

Abstract
Stavudine (d4T) was, until recently, one of the most widely prescribed antiretroviral drugs worldwide. While there has been a major shift away from d4T use in resource-limited countries, a large number of patients have previously received (or continue to receive) d4T, and many have developed peripheral neuropathy. The identification of genetic predictors of increased risk might suggest novel therapeutic targets for such patients. In AIDS Clinical Trials Group protocol 384, antiretroviral-naïve patients were randomized to d4T/didanosine (ddI)- or zidovudine/lamivudine-containing regimens. Data from d4T/ddI recipients were analyzed for genome-wide associations (approximately 1 million genetic loci) with new onset distal sensory peripheral neuropathy. Analyses involved 254 patients (49 % White, 34 % Black, 17 % Hispanic), comprising 90 peripheral neuropathy cases (32 grade 1, 35 grade 2, 23 grade 3) and 164 controls. After correcting for multiple comparisons, no polymorphism was consistently associated with neuropathy among all patients, among White, Black, and Hispanic patients analyzed separately, both in genome-wide analyses (threshold, P < 5.0 × 10−8) and focused on 46 neuropathy-associated genes (threshold, P < 3.5 × 10−5). In the latter analyses, the lowest P values were in KIF1A among Whites (rs10199388, P = 8.4 × 10−4), in LITAF among Blacks (rs13333308, P = 6.0 × 10−6), and in NEFL among Hispanics (rs17763685, P = 5.6 × 10−6). Susceptibility to d4T/ddI-associated neuropathy is not explained by a single genetic variant with a marked effect.
Electronic supplementary material
The online version of this article (doi:10.1007/s13365-014-0235-9) contains supplementary material, which is available to authorized users.
Keywords: Peripheral neuropathy, HIV-1, Stavudine, Didanosine, Genomics
Introduction
Antiretroviral therapy has greatly reduced human immunodeficiency virus (HIV)-1-associated morbidity and mortality. The dideoxynucleoside reverse transcriptase inhibitors didanosine (ddI) and stavudine (d4T) have been widely prescribed in multidrug antiretroviral regimens, but severe toxicities associated with these “D-drugs” have markedly curtailed their prescribing (Dalakas 2001). Such toxicities including peripheral neuropathy, hepatic steatosis, lactic acidosis, and peripheral lipoatrophy are likely the consequence of inhibition of mitochondrial DNA polymerase gamma (Lewis et al. 2001).
Until recently, d4T was one of the most widely prescribed antiretroviral drugs worldwide. Over the past several years, there has been a major shift from d4T use to tenofovir use in resource-limited countries, a change that had already occurred in most developed countries. Large numbers of patients, however, have previously received (or continue to receive) d4T for many months or years, and many have developed peripheral neuropathy. The identification of genetic predictor(s) of increased risk for peripheral neuropathy might suggest therapeutic targets for such patients.
Previous reports involving patients of European and African ancestry have suggested associations between heritable variations in mitochondrial DNA (so-called mitochondrial haplogroups) and increased risk for peripheral neuropathy with ddI/d4T-containing regimens (Canter et al. 2010; Hulgan et al. 2005). The present study examined genetic associations between nuclear variants and risk of developing peripheral neuropathy among HIV-infected patients who initiated d4T/ddI-containing regimens in a prospective clinical trial.
