

Abstract
Duchenne muscular dystrophy (DMD), a severe muscle-wasting disease, is caused by mutations in the DMD gene, which encodes for the protein dystrophin. Its regulation is of therapeutic interest as even small changes in expression of functional dystrophin can significantly impact the severity of DMD. While tissue-specific distribution and transcriptional regulation of several DMD mRNA isoforms has been well characterized, the post-transcriptional regulation of dystrophin synthesis is not well understood. Here, we utilize qRTPCR and a quantitative dual-luciferase reporter assay to examine the effects of isoform specific DMD 5’ UTRs and the highly conserved DMD 3’ UTR on mRNA abundance and translational control of gene expression in C2C12 cells. The 5’ UTRs were shown to initiate translation with low efficiency in both myoblasts and myotubes. Whereas, two large highly conserved elements in the 3’ UTR, which overlap the previously described Lemaire A and D regions, increase mRNA levels and enhance translation upon differentiation of myoblasts into myotubes. The results presented here implicate an important role for DMD UTRs in dystrophin expression and delineate the cis-acting elements required for the myotube-specific regulation of steady-state mRNA levels and translational enhancer activity found in the DMD 3’ UTR.
Keywords: DMD, Duchenne Muscular Dystrophy, 3’ UTR, 5’ UTR, translational control
Introduction
The DMD gene is the largest known human gene, spanning more than 2 MB and comprising 79 exons. Tissue-specific promoters drive the expression of several isoforms of dystrophin, including those expressed in brain and muscle tissue, which differ only in their first exon, and several truncated isoforms that are derived from promoters found within the DMD gene [1, 2]. The predominant muscle isoform, dp427m, has been shown to play a structural role in maintaining muscle fiber integrity by connecting the cytoskeleton of muscle fibers to the extracellular matrix via the dystrophin-associated glycoprotein complex (DGC), and to mediate signal transduction cascades through the C-terminal domain of dystrophin [3, 4]. Mutations in the DMD gene cause the dystrophinopathies, Duchenne Muscular Dystrophy (DMD) and Becker Muscular Dystrophy (BMD) [2, 5]. Patients with DMD exhibit severe muscle degeneration, development of cardiac hypertrophy, an association with neurological disorders, and a life expectancy into the second decade of life. BMD patients exhibit muscle degeneration that occurs at a much slower rate and typically survive into late adulthood. The more severe DMD phenotype is typically caused by mutations that severely reduce or eliminate synthesis of functional dystrophin protein, such as those that disrupt the reading frame [2, 6-8]. Whereas, the less severe BMD is caused by mutations that maintain the open reading frame and allow for residual expression of functional dystrophin protein [2, 6-8]. This observation and the finding that expression of even small amounts of dystrophin can lessen disease severity [9, 10] has increased interest in the mechanisms that control DMD expression and has also led to the development of several therapeutic approaches to treat DMD patients [11-13]. Examples of the latter include delivery of antisense molecules designed to induce skipping of mutated exons [14-20], premature stop codon suppression therapies [21-24] and the delivery and expression of minidystrophin gene constructs lacking non-essential DMD coding exons [25-33].
5’ UTRs have been shown to be important for determining translation efficiency, and, in some cases, mutations in this region can be pathogenic [34, 35]. The length, GC content and secondary structures of a 5’ UTR all have an impact on translation. mRNAs with high translation rates often contain 5’ UTRs that are short, with low GC content, and little secondary structure. Whereas, mRNAs with a low translation rate, or those that are regulated in a tissue-specific manner or involved in developmental processes, often contain 5’ UTRs that are longer, more GC rich, and contain more secondary structure [34, 36]. Most of the DMD 5’ UTRs are longer and have a lower GC content compared to the average 5’ UTR (Table 1) [37, 38]. Internal ribosome entry sites (IRES) contained within 5’ UTRs have also been shown to regulate gene expression. The 5’ UTR of utrophin, a homologue of dystrophin encoding a functionally-related protein, was shown to regulate translation during muscle regeneration through an IRES [39, 40]. Likewise, the retinal dystrophin 5’ UTR (dp260) was shown to contain a cryptic intron with IRES elements that regulates translation of the R-dystrophin transcript [41]. The functions of the 5’ UTRs for the remaining dystrophin isoforms are unknown, and may be involved in the post-transcriptional regulation of dystrophin synthesis in other tissues.
Table 1. Properties of DMD 5’ UTR isoforms.
The 5’ UTR length, percent GC content, and Kozak sequence is shown for the 5’ UTRs of several dystrophin isoforms along with tissues expressing each isoform. The average 5’ UTR length and percent GC content is shown for human 5’ UTRs along with the consensus Kozak sequence. The length of most DMD 5’ UTRs are longer than average and have a GC content that is less than the average human 5’ UTR. The Kozak sequence for each 5’ UTR isoform and exon 1 is shown with the -3 and +4 positions underlined. The DMD 5’ UTRs have several changes compared to the consensus sequence.
| Isoform | 5’ UTR Length | 5’ UTR %GC | Kozak Sequence | Isoform Expression |
|---|---|---|---|---|
| Average 5’ UTR | 210.2 [26] | 58.4% [27] | gccrccAUGG | N/A |
| Dp427m | 244 bp | 38.9% | uucaaaAUGC | muscle |
| Dp427c | 344 bp | 44.2% | gcuggcAUGG | brain |
| Dp427p1 | 263 bp | 45.2% | uuugaaAUGU | Purkinje cells |
| Dp427p2 | 703 bp | 42.7% | aauguaAUGA | Purkinje cells |
| Dp260-1 | 45 bp | 42.2% | auugcaAUGA | retina |
| Dp260-2 | 197 bp | 34.0% | agcugaAUGA | retina |
| Dp140 | 1041 bp | 43.4% | cuagaaAUGC | central nervous system, kidney |
| Dp116 | 61 bp | 49.2% | cugaaaAUGU | Schwann cells |
| Dp71 | 78 bp | 67.9% | gcagccAUGA | ubiquitous |
With the exception of the dp40 isoform, all identified DMD mRNA isoforms encode the essential C-terminal domain and are thought to share the same 3’ UTR. 3’ UTRs have been shown to regulate gene expression post-transcriptionally by altering mRNA stability, localizing mRNA, or directly affecting translation [42-45]. The 2.7 kb DMD 3’ UTR is longer than the average 3’ UTR length of ~1020 nt [37] in human transcripts, and contains several large, highly conserved regions that may be regulating the expression of DMD mRNA through these mechanisms (Figure 1). The high amount of conservation found within the DMD 3’ UTR was first described over two decades ago [46]. Two large, conserved regions were identified, named Lemaire A and Lemaire D, both of which are more highly conserved than the protein coding region (Figure 1) [46, 47]. The unusually high amount of conservation extends beyond the Lemaire sequences. For example, the sequence in the Lemaire A region spans 429 nucleotides with 97.4% sequence identity between human and mouse sequences (Figure 1). These regions are similar to the large, hyper or highly conserved elements (HCEs) identified in other 3’ UTRs [48, 49]. There is evidence that HCEs are involved in transcriptional and post-transcriptional gene regulation [48-51], however the mechanism of how HCEs regulate gene expression is still not understood. The extensive length and high conservation of these regions is unusual, and greater than what would be expected to retain a single binding site for RNA-binding proteins or miRNAs. Nevertheless, evidence for two potential regulatory elements within these regions of the DMD 3’ UTR have been previously reported. These include an miRNA-31 binding site within Lemaire A, thought to be involved in suppressing dystrophin synthesis in myoblasts prior to differentiation and in dystrophic muscle [52], and a binding site for the RNA-binding protein vigilin within Lemaire D [53]. Each of these elements spans only a small portion of the respective conserved domains and is consequently unlikely to explain the high degree of sequence conservation observed across hundreds of nucleotides in the DMD 3’ UTR (Figure 1).
Figure 1. Conservation of the DMD 3’ UTR.
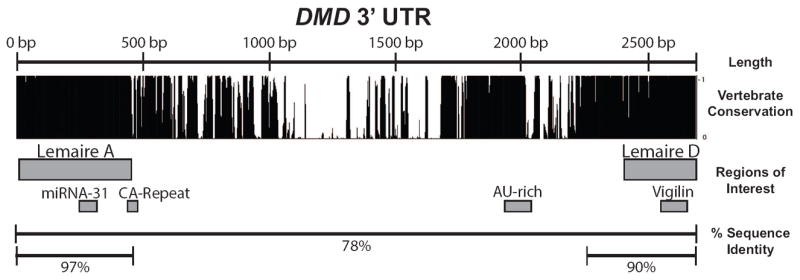
The conservation and regions of interest are shown for the DMD 3’ UTR. The 2.7 kb DMD 3’ UTR contains several large regions of high conservation. Conservation among vertebrates is shown using the PhastCons measurement of 100 vertebrate species (PhastCons track, UCSC Genome Browser). Regions of interest in the 3’ UTR include the highly conserved Lemaire A and Lemaire D regions, a previously described miRNA-31 and vigilin binding sites, a CA-repeat that is variable in size, and an AU-rich region. The percent sequence identity between human and mouse sequences are shown for the full 3’ UTR (78%), the Lemaire A region (97%), and Lemaire D region (90%) (See Methods and Materials). The percent sequence identity between human and mouse is 74.7% for the average human 3’ UTR and 84.7% for the coding region of a gene [27].
