAH patients with high dose CCBs, including a survival advantages, has been shown in several, non-randomized, non-controlled studies.[3,4,5,6] The 1, 3, and 5-year survival in CCBs treated patients was 94%, 94%, and 94% compared to 68%, 47%, and 38% in those who were classified as non-responders. In these studies, the control group, however, consisted of non-vasoreactive patients who may have an inherently poorer prognosis as compared to vasoreactive individuals.
Generally, less than 10-15% of PAH will meet the criteria for a positive acute vasoreactive response and only a subset of them will demonstrate clinical and hemodynamic long-term response to CCB treatment. A positive acute vasoreactive response (positive acute responders) is defined as a reduction of mean pulmonary artery pressure (mPAP) by >10 mmHg to reach an absolute value of mPAP <40 mmHg, with an increased or unchanged cardiac output.[3,4]
The characteristics of PAH patients, who benefit from long-term CCBs have been clearly identified.[7] These include vasoreactive PAH patients, who continue to be in modified NYHA functional class I or II after at least 1 year on CCBs monotherapy. Only a subset of vasoreactive patients (around 50%) shows such a long-term improvement. Children, however, were shown to have a better response to CCBs compared to adults.
The long-term outcome of CCB treatment in IPAH pediatric patients was evaluated in a cohort of 77 children.[8] Survival for all children treated with CCBs at 1, 5, and 10 years was 97%, 97%, and 81%, respectively.
CCBs with a significant negative inotropic effect, such as verapamil, should be avoided. Nifedipine, diltiazem, or amlodipine are used most frequently, with the choice often based on the heart rate at baseline (relative bradycardia favoring nifedipine, while relative tachycardia favoring diltiazem). The doses of these drugs that have shown efficacy in IPAH are relatively high (i.e. up to 120-240 mg/day for nifedipine and 240-720 mg/day for diltiazem).[5] While early recommendations seemed to favor starting with relatively high doses of CCBs, it is probably more advisable to start with low doses to be increased cautiously and progressively in the subsequent weeks to the maximal tolerated regimen.
Side effects and limiting factors for dose increase are usually systemic hypotension. Lower limb edema is another bothering side effect, but not a limiting one.
Clinical pearls
Only vasoreactive PAH patients are to be considered for CCB therapy. In contrast, patients with a negative vasoreactivity test should be treated with an alternative agent because CCBs have not been shown to be beneficial in these patients and may be harmful.
Patients should be followed up closely for both safety and efficacy, with an initial reassessment after 3 months of therapy to ensure that patients maintain the long-term response to CCBs therapy.
In those patients who fail to maintain the long-term response, addition of PAH specific drug therapy rather than replacement is recommended, as some patients show significant hemodynamic deterioration if CCBs are withdrawn.
Prostacyclin
Prostacyclin (PGI2) is naturally synthesized by vascular endothelium. It is a potent vasodilator of all vascular beds and has significant antiproliferative activities.[9] An imbalance of the prostacyclin metabolic pathways has been found in patients with PAH.[10] Such imbalance represents a convincing basis for the therapeutic use of prostacyclin in PAH patients.
Beside its role as a vasodilator, the long-term effect of prostacyclin is thought to relate to its anti-remodeling effects that result from its inhibitory effects on vascular growth, muscular hypertrophy and thrombotic obliteration.[11]
Epoprostenol (Flolan)
Class of recommendation: I & Level of evidence: A FDA Approval: 1995
Epoprostenol is a synthetic prostacyclin with a short half-life in the circulation (3-5 min). It is rapidly converted to stable breakdown products or metabolites and is stable at room temperature for only 8 hours. Because of these features, the drug is administered by a continuous IV infusion through a special pumps and permanent intravascular catheter.
The efficacy of epoprostenol has been confirmed in controlled clinical trials in IPAH patients[12,13,14] and in PAH associated with the scleroderma.[15] Such efficacy was reflected by improved quality of life, hemodynamics, and exercise capacity. Favorable results have also been shown in uncontrolled studies in other PAH subclasses, such as PAH associated with systemic lupus erythematosus and other CTD,[16,17] PAH associated with porto-pulmonary hypertension,[18] HIV infection,[19] and PAH associated with congenital heart defects (with or without systemic to pulmonary shunts).[20]
Survival advantage for epoprostenol has been also confirmed.[21,22] The estimated 1, 2, 3, and 5-year survival rates in the epoprostenol-treated patients are 85%, 70%, 63%, and 55% and significantly better than those of the historical group: 58%, 43%, 33%, and 28%, respectively. Epoprostenol survival was also evaluated in pediatric patients with PAH and showed encouraging results. In a cohort of 35 children diagnosed with IPAH and treated with epoprostenol, the survival at 1, 5, and 10 years was 94%, 81%, and 61%, respectively.[8]
Treatment with epoprostenol is usually started at a low dose, 1 to 2 ng/kg/min, and increased by 1 to 2 ng/kg/min every one to two days as tolerated. Once the dose of 6-10 ng/kg/min is achieved (usually within one to two weeks), most patients require dose increases of 1 to 2 ng/kg/min every two to four weeks to sustain the clinical effect. The optimal dose varies between individual patients ranging in the majority between 20 and 40 ng/kg/min. However, a maximal dose has not been established, and patients on chronic therapy may receive doses as high as 150 to 200 ng/kg per/min with sustained clinical and hemodynamic benefit.
Adverse effects of epoprostenol are usually mild and dose dependent (mainly initial dosing). Flushing, jaw pain, diarrhea, headache, backache, foot and leg pain, abdominal cramping, nausea, and rarely hypotension are among the most commonly observed side effects. Ascites has also been reported, which may be related to increased permeability of the peritoneal membrane induced by epoprostenol. Dose reduction is required only if the intensity of the side effect is moderate to severe. Recurrence of side effects is less likely to occur if the dose is increased slowly and usually is mild and self-limiting over time without dose changes.
Successful use of epoprostenol in pregnancy has been reported in case reports in women with PAH.[23] However, despite this anecdotal experience, it must be clearly emphasized that all young female patients should be strongly advised against pregnancy, given appropriate contraceptive recommendations, and, if necessary, undergo early termination of pregnancy (see the article “General Management of Pulmonary Hypertension").
Complications of the drug delivery system, such as pump malfunction, local cellulitis, catheter obstruction and sepsis are often serious and require prompt identification and management. In two large series, 0.14 and 0.19 episodes of sepsis per patients-year were reported, and 8 deaths (2.8%) out of a total of 340 subjects were directly related to catheter infections.[21,22]
Finally, any abrupt interruption of the epoprostenol infusion should be avoided by all means, as a rebound worsening of pulmonary hypertension (PH) with acute right heart failure and even death has been reported.
Clinical peals
Whenever possible, epoprostenol should be considered as the first-line agent in patients with severe disease (i.e., modified NYHA class IV).
Complications of the delivery system should be recognized and dealt with promptly.
Interruption of treatment infusion, even for a brief time, can be hazardous.
Treprostinil (Remodulin)
Class of recommendation: I (NYHA III) & IIa (NYHA IV). Level of evidence: B FDA Approval: 2002 (Subcutaneous) & 2004 (Intravenous)
Treprostinil is an analogue of epoprostenol that can be administered by intravenous (IV) or subcutaneous route. Micro-infusion pumps and small subcutaneous catheters are used to deliver via the subcutaneous route of treprostinil, and therefor, the complications of this delivery system are much less than the IV system used for epoprostenol treatment. However, the side effect may be greater, especially related to pain at the site of subcutaneous infusion.
The efficacy of continuous subcutaneous administration of treprostinil in PAH patients has been confirmed in a number of studies. Such beneficial effects include improvements in 6 minute walk test (6MWT), modified NYHA functional class, hemodynamics, and possibly survival.[24,25,26,27] The greatest improvement is observed in patients who are more compromised at baseline, and the benefits are dose dependent.[24]
IV treprostinil has also been studied and found to be effective in improving symptoms, exercise intolerance, and hemodynamics.[28] Inhalation form of treprostinil are also available with limited clinical use.
A number of advantages of parenteral treprostinil compared to epoprostenol have been noticed. Such advantages, which include the option of continuous subcutaneous delivery, the longer half-life that makes interruption of the infusion less immediately life threatening, and the stability at ambient temperature, allow more flexibility and easier administration. Furthermore, based upon preliminary results, patients who are already receiving IV epoprostenol can be safely transitioned to treprostinil (subcutaneous or intravenous) without a significant loss of clinical efficacy.[29,30]
The recommended starting dose of treprostinil is 1.25 ng/kg/min, which is increased by 1.25 ng/kg/min per week during the first 4 weeks, hen by 2.5 ng/kg/min per week until the desired effect is achieved. A more rapid dose escalation has been recommended in severely ill patients and found to be safe.