Methods
This retrospective, case-control genome-wide association study (GWAS) utilized data and specimens that had been prospectively collected from participants in AIDS Clinical Trials Group (ACTG) protocol 384, who consented for genetic analysis under protocol A5128. Study ACTG 384 was a multicenter randomized trial designed to compare antiretroviral drug regimens (ddI/d4T or zidovudine/lamivudine given with efavirenz, nelfinavir or both) as initial treatment for HIV-1 infection (Robbins et al. 2003). Patients in the present analyses enrolled in ACTG 384 in 1998 and 1999 in the USA and received ddI/d4T-containing regimens. Cases were patients who developed clinical signs or symptoms of distal sensory neuropathy within the first 96 weeks of initiating ddI/d4T. Controls were patients who did not develop such signs or symptoms within 96 weeks of initiating ddI/d4T. Patients with peripheral neuropathy documented prior to initiating ddI/d4T were excluded from these analyses. Peripheral neuropathy was assessed at each study visit and was categorized as grade 1 (asymptomatic with sensory alteration on exam or minimal paresthesia causing no or minimal interference with usual social and functional activities), grade 2 (sensory alteration or paresthesia causing greater than minimal interference with usual social and functional activities), and grade 3 (sensory alteration or paresthesia causing inability to perform usual social and functional activities).
Genotyping of DNA extracted from whole blood was by Illumina Human-1M Duo Beadchip (Illumina Inc., San Diego, CA, USA) and was available from a separate HIV immunogenomics project (Pereyra et al. 2010). Population ancestry was assessed by projecting genetic assay data onto HapMap Phase III data (Pereyra et al. 2010). Analyses were limited to nuclear polymorphisms. Hereafter, we refer to non-Hispanic White, non-Hispanic Black, and Hispanic patients as White, Black, and Hispanic, respectively.
Quality control was applied to genetic assay data as follows. Variants with missingness >2 % or minor allele frequencies <5 % were excluded. Data from samples with exceptionally high or low heterozygosity values (|F| > 0.1) or genotyping failure >2 % were excluded. A total of 936,149 polymorphisms passed quality control. Within each race/ethnicity group, associations were assessed for genome-wide significance (P < 5.0 × 10−8) by logistic regression. To examine associations across all three race/ethnicity groups together, meta-analyses were also performed. Further analyses focused on polymorphisms in the set of neuropathy-associated genes listed in the Inherited Peripheral Neuropathies Mutation Database (Timmermen 2011) and also in DNMT1 and KIF1A (list of genes provided in Supplemental On-line Table 1). For analyses involving neuropathy-associated gene, polymorphisms within 100 kb upstream and downstream of each gene were included, and the genome-wide significance threshold was P < 3.5 × 10−5. Analyses were performed separately for grade ≥1, grade ≥2, and grade 3 peripheral neuropathies. Quality control of genotype data and genetic association analyses were performed using PLINK v1.07 (Purcell et al. 2007). Additional statistical analyses were done with the R statistical package version 2.13.1.
Results
A total of 254 patients were included in these analyses, comprising 90 peripheral neuropathy cases (32 grade 1, 35 grade 2, and 23 grade 3) and 164 controls. There were 125 (49.0 %) White, 86 (34 %) Black, and 43 (17 %) Hispanic patients as assessed by principal components. In meta-analyses, involving all 254 White, Black, and Hispanic patients, that focused on the 90 grade ≥1 peripheral neuropathy cases, no polymorphism was genome-wide significant (Fig. 1). The lowest P value was intronic in KRR1 (rs11540407, odds ratio (OR) = 2.6; P = 9.6 × 10−6). Separate meta-analyses limited to the 58 grade ≥2 peripheral neuropathy cases and limited to the 23 grade 3 peripheral neuropathy cases did not similarly show any polymorphism to be genome-wide significant. Polymorphisms with the lowest P values were not consistent between analyses nor were they consistent between populations (Table 1).
Fig. 1.
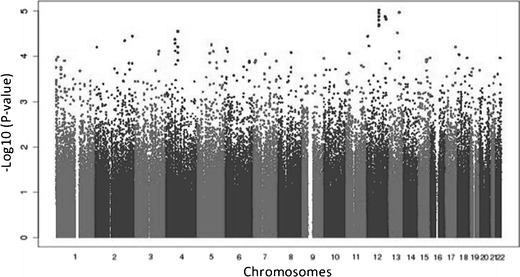
Manhattan plot of associations between genetic polymorphisms and grade ≥1 sensory peripheral neuropathy by meta-analysis. The −log10 of P values are shown. The genome-wide significance threshold is P = 5.0 × 10−8
Table 1.