The results presented here demonstrate that the 5’ UTRs for the full-length muscle and brain isoforms initiate translation with low efficiency in C2C12 cells and that, in differentiated myotubes, the highly conserved region overlapping Lemaire A increases both mRNA levels and translation efficiency, whereas the region overlapping Lemaire D primarily affects mRNA abundance. Directed mutagenesis further demonstrates that the elements responsible for increasing dystrophin expression during differentiation span the entire Lemaire A and D domains, and that the predicted binding of vigilin and miRNA-31 does not appear to be involved in this function of the DMD 3’ UTR in differentiated C2C12 myotubes.
Materials and Methods
Cell Culture
Mouse C2C12 myoblast cells were purchased from ATCC (CRL-1772). C2C12 myoblasts were cultured in growth medium consisting of Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies) supplemented with 10% fetal bovine serum (FBS) (Hyclone). Myogenic differentiation was induced by replacing the medium of the C2C12 myoblasts with DMEM/F-12 media supplemented with 2% horse serum (Sigma). In our studies, myoblasts were examined two days after being transfected, whereas myotubes were analyzed after six days in differentiation media unless otherwise stated. HEK 293 cells were obtained from ATCC (CRL-1573), and cultured in growth medium consisting of Minimum Essential Medium (MEM, Life Technologies) supplemented with 10% FBS (Hyclone), 1mM Sodium Pyruvate (Gibco), and 1x MEM Non-Essential Amino Acids (NEAA, Gibco). All cells were cultured at 37°C and 5% CO2.
Generation of DMD 5’ and 3’ UTR reporter constructs and mutagenesis
The full-length DMD 3’ UTR was amplified from a European human genomic DNA sample using PCR and the following specific primers GCCGCTTCCTAGGAGGAAGTCTTTTCCACATGGC and GCTCATGGGATCCGCATGTATTACCTATTTAAAAAGTAAGTAAGTAAG that include an overhang containing the sequence of AvrII and BamH1 restriction sites, respectively. The resulting 2,811 bp PCR product was digested with XbaI and AvrII and ligated into the XbaI and BamHI restriction sites of pHRL-CMV (Promega) immediately downstream of the Renilla Luciferase ORF. The resulting plasmid contains the human DMD 3’ UTR in place of the SV40 polyA region of the pHRL-CMV vector. The DMD 3’ UTR was inserted in the reverse orientation as described above except that the 3’ UTR from genomic DNA was amplified using a forward primer containing the BamH1 restriction site and the reverse primer containing the AvrII restriction site.
The dp427m, dp427c, and dp427p1 5’ UTRs and surrounding regions were amplified from a human European genomic DNA sample using PCR and primers specific to each region (Supplementary Table 1). The resulting PCR products were used as a template for a second PCR that amplified the dp427m, dp427c, and dp427p1 5 ‘UTRs using primers specific to each 5’ UTR and containing overhang sequences with the KpnI and XhoI cut sites in the forward and reverse primers, respectively (Supplementary Table 1). The pHRL-CMV vector containing the DMD 3’ UTR (3’ UTR construct) was amplified using primers that annealed at the beginning of the Renilla coding sequence containing an overhang with the KpnI cut site in the reverse primer, and an XhoI cut site and the dp427m, dp427c, or dp427p exon 1 in the forward primer (Supplementary Table 1). The amplified 5’ UTR products were digested with KpnI and XhoI and ligated into the amplified 3’ UTR vectors containing the exon 1 isoforms. The resulting XhoI cut site found between the DMD 5’ UTRs and the coding sequence was removed using the Phusion Site-Directed Mutagenesis Kit (Thermo Scientific), as specified by the manufacturer using phosphorylated primers specific for each construct (Supplementary Table 1).
Deletion constructs were made using the Phusion Site-Directed Mutagenesis Kit (Thermo Scientific), as specified by the manufacturer using the DMD 3’ UTR construct as a template and phosphorylated primers specific for each deletion (Supplementary Table 2). For all subsequent experiments, plasmid DNA was prepared using QIAGEN kits.
A DMD 3’ UTR construct containing six point mutations in the predicted vigilin binding site as previously described [53] was made by amplifying the 3’ UTR construct using PCR and two sets of primers containing the desired nucleotide changes. Specifically, the DMD 3’ UTR construct was first amplified using the primers CATACTTCACCAAGTATATGCCTTACTATTATATTATAGTACTG and ACTGAAGTTTACAAAAATAATTTGTAAATGTTACAGTGTTGG (desired mutations underlined), and the purified product was amplified a second time using the phosphorylated primers AACATATCATACTTCACCAAGTATATGCCTTACTATTA and GCGAAAATGCAGTAAAACTGAAGTTTAC (desired mutations underlined). The resulting PCR product was purified and ligated using T4 DNA Ligase (New England Biolabs) according to the manufacturer’s instructions. All constructs described in the materials and methods were sequence verified.
Dual-Luciferase Reporter Assay
C2C12 myoblasts (1×104 cells) or HEK 293 cells (4×104 cells) were transfected with 25 ng of the pHRL-CMV vector (Promega) containing either the full-length DMD 3’ UTR or a mutant 3’ UTR construct using 0.3uL of Lipofectamine 2000 reagent (Life Technologies) in 96-well half-area plates (Corning) with 0.16 cm2 growth area. All reactions were co-transfected with 6.25 ng of the pGL4.13 [luc2/SV40] Firefly Luciferase vector (Promega) as a transfection control. To measure protein expression in myoblasts, cells were harvested 48 hours after transfection by adding 14 uL of a passive lysis buffer (Promega). To measure protein levels in myotubes, the growth media for C2C12 myoblasts was replaced 48 hours after transfection with a low serum media to induce differentiation. The differentiated myotubes were harvested after 6 days in differentiation media by adding 14 uL of a passive lysis buffer (Promega). C2C12 myoblasts and myotubes underwent one freeze-thaw cycle to aid in lysis. The bioluminescence of Renilla and firefly luciferase was measured using the Dual-Luciferase Reporter Assay System (Promega) in a Veritas Microplate Luminometer (Turner Biosystems) to determine luciferase levels, and the Renilla luciferase signal was normalized to the pGL4.13 firefly luciferase signal in each well to account for variation in transfection efficiency. All data presented in the paper are expressed as the average +/- 1 standard deviation from a representative experiment with at least four biological replicates.
Determining mRNA levels of 3’ UTR constructs
To determine the relative amounts of mRNA levels for the DMD 3’ UTR construct and deletion constructs, C2C12 myoblasts (6×105 cells) were transfected with 750 ng of a 3’ UTR construct and 750 ng of the control pHRL construct using 18 uL of Lipofectamine 2000 in a 24-well tissue culture plate (Falcon) with 2 cm2 growth area. Total RNA was extracted from C2C12 cells two days after transfection (myoblasts) or after C2C12 cells had differentiated for six days in a low serum media (myotubes) using Trizol (Invitrogen) as described by the manufacturer. Each RNA sample was treated with DNAse I (Thermo Scientific) to remove any DNA contamination, and cDNA was synthesized using the SuperScript III First-Strand Synthesis Kit (Life Technologies) using random hexamers. Real-time PCR was performed using the Applied Biosystems 7900HT Real-time PCR system using Express SYBR GreenER qPCR SuperMix (Life Technologies) with primers optimized to amplify each construct according to the manufacturer’s protocols. Results were analyzed using SDS 2.3 software and calculated using the comparative CT (ΔΔCT) method by normalizing the transfected construct levels to the control pHRL co-transfected construct value at 100%. The forward and reverse primers used to amplify the cDNA of each construct are as follows: GCAACTACAACGCCTACCTT and TGGTTTGTCCAAACTCATCAA used to amplify the co-transfected pHRL vector; CTGAGGAGTTCGCTGCCTAC and GCCATGTGGAAAAGACTTCC to measure the full-length 3’ UTR, dl-M, and dl-D constructs; CTGAGGAGTTCGCTGCCTAC and CCCCACTCAGCTGACAGTTC to measure the dl-A construct.
Calculating percent sequence identity
To determine the percent sequence identity between human and mouse sequences in the DMD 3’ UTR, the human and mouse Lemaire A region, Lemaire D region, or the full-length 3’ UTR were aligned in ClustalW. The percent sequence identity was calculated as the percent of aligned nucleotides to the total number of nucleotides in the human sequence.
