

Abstract
Two recent papers in Nature, Ben-Zvi et al. (2014) and Nguyen et al. (2014), report a surprising dual role for the blood-brain barrier transporter MFSD2A in both establishing BBB integrity and in uptake of the fatty acid DHA.
Blood vessels in the brain are organized with surprising precision, patterned in parallel with the major brain circuits tasked with sensation, memory, and motion. The term “neurovascular unit” (NVU) has been coined to reflect this intimate connection between neuronal circuits and their associated blood vessels (Moskowitz et al., 2010; Zlokovic, 2011; Iadecola, 2013). This tight interrelationship may reflect key functional roles of blood vessels in neuronal normal function and disease.
The CNS vascular system is a highly dynamic multicellular structure capable of integrating and responding to both systemic and neural cues. At the core of its proper functionality is the intimately connected and highly coordinated NVU. The NVU comprises vascular cells such as endothelial cells and pericytes at the capillary level and vascular smooth muscle cells at the arterial level, glia (e.g., astrocytes, microglia, oligodendrocytes), and neurons (Figure 1, top). Proper signal transduction between these different, but equally important, cell types is essential for effective CNS functioning underlying cortical and subcortical information processing of motor functions as well as the higher integrative CNS functions associated with cognition, memory, and learning. Prior studies using genetically manipulated mouse lines have shown that disrupted crosstalk between endothelial cells and pericytes (Armulik et al., 2010; Daneman et al., 2010; Bell et al., 2010) or astrocytes and pericytes (Bell et al., 2012) leads to a loss of cerebrovascular integrity. This in turn leads to accumulation of neurotoxic blood-derived products in the CNS including hemoglobin, iron, thrombin, fibrin, or plasmin resulting in oxidative stress, intraneuronal accumulation of toxic proteins, and/or neuronal detachment from the extracellular matrix, respectively, causing neuronal dysfunction, injury, neurodegeneration, and neuronal loss (Bell et al., 2012; Winkler et al., 2014).
Figure 1. The Neurovascular Unit and Blood-Brain Barrier.
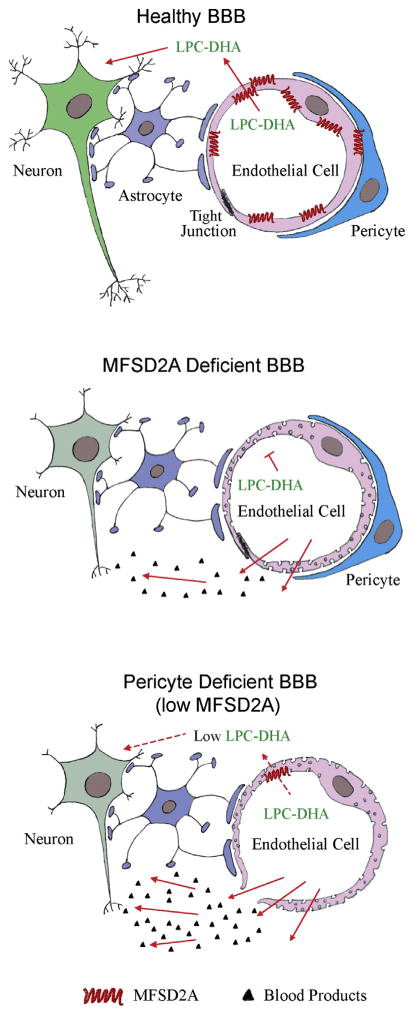
In the NVU, the endothelial cells form a continuous, highly specialized biological membrane around blood vessels underlying the blood-brain barrier (BBB), which limits the entry of plasma components, red blood cells, and leukocytes into the brain. Importantly, the BBB also regulates the delivery of energy metabolites and essential nutrients to the brain and maintains the removal from the brain to blood of neurotoxic molecules. This transport function of the BBB is controlled by highly specialized substrate-specific transport proteins expressed in brain endothelium, such as, for example, (1) the glucose transporter-1 (GLUT1) that directs influx of glucose across the BBB, providing a key energy substrate for neuronal metabolism; (2) the monocarboxylate transporter that mediates transport of lactate and other monocarboxylates including ketone bodies, as an alternative source of brain energy; (3) multiple nutrient amino acid transporters that maintain delivery of all essential and nonessential amino acids to neurons according to their respective blood-to-brain interstitial fluid (ISF) concentration gradients; and/or (4) ABC transporters that limit the penetration of many drugs into the brain by an “up-hill” energy-dependent efflux (Zlokovic, 2008a).