Beside the prostacyclin-type adverse effects, infusion site pain is the most common side effect of subcutaneous treprostinil and occurs in around 80% of patients, leading to discontinuation of the treatment in 6-8% of cases. It is primarily related to initiation of infusion and seems to be also related to the volume infused. In most cases, the pain is manageable and improves after several weeks. Relocation of the infusion site, cold or hot compresses, local analgesic ointments and administration of paracetamol or non-steroidal anti-inflammatory drugs are sometimes helpful. Short course of oral systemic steroid (Prednisolone 1-2 mg/kg/day) has been also found to be helpful, but occasionally, narcotic drugs might be necessary. In severe cases, shifting to IV treprostinil or epoprostenol is needed.
Inhaled iloprost (Ventavis)
Class of recommendation: I & Level of evidence: A FDA Approval: 2004
Iloprost is a chemically stable prostacyclin analogue available for IV in aerosol forms. Inhaled therapy for PAH is an attractive rout for drug delivery because of the theoretical advantage that the deposition of the drug is in predominantly well-ventilated alveoli, and hence improve the V/Q matching and oxygenation.
After a single dose of inhaled iloprost, 10-20% reduction of mPAP was observed and lasted up to one hour.[31,32] The short duration of action requires frequent inhalations (from 6 to 9 times daily) to obtain a long-lasting effect with chronic therapy. With jet nebulizers, the duration of each inhalation takes about 15 min; the inhalation time, however, can be shortened to about 5 min with ultrasound nebulizers.
Inhaled iloprost has been shown to be a safe, effective, and well-tolerated treatment for varying classes of severe PAH, including IPAH and CTD-APAH. It has also shown some efficacy in inoperable CTEPH patients. Such efficacy includes improved modified NYHA functional class, exercise tolerance, hemodynamics, clinical worsening, and probably survival.[33,34,35]
Finally, the use of inhaled Iloprost has been also reported in case series of pregnant women with PAH. Nebulized Iloprost was commenced as early as 8 weeks of gestation. All delivered children were free from congenital abnormalities, and there was no post-partum maternal or infant mortality.[36]
Sodium Beraprost (Dorner)
Class of recommendation: IIb & Level of evidence: B
Beraprost is an orally active prostacyclin analogue. It has a rapid absorption in fasting conditions and peak concentration is reached after 30 min.
Beraprost has been used extensively in PAH patients in the Far East countries, but has seen limited usage in other parts of the world. The drug has been evaluated in PAH patients in many studies and showed efficacy in improving 6MWT and slowing disease progression as defined as death, transplantation, epoprostenol rescue, or > 25% decrease in VO2max.[37,38] These beneficial effects were evident in the first 3 months of starting therapy but not at either 9 or 12 months.
Drug-related adverse events include the usual prostacyclin-related adverse effect. However, GI symptoms are more likely to develop compared to other forms of prostacyclin. A slow release form of Beraprost is available that was found to have less severe GI side effects that the regular form and allows for twice daily dosing.
Beraprost has been used only in few patients in Saudi Arabia, and so the local clinical experience is limited.
Endothelin-1 receptor antagonists (ERAs)
Endothelin-1 (ET-1) is a potent vasoconstrictor and a smooth-muscle mitogen that contributes to the increased vascular tone and to the pulmonary vascular hypertrophy associated with PAH. The activation of the ET-1 system has been clearly demonstrated in both plasma and lung tissues of PAH patients.[39,40] Two distinct endothelin-receptor types located in vascular smooth muscle cells have been identified, ETA and ETB.[41] Activation of these receptors facilitates vasoconstriction and proliferation of vascular smooth-muscle cells. ETB receptors are also present in the endothelial cells, where their activation principally promotes the clearance of endothelin in the vascular beds of the lung and kidney, and also cause vasodilation and NO release.[41]
Bosentan
Class of recommendation: I & Level of evidence: A FDA Approval: 2001
Bosentan is a non-specific oral dual ETA and ETB receptor antagonist.[42] The efficacy of bosentan in IPAH has been evaluated in many randomized clinical trial (RCTs) that have shown significant improvement in exercise capacity, modified NYHA functional class, hemodynamics, and time to clinical worsening.[43,44] One RCT enrolled exclusively milder patients with modified NYHA FC II and has confirmed the efficacy of bosentan in slowing down the disease progression at this early stage of the disease.[45]
The role of bosentan in PAH associated with congenital heart disease (CHD) was also evaluated. A multicenter, 16-week, double-blind, randomized, and placebo-controlled study of bosentan therapy in patients with Eisenmenger syndrome (BREATH-5) has reported a significant improvements in both hemodynamic parameters and exercise tolerance without worsening of oxygen saturation in this group of patients.[46]
The use of bosentan in children 4-17 years of age, with PAH was evaluated in an open-label, uncontrolled single and multiple-dose study (BREATHE -3), in which a significant improvement in hemodynamics was observed after 12 weeks of treatment either with bosentan alone or in combination with epoprostenol.[47]
The effect of bosentan on survival was also evaluated. Observed survival up to 3 years was reported as Kaplan-Meier estimates and compared with predicted survival as determined for each patient by the National Institutes of Health (NIH) Registry formula.
Kaplan-Meier bosentan survival estimates were 96% at 1 year and 89% at 2 years, while the predicted survival was 69% and 57%, respectively.[48]
A dose dependent hepatic toxicity is the most serious side effect of bosentan, and so the US Federal Drug Association requires that liver function tests be performed at least monthly in patients receiving bosentan. Also mild anemia is a frequently reported complication of long-term bosentan treatment and serial checking for hemoglobin level is recommended.
Women in childbearing age should be informed about the potential teratogenicity of the drug and proper contraceptive methods must be employed. Drug-drug interaction with contraceptive medication has been noticed with bosentan, and so oral contraceptive pills should not be used as a sole mechanism for contraception. Regular pregnancy testing is recommended in women of childbearing age. Finally, there is a concern that the endothelin antagonists as a class may cause testicular atrophy and male infertility. Younger males who may consider conceiving should be counseled regarding this potential side effect before taking these drugs.
The bosentan starting dose is 62.5 mg twice daily for 4 weeks, and if liver function tests are stable, then the dose should be increased to 125 mg twice daily.
Ambrisentan
Class of recommendation: I & Level of evidence: A FDA Approval: 2007
Two Phase III clinical trials (ARIES-1 & ARIES-2) have confirmed the efficacy of ambrisentan in the treatment of PAH patients.[49] The drug has shown significant efficacy in improving exercise capacity, modified NYHA functional class, hemodynamics, and the time to clinical worsening in this group of patients. Ambrisentan also has a possible survival benefit when compared with predictive survival based on the NIH registry data.
The most frequent side effect of ambrisentan is headache, which occurs in about 10% of the patients. Hepatic toxicity, however, has not been reported in ambrisentan-treated patients. Furthermore, ambrisentan has no apparent drug-drug interaction with sildenafil or warfarin. Finally, similar to bosentan, ambrisentan is contraindicated in pregnancy. There is a concern regarding the use of ambrisentan in patients with concurrent idiopathic pulmonary fibrosis (IPF) as a randomized placebo-controlled trial of patients with IPF found that ambrisentan was associated with an increased risk of disease progression and hospitalizations.[50]
Ambrisentan is used as a 5 or 10 mg single daily dose.
Sitaxsentan
Sitaxsentan was withdrawn from the market because of hepatic toxicity.
Macitentan
Class of recommendation: I & Level of evidence: A FDA Approval: 2013
Macitentan is a novel dual endothelin receptors antagonist with sustained receptor binding that holds great promise as a therapy for patients with PAH. The long-term efficacy and safety of macitentan in PAH patients has been recently reported in a large, multicenter, double-blinded, placebo-controlled, event-driven, phase III trial, the SERAPHIN study.[51] Macitentan at 2 different doses (3 mg and 10 mg once daily) was shown to significantly reduce both morbidity and mortality in PAH patients. Patients receiving the 10 mg dose had 45% reduction in morbidity and mortality compared to placebo. The drug was well tolerated and no hepatic toxicity was noted.