Results
DMD 3’ UTR regulates expression during myogenesis
To investigate whether the DMD 3’ UTR regulates gene expression, we designed a Renilla reporter construct containing the DMD 3’ UTR downstream of the Renilla coding sequence in pHRL-CMV (3’ UTR) (Figure 2a). This reporter construct along with controls containing either an SV40 derived 3’ UTR (pHRL) or the DMD 3’ UTR inserted in the reverse orientation (3’ UTR Rev) were transfected into mouse muscle C2C12 and human embryonic kidney HEK 293 cell lines with a co-transfected pGL4.13 firefly luciferase vector to normalize for variation in transfection efficiency. The undifferentiated C2C12 myoblasts and HEK 293 cells were subsequently lysed and the relative amounts of Renilla and Firefly protein were measured using a Renilla/Firefly dual-luciferase assay (Materials and Methods). Changes in gene expression were determined by comparing the Renilla luciferase activities expressed by each experimental construct normalized to the co-transfected Firefly luciferase control (Figure 2b, C2C12 Myoblasts). To determine whether the 3’ UTR regulates expression during myogenesis, transfected C2C12 cells were induced to differentiate for six days and relative expression levels were determined as described above (Figure 2b, C2C12 Myotubes). The DMD 3’ UTR increased expression by 350% and 200% compared to pHRL in C2C12 myotubes and HEK 293 cells, respectively, whereas in C2C12 myoblasts, relative expression levels decreased by 50% (Figure 2b). To monitor changes in expression of the pHRL-CMV (pHRL) control construct and the DMD 3’ UTR (3’ UTR) construct during differentiation, Renilla and firefly protein levels were measured every 24 hours for seven days as the cells differentiated (Supplementary Figure 1). The observed increase in expression of the 3’ UTR construct (3’ UTR) began two days after C2C12 cells were grown in differentiation media and continued to increase until the fifth day of differentiation (Supplementary Figure 1). This increase in expression of the 3’ UTR construct correlates with the morphology of the differentiating C2C12 cells with elongation of the myoblasts first being observed at Day 2 and large, mature myotubes being formed by Day 5 (data not shown). As a control, the DMD 3’ UTR was inserted in the reverse orientation with an intact DMD polyadenylation signal (3’ UTR Rev), and transfected into C2C12 cells or HEK 293 cells, which caused expression to decrease by more than 85% in all cell types (Figure 2b). These experiments show that the DMD 3’ UTR increases expression of the reporter gene in differentiating myoblast cell lines and correlates with the expected increase in the muscle isoform of dystrophin in differentiated myotubes.
Figure 2. The DMD 3’UTR regulates expression in C2C12 and HEK 293 cells.
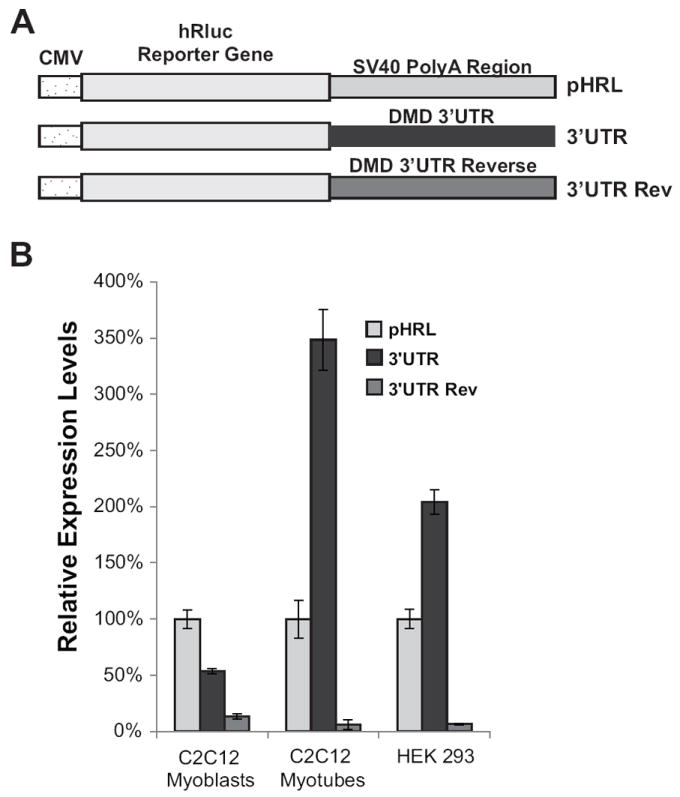
(A) Diagram of the control pHRL-CMV Renilla vector (pHRL) with the SV40 PolyA region, and the pHRL-CMV vector with the DMD 3’UTR replacing the SV40 polyA region in either the forward (3’UTR) or reverse orientation (3’UTR Rev). (B) Relative expression levels in C2C12 and HEK 293 cells are shown. Relative expression levels are normalized to the ratio of the control Renilla pHRL vector. The average of four biological transfection replicates is shown for each construct. Error bars equal +/- 1 standard deviation.
Conserved domains in the DMD 3’ UTR regulate gene expression
Having shown that the DMD 3’ UTR can regulate gene expression during myogenesis, we were interested in determining which regions of the DMD 3’ UTR were important for this regulation. We hypothesized that the domains responsible for gene regulation are most likely contained in one of the highly conserved regions of the DMD 3’ UTR. For this reason, we split the DMD 3’ UTR into three segments – the first segment (A) consisting of the first 626 nucleotides of the DMD 3’ UTR that includes Lemaire A, a CA-repeat domain, and surrounding highly conserved regions; a middle section (M) consisting of 1,542 nucleotides found in the middle of the DMD 3’ UTR that contains a few regions of high conservation but consists mostly of unconserved sequences; and a third segment (D) consisting of the last 431 nucleotides of the DMD 3’ UTR that contains Lemaire D and the surrounding highly conserved sequences (Figure 3a). We made three deletions in our DMD 3’ UTR construct corresponding to each of the major sections of the DMD 3’ UTR (dl-A, dl-M, and dl-D, Figure 3a while maintaining an intact polyadenylation site and transfected these constructs into C2C12 cells. After 48 hours, the media was replaced with differentiation media and the relative expression levels for each construct were measured each day for seven days (Figure 3b). As we saw before, the expression of the full-length 3’ UTR construct had a 6-7 fold increase in expression as the cells completed differentiation, but the constructs with the Lemaire A or Lemaire D regions deleted (dl-A, dl-D) had only about a 2-fold increase in expression, showing that the large increase in expression seen during C2C12 differentiation is dependent on the presence of both the Lemaire A and Lemaire D regions, whereas deleting the middle region of the 3’ UTR (dl-M) had a smaller impact on expression (Figure 3b).
Figure 3. The conserved Lemaire A and Lemaire D regions are necessary for the increase in expression during C2C12 myogenesis.
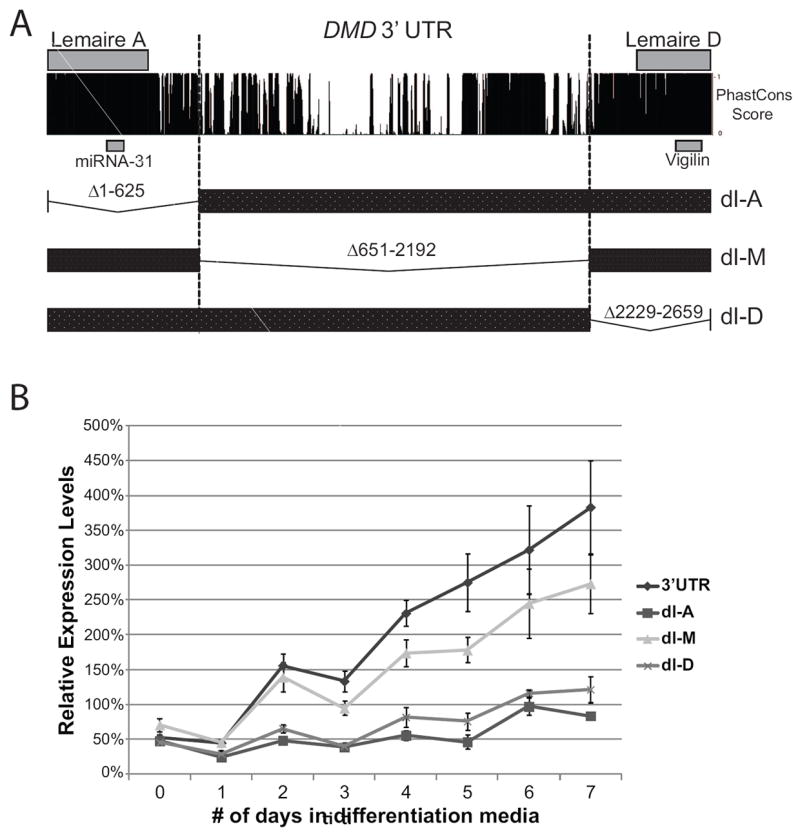
(A) Diagram showing the conservation of the DMD 3’UTR and the regions deleted in each 3’UTR construct. Conservation of the DMD 3’UTR across vertebrates is shown using the PhastCons score across the 3’UTR (100 Vertebrate Phastcons track, UCSC genome browser). Three deletion constructs were made in the DMD 3’UTR that included Lemaire A (dl-A), the middle portion of the 3’UTR (dl-M), and Lemaire D (dl-D). (B) Relative expression levels were measured in C2C12 cells transfected with the full-length DMD 3’UTR Renilla construct (3’UTR) or the deletion constructs (dl-A, dl-M, dl-D) during differentiation from myoblasts to myotubes. Expression levels were normalized to the expression of the control pHRL Renilla construct (pHRL) during each day of differentiation. The average of four biological transfection replicates is shown for each construct at each time point. Error bars equal +/- 1 standard deviation.