The diffusion distances from the neighboring capillaries to each neuronal cell body within the CNS are extremely short, i.e., ∼15–30 μm (Bell et al., 2010; Blinder et al., 2013). These short diffusion pathways allow rapid, almost instantaneous (<1 s) delivery of glucose, other nutrients, and/or small molecules to neurons as soon as they cross the BBB and enter the ISF. Most of these molecules and nutrients also cross the blood-cerebrospinal fluid (CSF) barrier via specialized transporters expressed in the choroid plexus epithelium. However, the diffusion of small polar molecules as well as large molecules from CSF to neurons via CSF-ISF diffusion pathway is several orders of magnitude slower than the BBB pathway. It would take hours or days for small and large molecules, respectively, to reach neurons from CSF because the CSF-to-neuron diffusion distances are several orders of magnitude greater than from the BBB sites (Pardridge, 1991; Zlokovic, 2011). Therefore, the BBB endothelial transporters play a crucial role in CNS health and disease both in the adult and aging brain and during CNS development.
In a recent paper in Nature, Ben-Zvi et al. (2014) report that an orphaned transporter, the major facilitator super family domain containing 2a (Mfsd2a), is selectively expressed in the BBB endothelium and is required for normal formation and maintenance of the BBB integrity. Utilizing an improved method to assess BBB integrity during mouse CNS development, in which a small volume of tracer is injected into embryonic liver to minimize changes in blood pressure, Ben-Zvi et al. (2014) show that the BBB becomes functional at embryonic day 15.5 (E15.5). By comparing the gene expression profiles of different transcripts for transporters, transcription factors, and secreted and transmembrane proteins that are enriched in the BBB endothelium (cortex) with the barrier properties compared to non-BBB (lung) endothelium, they found that at embryonic day E13.5 the levels of Mfsd2a transcripts are higher by ∼80-fold in the BBB endothelium. Utilizing in situ hybridization and immunostaining, they convincingly demonstrate that Mfsd2a is expressed exclusively in brain endothelium but is not expressed at detectable levels in pericytes, astrocytes, or neuronal cells, suggesting that it has an endothelial cell-specific role. Using transgenic Mfsd2a−/− mice, Ben-Zvi et al. (2014) show that genetic deletion of Mfsd2a leads to BBB defects in the developing CNS, whereas the patterning of vascular networks remains unaffected. They also found that the BBB breakdown in Mfsd2a−/− mice is due to an uninhibited bulk flow transcytosis across the endothelium that is comparable to that previously shown in pericyte-deficient Pdgfbret/ret mice (Armulik et al., 2010).
To determine how their findings in Mfsd2a−/− mice relate to previous findings in pericyte-deficient mice (Armulik et al., 2010; Bell et al., 2010), Ben-Zvi et al. (2014) reanalyzed the micro-array data in Pdgfbret/ret mice and found direct correlation between the reduction of Mfsd2a gene expression and the degree of pericyte coverage. Although Mfsd2a−/− mice had normal pericyte coverage, they found a significant decrease in MFSD2A protein in brain endothelial cells in pericyte-deficient Pdgfbret/ret mice particularly in the microvessels lacking pericyte coverage. Therefore, Ben-Zvi et al. (2014) concluded that an increased vesicular trafficking underlying Mfsd2a-dependent BBB breakdown (Figure 1, middle) could be, at least in part, regulated by pericytes as suggested by their data in pericyte-deficient mice showing strong downregulation of endothelial MFSD2A (Figure 1, bottom). As downregulation of tight junction proteins has also been demonstrated to contribute to BBB breakdown in pericyte-deficient mouse lines (Bell et al., 2010), collectively these data indicate that pericytes may regulate both Mfsd2a-dependent and Mfsd2a-independent BBB breakdown by controlling the levels of tight junction proteins. Future studies are needed, however, to identify the signal transduction pathways and transcriptional regulation between pericytes and endothelial cells that control Mfsd2a expression and the associated Mfsd2a-dependent changes in BBB permeability, as well as Mfsd2a-independent regulation of the tight junction proteins in the endothelium. Moreover, it would be interesting to find out whether ablation of pericytes in adulthood can also lead to BBB breakdown through downregulation of Mfsd2a expression and/or reductions in the tight junction proteins as found in models of inherited pericyte deficiency.