Type 5-phosphodiesterase inhibitors
The vasodilator effect of nitric oxide (NO) is dependent on its ability to sustain cGMP content in vascular smooth muscle. The half-life of intracellular cGMP is short secondary to the rapid degradation by phosphodiesterase enzymes; the most important is phosphodiesterase type 5 (PDE-5).[52,53] PDE-5 gene expression and activity are found to be increased in chronic PH and PDE-5 is strongly expressed in the lungs.[54,55]
Sildenafil
Class of recommendation: I & Level of evidence: A FDA Approval: 2005
Sildenafil is a highly specific PDE-5 inhibitor and has been shown in numerous studies to have favorable effects in many forms of PAH.[56,57,58] A pivotal RCT (SUPER-1) evaluated the efficacy and safety of oral sildenafil 20 mg, 40 mg, and 80 mg 3 times daily versus placebo, for 12 weeks, in the treatment 278 WHO functional class II and III PAH patients (≥18 years of age and have either idiopathic or associated with connective tissue disease or with repaired congenital systemic-to-pulmonary shunts).[59] Compared to placebo, all sildenafil doses led to significant improvement in symptoms, 6MWT, and hemodynamic parameters at week 12. Although all doses showed clear short-term efficacy, the 1-year durability effect has been demonstrated only with the higher dose of 80 mg. However, recent study has raised concern about the safety of high dose sildenafil in pediatric PAH patients,[60] and subsequently the FDA has released a warning letter regarding using sildenafil in this group of patients. Despite the FDA warning, both the European Medicine Evaluation (EMEA) and the Saudi Association for Pulmonary Hypertension (SAPH) have released a safety letter concerning the use of moderate to high doses of sildenafil in pediatric PAH patients, and as such most pediatric PH specialist in Europe and the Middle East have continued using sildenafil in pediatric populations, although the lowest effective dose is always chosen.
From a broader perspective, the acute hemodynamic effect of sildenafil has been shown to be comparable to that of inhaled NO (iNO), while sildenafil plus iNO was more effective than iNO alone.[61] Finally, sildenafil has also been shown to inhibit high altitude-induced pulmonary hypertension.[62] In this study, sildenafil was started (at the dose of 40 mg T.I.D) 6 to 8 hours after arrival to high altitude and maintained for the whole duration.
In general, sildenafil treatment is well tolerated. Headache, nasal congestion, flushing, and visual disturbances are the most widely reported side effects and related to the vasodilatation.
Tadalafil
Class of recommendation: I & Level of evidence: A FDA Approval: 2009
Tadalafil is another selective phosphodiesterase type-5 inhibitor that shows efficacy in PAH management.
A large RCT (PHIRST) on 406 PAH patients (approximately 50% were on background bosentan therapy) treated with tadalafil 5, 10, 20, or 40 mg once daily has shown favorable results on exercise capacity, modified NYHA functional class, hemodynamics, and time to clinical worsening at the largest dose.[63] The one-year durability of the effect has also been shown in an open extension study.
Tadalafil is used as a single daily dose of 20-40 mg and the side effect profile is similar to that of sildenafil.
Guanylate cyclase stimulators
Stimulators of the nitric oxide receptor, soluble guanylate cyclase (sGC), have a dual mode of action. They increase the sensitivity of sGC to endogenous NO and they also directly stimulate the receptor by a mechanism, which is independently of NO.
Riociguat
Class of recommendation: I & Level of evidence: A FDA Approval: 2013
Riociguat is a novel oral sGC stimulant. In a recently published large RCT (PATENT) in PAH patients, riociguat has been shown to improve the 6MWT both in patients who were treatment naïve for the disease and in those who were receiving ERAs or prostanoids. Riociguat also improved hemodynamics, NT-pro BNP levels, modified NYHA functional class, time to clinical worsening, and Borg dyspnea score.[64]
Furthermore, riociguat is the first pharmacological therapy to show significant and sustained benefits in patients with inoperable chronic thromboembolic pulmonary hypertension (CTEPH). In a recently published RCT (CHEST) in inoperable CTEPH patients, riociguat significantly improved 6MWT in this group of patients. Pulmonary vascular resistance (PVR) decreased by 226 dyn/sec/cm-5 in the riociguat group compared to placebo, while NT-pro BNP and modified NYHA functional class significantly improved.[65]
In both studies, riociguat was tested in 2 different doses, 1.5 mg and 2.5 mg 3 times per day. The drug was well tolerated and the most common side effects were dizziness, hypotension, and syncope that were not significantly different compared to placebo. Hemoptysis was noticed in few patients and warrants further evaluation. Of note, riociguat should not be used along with PDE-5 inhibitors due to a risk of hypotension.
Tyrosine kinase inhibitors
A cancer biology theory has been proposed in PAH. Undoubtedly, PAH has many bio-pathological neoplastic features, including anti-apoptosis, upregulation of many growth factors, such as platelets derived growth factor, and a selective down-regulation of the potassium channels that result in increase in intracellular free Ca2+ and K+ concentrations, al of which play an important role in pulmonary artery smooth muscle cells (PASMC) contraction, proliferation and resistance to apoptosis.[66] All of these active “neoplastic”-like features suggested antineoplastic therapy could be a very attractive tool in the management of PAH patients.
Imatinib
Class of recommendation: IIb & Level of evidence: B FDA Approval: Not approved
Imatinib, a tyrosine kinase inhibitor, has shown good efficacy in improving clinical symptoms, exercise tolerance, and hemodynamics in phase II trials.[67]
However, a double-blinded placebo-controlled, randomized phase III trial (IMPRES) resulted in major concerns with using this drug in PAH patients. Despite the fact that patients treated with imatinib experienced better exercise tolerance (the primary endpoint) and significant improvement in hemodynamics, the time to clinical worsening and mortality were not different between the 2 groups.[68]
Furthermore, the side effects profile was a major concern in imatinib-treated patients (mainly cerebral hematoma). Certainly, more studies are needed before this group of drugs can be routinely considered in the management of PAH patients.
The class of recommendations and level of evidence for all specific drugs are summarized in table 11 in the main guideline.
Future treatment
New therapeutic strategies targeting different pathobiologic pathways are under phase II and III clinical trials.
The proliferation of pulmonary artery smooth muscle cells (PASMCs) in PAH is stimulated by serotonin due to increased expression of the serotonin transporter (5-HTT).[69] 5-HTT inhibitors may thus prove to be therapeutically efficacious in the management of PAH.
Thromboxane A2 is a powerful vasoconstrictor, platelet aggregant, and smooth muscle mitogen. Terbogrel, a potent orally active thromboxane synthetase inhibitor and thromboxane receptor antagonist, was effective from a pharmacologic standpoint in reducing thromboxane metabolites in PAH patients by as much as 98%.[70] However, the study had early termination because of the side effects in terbogrel group.
Selexipag is an oral prostacyclin (PGI2) receptor agonist that is clinically distinct from PGI2. Results of placebo-controlled, double blind, phase II study showed a statistically significant reduction in PVR (primary parameter for the study).[71] Results also showed an encouraging numerical improvement in 6MWT. Selexipag was well tolerated and the safety profile was in line with the expected pharmacologic effect. Selexipag is being evaluated in the Phase III GRIPHON trial. Final study results of this event-driven study are expected by mid-2014.
Additional compounds are at different stages of development: Inhaled vasoactive intestinal peptide, rho-kinase inhibitors, vascular endothelial growth factor receptor inhibitors, angiopoietin-1 inhibitors, and elastase inhibitors.
Gene therapy has been tested in different animal models. Stem cell therapy has proven to be effective in the monocrotalin rat model and is currently being evaluated in proof-of-concept and dose finding studies in PAH patients.
Combination therapy
Combination therapy refers to the simultaneous use of more than one PAH-specific class of drugs. Despite the absence of long-term data confirming the efficacy and safety of combination therapy, this strategy has become the standard of care in most PAH centers.
Combination therapy has been suggested in two different strategies, sequential or upfront combination strategy.
Sequential combination strategy
Class of recommendation: IIa & Level of evidence: B
A number of case series have suggested the efficacy and safety of sequential combination strategy in PAH patients who are not controlled on monotherapy. In one series, a step-wise use of combination therapy according to predefined treatment goals (so called “Goal oriented strategy”) was associated with an improved outcome compared with a historical control.[72]
The following combinations have been evaluated:
-
Prostacyclin and ERAs: The efficacy and safety of the concurrent initiation of bosentan and prostacyclin were investigated in 22 modified NYHA class III and IV PAH randomized to receive either epoprostenol and placebo or epoprostenol and bosentan (BREATHE-2).[73] There was a trend towards a greater improvement in hemodynamic parameters in the combination group but that did not reach statistical significance.
However, it has been concluded that the study was underpowered to allow for definite conclusions.[74] Another open-label study has addressed the combination of non-parenteral prostanoids and bosentan in 20 patients with IPAH.[75] Bosentan was used as an add-on medication because of insufficient response to prostanoid treatment. There was a significant increase in the 6 MWT distance by 58 ±43m and significant improvement in other secondary endpoints. Adding prostacyclin treatment to bosentan was also evaluated. The addition of inhaled treprostinil was evaluated in 12 patients with symptomatic PAH despite bosentan treatment.[76] Six-MWD, functional class, and hemodynamics were assessed at baseline and 12 weeks. Combination therapy was associated with an increase in 6MWD and significant improvement in both functional class and hemodynamics. Adding inhaled Iloprost to Bosentan (STEP 1) was tested in a small randomized, multicenter, double-blind trial.[77] Although this was primarily a safety study, efficacy data were also available for analysis.