Polymorphisms with the lowest P values for association with sensory peripheral neuropathy
| Case/control | Entire genomea | Neuropathy-associated genesa,b | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | Polymorphism | Odds ratio | P value | Gene | Polymorphism | Odds ratio | P value | ||
| Grade ≥1 | 90/164 | KRR1 | rs11540407 | 2.6 | 9.6 × 10−6 | FGD4 | rs1909509 | 2.1 | 9.6 × 10−4 |
| Intergenic | rs9518599 | 2.5 | 1.1 × 10−5 | FGD4 | rs7298165 | 1.9 | 1.8 × 10−3 | ||
| KRR1 | rs1552039 | 2.6 | 1.1 × 10−5 | FGD4 | rs6488066 | 1.9 | 2.2 × 10−3 | ||
| ACE2 | rs1514280 | 3.6 | 1.2 × 10−5 | RAD52 | rs1051669 | 1.8 | 3.4 × 10−3 | ||
| KRR1 | rs2070162 | 2.6 | 1.3 × 10−5 | NDRG1 | rs2930004 | 1.7 | 4.3 × 10−3 | ||
| Grade ≥2 | 58/164 | SASH1 | rs8641 | 3.1 | 5.8 × 10−6 | FAM134B c | rs149511 | 0.4 | 4.7 × 10−3 |
| SASH1 c | rs208740 | 2.9 | 9.0 × 10−6 | FGD4 | rs1909509 | 2.1 | 5.4 × 10−3 | ||
| ST8SIA2 c | rs12913269 | 3.4 | 1.3 × 10−5 | WISP1 | rs2929965 | 0.5 | 5.6 × 10−3 | ||
| ST8SIA2 c | rs2129797 | 3.4 | 1.4 × 10−5 | LITAF c | rs12595973 | 0.5 | 7.7 × 10−3 | ||
| NTF3 | rs6332 | 2.7 | 1.7 × 10−5 | KIF1A c | rs4234121 | 0.4 | 9.2 × 10−3 | ||
| Grade 3 | 23/164 | ST8SIA2 c | rs12913269 | 6.5 | 3.2 × 10−7 | KIF1B c | rs11587309 | 2.7 | 5.1 × 10−3 |
| ST8SIA2 c | rs2129797 | 6.4 | 3.5 × 10−7 | KIF1A | rs10198394 | 2.4 | 7.3 × 10−3 | ||
| Intergenic | rs4397851 | 5.3 | 1.5 × 10−6 | NDRG1 | rs10505606 | 2.4 | 7.7 × 10−3 | ||
| ADAMTS2 | rs6873892 | 6.1 | 2.1 × 10−6 | NDRG1 | rs2233326 | 2.4 | 1.1 × 10−2 | ||
| ST8SIA2 c | rs12910290 | 5.1 | 2.8 × 10−6 | KIF1B | rs10492970 | 7.1 | 1.1 × 10−2 | ||
aMeta-analyses were performed to combine results from White, Black, and Hispanic groups
bThe neuropathy-associated genes from the Inherited Peripheral Neuropathies Mutation Database plus DNMT1 and KIF1A are listed in Supplemental On-line Table 1
cThese polymorphisms are within 100 kb of the named gene
Using whole genome data, separate analyses were performed in White, Black, and Hispanic patients and separately for grade ≥1, grade ≥2, and grade 3 peripheral neuropathies (i.e., nine separate analyses) (Supplemental on-line Table 2). Polymorphisms with the lowest P values are as follows. In White patients with grade 3 peripheral neuropathy, the polymorphism with the lowest P value was intronic in IL2RA (rs12722486, OR = 38.2; 95 % confidence interval (CI) = 6.6–219.6; P = 1.5 × 10−9). In Black patients with grade 3 peripheral neuropathy, the polymorphism with the lowest P value was intronic in ZNF648 (rs7554128, OR = 48.5; 95 % CI = 5.7–414; P = 5.7 × 10−8). In Hispanic patients with grade ≥2 peripheral neuropathy, the polymorphism with the lowest P value was within 100 kb of RSP04 (rs502716, OR = 28; 95 % CI = 6.8–114.7; P = 3.2 × 10−8)
Further analyses were limited to 46 neuropathy-associated genes and comprised 1,395 polymorphisms. In a meta-analysis, involving White, Black, and Hispanic patients, that focused on all 90 grade ≥1 peripheral neuropathy cases, no polymorphism was genome-wide significant at P < 3.5 × 10−5. The polymorphism with the lowest P value was intronic in FGD4 (rs1909509, OR = 2.1; P = 9.6 × 10−4). Separate meta-analyses limited to grade ≥2 as well as grade 3 peripheral neuropathy cases did not show any polymorphism to be genome-wide significant.
