

Abstract
A SAR translation strategy adopted for the discovery of tetrahydroisoquinolinone (THIQ)-based steroidomimetic microtubule disruptors has been extended to dihydroisoquinolinone (DHIQ)-based compounds. A steroid A,B-ring-mimicking DHIQ core was connected to methoxyaryl D-ring mimics through methylene, carbonyl, and sulfonyl linkers, and the resulting compounds were evaluated against two cancer cell lines. The carbonyl-linked DHIQs in particular exhibit significant in vitro antiproliferative activities (e.g., 6-hydroxy-7-methoxy-2-(3,4,5-trimethoxybenzoyl)-3,4-dihydroisoquinolin-1(2H)-one (16 g): GI50 51 nm in DU-145 cells). The broad anticancer activity of DHIQ 16 g was confirmed in the NCI 60-cell line assay giving a mean activity of 33 nm. Furthermore, 6-hydroxy-2-(3,5-dimethoxybenzoyl)-7-methoxy-3,4-dihydroisoquinolin-1(2H)-one (16 f) and 16 g and their sulfamate derivatives 17 f and 17 g (2-(3,5-dimethoxybenzoyl)-7-methoxy-6-sulfamoyloxy-3,4-dihydroisoquinolin-1(2H)-one and 7-methoxy-2-(3,4,5-trimethoxybenzoyl)-6-sulfamoyloxy-3,4-dihydroisoquinolin-1(2H)-one, respectively) show excellent activity against the polymerization of tubulin, close to that of the clinical combretastatin A-4, and bind competitively at the colchicine binding site of tubulin. Compounds 16 f and 17 f were also shown to demonstrate in vitro anti-angiogenic activity. Additionally, X-ray and computational analyses of 17 f reveal that electrostatic repulsion between the two adjacent carbonyl groups, through conformational biasing, dictates the adoption of a “steroid-like” conformation that may partially explain the excellent in vitro activities.
Keywords: colchicine, dihydroisoquinolinones, electrostatic repulsion, microtubules, tubulin
Introduction
In previous studies we optimised sulfamoylated estratrienes 1 a–b (Figure 1) as anticancer agents.[1–6] These compounds exhibit antiproliferative activity against a range of human cancer cell lines and are also capable of inhibiting angiogenesis. This dual mechanism of action can be ascribed to their ability to inhibit normal microtubule dynamics and, in addition to good oral bioavailability and excellent in vivo activity, they proved capable of inhibiting the growth of cell lines resistant to existing microtubule disruptors such as the taxanes. To develop further series of compounds that share this mechanism of action we were drawn to investigate whether, by translating key pharmacophoric elements from the steroidal series into nonsteroidal motifs, we could generate new microtubule disruptors with further enhanced activity and/or physicochemical properties. In initial studies[7, 8] we used a tetrahydroisoquinoline (THIQ) decorated at C6 and C7 to mimic the steroidal A,B-ring of the estratrienes tethered through N2 to a D-ring mimic, initially a benzyl group, which projects a hydrogen bond acceptor into the appropriate region of space to address the pharmacophore for antiproliferative activity in that region. This delivered a series of microtubule disruptors with antiproliferative activity in the micromolar range that could be further optimised by introducing a substituent at C3 to sterically inhibit the free rotation of the N-benzyl group and thus favour conformational populations in which the N-benzyl group mimics the steroidal D-ring. In this manner compounds displaying nanomolar activity (equivalent to that of the steroid derivatives upon which their design was based) were elaborated.[9] In tandem, chimeric microtubule disruptors built from the THIQ core and the trimethoxy aryl motif common to many colchicine site binders were constructed.[10, 11]
Figure 1.
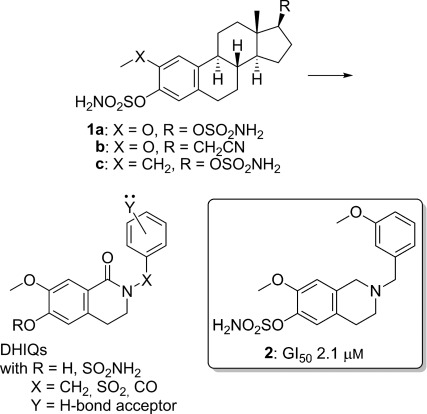
Design of DHIQ-based steroidomimetic microtubule disruptors.