Is MFSD2A a real orphan transporter? The accompanying paper in Nature by Nguyen et al. (2014) was focused on the transport function of Mfsd2a, asking whether MSFD2A has a role in BBB influx of substrates essential for normal neuronal function. Besides confirming exclusive MSFD2A expression in BBB endothelium as they show in the young adult mouse brain, Nguyen et al. (2014) performed a series of in vivo and in vitro experiments and found that MFSD2A is the major transporter mediating brain uptake of docosahexaenoic acid (DHA), an omega-3 fatty acid essential for normal brain growth and cognitive function (Figure 1, top). Using lipidomic analysis, Nguyen et al. (2014) show that Mfsd2a−/− mice have significantly reduced levels of DHA in brain accompanied with neuronal cell loss in the hippocampus and cerebellum, neurological and behavioral deficits, and reduced brain size (Figure 1 middle). Using cell-based assays and brain uptake studies in Mfsd2a−/− mice, they next show that Mfsd2a transports DHA and fatty acids into the brain across the BBB only in the form of esters with lysophosphatidylcholines (LPCs), but not as free unesterified fatty acids. They also show that MSFD2A transport protein prefers long-chain fatty acids such as LPC-oleate and LPC-palmitate but does not transport LPCs with less than a 14-carbon acyl chain. Their study demonstrates that LPCs phosphor-zwitterionic headgroup is critical for MFSD2A-mediated transport and that Mfsd2a−/− mice have substantially reduced uptake of labeled LPC-DHA and other LPCs from plasma into the brain.
These two studies, one demonstrating a transport role of MFSD2A mediating BBB uptake of an essential omega-3 fatty acid, and the other showing its critical role in the formation of the BBB integrity, raise several interesting questions. For example, to what extent the neuronal phenotype as seen by Nguyen et al. (2014) in Mfsd2a−/− mice is related to a shortfall in DHA transport as suggested by the authors, as opposed to nonmetabolic neuronal injury and loss mediated by unrestricted influx of potentially toxic blood-derived products into the brain across the defective BBB (Figure 1, middle), as previously shown in other models of a chronic BBB breakdown including pericyte-deficient mice (Bell et al., 2010), apolipoprotein E4-expressing mice (Bell et al., 2012), and/or after experimentally induced BBB breakdown in a mouse model of amyotrophic lateral sclerosis (ALS) (Winkler et al., 2014). It would be quite intriguing to find out whether pericytes can regulate MFSD2A-mediated DHA BBB transport across brain endothelium as they regulate the BBB permeability, as suggested by Ben-Zvi et al. (2014) (Figure 1, bottom). Finally, in regards to significance of these two new studies to the field of clinical neurosciences, one can speculate that the present findings might have even broader implications for better understanding of neurological diseases associated with neurovascular dysfunction, particularly the BBB breakdown and pericyte degeneration and/or loss, as seen in Alzheimer's disease, ALS, and/or after an ischemic CNS injury and stroke (Zlokovic, 2008b, 2011; Moskowitz et al., 2010; Carmeliet and Ruiz de Almodovar, 2013; Iadecola, 2013).
A discovery that a transport protein can play a dual role at the BBB by regulating brain uptake of an essential substrate and by controlling at the same time the BBB integrity is provocative and important. Here, MFSD2A joins the club of GLUT1 glucose transporter, which has previously been demonstrated to be essential for BBB formation in the developing CNS in zebrafish (Zheng et al., 2010) in addition to its established role in regulating glucose uptake by the brain. Whether MFSD2A and GLUT1 are two exceptional transport proteins that can play this dual role at the BBB, or whether they are only the first two transporters discovered of a more common class of transport proteins with a dual role at the BBB, remains to be seen!
References
- Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, et al. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, et al. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, Gu C. Nature. 2014 doi: 10.1038/nature13324. Published online May 14, 2014 http://dx.doi.org/10.1038/nature13324. [DOI] [PMC free article] [PubMed]
- Blinder P, Tsai PS, Kaufhold JP, Knutsen PM, Suhl H, Kleinfeld D. Nat Neurosci. 2013;16:889–897. doi: 10.1038/nn.3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Ruiz de Almodovar C. Cell Mol Life Sci. 2013;70:1763–1778. doi: 10.1007/s00018-013-1283-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. Neuron. 2013;80:844–866. doi: 10.1016/j.neuron.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LN, Ma D, Shui G, Wong P, Cazenave-Gassiot A, Zhang X, Wenk MR, Goh ELK, Silver DL. Nature. 2014 doi: 10.1038/nature13241. Published online May, 2014 http://dx.doi.org/10.1038/nature13241. [DOI] [PubMed]
- Pardridge WM. Peptide Drug Delivery to the Brain. New York: Raven Press; 1991. [Google Scholar]
- Winkler EA, Sengillo JD, Sagare AP, Zhao Z, Ma Q, Zuniga E, Wang Y, Zhong Z, Sullivan JS, Griffin JH, et al. Proc Natl Acad Sci USA. 2014;111:E1035–E1042. doi: 10.1073/pnas.1401595111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng PP, Romme E, van der Spek PJ, Dirven CMF, Willemsen R, Kros JM. Ann Neurol. 2010;68:835–844. doi: 10.1002/ana.22318. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neuron. 2008a;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurotherapeutics. 2008b;5:409–414. doi: 10.1016/j.nurt.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]