Considering only the 46 neuropathy-associated genes, separate analyses were conducted in White, Black, or Hispanic patients and for grade ≥1, grade ≥2, and grade 3 peripheral neuropathies (Supplemental on-line Table 3). Polymorphisms with the lowest P values were as follows. In White patients with grade 3 peripheral neuropathy, the lowest P value was within 100 kb of KIF1A (rs10199388, OR = 5.3; 95 % CI = 1.8–15.3; P = 8.4 × 10−4). In Black patients with grade ≥2 peripheral neuropathy, the polymorphism with the lowest P value was within 100 kb of LITAF (rs13333308, OR = 17.1; 95 % CI = 2.1–138.9; P = 4.6 × 10−4). This polymorphism also had the lowest P value in an analysis limited to 23 grade 3 peripheral neuropathy cases (rs13333308, OR = 32.3; 95 % CI = 3.7–284.9; P = 6.0 × 10−6). In Hispanic patients with grade ≥2 peripheral neuropathy, the polymorphism with the lowest P value was within 100 kb of NEFL (rs6557786, OR = 23.2; 95 % CI = 4.3–125.6; P = 4.7 × 10−6).
Discussion
Among patients exposed to ddI/d4T-containing regimens in ACTG 384, who were evaluable for genetic associations with new onset sensory peripheral neuropathy within the first 96 weeks of treatment, no polymorphism was consistently genome-wide significant. This was so in analyses of polymorphisms across the entire nuclear genome, and in analyses focused on polymorphisms in neuropathy-associated genes. This was also so in analyses that included all grade ≥1 peripheral neuropathy cases and in analyses limited to grade ≥2 and grade 3 peripheral neuropathy cases. Furthermore, polymorphisms with the lowest P values were not consistent across analyses limited to grade ≥1, grade ≥2, and grade 3 peripheral neuropathies. The fact that associations did not reproduce across populations nor across grades of neuropathy suggests that such associations may not also reproduce in other cohorts.
In White patients with grade 3 peripheral neuropathy, the polymorphism with the lowest P value was in IL2RA. The interleukin-2 receptor alpha (IL2RA) gene encodes a regulator of immune responses (Maier et al. 2009) but has no clear relevance to neuronal physiology. In Black patients with grade 3 peripheral neuropathy, the polymorphism with the lowest P value was in ZNF648, which may be involved in transcriptional regulation. In Hispanic patients with grade ≥2 peripheral neuropathy, the polymorphism with the lowest P value was near RSPO4 (which encodes R-spondin 4, possibly involved in Wnt/beta-catenin signaling pathways) (Blaydon et al. 2006) and ANGPT4 (which encodes angiopoietin 4, involved in vascular development and angiogenesis) (Olsen et al. 2006), genes with no specific relevance to neuronal physiology.