In the present work we sought to develop new microtubule disruptors through integration of lessons learned in work on the estratrienes and the THIQ-based systems. By using an alternate heterocyclic motif, the dihydroisoquinolinone (DHIQ) to mimic the steroidal A,B-ring system, we envisaged that some conformational pre-organisation could be achieved through electrostatic repulsion. Tethering the D-ring-mimicking aryl group through a carbonyl linkage, repulsion of the two carbonyl groups of the imide should cause adoption of a dihedral angle in which they minimise their electrostatic clash; this should deliver a conformation in which the D-ring mimic projects into a region of space broadly analogous to a steroidal D-ring, thus decreasing rotation to achieve optimal binding. To assess this strategy a series of 2′-, 3′- and 4′-methoxybenzoyl DHIQs was synthesised alongside corresponding N-benzyl and N-arylsulfonyl DHIQ series for which no positive conformational biasing is envisaged (Figure 1).
Results and Discussion
Chemistry
The synthetic strategy to access the dihydroisoquinolinone core structure is outlined below (Scheme 1). 5,6-Dimethoxyindan-1-one 3 was selectively demethylated in aqueous piperidine and the product subsequently reacted with benzyl bromide to furnish compound 5 in good yield. Reaction of 5 with sodium azide and methanesulfonic acid in dichloromethane gave dihydroisoquinolinone 6 a. Some of the material was transformed into the corresponding unprotected phenol 6 b, by treatment with hydrogen and palladium on charcoal, that was subsequently protected with TIPSCl in dichloromethane to afford compound 6 c (Scheme 1).
Scheme 1.
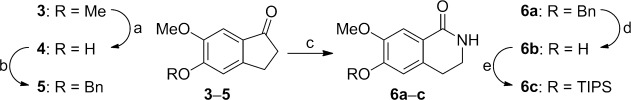
Synthesis of the DHIQ core structure. Reagents and conditions: a) piperidine/H2O, 140 °C; b) BnBr, K2CO3, DMF, RT; c) NaN3, CH3SO3H, CH2Cl2, 0 °C→RT; d) H2, Pd/C, THF/MeOH, RT; e) TIPSCl, imidazole, CH2Cl2, RT.
N-Benzylation was then performed on compound 6 c. Under the conditions applied in these reactions TIPS-deprotection and subsequent benzylation of the unprotected phenol was also observed to give compounds 7 a–c. In two further experiments the more stable benzyl-protected compound 6 a was reacted in the same manner to afford compounds 7 d–e. These were transformed into the corresponding unprotected phenols 8 a–e either by treatment with hydrogen and Pd/C or with TBAF (only for 7 b) in good yields. Treatment of 8 a–e with sulfamoyl chloride in DMA[12] gave the corresponding sulfamates 9 a–e (Scheme 2).
Scheme 2.
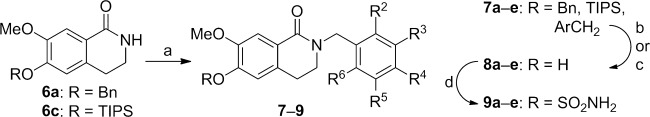
Synthesis of functionalised N-benzyl-substituted DHIQs. Reagents and conditions: a) NaH, 0 °C, DMF, then benzyl halide, RT; b) H2, Pd/C, THF/MeOH, RT; c) TBAF, THF, RT; d) H2NSO2Cl, DMA, RT.
A second compound set was produced by N-sulfonylating 6 a with various arylsulfonyl chlorides. Compounds 10 a–e were transformed into the corresponding unprotected phenols 11 a–e, by treatment with hydrogen and Pd/C, that were then reacted with sulfamoyl chloride in DMA to afford the corresponding sulfamates 12 a–e (Scheme 3).
Scheme 3.
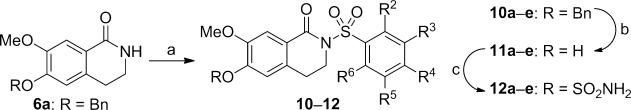
Synthesis of functionalised N-arylsulfonyl-substituted DHIQs. Reagents and conditions: a) NaH, DMF, 50 °C, then ArSO2Cl, DMF, RT or 40 °C; b) H2, Pd/C, THF/EtOH, RT; c) H2NSO2Cl, DMA, RT.