To determine whether the Lemaire A and Lemaire D regions were regulating expression by directly altering translation of the mRNA or by altering steady-state mRNA levels, we measured mRNA and protein levels for each transfection in both C2C12 myoblasts and myotubes (Figure 4). We measured Renilla steady-state mRNA levels using qPCR relative to a co-transfected pHRL control vector, and measured Renilla protein levels for each construct using the Firefly/Renilla dual-luciferase assay described above. Consistent with our previous experiment, deleting Lemaire A or Lemaire D did not have a significant effect on mRNA or protein levels in C2C12 myoblasts (Figure 4). Surprisingly, deleting the middle section (dl-M) resulted in a ~40-50% increase in mRNA and protein levels in myoblasts compared to the full-length 3’ UTR construct (3’ UTR). In myotubes, deleting any region of the 3’ UTR resulted in a significant decrease in protein expression compared to the full-length 3’ UTR construct, with the largest effect on expression observed when the conserved Lemaire A or Lemaire D regions were deleted. When the Lemaire A region was deleted (dl-A), steady state mRNA levels decreased by ~50% whereas protein levels decreased by ~75% showing that this region is critical for the increased expression seen in C2C12 myotubes by affecting both translation and steady-state mRNA levels of the reporter mRNA (Figure 4). Deleting the Lemaire D region (dl-D) resulted in a ~50% decrease in steady-state mRNA levels along with ~65% decrease in protein expression (Figure 4) showing that this region has a larger impact on maintaining mRNA steady-state levels in C2C12 myotubes than on translation.
Figure 4. The conserved regions in the DMD 3’UTR increase translation in C2C12 myotubes.
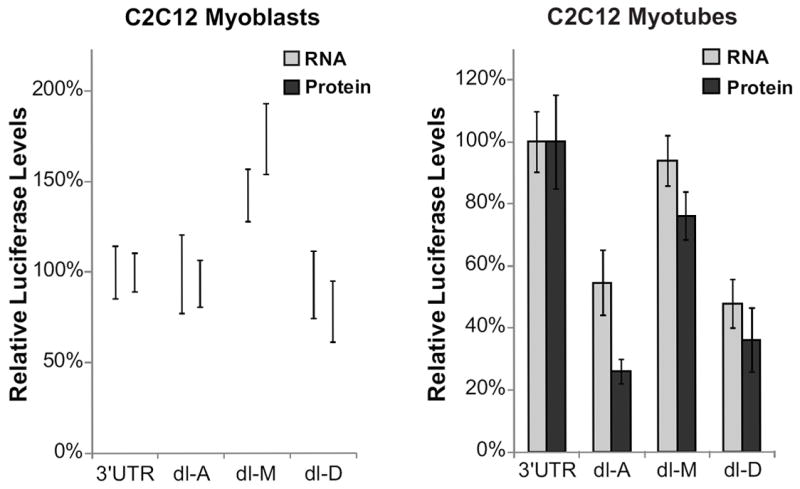
Renilla mRNA and protein levels for C2C12 myoblasts and myotubes transfected with the full-length 3’UTR construct (3’UTR) or deletion constructs (dl-A, dl-M, dl-D) are shown. mRNA levels were measured using qPCR relative to a co-transfected pHRL control. Protein levels were measured using a dual-luciferase assay as previously described. mRNA and protein levels were normalized to the amount of mRNA and protein levels of the full-length 3’UTR. The average of at least three biological transfection replicates is shown for each construct. Error bars equal +/- 1 standard deviation.
There is evidence that the length of a 3’ UTR can affect translational efficiency and mRNA stability [54, 55]. To determine whether the effect of deleting regions of the DMD 3’ UTR were due to changing the length of the 3’ UTR or the absence of the region deleted, we made DMD 3’ UTR constructs with the Lemaire A section (A) or middle section (M) of the 3’ UTR reversed and transfected these constructs into C2C12 cells. Expression decreased by the same amount in C2C12 myotubes whether these regions were deleted or reversed in our constructs (data not shown).
Lemaire A and Lemaire D regions of the DMD 3’ UTR are necessary for increasing expression during myogenesis
It was shown previously that Lemaire A contains a predicted miRNA-31 binding site and that overexpression of miRNA-31 can inhibit dystrophin expression in cultured cells [52]. An unrelated analysis of the conserved regions in the DMD 3’ UTR predicted the formation of a conserved 27-bp stem-loop structure within the Lemaire A region [47]. To experimentally address the presence of discrete regulatory elements within the 626 nucleotide Lemaire A region, the entire region was divided into four parts of ~150 base pairs each (Aa, Ab, Ac and Ad) (Figure 5a). The first three regions (Aa, Ab, and Ac) overlap the previously described Lemaire A, with the predicted miRNA-31 binding site lying within the Ac region (Figure 5a). The last region (Ad) contains the CA repeat and conserved sequences downstream of Lemaire A. Each of these smaller regions was deleted from the DMD 3’ UTR to create the reporter constructs dl-Aa, dl-Ab, dl-Ac, dl-Ad (Figure 5a), and these were transfected into C2C12 cells. After two days in growth media (myoblasts) or six days in differentiation media (myotubes), relative expression levels were measured using the luciferase assay as previously described. In myotubes, we found that deleting any portion of Lemaire A (dl-Aa, dl-Ab, and dl-Ac) decreased expression as much as deleting the entire Lemaire A region (dl-A) (Figure 5b), showing that all sections of the conserved Lemaire A region must be present to increase expression in myotubes. Deleting the portion of the 3’ UTR containing the CA-repeat and downstream conserved sequences (dl-Ad) decreased expression by ~50% (Figure 5b). Consistent with our previous experiment, deleting any portion of the Lemaire A region had little impact on expression in C2C12 myoblasts compared to the full-length 3’ UTR construct (Supplementary Figure 2).
Figure 5. Lemaire A is necessary for high expression in C2C12 myotubes.
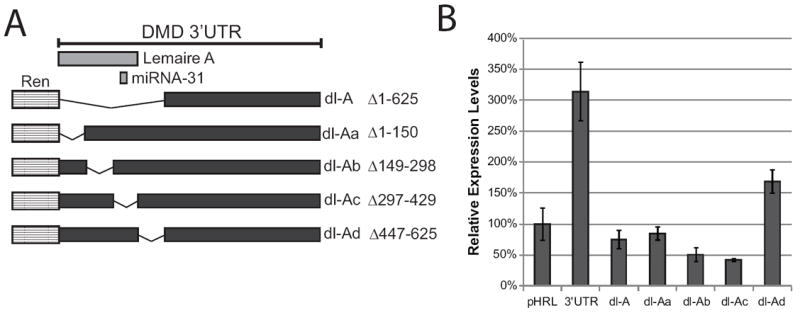
(A) Diagram of the pHRL 3’UTR constructs with ~ 150 nucleotide portions of the Lemaire A region deleted (dl-Aa, dl-Ab, dl Ac, and dl-Ad). Lemaire A overlaps the sequences deleted in the dl-Aa, dl-Ab, and dl-Ac constructs. The miRNA-31 binding site lies within the Ac region. (B) Renilla protein expression in day six C2C12 myotubes transfected with the Lemaire A deletion constructs is shown. Relative expression levels for each construct were measured using a dual-luciferase assay as previously described. The average of four biological transfection replicates is shown for each construct. Error bars equal +/- 1 standard deviation.
To determine if the entire Lemaire D region was required for its effects on expression, we used deletion mutagenesis to make DMD 3’ UTR constructs with ~150 bp sections of the Lemaire D region deleted (dl-Da, dl-Db, dl-Dc) (Figure 6a), and transfected these constructs into C2C12 cells. Deleting the entire Lemaire D region (dl-D) resulted in 65% decrease in expression compared to the full-length 3’ UTR construct in differentiated myotubes (Figure 6b). Deleting any portion of the conserved Lemaire D region (dl-Da, dl-Db, dl-Dc) resulted in a similar decrease in expression compared to deleting the entire Lemaire D region (50%, 57%, and 43% respectively) (Figure 6b) demonstrating that, similar to Lemaire A, the entire Lemaire D region is required for full expression in myotubes. Previously, it was shown that the RNA-binding protein vigilin can bind in vitro to a predicted binding site found within Lemaire D of the DMD 3’ UTR (overlapping the Dc region) (Figure 6a). This binding did not occur when six point mutations were introduced into the DMD 3’ UTR transcript that disrupted the predicted secondary structure of the vigilin-binding site [53]. To determine whether vigilin may be regulating dystrophin expression in muscle cells, we recreated the previously described DMD 3’ UTR mutant construct containing these six point mutations and transfected this construct into C2C12 cells. We found that mutating the vigilin-binding site did not alter expression in either myoblasts (Supplementary Figure 2) or myotubes (Figure 6b), suggesting that vigilin is not regulating dystrophin expression in cultured muscle cells through binding to this site.
Figure 6. Lemaire D region is necessary for high expression in C2C12 myotubes.