In analyses limited to neuropathy-associated genes, a meta-analysis considering all grades of peripheral neuropathy showed the lowest P value for a polymorphism in FGD4. This gene encodes frabin which is involved in peripheral nerve myelination, and FGD4 mutations have been associated with autosomal recessive Charcot-Marie-Tooth Type 4H (Charcot-Marie-Tooth disease comprises of a clinically and genetically diverse group of inherited disorders of peripheral nerves) (Delague et al. 2007). Meta-analysis of grade 3 peripheral neuropathy showed the lowest P value for a polymorphism near KIF1B. In White patients, the polymorphism with the lowest P value for association with all grades of peripheral neuropathy and with grade 3 peripheral neuropathy was in KIF1A, while in Black patients, the lowest P values were for polymorphisms in LITAF. KIF1A encodes a protein involved in anterograde transport of organelles along axonal microtubules, and KIF1A mutations have been associated with spastic paraplegia and hereditary sensory neuropathy (Klebe et al. 2012; Riviere et al. 2011), while KIF1B encodes for a protein involved in apoptosis and axonal transport of membranous organelles. Mutations to KIF family genes have been associated with Charcot-Marie-Tooth disease type 2A, while LITAF mutations have been associated with Charcot-Marie-Tooth disease type 1C (CMT1C) (Street et al. 2003). In Hispanic patients, the polymorphism with the lowest P value for association with all grades of peripheral neuropathy was in NEFL, which encodes the neurofilament light polypeptide. Mutations in NEFL have been associated with Charcot-Marie-Tooth disease types 2E and 1F (Jordanova et al. 2003; Shin et al. 2008)
There were limitations to this study. The sample size was modest, so only polymorphisms with marked effects could have achieved genome-wide significance. In addition, peripheral neuropathy in ACTG 384 was based on signs and symptoms, rather than formal neurologic evaluation assessments.
In summary, among patients exposed to ddI/d4T-containing regimens in the ACTG 384, no genetic polymorphism was consistently associated with new onset distal sensory peripheral neuropathy at genome-wide significance. Polymorphisms identified herein (e.g., in KIF1A, LITAF, and NEFL) warrant replication in other cohorts of HIV-infected patients who have developed D-drug-associated peripheral neuropathy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(DOC 97 kb)
(DOC 289 kb)
(DOC 301 kb)
Acknowledgments
The authors are grateful to the many persons with HIV infection who volunteered for ACTG 384 and A5128. In addition, they acknowledge the contributions of study teams and site staff for protocols ACTG 384 and A5128. This project was supported by grant AI-077505 (DWH). The project described was also supported by award number U01 AI-068636 from the National Institute of Allergy and Infectious Diseases (NIAID) and supported by National Institute of Mental Health (NIMH) and National Institute of Dental and Craniofacial Research (NIDCR). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. Additional grant support included AI-038858, AI-068634, AI-038855, GM-80178, AI-062435, AI-069415, AI-069419, AI-069423, AI-069424, AI-069428, AI-069432, AI-069434, AI-069450, AI-069452, AI-069465, AI-069471, AI-069472, AI-069474, AI-069477, AI-069484, AI-069495, AI-069501, AI-069502, AI-069511, AI-069513, AI-069532, AI-069556, AI-25859, AI-27661, AI-34835, AI-34853, AI-38844, AI-46370, AI-54999, AI-69467, AI-69495, AL-32782, HL-087726, NS-059330, and TR-000445.
Conflict of interest
David W. Haas has been the principal investigator on research grants to Vanderbilt University from Boehringer Ingelheim, Merck, and Gilead Sciences and has been a consultant to Merck. Gregory K. Robbins received research support from Gilead Sciences and Schering-Plough and received royalties from Wolters Kluwer. David B. Clifford has served on Data Safety Boards for Millennium, Pfizer, Genzyme, and Amgen and has been a consultant to Biogen Idec, Millennium, Bristol Myers Squibb, Pfizer, Genzyme, Amgen, and Quintiles. For the remaining authors, none were declared.