Beside confirmation of safety, the combination of inhaled iloprost and bosentan showed efficacy in improving functional class, hemodynamics, and time to clinical worsening. The placebo-adjusted difference in 6MWT was not statistically significant (p = 0.051).
Another 2 studies evaluated the adding of treprostinil to either bosentan or sildenafil. The first study, TRIUMPH, tested the addition of inhaled treprostinil to modified NYHA functional class III or IV despite bosentan or sildenafil monotherapy.[78] The combination group had a larger improvement in the 6MWT and quality of life, but there were no differences in the time to clinical worsening, dyspnea, or modified NYHA functional class. The second study, FREEDOM-C, tested the addition of oral treprostinil to symptomatic PAH patients despite of using monotherapy of either bosentan or sildenafil.[79] The dose of treprostinil was increased to a median dose of 3 mg twice daily. Discontinuation rate of 22% was noted in the treprostinil group secondary to the high incidence (> 40%) of side effects of headache, nausea, vomiting, diarrhea, flushing, and jaw pain. However, significant improvements were noted in the secondary end-points of median dyspnea fatigue index score and combined 6MWD and Borg dyspnea score. It is important to mention, however, that oral treprostinil is an experimental compound that has not yet been proven efficacy as monotherapy and does not have a current clinical application.
Prostacyclin and phosphodiesterase-5 inhibitors: The benefit of adding sildenafil to inhaled iloprost in PAH patients has been evaluated and found to have a positive impact on exercise tolerance (6MWT) and hemodynamics.[80] A multi-national, multi-center, randomized, double-blind, placebo-controlled, parallel-group efficacy study (PACES-1) of sildenafil used in combination with IV prostacyclin to treat PAH has shown that the combination therapy improves exercise capacity, hemodynamic measurements, time to clinical worsening, and quality of life, but not Borg dyspnea score.[81] Finally, 30 patients with severe PAH (n = 16), CTEPH (n = 13), or PH due to aplasia of the left pulmonary artery (n = 1) (all classified as modified NYHA class III or IV) were evaluated in a randomized, controlled, open-label trial.[82] Patients were randomly assigned to receive 12.5 mg of oral sildenafil, 50 mg of sildenafil, combination of 12.5 mg of sildenafil plus inhaled iloprost, or combination 50 mg of sildenafil plus inhaled iloprost. Sildenafil 50 mg plus iloprost was the most effective “vasodilator regimen” as reflected by maximum reduction of pulmonary vascular resistance and increase in cardiac index, followed by 12.5 mg of sildenafil plus iloprost. Monotherapy with either drug was less potent than the combination regimens.
-
ERAs and phosphodiesterase-5 inhibitors: The combination of ERAs and PDE-5 inhibitors has been evaluated in few studies. In one study of severe IPAH patients on bosentan therapy, the addition of sildenafil was associated with improvement of exercise tolerance (both 6MWT VO2max as measured by CPET).[83] A different study looked at the benefit for adding sildenafil in patients with PAH who failed monotherapy with bosentan.[84]
Significant improvement was achieved after the combination, as measured by symptoms, exercise capacity, and modified NYHA functional class. Of note, the improvement was more prominent in IPAH patients, compared to patients with scleroderma-associated PAH.
In contrast to the other combination regimens, co-administration of bosentan and sildenafil may be associated with relevant pharmacokinetic interactions.[85] Sildenafil has inhibitory effects on cytochrome P450 3A4 (CYP3A4) activity, which may lead to increased plasma concentrations of bosentan; [Table 1]. Bosentan may exert hepatotoxic effects and there is always a concern about a higher risk of liver damage with combined administration of bosentan and sildenafil. None of the patients in the studies previously described experienced elevations in hepatic aminotransferases when sildenafil was added to bosentan, but the small number of patients precludes any meaningful safety analysis. On the other hand, induction of CYP3A4 activity by bosentan may accelerate the metabolism of sildenafil, which may decrease the plasma concentrations of sildenafil by as much as 60%.
Table 1.
PAH drugs dosing, contraindications, interactions, and precautions
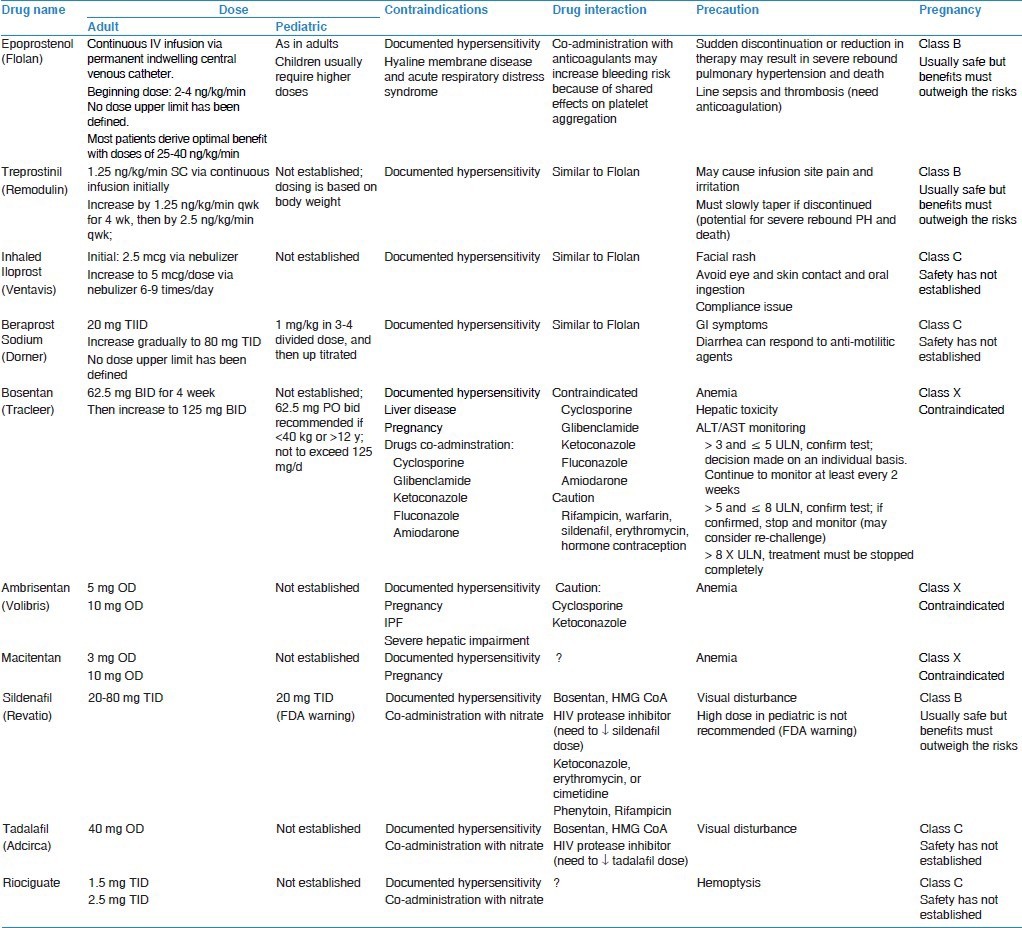
Upfront combination strategy
Class of recommendation: IIb & Level of evidence: C
The upfront combination strategy has gained recent interest among PH experts, especially in treating advanced PAH patients, who are known to have very poor prognosis. Few studies have addressed this new strategy. In one study, 2-drugs upfront combination therapy (ERAs + PDE-5 inhibitor or PDE-5 inhibitors + prostanoids) was started in PAH patients with modified NYHA functional class III or IV.[86] Most patients showed significant improvement in symptoms scoring, 6MWT, and hemodynamics parameters, without significant side effects. Another prospective observational analysis of the efficacy and safety of upfront triple combination therapy of IV epoprostenol, Bosentan, and sildenafil in severe idiopathic and heritable PAH patients has shown marked functional and hemodynamic improvement among most patients treated with this strategy.[87] The mean fall in PVR was 71% relative to baseline values, and most patients had a significant improvement in their functional class. The AMBITION study is a phase III trial comparing the upfront combination therapy of ambrisentan and tadalafil versus first-line monotherapy of either drug in subjects with PAH. The results of this study are expected in 2015 and should further help in evaluating the benefit of upfront combination strategy.
Atrial septostomy
Class of Recommendation = IIa (for NYHA FC IV); Level of Evidence = C
Many studies have suggested that atrial septostomy (AS) might be beneficial in the management of severe PH.[88,89] AS would allow right-to-left shunting and subsequently “decompress” the right ventricle. Furthermore, it should improve left heart filling leading to increased left ventricular “systemic” cardiac output. In selected patients, the net result should be towards improving oxygen delivery despite the fall in systemic arterial oxygen saturation.