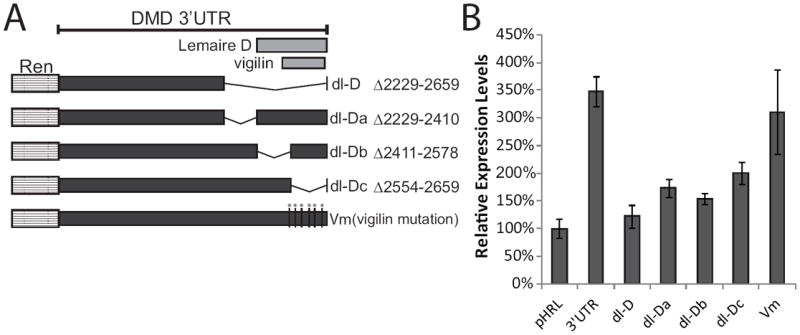
(A) Diagram of the pHRL 3’UTR constructs with portions of the Lemaire D region deleted (dl-D, dl-Da, dl-Db, dl-Dc). Lemaire D and the vigilin binding site overlap the Db and Dc regions. A construct containing six point mutations in the vigilin binding site was made (Vm). (B) Relative expression levels from day six C2C12 myotubes transfected with the pHRL Renilla construct (pHRL), the full-length 3’UTR construct (3’UTR), or Lemaire D mutant constructs (dl-Da, dl-Db, dl-Dc, and Vm containing 6 point mutations in the vigilin binding site) is shown. Relative expression levels for each construct were measured using a dual-luciferase assay as previously described. The average of four biological transfection replicates is shown for each construct. Error bars equal +/- 1 standard deviation.
An AU-rich region increases expression in myotubes
The middle portion of the DMD 3’ UTR contains only limited regions of conservation, but protein and mRNA levels significantly increased in C2C12 myoblasts when this region was removed, suggesting that this region may inhibit expression in myoblasts (Figure 4). In contrast, protein expression decreased by ~25% in myotubes when the middle region of the 3’ UTR was removed (Figure 4). After analyzing the sequence in the middle region of the 3’ UTR, we discovered a highly conserved AU-rich region at the distal end of the middle section (Figure 1). AU-rich elements in 3’ UTRs have been shown to regulate mRNA stability and translation of mRNA transcripts [56-58]. To investigate whether this sequence contained a functional AU-rich element, C2C12 cells were transfected with a 3’ UTR construct that contained a 262 base pair deletion spanning the conserved AU-rich sequence in the middle section of the 3’ UTR (dl-ARE). Deleting the AU-rich sequence had no effect on protein expression of the Renilla reporter construct in myoblasts, but, in myotubes, protein expression decreased to 64% compared to the full-length 3’ UTR construct, similar to the protein levels seen when the entire middle region is deleted (Figure 7). The increase in mRNA and protein levels seen in myoblasts when the entire middle region is deleted does not appear to be due to an effect of the conserved AU-rich sequence.
Figure 7. An AU-rich region in the DMD 3’UTR decreases expression in myotubes.
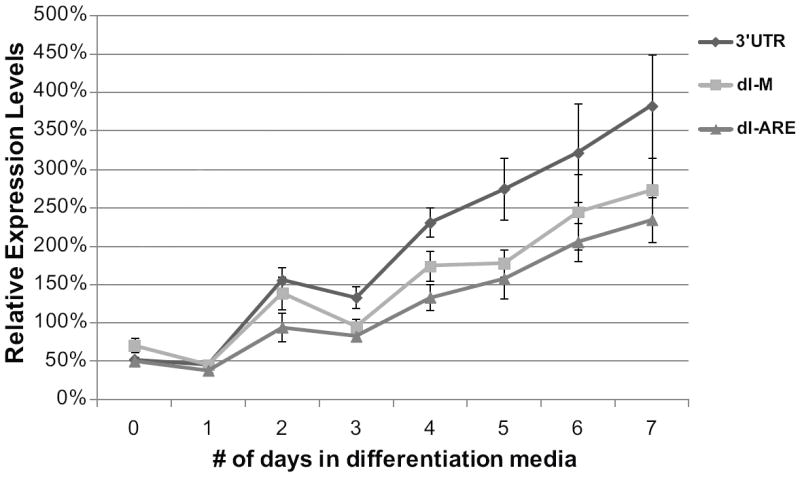
Relative expression levels for C2C12 cells transfected with either the Renilla reporter construct containing the full-length DMD 3’UTR (3’UTR), a deletion construct deleting the middle region of the DMD 3’UTR (dl-M), or a construct with a 262 base pair region containing an AU-rich sequence found in the middle region of the 3’UTR deleted (dl-ARE) are shown for seven days of C2C12 cell differentiation. Expression is normalized to the expression of a control Renilla construct (pHRL) at each time point. The average of four biological transfection replicates is shown for each construct at each time point. Error bars equal +/- 1 standard deviation.
mRNAs containing DMD 5’ UTRs initiate translation with low efficiency
To determine the effect the dp427m 5’ UTR has on expression in muscle cells, we inserted the 5’ UTR sequence from the muscle isoform of dystrophin immediately upstream of the Renilla coding sequence in our 3’ UTR construct to create a Renilla construct with both the DMD 5’ and 3’ UTR (5’ UTRM) (Figure 8a). To maintain the initiation codon sequence context for the muscle isoform of dystrophin, we also added the three amino acids of the dp427m exon 1 transcript to the beginning of the Renilla sequence. We transfected this construct into C2C12 cells and analyzed relative expression levels in myoblasts and in myotubes that were differentiated for six days in low serum media. We saw a significant decrease in expression when the dp427m 5’ UTR was added to the Renilla construct with 2.2% protein expression in myoblasts and 11.4% protein expression in myotubes compared to the control pHRL-CMV reporter (pHRL) (Figure 8b). The relative increase in expression between transfected myoblasts and myotubes is the same when the Renilla construct contains the dp427m 5’ UTR (5’ UTRM) compared to the 3’ UTR construct lacking the 5’ UTR (3’ UTR), thus the difference between myoblasts and myotube expression levels appears to be solely due to the presence of the 3’ UTR. To determine if there were any differences between the different 5’ UTR isoforms, we also made constructs containing the 5’ UTRs from the two full-length brain dystrophin isoforms (dp427p, dp427c) (Figure 8a). Similar to the results obtained for the dp427m 5’ UTR, we found a large reduction in expression in both C2C12 myoblasts and myotubes with the brain dp427p 5’ UTR 8% in myoblasts and 28% in myotubes) and dp427c 5’ UTR (5% in myoblasts and 13% in myotubes) compared to the control pHRL-CMV vector (Figure 8c). To determine whether the Lemaire A and Lemaire D regions had similar effects on expression in the presence of the muscle DMD 5’ UTR, we used deletion mutagenesis to delete the Lemaire A and Lemaire D regions (dl-A and dl-D) in a construct containing the dp427m 5’ UTR, and transfected these constructs into C2C12 cells. The relative changes in expression when these regions were deleted was independent of whether the 5’ UTR was present or absent in C2C12 myoblasts and myotubes (Supplementary Figure 3).
Figure 8. The DMD 5’UTRs sequences cause suboptimal translation initiation.
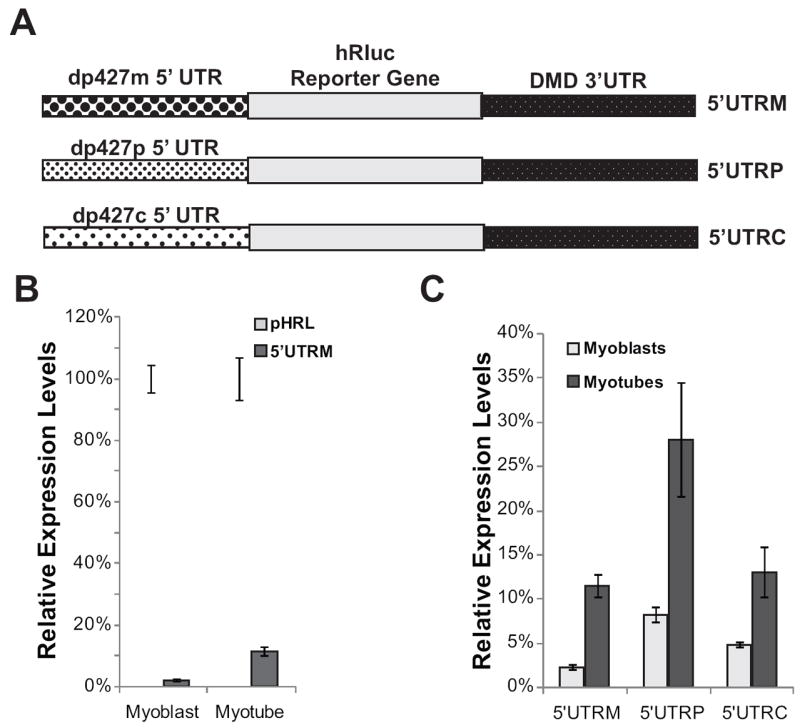
(A) Diagram of pHRL Renilla vectors containing the DMD 3’UTR and the dp427m muscle 5’UTR (5’UTRM), the dp427p brain 5’UTR (5’UTRP), or the dp427c brain 5’UTR (5’UTRC) inserted immediately upstream of the hRluc Renilla coding sequence in the pHRL-CMV vector. (B) Relative expression levels for C2C12 myoblasts or myotubes transfected with either the control Renilla reporter construct (pHRL) or a Renilla reporter construct containing the dp427m 5’UTR and the full-length DMD 3’UTR (5’UTRM) are shown. (C) Relative expression levels in C2C12 myoblasts or myotubes transfected with a reporter construct containing either the dp427m muscle 5’UTR (5’UTRM) or the 5’UTRs of the full-length dp427p and dp427c brain isoforms (5’UTRP, 5’UTRC) are shown. The average of four biological transfection replicates is shown for each construct at each time point. Error bars equal +/- 1 standard deviation.