For the synthesis of N-benzoylated compounds a direct strategy starting from 6 a proved not very successful. Therefore, an alternate approach using benzyl-protected THIQ 13[8] as starting material was adopted. N-Benzoylation using various benzoyl chlorides followed by permanganate oxidation gave access to the desired N-benzoylated dihydroisoquinolines 15 a–g. These were reacted with hydrogen and Pd/C and the product treated with sulfamoyl chloride in DMA to give sulfamates 17 a–g. Additionally, a set of control compounds without the carbonyl at C1 was achieved by hydrogenation of compounds 14 e–g with Pd/C to give phenols 18 a–c. Treatment with sulfamoyl chloride in DMA then afforded sulfamates 19 a–c (Scheme 4).[8]
Scheme 4.
Synthesis of functionalised N-benzoyl-substituted DHIQs and THIQs. Reagents and conditions: a) ArCOCl, Et3N, CH2Cl2, RT; b) KMnO4, 18-crown-6, CHCl3, RT; c) H2, Pd/C, THF/MeOH, RT; d) H2NSO2Cl, DMA, RT.
Biology
The compounds were then evaluated in vitro for their ability to inhibit the proliferation of DU-145 human prostate cancer cells and MDA MB-231 breast cancer cells. As can be observed in Table 1, the N-benzyl derivatives, regardless of the nature of substitution of the benzyl motif, prove to be uniformly inactive in contrast to the corresponding THIQ series in which the 3′-methoxy compound 2 (Figure 1) exhibits a GI50 value of 2.1 μm.[7, 8] The addition of a carbonyl at C1 in this case is clearly detrimental, and it seems reasonable to suggest that this group, through steric considerations, disfavours adoption of a conformation in which the benzyl group can potentially mimic the steroid D-ring. Likewise, the series of N-arylsulfonyl derivatives prove to be inactive. The corresponding series of N-arylsulfonyl THIQs also fails to display significant activity, a fact that we ascribe to the likely unacceptable steric bulk of the sulfonyl group at the site of binding.[8] The N-benzoyl DHIQs, in contrast, provide interesting activity in both the 6-hydroxy and 6-sulfamoyloxy series. Here, both the 2′- and 3′-methoxy compounds give low micromolar GI50 values against the proliferation of both DU-145 and MDA MB-231 cells. It is notable that the 6-hydroxy compounds 16 a–b display superior activity to the 6-O-sulfamates 17 a–b and that, as found for the N-benzyl THIQs (with which we believed they should act in an analogous manner), the 3′-substituted derivative displays the highest activity. In contrast, as found in the N-benzyl THIQ series, the 4′-substituted compounds 16 d and 17 d are inactive. Several polymethoxylated compounds were also evaluated revealing that, although 4′-substitution alone did not afford activity, it is tolerated as the 3′,4′-dimethoxy compounds 16 e and 17 e display good activity and are more active than the 3′-methoxy derivatives 16 b and 17 b. The major step forward in activity was realised when the 3′,5′-dimethoxy derivative 16 f and its sulfamate 17 f were evaluated, with roughly equivalent activity (GI50 values of 215 and 179 nm for DU-145, respectively). Compounds 16 f and 17 f are >10-fold more active than the 3′-methoxy derivatives 16 b and 17 b. We postulate that this symmetric substitution adds a further entropic advantage in combination with the pre-organisation we believed would derive from the repulsion of the carbonyls of the imide discussed above. In any case, the activity of 16 f and 17 f is equal to that of both the conformationally biased THIQs and the steroidal derivatives from which the nonsteroidal design was conceived.
Table 1.