The efficacy of AS in the treatment of PAH patients has been evaluated in small series.[90,91] In addition to symptomatic and hemodynamic improvement, an increase in survival as compared with historical control groups has also been reported.[92] At present, only patients with “severe PAH” who fail medical treatment should be considered as potential candidates for the procedure.
Procedural mortality has been reported to range from 5 to 50%. Appropriate selection of patients for AS procedure is crucial. Patients with markedly elevated PVR, low arterial oxygen saturations (<80% at rest), and those with severe right-heart failure appear more likely to have a higher complication rate and mortality following the procedure.[93]
AS should only be performed in specialized centers with adequate experience in this procedure.
Lung transplantation
Class of Recommendation = I (for NYHA FC IV); Level of Evidence = C
Heart-lung transplantation (HLT) and lung transplant (LT) were developed in the 1980s as a cure for adults with severe pulmonary vascular disease, specifically idiopathic PAH (IPAH) and complex cyanotic congenital heart disease (CHD). The LT or HLT is offered to the patients with PAH that have failed the maximum medical therapy. As the transplant procedure carries a substantial morbidity and mortality, it is only offered to the patients in whom the survival after LT is expected to be better than the survival without the transplant.
Timing of listing for lung transplant
Although the patients in PAH group share similarities in clinical presentation, pathobiology, and therapeutic approaches, they differ in long-term survival. This difference is persistent even with the administration of advanced therapies.[94] The CHD associated with PAH (CHD-APAH) patients have the best survival among the group,[95] while the connective tissue diseases associated with PAH (CTD-APAH) patients have the worst long term survival.[96] The HIV-APAH will not be discussed here as the HIV is considered a contraindication for lung transplant by the majority of centers. Venoocclusive disease is also an exception as there is no effective medical treatment available for this disease.
Table 2 summarizes the contraindication for LT in PAH patients.
Table 2.
Contraindication for lung transplantation in PAH
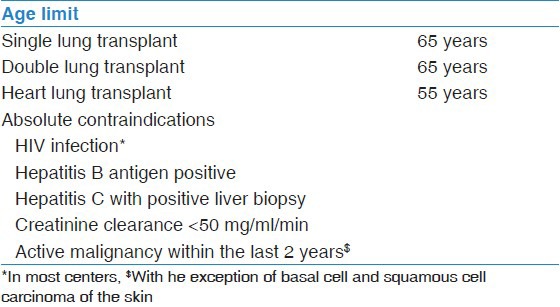
The ISHLT guidelines have given the following recommendations regarding LT in PAH:
-
IPAH:
-
Guidelines for transplantation referral:
- NYHA functional class III or IV, irrespective of treatment.
- Rapidly progressive disease.
-
Guidelines for listing/transplantation:
- NYHA FC III or IV despite optimal pulmonary vasodilator therapy, including intravenous prostanoid.
- Low (<350 m) or declining 6-min walk test (6MWT).
- Cardiac Index (CI) of < 2 l/min/m2.
- Mean right atrial pressure (mRAP) >15 mmHg.
-
-
CTD-APAH:
The guidelines recommendations are similar to IPAH; however, given the inferior long-term survival especially with scleroderma, an early referral is encouraged.[15]
-
CHD-APAH:
The decision for LT or HLT is more difficult in CHD-APAH patients, as this group has the best survival among all PAH groups. PAH targeted therapy has further improved survival in this group and the long survival especially after HLT remains inferior.
These patients generally have a slowly declining functional status with estimated survival of over twenty years in up to 80% of such patients. The median survival on the other hand after HLT is only 3.2 years.[97] As the transplant is ideally only offered to gain a survival advantage, it is difficult to decide the best timing to list these patients for transplant. There is general agreement however to list these patients for transplant if there is:
- Accelerated decline in functional status.
- More frequent hospitalizations.
- Recurrent massive hemoptysis.
-
Venoocclusive disease:
As there is no effective available medical treatment for venoocclusive disease, those patients should be referred early for LT.
Procedure of choice
The LT for pulmonary hypertension (PH) began with HLT. However with the improvements in the technique and the experience with bilateral (BLT) and single lung transplant (SLT), it was soon recognized that the right ventricular function recovers after the LT in patients with advanced (PH). Conte and colleagues reviewed the outcome of all SLT and BLT for idiopathic (primary) PAH or CTD-APAH or PH due to primary lung disease. In their review the patients with IPAH receiving BLT had better survival than the patients with SLT.[98] Most centers now offer BLT as the procedure of choice for patients with PAH. HLT is reserved only for patients with complex CHD where the cardiac defect cannot be repaired.
Lung transplant outcome for patients with PAH
The response of right ventricle to LT is immediate and remarkable. While the patients no longer require their PAH medications, they may require some support of the right ventricle as it recovers. The immediate postoperative course after LT for PAH can be stormy with significant morbidity and mortality in the first three months after the transplant.[99,100] This is judged by the difference in the overall median survival and the conditional survival after LT for PAH.[101] The median survival after lung transplant for PAH is 5.2 years.[101] On the other hand the conditional survival for the IPAH patients surviving the first three months after transplant is 8.9 years.[102]
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Galiè N, Manes A, Negro L, Palazzini M, Bacchi-Reggiani ML, Branzi A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J. 2009;30:394–403. doi: 10.1093/eurheartj/ehp022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Idrees M. Pulmonary hypertension: Another light in the dark tunnel. Learning the lesson from cancer. Ann Thorac Med. 2013;8:69–70. doi: 10.4103/1817-1737.109813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sitbon O, Humbert M, Ioos V, Jais X, Parent F, Garcia G, et al. Who benefits from longterm calcium-channel blocker therapy in primary pulmonary hypertension? Am J Respir Crit Care Med. 2003;167:A440. [Google Scholar]
- 4.Raffy O, Azarian R, Brenot F, Parent F, Sitbon O, Petitpretz P, et al. Clinical significance of the pulmonary vasodilator response during short-term infusion of prostacyclin in primary pulmonary hypertension. Circulation. 1996;93:484–8. doi: 10.1161/01.cir.93.3.484. [DOI] [PubMed] [Google Scholar]
- 5.Rich S, Kaufmann E, Levy PS. The effect of high doses of calciumchannel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327:76–81. doi: 10.1056/NEJM199207093270203. [DOI] [PubMed] [Google Scholar]
- 6.Rich S, Brundage BH. High-dose calcium channel-blocking therapy for primary pulmonary hypertension: Evidence for longterm reduction in pulmonary arterial pressure and regression of right ventricular hypertrophy. Circulation. 1987;76:135–41. doi: 10.1161/01.cir.76.1.135. [DOI] [PubMed] [Google Scholar]
- 7.Sitbon O, Humbert M, Jaïs X, Ioos V, Hamid AM, Provencher S, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111:3105–11. doi: 10.1161/CIRCULATIONAHA.104.488486. [DOI] [PubMed] [Google Scholar]
- 8.Yung D, Widlitz AC, Rosenzweig EB, Kerstein D, Maislin G, Barst RJ. Outcomes in children with idiopathic pulmonary arterial hypertension. Circulation. 2004;110:660–5. doi: 10.1161/01.CIR.0000138104.83366.E9. [DOI] [PubMed] [Google Scholar]
- 9.Jones DA, Benjamin CW, Linseman DA. Activation of thromboxane and prostacyclin receptors elicits opposing effects on vascular smooth muscle cell growth and mitogen-activated protein kinase signaling cascades. Mol Pharmacol. 1995;48:890–6. [PubMed] [Google Scholar]