Discussion
5’ and 3’ UTRs regulate gene expression and disease causing mutations in a number of genetic disorders have been linked to mutations in these regions [35, 59], including several DMD patients with mutations in the 5’ and 3’ UTR of the DMD gene. An L1 insertion in the dp427m 5’ UTR was identified in a patient with X-Linked Dilated Cardiomyopathy (XLDC) with no apparent muscular atrophy or weakness [60]. Because no mRNA was detected in these patients, it’s speculated that the insertion either disrupts transcription or stability of the muscle mRNA isoform [60] and that alternate isoforms may rescue dystrophin expression and function in skeletal but not cardiac muscle. There are two examples of BMD patients with mutations in the DMD 3’ UTR [47, 61]. However, these deletions extend into the coding region of the DMD gene or delete other neighboring genes, and mutations contained solely within the 3’ UTR have yet to be identified. Although it has been shown that altered expression levels can impact disease severity [9, 10] and evidence presented here supports a role for the DMD 3’ UTR in regulating expression, the identification of these mutations suggests that if pathogenic 3’ UTR mutations exist, they would most likely cause the milder BMD phenotype. Another possibility is that mutations in these regions cause non-muscular phenotypes and have been overlooked as disease causing. For example, the dystrophin glycoprotein complex is involved in brain development and function [62, 63] and approximately one third of DMD patients have cognitive impairments such as reduced intelligence quotient scores [64], autism spectrum disorder (ASD) and attention deficit hyperactivity disorder (ADHD) [65, 66]. Recently, a 3 base pair deletion in the coding region of the DMD gene was shown to cause intellectual disability, but causing no muscle phenotype [67], showing that such mutations do exist in the DMD gene. Mutations in either the DMD 5’ or 3’ UTR that alters expression of one or more dystrophin isoforms could have tissue-specific consequences that are not currently appreciated.
Examination of the 5’ UTR of the muscle (dp427m) and brain (dp427c and dp427p) isoforms of DMD revealed that these sequences reduced expression of a reporter construct 75-90% in both myoblasts and myotubes. Several factors may affect the efficiency of translation initiation, including the presence of upstream open reading frames or secondary structures that can limit the number of scanning pre-initiation ribosomes that access the initiation codon. Analysis of the DMD 5’ UTRs reveal several upstream AUGs in the 5’ UTRs of the dp427c and dp427p1 brain isoforms (11 and 4 AUGs respectively) that could be altering translation initiation, although there are no AUG sequences contained in the muscle dp427m 5’ UTR. Once the ribosome engages the initiation codon, positions upstream and downstream of the initiation codon determine the efficiency of productive translation initiation. The consensus translation initiation context in mammals is GCCRCCaugG, originally identified by Marilyn Kozak, and often referred to as the Kozak consensus sequence [68-70]. Deviations from this consensus, in particular at the -3 or +4 position, have been shown to reduce the efficiency of translation initiation [68, 70]. The control pHRL-CMV vector matches the Kozak consensus sequence, whereas the DMD 5’ UTR and exon 1 isoforms alter several key positions (Table 1), an observation that likely explains, at least in part, the low levels of translation initiation observed when the isoform-specific DMD 5’ UTRs, initiation codon, and exon 1 were placed upstream of the luciferase reporter gene.
We show that the DMD 3’ UTR, which is common to all but one mRNA isoform, regulates expression during myogenesis. Specifically, we show that the conserved Lemaire A regulates expression by increasing mRNA abundance and translation in differentiated myotubes, and that Lemaire D and the surrounding region primarily affects mRNA abundance. Deletion mutagenesis of Lemaire A and Lemaire D shows that the entire regions are necessary for the increase in expression observed during differentiation of myoblasts into myotubes. This result is consistent with the high level of conservation that spans these regions. The average sequence identity of 3’ UTRs between human and mouse sequences is 74.7% [38]. Lemaire A has the largest effect on protein expression in C2C12 cells (the Aa, Ab, and Ac regions), and spans 429 base pairs with 97.4% sequence identity, and the Lemaire D region contains a 459 base pair sequence with 90.2% sequence identity. These regions are similar to the previously described HCEs [48, 49]. HCEs are found across the genome in both coding and non-coding regions of genes [48-50, 71]. HCEs are associated with the 3’ UTRs of regulatory genes, and may be involved in post-transcriptional gene regulation, alternative splicing, and polyadenylation [48-51]. However, the mechanism of how HCEs regulate gene expression is still not understood, and why the extensive length and high conservation of these regions must remain intact is unknown. Although the existence of multiple adjacent regulatory elements that act cooperatively to ensure proper expression could conserve a larger sequence than a single regulatory element, it is unlikely to explain the conservation seen in HCEs that can span thousands of nucleotides. Long noncoding RNAs are typically less conserved than protein coding genes [72], and the complementary binding of antisense RNAs to these regions are also unlikely to account for the degree of conservation observed. Some unknown function yet to be identified within the primary sequence may exist.
It was shown previously that miRNA-31 could inhibit dystrophin expression by binding to a predicted miRNA-31 binding site in the DMD 3’ UTR [52]. miRNA-31 is highly expressed in undifferentiated myoblasts where it inhibits expression of proteins involved in myogenesis, and is decreased in expression as the cells differentiate into myotubes [52, 73]. In our experiments, the deletions that spanned the predicted miRNA-31 site resulted in a decrease in protein expression in C2C12 myotubes (Figure 5b). To determine whether the miRNA-31 binding site itself has an effect on expression, we made a 3’ UTR deletion construct with only the miRNA-31 binding site deleted, but saw no increase in expression as a result in C2C12 myotubes (data not shown) suggesting that miRNA-31 does not repress dystrophin expression in C2C12 myotubes, and may only be repressing dystrophin expression in myoblasts and in disease tissue [52] where miRNA-31 is highly expressed. In contrast, the regulatory factors that increase expression appear to be only active in differentiated myotubes. In vitro experimental evidence has also been presented for binding of the protein vigilin to the Lemaire D region of the DMD 3’ UTR [53]. However, in our experiments, we saw no difference in expression when we introduced mutations shown previously to prevent vigilin binding [53], suggesting that vigilin is not regulating dystrophin expression in cultured muscle cells. While our experiments do not rule out the possibility that vigilin or miRNA-31 may be regulating dystrophin expression in other tissues or during other developmental stages, we find no evidence that either of these predicted regulators of DMD are functioning through the 3’ UTR in differentiated C2C12 myotubes.
The involvement of lncRNAs in myogenic differentiation and the potential role they have in muscular dystrophies has become apparent in recent studies [74-77]. Several long non-coding RNAs transcribed from within the DMD gene and expressed during myogenic differentiation were shown to suppress endogenous dystrophin mRNA levels in Rhabdomyosarcoma and neuroblastoma cells when overexpressed [75]. In addition, several of these lncRNAs suppressed the expression of a luciferase reporter driven by the Dp427m promoter in C2C12 myoblasts [75]. A 1,872 bp lncRNA transcribed in the antisense orientation from the middle region of the DMD 3’ UTR was identified. While this lncRNA did not suppress dystrophin mRNA expression in these cell types [75], we find that deleting the middle region of the DMD 3’ UTR increases mRNA and protein levels by ~40-50% in undifferentiated C2C12 myoblasts, consistent with the prediction that this lncRNA could inhibit dystrophin expression in myoblasts by annealing to the complementary region of the 3’ UTR. It should be noted that this effect is only observed in undifferentiated myoblasts, and that deletions within the middle region either had no effect or reduced expression in differentiated myotubes.
DMD has been a model for developing genetic therapies to treat inherited genetic mutations, including approaches using antisense oligonucleotides to cause exon-skipping of dystrophin to restore the reading frame of a mutant transcript, treating patients with drugs that promote read-through to suppress premature stop codon mutations, and expression of minidystrophin gene constructs lacking non-essential DMD coding exons [11-13]. Clinical trials using antisense oligonucleotides, or compounds that suppress premature stop codon mutations, have shown that these compounds can restore dystrophin expression in the majority of muscle fibers of treated muscle [14, 15, 17, 19]. However, the total amount of dystrophin expressed in muscle tissue of these patients are highly variable between patients and typically result in less than 20% dystrophin levels compared to normal controls [14-17]. Sequence variations in genomic regions regulating dystrophin expression, such as the DMD UTRs, could account for some of this variability. It has also been shown that stability of the DMD transcript has a large impact on dystrophin protein levels in BMD patients and increasing stability could improve the outcome of therapies such as exon-skipping [78]. We have shown evidence that the DMD 3’ UTR has an effect on mRNA stability and could have an impact on these developing therapies. In addition, restoring dystrophin expression by viral gene therapy has shown promise, but has been challenging because the 14 kb dystrophin transcript is too large to insert into viral vectors. To overcome this, mini-dystrophin constructs (~3-8 kb) have been designed that contain the most essential domains of the DMD coding sequence that would express functional dystrophin [25-33]. Our results suggest that inclusion of the regulatory elements of the DMD 3’ UTR into these minidystrophin constructs could increase expression in differentiated muscle cells. From our experiments, we estimate that the smallest DMD 3’ UTR that could be utilized without significantly decreasing expression would be approximately 1.2 kb and contain the highly conserved Lemaire A and D regions of the 3’ UTR.