Antiproliferative activity of DHIQs against DU-145 human prostate cancer cells and MDA MB-231 human breast cancer cells in vitro
| Compd |  |
GI50 [μm][a] | ||||||
|---|---|---|---|---|---|---|---|---|
| R1 | X | R2 | R3 | R4 | R5 | DU-145 | MDA MB-231 | |
| 8 a | H | CH2 | OMe | H | H | H | >100 | ND[b] |
| 9 a | SO2NH2 | CH2 | OMe | H | H | H | >100 | ND[b] |
| 8 b | H | CH2 | H | OMe | H | H | >100 | ND[b] |
| 9 b | SO2NH2 | CH2 | H | OMe | H | H | >100 | ND[b] |
| 8 c | H | CH2 | H | H | OMe | H | >100 | ND[b] |
| 9 c | SO2NH2 | CH2 | H | H | OMe | H | >100 | ND[b] |
| 7 d | Bn | CH2 | H | OMe | H | OMe | >100 | ND[b] |
| 8 d | H | CH2 | H | OMe | H | OMe | >100 | ND[b] |
| 9 d | SO2NH2 | CH2 | H | OMe | H | OMe | >100 | ND[b] |
| 7 e | Bn | CH2 | H | OMe | OMe | OMe | >100 | ND[b] |
| 8 e | H | CH2 | H | OMe | OMe | OMe | >100 | ND[b] |
| 9 e | SO2NH2 | CH2 | H | OMe | OMe | OMe | >100 | ND[b] |
| 10 a | Bn | SO2 | OMe | H | H | H | >100 | >100 |
| 11 a | H | SO2 | OMe | H | H | H | >100 | 13.4 |
| 12 a | SO2NH2 | SO2 | OMe | H | H | H | >100 | >100 |
| 10 b | Bn | SO2 | H | OMe | H | H | >100 | >100 |
| 11 b | H | SO2 | H | OMe | H | H | >100 | >100 |
| 12 b | SO2NH2 | SO2 | H | OMe | H | H | >100 | >100 |
| 11 c | H | SO2 | H | H | OMe | H | >100 | >100 |
| 12 c | SO2NH2 | SO2 | H | H | OMe | H | >100 | >100 |
| 10 d | Bn | SO2 | H | Cl | H | H | >100 | >100 |
| 11 d | H | SO2 | H | Cl | H | H | >100 | >100 |
| 12 d | SO2NH2 | SO2 | H | Cl | H | H | >100 | >100 |
| 11 e | H | SO2 | CO2Me | H | H | H | >100 | >100 |
| 12 e | SO2NH2 | SO2 | CO2Me | H | H | H | >100 | 13 |
| 15 a | Bn | CO | OMe | H | H | H | >100 | 43.6 |
| 16 a | H | CO | OMe | H | H | H | 6.42 | 5.6 |
| 17 a | SO2NH2 | CO | OMe | H | H | H | 9.1 | 7.1 |
| 16 b | H | CO | H | OMe | H | H | 3.63 | 1.89 |
| 17 b | SO2NH2 | CO | H | OMe | H | H | 5.3 | 3.5 |
| 15 c | Bn | CO | H | CN | H | H | >100 | >100 |
| 16 c | H | CO | H | CN | H | H | 70 | 37.4 |
| 17 c | SO2NH2 | CO | H | CN | H | H | 25 | >100 |
| 15 d | Bn | CO | H | H | OMe | H | >100 | ND[b] |
| 16 d | H | CO | H | H | OMe | H | >100 | ND[b] |
| 17 d | SO2NH2 | CO | H | H | OMe | H | >100 | ND[b] |
| 15 e | Bn | CO | H | OMe | OMe | H | >100 | ND[b] |
| 16 e | H | CO | H | OMe | OMe | H | 2.39 | 1.6 |
| 17 e | SO2NH2 | CO | H | OMe | OMe | H | 3.4 | 2.08 |
| 15 f | Bn | CO | H | OMe | H | OMe | >100 | ND[b] |
| 16 f | H | CO | H | OMe | H | OMe | 0.215 | 0.161 |
| 17 f | SO2NH2 | CO | H | OMe | H | OMe | 0.179 | 0.118 |
| 15 g | Bn | CO | H | OMe | OMe | OMe | >100 | 77.5 |
| 16 g | H | CO | H | OMe | OMe | OMe | 0.051 | 0.071 |
| 17 g | SO2NH2 | CO | H | OMe | OMe | OMe | 0.166 | 0.275 |
[a] Results are the mean of three determinations. [b] Not determined.
Finally, 3′,4′,5′-trimethoxybenzoyl DHIQ derivatives 15 g, 16 g and 17 g were also evaluated. From our understanding drawn from experience with the THIQ systems these compounds could well act as chimeras addressing both the steroidal A-ring binding area and the trimethoxyaryl binding area of the colchicine binding site on tubulin. Phenol 16 g and its sulfamate 17 g prove to be the most active compounds evaluated, with 16 g displaying respective GI50 values of 51 and 71 nm against the proliferation of DU-145 and MDA MB-231 cells.