- 10.Galiè N, Manes A, Branzi A. Prostanoids for pulmonary arterial hypertension. Am J Respir Med. 2003;2:123–37. doi: 10.1007/BF03256644. [DOI] [PubMed] [Google Scholar]
- 11.Clapp LH, Finney P, Turcato S, Tran S, Rubin LJ, Tinker A. Differential effects of stable prostacyclin analogs on smooth muscle proliferation and cyclic AMP generation in human pulmonary artery. Am J Respir Cell Mol Biol. 2002;26:194–201. doi: 10.1165/ajrcmb.26.2.4695. [DOI] [PubMed] [Google Scholar]
- 12.Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334:296–301. doi: 10.1056/NEJM199602013340504. [DOI] [PubMed] [Google Scholar]
- 13.McLaughlin VV, Genthner DE, Panella MM, Rich S. Reduction in pulmonary vascular resistance with long-term epoprostenol (prostacyclin) therapy in primary pulmonary hypertension. N Engl J Med. 1998;338:273–7. doi: 10.1056/NEJM199801293380501. [DOI] [PubMed] [Google Scholar]
- 14.Rubin LJ, Mendoza J, Hood M, McGoon M, Barst R, Williams WB, et al. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann Intern Med. 1990;112:485–91. doi: 10.7326/0003-4819-112-7-485. [DOI] [PubMed] [Google Scholar]
- 15.Badesch DB, Tapson VF, McGoon MD, Brundage BH, Rubin LJ, Wigley FM, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med. 2000;132:425–34. doi: 10.7326/0003-4819-132-6-200003210-00002. [DOI] [PubMed] [Google Scholar]
- 16.Robbins IM, Gaine SP, Schilz R, Tapson VF, Rubin LJ, Loyd JE. Epoprostenol for treatment of pulmonary hypertension in patients with systemic lupus erythematosus. Chest. 2000;117:14–8. doi: 10.1378/chest.117.1.14. [DOI] [PubMed] [Google Scholar]
- 17.McLaughlin VV, Genthner DE, Panella MM, Hess DM, Rich S. Compassionate use of continuous prostacyclin in the management of secondary pulmonary hypertension: A case series. Ann Intern Med. 1999;130:740–3. doi: 10.7326/0003-4819-130-9-199905040-00014. [DOI] [PubMed] [Google Scholar]
- 18.Kuo PC, Johnson LB, Plotkin JS, Howell CD, Bartlett ST, Rubin LJ. Continuous intravenous infusion of epoprostenol for the treatment of portopulmonary hypertension. Transplantation. 1997;63:604–6. doi: 10.1097/00007890-199702270-00020. [DOI] [PubMed] [Google Scholar]
- 19.Aguilar RV, Farber HW. Epoprostenol (prostacyclin) therapy in HIV-associated pulmonary hypertension. Am J Respir Crit Care Med. 2000;162:1846–50. doi: 10.1164/ajrccm.162.5.2004042. [DOI] [PubMed] [Google Scholar]
- 20.Rosenzweig EB, Kerstein D, Barst RJ. Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation. 1999;99:1858–65. doi: 10.1161/01.cir.99.14.1858. [DOI] [PubMed] [Google Scholar]
- 21.McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: The impact of epoprostenol therapy. Circulation. 2002;106:1477–82. doi: 10.1161/01.cir.0000029100.82385.58. [DOI] [PubMed] [Google Scholar]
- 22.Sitbon O, Humbert M, Nunes H, Parent F, Garcia G, Hervé P, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: Prognostic factors and survival. J Am Coll Cardiol. 2002;40:780–8. doi: 10.1016/s0735-1097(02)02012-0. [DOI] [PubMed] [Google Scholar]
- 23.Geohas C, McLaughlin VV. Successful management of pregnancy in a patient with eisenmenger syndrome with epoprostenol. Chest. 2003;124:1170–3. doi: 10.1378/chest.124.3.1170. [DOI] [PubMed] [Google Scholar]
- 24.Simonneau G, Barst RJ, Galie N, Naeije R, Rich S, Bourge RC, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: A double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165:800–4. doi: 10.1164/ajrccm.165.6.2106079. [DOI] [PubMed] [Google Scholar]
- 25.Lang I, Gomez-Sanchez M, Kneussl M, Naeije R, Escribano P, Skoro-Sajer N, et al. Efficacy of long-term subcutaneous treprostinil sodium therapy in pulmonary hypertension. Chest. 2006;129:1636–43. doi: 10.1378/chest.129.6.1636. [DOI] [PubMed] [Google Scholar]
- 26.Barst RJ, Galie N, Naeije R, Simonneau G, Jeffs R, Arneson C, et al. Long-term outcome in pulmonary arterial hypertension patients treated with subcutaneous treprostinil. Eur Respir J. 2006;28:1195–203. doi: 10.1183/09031936.06.00044406. [DOI] [PubMed] [Google Scholar]
- 27.Benza RL, Rayburn BK, Tallaj JA, Pamboukian SV, Bourge RC. Treprostinil-based therapy in the treatment of moderate-tosevere pulmonary arterial hypertension: Long-term efficacy and combination with bosentan. Chest. 2008;134:139–45. doi: 10.1378/chest.07-2111. [DOI] [PubMed] [Google Scholar]
- 28.Tapson VF, Gomberg-Maitland M, McLaughlin VV, Benza RL, Widlitz AC, Krichman A, et al. Safety and efficacy of IV treprostinil for pulmonary arterial hypertension: A prospective, multicenter, open-label, 12-week trial. Chest. 2006;129:683–8. doi: 10.1378/chest.129.3.683. [DOI] [PubMed] [Google Scholar]
- 29.Vachiéry JL, Hill N, Zwicke D, Barst R, Blackburn S. Naeije Transitioning from i.v. epoprostenol to subcutaneous treprostinil in pulmonary arterial hypertension. Chest. 2002;121:1561–5. doi: 10.1378/chest.121.5.1561. [DOI] [PubMed] [Google Scholar]
- 30.Gomberg-Maitland M, Tapson VF, Benza RL, McLaughlin VV, Krichman A, Widlitz AC, et al. Transition from intravenous epoprostenol to intravenous treprostinil in pulmonary hypertension. Am J Respir Crit Care Med. 2005;172:1586–9. doi: 10.1164/rccm.200505-766OC. [DOI] [PubMed] [Google Scholar]
- 31.Hoeper MM, Olschewski H, Ghofrani HA, Wilkens H, Winkler J, Borst MM, et al. A comparison of the acute hemodynamic effects of inhaled nitric oxide and aerosolized iloprost in primary pulmonary hypertension. German PPH Study Group. J Am Coll Cardiol. 2000;35:176–82. doi: 10.1016/s0735-1097(99)00494-5. [DOI] [PubMed] [Google Scholar]
- 32.Idrees MM, Batubara E, Kashour T. Novel approach for the management of sub-massive pulmonary embolism. Ann Thorac Med. 2012;7:157–61. doi: 10.4103/1817-1737.98850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olschewski H, Simonneau G, Galiè N, Higenbottam T, Naeije R, Rubin LJ, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347:322–9. doi: 10.1056/NEJMoa020204. [DOI] [PubMed] [Google Scholar]
- 34.Hoeper MM, Schwarze M, Ehlerding S, Adler-Schuermeyer A, Spiekerkoetter E, Niedermeyer J, et al. Long-term treatment of primary pulmonary hypertension with aerosolized iloprost, a prostacyclin analogue. N Engl J Med. 2000;342:1866–70. doi: 10.1056/NEJM200006223422503. [DOI] [PubMed] [Google Scholar]
- 35.Opitz CF, Wensel R, Winkler J, Halank M, Bruch L, Kleber FX, et al. Clinical efficacy and survival with first-line inhaled iloprost therapy in patients with idiopathic pulmonary arterial hypertension. Eur Heart J. 2005;26:1895–902. doi: 10.1093/eurheartj/ehi283. [DOI] [PubMed] [Google Scholar]
- 36.Elliot CA, Stewart P, Webster VJ, Mills GH, Hutchinson SP, Howarth ES, et al. The use of iloprost in early pregnancy in patients with pulmonary arterial hypertension. Eur Respir J. 2005;26:168–73. doi: 10.1183/09031936.05.00128504. [DOI] [PubMed] [Google Scholar]
- 37.Galiè N, Humbert M, Vachiéry JL, Vizza CD, Kneussl M, Manes A, et al. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: A randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2002;39:1496–502. doi: 10.1016/s0735-1097(02)01786-2. [DOI] [PubMed] [Google Scholar]
- 38.Barst RJ, McGoon M, McLaughlin V, Tapson V, Rich S, Rubin L, et al. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2003;41:2119–25. doi: 10.1016/s0735-1097(03)00463-7. [DOI] [PubMed] [Google Scholar]
- 39.Stewart DJ, Levy RD, Cernacek P, Langleben D. Increased plasma endothelin-1 in pulmonary hypertension: Marker or mediator of disease? Ann Intern Med. 1991;114:464–9. doi: 10.7326/0003-4819-114-6-464. [DOI] [PubMed] [Google Scholar]
- 40.Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328:1732–9. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- 41.Benigni A, Remuzzi G. Endothelin antagonists. Lancet. 1999;353:133–8. doi: 10.1016/S0140-6736(98)09423-9. [DOI] [PubMed] [Google Scholar]