In summary, we show the ability of the DMD 5’ and 3’ UTR to regulate gene expression during myogenesis. Patient-specific variations in these regulatory elements may influence DMD expression and as a consequence explain some of the variation observed in disease phenotypes as well as the variable effectiveness of therapeutic approaches designed to restore endogenous dystrophin synthesis. Further, we propose that regulatory elements in the DMD 5’ and 3’ UTRs may be exploited for therapeutic purposes. For example, increasing the inherent low translation initiation efficiency imparted by the DMD 5’ UTRs or using the regulatory elements found within the 3’ UTR to increase expression in gene replacement therapy may be promising new approaches to increase dystrophin synthesis.
Supplementary Material
HIGHLIGHTS.
The effects of the DMD UTRs on dystrophin expression were examined in C2C12 cells.
DMD 5’ UTRs initiate translation with low efficiency in C2C12 cells.
The DMD 3’ UTR contains highly conserved elements (HCEs).
The DMD 3’ UTR HCEs increase mRNA levels and translation during differentiation.
Acknowledgments
We thank Chris Anderson and Norma Wills for technical assistance, and Drs. Kevin Flanigan (Ohio State University) and Robert Weiss (University of Utah) for comments and suggestions on this work. This work was supported in part by the National Institutes of Health NS083884 to MTH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–28. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 2.Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2:731–40. doi: 10.1016/s1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- 3.Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 4.Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23:1456–71. doi: 10.1002/1097-4598(200010)23:10<1456::aid-mus2>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 5.Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–17. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- 6.Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2:90–5. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- 7.Flanigan KM, Dunn DM, von Niederhausern A, et al. Nonsense mutation-associated Becker muscular dystrophy: interplay between exon definition and splicing regulatory elements within the DMD gene. Hum Mutat. 2011;32:299–308. doi: 10.1002/humu.21426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flanigan KM, Dunn DM, von Niederhausern A, et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30:1657–66. doi: 10.1002/humu.21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Putten M, Hulsker M, Nadarajah VD, et al. The effects of low levels of dystrophin on mouse muscle function and pathology. PLoSOne. 2012;7:e31937. doi: 10.1371/journal.pone.0031937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Putten M, Hulsker M, Young C, et al. Low dystrophin levels increase survival and improve muscle pathology and function in dystrophin/utrophin double-knockout mice. FASEB J. 2013;27:2484–95. doi: 10.1096/fj.12-224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pichavant C, Aartsma-Rus A, Clemens PR, et al. Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol Ther. 2011;19:830–40. doi: 10.1038/mt.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fairclough RJ, Wood MJ, Davies KE. Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nat Rev Genet. 2013;14:373–8. doi: 10.1038/nrg3460. [DOI] [PubMed] [Google Scholar]
- 13.Muir LA, Chamberlain JS. Emerging strategies for cell and gene therapy of the muscular dystrophies. Expert Rev Mol Med. 2009;11:e18. doi: 10.1017/S1462399409001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kinali M, Arechavala-Gomeza V, Feng L, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8:918–28. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Deutekom JC, Janson AA, Ginjaar IB, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;357:2677–86. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- 16.Cirak S, Arechavala-Gomeza V, Guglieri M, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goemans NM, Tulinius M, van den Akker JT, et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med. 2011;364:1513–22. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- 18.Lu QL, Rabinowitz A, Chen YC, et al. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc Natl Acad Sci U S A. 2005;102:198–203. doi: 10.1073/pnas.0406700102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mendell JR, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74:637–47. doi: 10.1002/ana.23982. [DOI] [PubMed] [Google Scholar]
- 20.Gurvich OL, Tuohy TM, Howard MT, et al. DMD pseudoexon mutations: splicing efficiency, phenotype, and potential therapy. Ann Neurol. 2008;63:81–9. doi: 10.1002/ana.21290. [DOI] [PubMed] [Google Scholar]
- 21.Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest. 1999;104:375–81. doi: 10.1172/JCI7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malik V, Rodino-Klapac LR, Viollet L, et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol. 2010;67:771–80. doi: 10.1002/ana.22024. [DOI] [PubMed] [Google Scholar]
- 23.Finkel RS. Read-through strategies for suppression of nonsense mutations in Duchenne/ Becker muscular dystrophy: aminoglycosides and ataluren (PTC124) J Child Neurol. 2010;25:1158–64. doi: 10.1177/0883073810371129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Howard MT, Anderson CB, Fass U, et al. Readthrough of dystrophin stop codon mutations induced by aminoglycosides. Ann Neurol. 2004;55:422–6. doi: 10.1002/ana.20052. [DOI] [PubMed] [Google Scholar]
- 25.Wang B, Li J, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci U S A. 2000;97:13714–9. doi: 10.1073/pnas.240335297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Z, Kuhr CS, Allen JM, et al. Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol Ther. 2007;15:1160–6. doi: 10.1038/sj.mt.6300161. [DOI] [PubMed] [Google Scholar]
- 27.Rodino-Klapac LR, Montgomery CL, Bremer WG, et al. Persistent expression of FLAG-tagged micro dystrophin in nonhuman primates following intramuscular and vascular delivery. Mol Ther. 2010;18:109–17. doi: 10.1038/mt.2009.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Storb R, Halbert CL, et al. Successful regional delivery and long-term expression of a dystrophin gene in canine muscular dystrophy: a preclinical model for human therapies. Mol Ther. 2012;20:1501–7. doi: 10.1038/mt.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foster H, Popplewell L, Dickson G. Genetic therapeutic approaches for Duchenne muscular dystrophy. Hum Gene Ther. 2012;23:676–87. doi: 10.1089/hum.2012.099. [DOI] [PubMed] [Google Scholar]
- 30.Fabb SA, Wells DJ, Serpente P, Dickson G. Adeno-associated virus vector gene transfer and sarcolemmal expression of a 144 kDa micro-dystrophin effectively restores the dystrophin-associated protein complex and inhibits myofibre degeneration in nude/mdx mice. Hum Mol Genet. 2002;11:733–41. doi: 10.1093/hmg/11.7.733. [DOI] [PubMed] [Google Scholar]
- 31.Foster H, Sharp PS, Athanasopoulos T, et al. Codon and mRNA sequence optimization of microdystrophin transgenes improves expression and physiological outcome in dystrophic mdx mice following AAV2/8 gene transfer. Mol Ther. 2008;16:1825–32. doi: 10.1038/mt.2008.186. [DOI] [PubMed] [Google Scholar]
- 32.Koo T, Malerba A, Athanasopoulos T, et al. Delivery of AAV2/9-microdystrophin genes incorporating helix 1 of the coiled-coil motif in the C-terminal domain of dystrophin improves muscle pathology and restores the level of alpha1-syntrophin and alpha-dystrobrevin in skeletal muscles of mdx mice. Hum Gene Ther. 2011;22:1379–88. doi: 10.1089/hum.2011.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koo T, Okada T, Athanasopoulos T, Foster H, Takeda S, Dickson G. Long-term functional adeno-associated virus-microdystrophin expression in the dystrophic CXMDj dog. J Gene Med. 2011;13:497–506. doi: 10.1002/jgm.1602. [DOI] [PubMed] [Google Scholar]
- 34.Pickering BM, Willis AE. The implications of structured 5’ untranslated regions on translation and disease. Semin Cell Dev Biol. 2005;16:39–47. doi: 10.1016/j.semcdb.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 35.Chatterjee S, Pal JK. Role of 5’- and 3’-untranslated regions of mRNAs in human diseases. Biol Cell. 2009;101:251–62. doi: 10.1042/BC20080104. [DOI] [PubMed] [Google Scholar]
- 36.Kochetov AV, Ischenko IV, Vorobiev DG, et al. Eukaryotic mRNAs encoding abundant and scarce proteins are statistically dissimilar in many structural features. FEBS Lett. 1998;440:351–5. doi: 10.1016/s0014-5793(98)01482-3. [DOI] [PubMed] [Google Scholar]
- 37.Mignone F, Gissi C, Liuni S, Pesole G. Untranslated regions of mRNAs. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-3-reviews0004. REVIEWS0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waterston RH, Lindblad-Toh K, Birney E, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–62. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 39.Miura P, Thompson J, Chakkalakal JV, Holcik M, Jasmin BJ. The utrophin A 5’-untranslated region confers internal ribosome entry site-mediated translational control during regeneration of skeletal muscle fibers. J Biol Chem. 2005;280:32997–33005. doi: 10.1074/jbc.M503994200. [DOI] [PubMed] [Google Scholar]