Comparing these results with those for related N-benzoylated THIQs (Table 2) it is evident that omitting the carbonyl at C1 is in general detrimental towards in vitro antiproliferative activity. Only polymethoxylated compounds are displayed here. The series of 3′,4′-dimethoxy THIQ compounds shows a different trend to the corresponding DHIQ series, because here benzyl-protected compound 14 e shows the best, but still relatively modest, activity (GI50 26.7 μm in DU-145) with phenol 18 a and sulfamate 19 a being surprisingly completely inactive. The 3′,5′-dimethoxy and 3′,4′,5′-trimethoxy derivatives were also evaluated and show a similar trend as their DHIQ counterparts, with phenols 18 b–c showing low micromolar activity that is slightly better than that of the corresponding sulfamates 19 b–c, while benzyl-protected compounds 14 f–g display only modest activity. However, the most active compounds 18 c and 19 c are still about 10- to 20-fold less active than DHIQs 16 g and 17 g, showing the importance of the combination of two carbonyl groups (at C1 and in the linker to the D-ring mimic) to achieve compounds with low nanomolar activity.
Table 2.
Antiproliferative activity of polymethoxylated N-benzoyl THIQs against DU-145 human prostate cancer cells and MDA MB-231 human breast cancer cells in vitro
| Compd |  |
GI50 [μm][a] | ||||
|---|---|---|---|---|---|---|
| R1 | R3 | R4 | R5 | DU-145 | MDA MB-231 | |
| 14 e | Bn | OMe | OMe | H | 26.7 | ND[b] |
| 18 a | H | OMe | OMe | H | >100 | ND[b] |
| 19 a | SO2NH2 | OMe | OMe | H | >100 | ND[b] |
| 14 f | Bn | OMe | H | OMe | 46.8 | ND[b] |
| 18 b | H | OMe | H | OMe | 6.4 | ND[b] |
| 19 b | SO2NH2 | OMe | H | OMe | 9 | ND[b] |
| 14 g | Bn | OMe | OMe | OMe | 62.7 | 18.9 |
| 18 c[c] | H | OMe | OMe | OMe | 0.921 | 0.733 |
| 19 c[c] | SO2NH2 | OMe | OMe | OMe | 1.81 | 1.72 |
[a] Results are the mean of three determinations. [b] Not determined. [c] Data for 18 c and 19 c are taken from the literature.[8]
The most active compound 16 g was also tested in the NCI 60-cell line assay (Table 3) that allows activity across a wide range of cancer types to be assessed. Data from seven cell lines are presented along with the mean activity across the whole panel (MGM value). Screening was conducted at concentrations ranging from 10 nm upwards, with maximal activity (where a 50 % growth inhibition was obtained in all cell lines at a concentration of 10 nm) being indicated by an MGM value of 10 nm. The data obtained in the assay confirm the very high potency of 16 g against a broad range of cancer phenotypes.
Table 3.
GI50 and MGM values of compound 16 g obtained for representative cell lines in the NCI-60 screening panel
| Cell Line | GI50 [μm][a] | Cell Line | GI50 [μm][a] |
|---|---|---|---|
| Leukemia CCRF-CEM | 0.0171 | Melanoma UACC-62 | <0.01 |
| Lung HOP-62 | 0.0295 | Ovarian OVCAR-3 | <0.01 |
| Colon HCT-116 | 0.0216 | Renal SN12-C | 0.056 |
| CNS SF-539 | 0.0102 | MGM | 0.033 |
[a] Results are the mean of three determinations. The MGM value (in italics) represents the mean concentration that caused 50 % growth inhibition in all 60 cell lines.
As with sulfamates in the tetrahydroisoquinoline series, sulfamate 17 f also inhibits carbonic anhydrase (IC50 890 nm), which may enhance the bioavailability of this compound.[13] Compounds 16 f and 17 f were also assayed for anti-angiogenic activity in an in vitro model of angiogenesis, wherein endothelial cells co-cultured in a matrix of human dermal fibroblasts are used to assess anti-angiogenic potential (Figure 2). When stimulated with VEGF the endothelial cells proliferate and migrate through the matrix to form tubule-like structures. The extent of tubule formation was quantified after 11 days as described elsewhere.[14] Treatment with 25 nm and 50 nm concentrations of 16 f and 17 f almost completely inhibits the formation of tubule-like structures (Figure 2). Quantification carried out by calculating total pixel length reflects the lack of tubule formation. Both phenol 16 f and its sulfamate 17 f are highly potent.