- 42.Clozel M, Breu V, Gray GA, Löffler BM. In vivo pharmacology of Ro 46-2005, the first synthetic nonpeptide endothelin receptor antagonist: Implications for endothelin physiology. J Cardiovasc Pharmacol. 1993;22(Suppl 8):S377–9. doi: 10.1097/00005344-199322008-00099. [DOI] [PubMed] [Google Scholar]
- 43.Galiè N, Hinderliter AL, Torbicki A, Fourme T, Simonneau G, Pulido T, et al. Effects of the oral endothelin-receptor antagonist bosentan on echocardiographic and doppler measures in patients with pulmonary arterial hypertension. J Am Coll Cardiol. 2003;41:1380–6. doi: 10.1016/s0735-1097(03)00121-9. [DOI] [PubMed] [Google Scholar]
- 44.Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903. doi: 10.1056/NEJMoa012212. [DOI] [PubMed] [Google Scholar]
- 45.Galiè N, Rubin LJ, Hoeper M, Jansa P, Al-Hiti H, Meyer G, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): A doubleblind, randomised controlled trial. Lancet. 2008;371:2093–100. doi: 10.1016/S0140-6736(08)60919-8. [DOI] [PubMed] [Google Scholar]
- 46.Galiè N, Beghetti M, Gatzoulis MA, Granton J, Berger RM, Lauer A, et al. Bosentan therapy in patients with Eisenmenger syndrome: A multicenter, double-blind, randomized, placebocontrolled study. Circulation. 2006;114:48–54. doi: 10.1161/CIRCULATIONAHA.106.630715. [DOI] [PubMed] [Google Scholar]
- 47.Barst RJ, Ivy D, Dingemanse J, Widlitz A, Schmitt K, Doran A, et al. Pharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertension. Clin Pharmacol Ther. 2003;73:372–82. doi: 10.1016/s0009-9236(03)00005-5. [DOI] [PubMed] [Google Scholar]
- 48.McLaughlin VV, Sitbon O, Badesch DB, Barst RJ, Black C, Galiè N, et al. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J. 2005;25:244–9. doi: 10.1183/09031936.05.00054804. [DOI] [PubMed] [Google Scholar]
- 49.Oudiz R, Torres F, Frost A, Badesch DB, Olschewski H, Galie N, et al. ARIES-1: A placebo-controlled efficacy and safety study of ambrisentan in patients with pulmonary arterial hypertension. Chest. 2006;130:121S. [Google Scholar]
- 50.Raghu G, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: A parallel, randomized trial. Ann Intern Med. 2013;158:641–9. doi: 10.7326/0003-4819-158-9-201305070-00003. [DOI] [PubMed] [Google Scholar]
- 51.Pulido T, Adzerikho I, Channick RN, Delcroix M, Galiè N, Ghofrani HA, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–18. doi: 10.1056/NEJMoa1213917. [DOI] [PubMed] [Google Scholar]
- 52.Beavo JA, Reifsnyder DH. Primary sequence of cyclic nucleotide phosphodiesterase isozymes and the design of selective inhibitors. Trends Pharmacol Sci. 1990;11:150–5. doi: 10.1016/0165-6147(90)90066-H. [DOI] [PubMed] [Google Scholar]
- 53.Ahn HS, Foster M, Cable M, Pitts BJ, Sybertz EJ. Ca/CaMstimulated and cGMP-specific phosphodiesterases in vascular and non-vascular tissues. Adv Exp Med Biol. 1991;308:191–7. doi: 10.1007/978-1-4684-6015-5_15. [DOI] [PubMed] [Google Scholar]
- 54.Hanson KA, Burns F, Rybalkin SD, Miller JW, Beavo J, Clarke WR. Developmental changes in lung cGMP phosphodiesterase-5 activity, protein, and message. Am J Respir Crit Care Med. 1998;158:279–88. doi: 10.1164/ajrccm.158.1.9711042. [DOI] [PubMed] [Google Scholar]
- 55.Hanson KA, Ziegler JW, Rybalkin SD, Miller JW, Abman SH, Clarke WR. Chronic pulmonary hypertension increases fetal lung cGMP phosphodiesterase activity. Am J Physiol. 1998;275:L931–41. doi: 10.1152/ajplung.1998.275.5.L931. [DOI] [PubMed] [Google Scholar]
- 56.Prasad S, Wilkinson J, Gatzoulis MA. Sildenafil in primary pulmonary hypertension. N Engl J Med. 2000;343:1342. doi: 10.1056/NEJM200011023431814. [DOI] [PubMed] [Google Scholar]
- 57.Bhatia S, Frantz RP, Severson CJ, Durst LA, McGoon MD. Immediate and long-term hemodynamic and clinical effects of sildenafil in patients with pulmonary arterial hypertension receiving vasodilator therapy. Mayo Clin Proc. 2003;78:1207–13. doi: 10.4065/78.10.1207. [DOI] [PubMed] [Google Scholar]
- 58.Michelakis ED, Tymchak W, Noga M, Webster L, Wu XC, Lien D, et al. Long-term treatment with oral sildenafil is safe and improves functional capacity and hemodynamics in patients with pulmonary arterial hypertension. Circulation. 2003;108:2066–9. doi: 10.1161/01.CIR.0000099502.17776.C2. [DOI] [PubMed] [Google Scholar]
- 59.Galiè N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–57. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 60.Barst RJ, Ivy DD, Gaitan G, Szatmari A, Rudzinski A, Garcia AE, et al. A randomized, double-blind, placebo-controlled, dose-ranging study of oral sildenafil citrate in treatment-naive children with pulmonary arterial hypertension. Circulation. 2012;125:324–34. doi: 10.1161/CIRCULATIONAHA.110.016667. [DOI] [PubMed] [Google Scholar]
- 61.Michelakis E, Tymchak W, Lien D, Webster L, Hashimoto K, Archer S. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: Comparison with inhaled nitric oxide. Circulation. 2002;105:2398–403. doi: 10.1161/01.cir.0000016641.12984.dc. [DOI] [PubMed] [Google Scholar]
- 62.Richalet JP, Gratadour P, Robach P, Pham I, Déchaux M, Joncquiert-Latarjet A, et al. Sildenafil inhibits altitude-induced hypoxemia and pulmonary hypertension. Am J Respir Crit Care Med. 2005;171:275–81. doi: 10.1164/rccm.200406-804OC. [DOI] [PubMed] [Google Scholar]
- 63.Galiè N, Brundage BH, Ghofrani HA, Oudiz RJ, Simonneau G, Safdar Z, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119:2894–903. doi: 10.1161/CIRCULATIONAHA.108.839274. [DOI] [PubMed] [Google Scholar]
- 64.Ghofrani HA, Galiè N, Grimminger F, Grünig E, Humbert M, Jing ZC, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369:330–40. doi: 10.1056/NEJMoa1209655. [DOI] [PubMed] [Google Scholar]
- 65.Ghofrani HA, D’Armini AM, Grimminger F, Hoeper MM, Jansa P, Kim NH, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013;369:319–29. doi: 10.1056/NEJMoa1209657. [DOI] [PubMed] [Google Scholar]
- 66.Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, et al. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A. 2007;104:11418–23. doi: 10.1073/pnas.0610467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ghofrani HA, Morrell NW, Hoeper MM, Olschewski H, Peacock AJ, Barst RJ, et al. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am J Respir Crit Care Med. 2010;182:1171–7. doi: 10.1164/rccm.201001-0123OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galié N, et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: Results of the randomized IMPRES study. Circulation. 2013;127:1128–38. doi: 10.1161/CIRCULATIONAHA.112.000765. [DOI] [PubMed] [Google Scholar]
- 69.Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F, et al. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest. 2001;108:1141–50. doi: 10.1172/JCI12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langleben D, Christman BW, Barst RJ, Dias VC, Galiè N, Higenbottam TW, et al. Effects of the thromboxane synthetase inhibitor and receptor antagonist terbogrel in patients with primary pulmonary hypertension. Am Heart J. 2002;143:E4. doi: 10.1067/mhj.2002.121806. [DOI] [PubMed] [Google Scholar]
- 71.Simonneau G, Lang I, Torbicki A, Lang I, Hoeper MM, Delcroix M, Karlocai K, et al. Efficacy, Safety And Tolerability Of ACT-293987, A Novel Oral, Non-prostanoid, Prostaglandin I2 (IP) Receptor Agonist: Results From. Am J Respir Crit Care Med. 2010;181:A2515. [Google Scholar]
- 72.Hoeper MM, Markevych I, Spiekerkoetter E, Welte T, Niedermeyer J. Goal-oriented treatment and combination therapy for pulmonary arterial hypertension. Eur Respir J. 2005;26:858–63. doi: 10.1183/09031936.05.00075305. [DOI] [PubMed] [Google Scholar]