- 40.Miura P, Coriati A, Belanger G, et al. The utrophin A 5’-UTR drives cap-independent translation exclusively in skeletal muscles of transgenic mice and interacts with eEF1A2. Hum Mol Genet. 2010;19:1211–20. doi: 10.1093/hmg/ddp591. [DOI] [PubMed] [Google Scholar]
- 41.Kubokawa I, Takeshima Y, Ota M, et al. Molecular characterization of the 5’-UTR of retinal dystrophin reveals a cryptic intron that regulates translational activity. Mol Vis. 2010;16:2590–7. [PMC free article] [PubMed] [Google Scholar]
- 42.Andreassi C, Riccio A. To localize or not to localize: mRNA fate is in 3’UTR ends. Trends Cell Biol. 2009;19:465–74. doi: 10.1016/j.tcb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 43.Mazumder B, Seshadri V, Fox PL. Translational control by the 3’-UTR: the ends specify the means. Trends Biochem Sci. 2003;28:91–8. doi: 10.1016/S0968-0004(03)00002-1. [DOI] [PubMed] [Google Scholar]
- 44.Matoulkova E, Michalova E, Vojtesek B, Hrstka R. The role of the 3’ untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol. 2012;9:563–76. doi: 10.4161/rna.20231. [DOI] [PubMed] [Google Scholar]
- 45.Gramolini AO, Belanger G, Jasmin BJ. Distinct regions in the 3’ untranslated region are responsible for targeting and stabilizing utrophin transcripts in skeletal muscle cells. J Cell Biol. 2001;154:1173–83. doi: 10.1083/jcb.200101108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lemaire C, Heilig R, Mandel JL. The chicken dystrophin cDNA: striking conservation of the C-terminal coding and 3’ untranslated regions between man and chicken. EMBO J. 1988;7:4157–62. doi: 10.1002/j.1460-2075.1988.tb03311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Greener MJ, Sewry CA, Muntoni F, Roberts RG. The 3’-untranslated region of the dystrophin gene - conservation and consequences of loss. Eur J Hum Genet. 2002;10:413–20. doi: 10.1038/sj.ejhg.5200822. [DOI] [PubMed] [Google Scholar]
- 48.Dassi E, Zuccotti P, Leo S, et al. Hyper conserved elements in vertebrate mRNA 3’-UTRs reveal a translational network of RNA-binding proteins controlled by HuR. Nucleic Acids Res. 2013;41:3201–16. doi: 10.1093/nar/gkt017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siepel A, Bejerano G, Pedersen JS, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–50. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sathirapongsasuti JF, Sathira N, Suzuki Y, Huttenhower C, Sugano S. Ultraconserved cDNA segments in the human transcriptome exhibit resistance to folding and implicate function in translation and alternative splicing. Nucleic Acids Res. 2011;39:1967–79. doi: 10.1093/nar/gkq949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ho ES, Gunderson SI. Long conserved fragments upstream of Mammalian polyadenylation sites. Genome Biol Evol. 2011;3:654–66. doi: 10.1093/gbe/evr053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cacchiarelli D, Incitti T, Martone J, et al. miR-31 modulates dystrophin expression: new implications for Duchenne muscular dystrophy therapy. EMBO Rep. 2011;12:136–41. doi: 10.1038/embor.2010.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanamori H, Dodson RE, Shapiro DJ. In vitro genetic analysis of the RNA binding site of vigilin, a multi-KH-domain protein. Mol Cell Biol. 1998;18:3991–4003. doi: 10.1128/mcb.18.7.3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanguay RL, Gallie DR. Translational efficiency is regulated by the length of the 3’ untranslated region. Mol Cell Biol. 1996;16:146–56. doi: 10.1128/mcb.16.1.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hogg JR, Goff SP. Upf1 senses 3’UTR length to potentiate mRNA decay. Cell. 2010;143:379–89. doi: 10.1016/j.cell.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gingerich TJ, Feige JJ, LaMarre J. AU-rich elements and the control of gene expression through regulated mRNA stability. Anim Health Res Rev. 2004;5:49–63. doi: 10.1079/ahr200460. [DOI] [PubMed] [Google Scholar]
- 57.Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles? . Nucleic Acids Res. 2005;33:7138–50. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chakkalakal JV, Miura P, Belanger G, Michel RN, Jasmin BJ. Modulation of utrophin A mRNA stability in fast versus slow muscles via an AU-rich element and calcineurin signaling. Nucleic Acids Res. 2008;36:826–38. doi: 10.1093/nar/gkm1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barrett LW, Fletcher S, Wilton SD. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell Mol Life Sci. 2012;69:3613–34. doi: 10.1007/s00018-012-0990-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoshida K, Nakamura A, Yazaki M, Ikeda S, Takeda S. Insertional mutation by transposable element, L1, in the DMD gene results in X-linked dilated cardiomyopathy. Hum Mol Genet. 1998;7:1129–32. doi: 10.1093/hmg/7.7.1129. [DOI] [PubMed] [Google Scholar]
- 61.McCabe ER, Towbin J, Chamberlain J, et al. Complementary DNA probes for the Duchenne muscular dystrophy locus demonstrate a previously undetectable deletion in a patient with dystrophic myopathy, glycerol kinase deficiency, and congenital adrenal hypoplasia. J Clin Invest. 1989;83:95–9. doi: 10.1172/JCI113890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Waite A, Brown SC, Blake DJ. The dystrophin-glycoprotein complex in brain development and disease. Trends Neurosci. 2012;35:487–96. doi: 10.1016/j.tins.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 63.Waite A, Tinsley CL, Locke M, Blake DJ. The neurobiology of the dystrophin-associated glycoprotein complex. Ann Med. 2009;41:344–59. doi: 10.1080/07853890802668522. [DOI] [PubMed] [Google Scholar]
- 64.Cotton SM, Voudouris NJ, Greenwood KM. Association between intellectual functioning and age in children and young adults with Duchenne muscular dystrophy: further results from a meta-analysis. Dev Med Child Neurol. 2005;47:257–65. doi: 10.1017/s0012162205000496. [DOI] [PubMed] [Google Scholar]
- 65.Wu JY, Kuban KC, Allred E, Shapiro F, Darras BT. Association of Duchenne muscular dystrophy with autism spectrum disorder. J Child Neurol. 2005;20:790–5. doi: 10.1177/08830738050200100201. [DOI] [PubMed] [Google Scholar]
- 66.Hendriksen JG, Vles JS. Neuropsychiatric disorders in males with duchenne muscular dystrophy: frequency rate of attention-deficit hyperactivity disorder (ADHD), autism spectrum disorder, and obsessive--compulsive disorder. J Child Neurol. 2008;23:477–81. doi: 10.1177/0883073807309775. [DOI] [PubMed] [Google Scholar]
- 67.de Brouwer AP, Nabuurs SB, Verhaart IE, et al. A 3-base pair deletion, c.9711_9713del, in DMD results in intellectual disability without muscular dystrophy. Eur J Hum Genet. 2014;22:480–5. doi: 10.1038/ejhg.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–92. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- 69.Kozak M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J Mol Biol. 1987;196:947–50. doi: 10.1016/0022-2836(87)90418-9. [DOI] [PubMed] [Google Scholar]
- 70.Kozak M. Recognition of AUG and alternative initiator codons is augmented by G in position +4 but is not generally affected by the nucleotides in positions +5 and +6. EMBO J. 1997;16:2482–92. doi: 10.1093/emboj/16.9.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Forest D, Nishikawa R, Kobayashi H, Parton A, Bayne CJ, Barnes DW. RNA expression in a cartilaginous fish cell line reveals ancient 3’ noncoding regions highly conserved in vertebrates. Proc Natl Acad Sci U S A. 2007;104:1224–9. doi: 10.1073/pnas.0610350104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnsson P, Lipovich L, Grander D, Morris KV. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim Biophys Acta. 2014;1840:1063–71. doi: 10.1016/j.bbagen.2013.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Daubas P, Crist CG, Bajard L, et al. The regulatory mechanisms that underlie inappropriate transcription of the myogenic determination gene Myf5 in the central nervou system. Dev Biol. 2009;327:71–82. doi: 10.1016/j.ydbio.2008.11.031. [DOI] [PubMed] [Google Scholar]
- 74.Erriquez D, Perini G, Ferlini A. Non-Coding RNAs in Muscle Dystrophies. Int J Mol Sci. 2013;14:19681–704. doi: 10.3390/ijms141019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bovolenta M, Erriquez D, Valli E, et al. The DMD locus harbours multiple long non-coding RNAs which orchestrate and control transcription of muscle dystrophin mRNA isoforms. PLoS One. 2012;7:e45328. doi: 10.1371/journal.pone.0045328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hube F, Velasco G, Rollin J, Furling D, Francastel C. Steroid receptor RNA activator protein binds to and counteracts SRA RNA-mediated activation of MyoD and muscle differentiation. Nucleic Acids Res. 2011;39:513–25. doi: 10.1093/nar/gkq833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cesana M, Cacchiarelli D, Legnini I, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147:358–69. doi: 10.1016/j.cell.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Spitali P, van den Bergen JC, Verhaart IE, et al. DMD transcript imbalance determines dystrophin levels. FASEB J. 2013;27:4909–16. doi: 10.1096/fj.13-232025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.