References
- Blaydon DC, Ishii Y, O’Toole EA, Unsworth HC, Teh MT, Ruschendorf F, Sinclair C, Hopsu-Havu VK, Tidman N, Moss C, Watson R, de Berker D, Wajid M, Christiano AM, Kelsell DP. The gene encoding R-spondin 4 (RSPO4), a secreted protein implicated in Wnt signaling, is mutated in inherited anonychia. Nat Genet. 2006;38:1245–1247. doi: 10.1038/ng1883. [DOI] [PubMed] [Google Scholar]
- Canter JA, Robbins GK, Selph D, Clifford DB, Kallianpur AR, Shafer R, Levy S, Murdock DG, Ritchie MD, Haas DW, Hulgan T. African mitochondrial DNA subhaplogroups and peripheral neuropathy during antiretroviral therapy. J Infect Dis. 2010;201:1703–1707. doi: 10.1086/652419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalakas MC. Peripheral neuropathy and antiretroviral drugs. J Peripher Nerv Syst : JPNS. 2001;6:14–20. doi: 10.1046/j.1529-8027.2001.006001014.x. [DOI] [PubMed] [Google Scholar]
- Delague V, Jacquier A, Hamadouche T, Poitelon Y, Baudot C, Boccaccio I, Chouery E, Chaouch M, Kassouri N, Jabbour R, Grid D, Megarbane A, Haase G, Levy N. Mutations in FGD4 encoding the Rho GDP/GTP exchange factor FRABIN cause autosomal recessive Charcot-Marie-Tooth type 4H. Am J Hum Genet. 2007;81:1–16. doi: 10.1086/518428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulgan T, Haas DW, Haines JL, Ritchie MD, Robbins GK, Shafer RW, Clifford DB, Kallianpur AR, Summar M, Canter JA. Mitochondrial haplogroups and peripheral neuropathy during antiretroviral therapy: an adult AIDS clinical trials group study. AIDS. 2005;19:1341–1349. doi: 10.1097/01.aids.0000180786.02930.a1. [DOI] [PubMed] [Google Scholar]
- Jordanova A, De Jonghe P, Boerkoel CF, Takashima H, De Vriendt E, Ceuterick C, Martin JJ, Butler IJ, Mancias P, Papasozomenos S, Terespolsky D, Potocki L, Brown CW, Shy M, Rita DA, Tournev I, Kremensky I, Lupski JR, Timmerman V. Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease. Brain. 2003;126:590–597. doi: 10.1093/brain/awg059. [DOI] [PubMed] [Google Scholar]
- Klebe S, Lossos A, Azzedine H, Mundwiller E, Sheffer R, Gaussen M, Marelli C, Nawara M, Carpentier W, Meyer V, Rastetter A, Martin E, Bouteiller D, Orlando L, Gyapay G, El-Hachimi KH, Zimmerman B, Gamliel M, Misk A, Lerer I, Brice A, Durr A, Stevanin G. KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: distinct phenotypes according to the nature of the mutations. Eur J Hum Genet. 2012;20:645–649. doi: 10.1038/ejhg.2011.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis W, Copeland WC, Day BJ. Mitochondrial dna depletion, oxidative stress, and mutation: mechanisms of dysfunction from nucleoside reverse transcriptase inhibitors. Lab Invest. 2001;81:777–790. doi: 10.1038/labinvest.3780288. [DOI] [PubMed] [Google Scholar]