Figure 2.
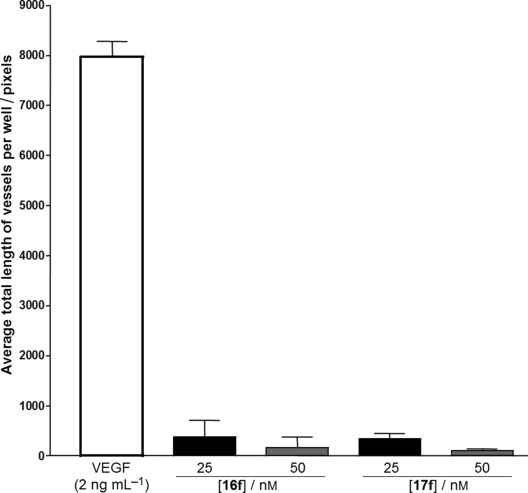
Activity of 16 f and 17 f in an in vitro model of angiogenesis.
With these excellent in vitro data in hand, we decided to explore further the microtubule disruptor activity in particular of 16 f–g and 17 f–g alongside the established potent agent combretastatin A-4 (CA-4), currently in clinical trials. All the tested novel compounds show excellent activity as inhibitors of tubulin assembly, with IC50 values of 1.2–1.5 μm similar to that of CA-4 (IC50 1.1 μm). Ultimately, as observed before, the concentration required in tubulin-based assays far exceeds the antiproliferative dose, and most likely it suffices to disrupt microtubule dynamics to arrest the cell cycle rather than cause a catastrophic depolymerisation event. It should also be stressed that the nominal compound concentration in antiproliferative assays is that of agent added to the culture medium, rather than the actual concentration within cells, and drug transport can also be a key determinant in reaching the concentration required to achieve an effect. We also determined that 16 f–g and 17 f–g inhibit colchicine binding to tubulin very effectively. Compound 17 f, which is the most active (89 % inhibition at 5 μm), was also tested at lower concentration and shows 65 % inhibition at 1 μm, approaching again the activity of CA-4 (99 and 90 %). It is thus reasonable to suggest that the activity of the novel DHIQ derivatives can at least partially be ascribed to their ability to disrupt the normal dynamic polymerisation of tubulin by interaction at, or around, the colchicine binding site (Table 4).
Table 4.
Activity of selected DHIQs as inhibitors of tubulin polymerisation (TP) and colchicine binding (CB) to tubulin
| Compd | TP IC50 [μm][a] | CB [% inhib.][a] | |
|---|---|---|---|
| 5 μm inhibitor | 1 μm inhibitor | ||
| CA-4 | 1.1±0.1 | 99±0.6 | 90±0.2 |
| 1 a[c] | 2.2±0.3 | 28±3 | ND[b] |
| 1 b[c] | 1.3±0.08 | 78±0.9 | ND[b] |
| 1 c[c] | 1.3±0.01 | 45±4 | ND[b] |
| 16 f | 1.5±0.1 | 81±0.7 | ND[b] |
| 16 g | 1.3±0.1 | 83±2 | ND[b] |
| 17 f | 1.3±0.01 | 89±0.1 | 65±3 |
| 17 g | 1.2±0.03 | 85±2 | ND[b] |
[a] Values are the mean ±SD of at least two determinations. [b] Not determined. [c] Data for 1 a–c are taken from the literature.[3]
Finally, an X-ray crystal structure of 17 f was obtained to explore conformational effects (Figure 3).[15] The crystal structure shows that in the solid state the molecule adopts a “steroid-like” conformation as planned with a dihedral angle of 148.4° between the two carbonyl groups (Figure 3 a). In the single crystal this fixed angle also appears as a result of positive intermolecular interactions (Figure 3 b). The sulfamate group plays a key role to stabilise the framework of the lattice structure by acting as a hydrogen donor to the C3′-methoxy group of a second molecular unit that is part of the same string and to the carbonyl group at C1 of a third molecular unit that is part of a second string running in the opposite direction. The angles in the crystal structure do not necessarily reflect conditions in solution. However, the excellent in vitro antiproliferative activities obtained for this class of compound suggest that adoptable conformations might be very restricted in solution as well, mainly as a result of electrostatic repulsion between the two carbonyl groups, and somewhere around the angle observed in the crystal. Control compounds like 8 d–e and 9 d–e without the carbonyl group in the linker to the D-ring mimic and 18 b–c and 19 b–c without the carbonyl at C1 do not have this favourable restriction and therefore show far less potency than their counterparts 16 f–g and 17 f–g that have both carbonyl groups present.