- 73.Humbert M, Barst RJ, Robbins IM, Channick RN, Galiè N, Boonstra A, et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J. 2004;24:353–9. doi: 10.1183/09031936.04.00028404. [DOI] [PubMed] [Google Scholar]
- 74.Hoeper MM, Dinh-Xuan AT. Combination therapy for pulmonary arterial hypertension: Still more questions than answers. Eur Respir J. 2004;24:339–40. doi: 10.1183/09031936.04.00072104. [DOI] [PubMed] [Google Scholar]
- 75.Hoeper MM, Taha N, Bekjarova A, Gatzke R, Spiekerkoetter E. Bosentan treatment in patients with primary pulmonary hypertension receiving nonparenteral prostanoids. Eur Respir J. 2003;22:330–4. doi: 10.1183/09031936.03.00008003. [DOI] [PubMed] [Google Scholar]
- 76.Channick RN, Olschewski H, Seeger W, Staub T, Voswinckel R, Rubin LJ. Safety and efficacy of inhaled treprostinil as add-on therapy to bosentan in pulmonary arterial hypertension. J Am Coll Cardiol. 2006;48:1433–7. doi: 10.1016/j.jacc.2006.05.070. [DOI] [PubMed] [Google Scholar]
- 77.McLaughlin VV, Oudiz RJ, Frost A, Tapson VF, Murali S, Channick RN, et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174:1257–63. doi: 10.1164/rccm.200603-358OC. [DOI] [PubMed] [Google Scholar]
- 78.McLaughlin VV, Benza RL, Rubin LJ, Channick RN, Voswinckel R, Tapson VF, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: A randomized controlled clinical trial. J Am Coll Cardiol. 2010;55:1915–22. doi: 10.1016/j.jacc.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 79.Tapson VF, Torres F, Kermeen F, Keogh AM, Allen RP, Frantz RP, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): A randomized controlled trial. Chest. 2012;142:1383–90. doi: 10.1378/chest.11-2212. [DOI] [PubMed] [Google Scholar]
- 80.Ghofrani HA, Rose F, Schermuly RT, Olschewski H, Wiedemann R, Kreckel A, et al. Oral sildenafil as long-term adjunct therapy to inhaled iloprost in severe pulmonary arterial hypertension. J Am Coll Cardiol. 2003;42:158–64. doi: 10.1016/s0735-1097(03)00555-2. [DOI] [PubMed] [Google Scholar]
- 81.Simonneau G, Rubin LJ, Galiè N, Barst RJ, Fleming TR, Frost AE, et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: A randomized trial. Ann Intern Med. 2008;149:521–30. doi: 10.7326/0003-4819-149-8-200810210-00004. [DOI] [PubMed] [Google Scholar]
- 82.Ghofrani HA, Wiedemann R, Rose F, Olschewski H, Schermuly RT, Weissmann N, et al. Combination therapy with oral sildenafil and inhaled iloprost for severe pulmonary hypertension. Ann Intern Med. 2002;136:515–22. doi: 10.7326/0003-4819-136-7-200204020-00008. [DOI] [PubMed] [Google Scholar]
- 83.Hoeper MM, Faulenbach C, Golpon H, Winkler J, Welte T, Niedermeyer J. Combination therapy with bosentan and sildenafil in idiopathic pulmonary arterial hypertension. Eur Respir J. 2004;24:1007–10. doi: 10.1183/09031936.04.00051104. [DOI] [PubMed] [Google Scholar]
- 84.Mathai SC, Girgis RE, Fisher MR, Champion HC, Housten-Harris T, Zaiman A, et al. Addition of sildenafil to bosentan monotherapy in pulmonary arterial hypertension. Eur Respir J. 2007;29:469–75. doi: 10.1183/09031936.00081706. [DOI] [PubMed] [Google Scholar]
- 85.Paul GA, Gibbs JS, Boobis AR, Abbas A, Wilkins MR. Bosentan decreases the plasma concentration of sildenafil when coprescribed in pulmonary hypertension. Br J Clin Pharmacol. 2005;60:107–12. doi: 10.1111/j.1365-2125.2005.02383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bachetti C, Palazzini M, Monti E, Albini A, Dardi F, Rinaldi A, et al. Efficacy and safety of upfront combination therapy. Am J Respir Crit Care Med. 2013;187:A3277. [Google Scholar]
- 87.Sitbon O, Jais X, Savale L, Dauphin C, Natali D, O’Callaghan D, et al. Upfront triple combination therapy of IV epoprostenol with oral Bosentan and sildenafil in idiopathic and heritable pulmonary arterial hypertension. Am J Respir Crit Care Med. 2011;183:A5910. [Google Scholar]
- 88.Austen WG, Morrow AG, Berry WB. Experimental studies of the surgical treatment of primary pulmonary hypertension. J Thorac Cardiovasc Surg. 1964;48:448–455. [PubMed] [Google Scholar]
- 89.Rozkovec A, Montanes P, Oakley CM. Factors that influence the outcome of primary pulmonary hypertension. Br Heart J. 1986;55:449–458. doi: 10.1136/hrt.55.5.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sandoval J, Rothman A, Pulido T. Atrial septostomy for pulmonary hypertension. Clin Chest Med. 2001;22:547–560. doi: 10.1016/s0272-5231(05)70291-4. [DOI] [PubMed] [Google Scholar]
- 91.Klepetko W, Mayer E, Sandoval J, Trulock EP, Vachiery JL, Dartevelle P, et al. Interventional and surgical modalities of treatment for pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:S73–S80. doi: 10.1016/j.jacc.2004.02.039. [DOI] [PubMed] [Google Scholar]
- 92.Peacock A, Naeije R, Galie N, Reeves JT. End-points for clinical trials in pulmonary arterial hypertension. Eur Respir J. 2004;23:947–953. doi: 10.1183/09031936.04.00122204. [DOI] [PubMed] [Google Scholar]
- 93.Moscucci M, Dairywala IT, Chetcuti S, Mathew B, Li P, Rubenfire M, et al. Balloon atrial septostomy in end-stage pulmonary hypertension guided a novel intracarduiac echocardiographic transducer. Catheter Cardiovasc Interv. 2001;52:530–4. doi: 10.1002/ccd.1116. [DOI] [PubMed] [Google Scholar]
- 94.Dimopoulos K, Inuzuka R, Goletto S, Giannakoulas G, Swan L, Wort SJ, et al. Improved survival among patients with Eisenmenger syndrome receiving advanced therapy for pulmonary arterial hypertension. Circulation. 2010;121:20–5. doi: 10.1161/CIRCULATIONAHA.109.883876. [DOI] [PubMed] [Google Scholar]
- 95.Kidd L, Driscoll DJ, Gersony WM, Hayes CJ, Keane JF, O’Fallon WM, et al. Second natural history study of congenital heart defects. Results of treatment of patients with ventricular septal defects. Circulation. 1993;87:I38–51. [PubMed] [Google Scholar]
- 96.Fisher MR, Mathai SC, Champion HC, Girgis RE, Housten-Harris T, Hummers L, et al. Clinical differences between idiopathic and scleroderma-related pulmonary hypertension. Arthritis Rheum. 2006;54:3043–50. doi: 10.1002/art.22069. [DOI] [PubMed] [Google Scholar]
- 97.Trulock EP. Lung and heart-lung transplantation: Overview of results. Semin Respir Crit Care Med. 2001;22:479–88. doi: 10.1055/s-2001-18420. [DOI] [PubMed] [Google Scholar]
- 98.Conte JV, Borja MJ, Patel CB, Yang SC, Jhaveri RM, Orens JB. Lung transplantation for primary and secondary pulmonary hypertension. Ann Thorac Surg. 2001;72:1673–9. doi: 10.1016/s0003-4975(01)03081-8. [DOI] [PubMed] [Google Scholar]
- 99.Ritchie M, Waggoner AD, Dávila-Román VG, Barzilai B, Trulock EP, Eisenberg PR. Echocardiographic characterization of the improvement in right ventricular function in patients with severe pulmonary hypertension after single-lung transplantation. J Am Coll Cardiol. 1993;22:1170–4. doi: 10.1016/0735-1097(93)90433-2. [DOI] [PubMed] [Google Scholar]
- 100.Katz WE, Gasior TA, Quinlan JJ, Lazar JM, Firestone L, Griffith BP, et al. Immediate effects of lung transplantation on right ventricular morphology and function in patients with variable degrees of pulmonary hypertension. J Am Coll Cardiol. 1996;27:384–91. doi: 10.1016/0735-1097(95)00502-1. [DOI] [PubMed] [Google Scholar]
- 101.Trulock EP, Edwards LB, Taylor DO, Boucek MM, Mohacsi PJ, Keck BM, et al. The Registry of the International Society for Heart and Lung Transplantation: Twentieth official adult lung and heart-lung transplant report - 2003. J Heart Lung Transplant. 2003;22:625–35. doi: 10.1016/s1053-2498(03)00182-7. [DOI] [PubMed] [Google Scholar]
- 102.Yusen RD, Christie JD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, et al. The Registry of the International Society for Heart and Lung Transplantation: Thirtieth adult lung and heart-lung transplant report - 2013; focus theme: Age. J Heart Lung Transplant. 2013;32:965–78. doi: 10.1016/j.healun.2013.08.007. [DOI] [PubMed] [Google Scholar]