- Maier LM, Lowe CE, Cooper J, Downes K, Anderson DE, Severson C, Clark PM, Healy B, Walker N, Aubin C, Oksenberg JR, Hauser SL, Compston A, Sawcer S, De Jager PL, Wicker LS, Todd JA, Hafler DA. IL2RA genetic heterogeneity in multiple sclerosis and type 1 diabetes susceptibility and soluble interleukin-2 receptor production. PLoS Genet. 2009;5:e1000322. doi: 10.1371/journal.pgen.1000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen MW, Ley CD, Junker N, Hansen AJ, Lund EL, Kristjansen PE. Angiopoietin-4 inhibits angiogenesis and reduces interstitial fluid pressure. Neoplasia. 2006;8:364–372. doi: 10.1593/neo.06127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, Ripke S, Brumme CJ, Pulit SL, Carrington M, Kadie CM, Carlson JM, Heckerman D, Graham RR, Plenge RM, Deeks SG, Gianniny L, Crawford G, Sullivan J, Gonzalez E, Davies L, Camargo A, Moore JM, Beattie N, Gupta S, Crenshaw A, Burtt NP, Guiducci C, Gupta N, Gao X, Qi Y, Yuki Y, Piechocka-Trocha A, Cutrell E, Rosenberg R, Moss KL, Lemay P, O’Leary J, Schaefer T, Verma P, Toth I, Block B, Baker B, Rothchild A, Lian J, Proudfoot J, Alvino DM, Vine S, Addo MM, Allen TM, Altfeld M, Henn MR, Le Gall S, Streeck H, Haas DW, Kuritzkes DR, Robbins GK, Shafer RW, Gulick RM, Shikuma CM, Haubrich R, Riddler S, Sax PE, Daar ES, Ribaudo HJ, Agan B, Agarwal S, Ahern RL, Allen BL, Altidor S, Altschuler EL, Ambardar S, Anastos K, Anderson B, Anderson V, Andrady U, Antoniskis D, Bangsberg D, Barbaro D, Barrie W, Bartczak J, Barton S, Basden P, Basgoz N, Bazner S, Bellos NC, Benson AM, Berger J, Bernard NF, Bernard AM, Birch C, Bodner SJ, Bolan RK, Boudreaux ET, Bradley M, Braun JF, Brndjar JE, Brown SJ, Brown K, Brown ST, et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riviere JB, Ramalingam S, Lavastre V, Shekarabi M, Holbert S, Lafontaine J, Srour M, Merner N, Rochefort D, Hince P, Gaudet R, Mes-Masson AM, Baets J, Houlden H, Brais B, Nicholson GA, Van Esch H, Nafissi S, De Jonghe P, Reilly MM, Timmerman V, Dion PA, Rouleau GA. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet. 2011;89:219–230. doi: 10.1016/j.ajhg.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins GK, De GV, Shafer RW, Smeaton LM, Snyder SW, Pettinelli C, Dube MP, Fischl MA, Pollard RB, Delapenha R, Gedeon L, van der Horst C, Murphy RL, Becker MI, D’Aquila RT, Vella S, Merigan TC, Hirsch MS. Comparison of sequential three-drug regimens as initial therapy for HIV-1 infection. N Engl J Med. 2003;349:2293–2303. doi: 10.1056/NEJMoa030264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JS, Chung KW, Cho SY, Yun J, Hwang SJ, Kang SH, Cho EM, Kim SM, Choi BO. NEFL Pro22Arg mutation in Charcot-Marie-Tooth disease type 1. J Hum Genet. 2008;53:936–940. doi: 10.1007/s10038-008-0333-8. [DOI] [PubMed] [Google Scholar]
- Street VA, Bennett CL, Goldy JD, Shirk AJ, Kleopa KA, Tempel BL, Lipe HP, Scherer SS, Bird TD, Chance PF. Mutation of a putative protein degradation gene LITAF/SIMPLE in Charcot-Marie-Tooth disease 1C. Neurology. 2003;60:22–26. doi: 10.1212/WNL.60.1.22. [DOI] [PubMed] [Google Scholar]
- Timmerman V (2011). Inherited Peripheral Neuropathy Mutation Database; Department of Molecular Genetics, University of Antwerp, Belgium. Accessed August 30, 2013. Available at: http://www.molgen.ua.ac.be/CMTMutations/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 97 kb)
(DOC 289 kb)
(DOC 301 kb)