Figure 3.
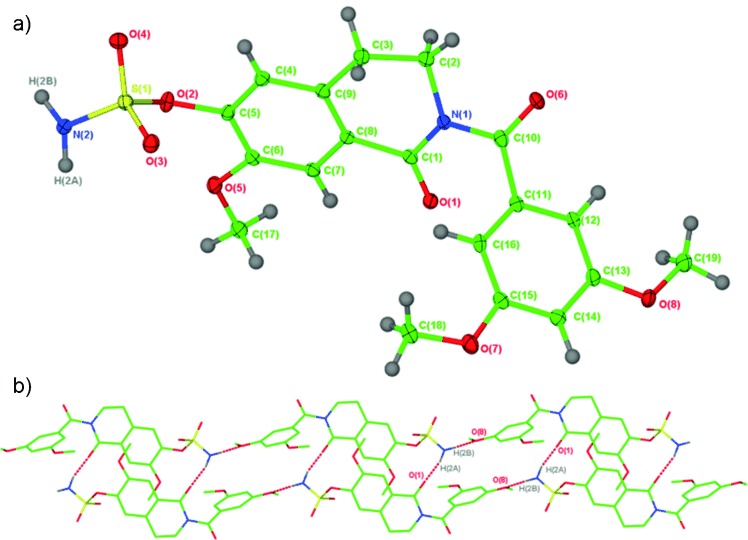
a) Single-crystal X-ray structure of 17 f. Ellipsoids are depicted at 30 % probability. b) Part of the crystal lattice packing diagram of 17 f to illustrate the hydrogen-bonded linear arrangements present in the gross structure.
Molecular structure 17 f was treated with a computational energy minimisation procedure. These calculations showed, in the minimal energy conformation, that a dihedral angle of 151.0° exists between the two carbonyl groups that is in good agreement with the observed angle of 148.4° in the crystal structure. As we postulated, the 3′,5′-dimethoxybenzoyl group is projected into the area of space that is occupied by the steroidal D ring and the C18 methyl group in the estratriene series. This is illustrated in Figure 4 where the minimum energy state of 17 f is overlaid with energy-minimised 1 a. Additionally, the D-ring is rotated 47.2° out of plane of the A,B-ring system leading to one methoxy group being projected into the space of the sulfamate group and the other into the space of the C18 methyl group of 1 a. Although not shown here, the most potent DHIQs were subjected to the same conformational analysis and the results (angles) obtained were unsurprisingly very similar (16 g: 149.8° and 46.9°; 17 g: 150.5° and 47.1°) to the ones for compound 17 f. These calculations support our postulate that the presence of the imide sub-structure favours adoption of a “steroid-like” conformation. It seems reasonable to propose that the positive effects on activity can, to some degree, be ascribed to this conformational biasing.
Figure 4.
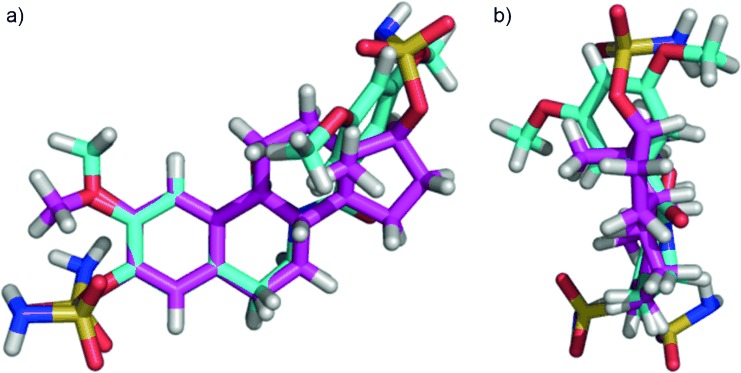
Overlay of the minimum energy conformation of DHIQ 17 f (cyan) with steroid 1 a (pink) viewed from two perspectives.
Conclusions
In summary, dihydroisoquinolinone derivatives including their sulfamate esters are compounds with activities in the low micromolar and nanomolar range against the proliferation of prostate and breast cancer cells. The most potent DHIQ derivative synthesised 16 g, is also confirmed as having very high potency against a broad range of cancer phenotypes in the NCI 60-cell line assay. As anticipated, these compounds also display anti-angiogenic activity in an in vitro angiogenesis assay and appear to function as microtubule disruptors, as evidenced by their ability to inhibit polymerisation of tubulin and compete at the colchicine binding site. The most active DHIQs 16 f–g and their sulfamates 17 f–g are nearly equipotent to combretastatin A-4 (CA-4) as inhibitors of tubulin assembly and colchicine binding. Additionally, X-ray analysis of 17 f reveals that electrostatic repulsion between the two adjacent carbonyl groups might partially explain the excellent in vitro activities of this novel series of microtubule disruptors. This conclusion is further supported by molecular modelling studies. Thus, compounds derived from the DHIQ series are worthy of in vivo investigation and potential further pre-clinical development.
Experimental Section
In vitro studies
Cell lines: DU-145 (brain metastasis carcinoma of the prostate) and MDA MB-231 (metastatic pleural effusion of breast adenocarcinoma) established human cell lines were obtained from ATCC Global Bioresource Center. Cells were maintained in a 5 % CO2 humidified atmosphere at 37 °C in RPMI-1640 medium, supplemented with 10 % fetal bovine serum, penicillin, (100 U mL−1), and streptomycin (0.1 mg mL−1). The effect of drugs on tubule formation was assessed using an angiogenesis kit (TCS-Cellworks).
Antiproliferative assays: DU-145 and MDA MB-231 cells were seeded into 96-well microtitre plates (5000 cells per well) and treated with compound (10−9–10−4 m) or with vehicle control. At 96 h post-treatment, live cell counts were determined by WST-1 cell proliferation assay (Roche, Penzberg, Germany), as per the manufacturer’s instructions. Viability results were expressed as a percentage of mean control values resulting in the calculation of the 50 % growth inhibition (GI50). All experiments were performed in triplicate.
Anti-angiogenic assays: HUVECs were cultured in a 24-well plate within a matrix of human diploid fibroblasts of dermal origin in optimised medium supplied by the company. The co-cultured cells were incubated throughout the experiment at 37 °C under 5 % CO2 in a humidified incubator. On day 1, the culture medium was removed and replaced with medium containing the drug under investigation. On days 4, 7 and 9 the medium was replaced with fresh medium containing the drugs. Suramin (20 μL) was used as a positive anti-angiogenic control and vascular endothelial growth factor (VEGF, 2 ng mL−1) as a pro-angiogenic control. Each compound was tested in triplicate. On day 11, the cells were washed (PBS) and 70 % EtOH (1 mL) added to each well for 30 min to fix the cells. After fixation the cells were washed with blocking buffer (1 mL, PBS 1 % BSA; Sigma, UK) and stained for von Willebrand’s factor (manual scoring) for CD31 (computer analysis) in accordance with the manufacturer’s instructions (TCS-Cellworks). The extent of tubule formation was then quantified, either manually by eye or using free software available through the National Institutes of Health website (NIH image [Mac version] from http://rsb.info.nih.gov/nih-image/download.html). For manual scoring a grid was drawn on the back of the plate using a fine marker pen. A 25-point Chalkley eyepiece graticule (Pyser SGI Ltd, UK) was fitted to the microscope and 10x magnification was used to count the tubules within five viewing fields (where the grid lines intersect), the average and SE were then calculated. For computer analysis, eight fixed fields of view for each well (10x magnification) were digitally photographed using a DC120 camera (Kodak, Rochester, NY, USA) with the Photoshop (Adobe, USA) plug-in MDS 120 software (Kodak). The pictures were copied to the NIH image software using Quicktime (Apple, USA) translation. After correcting for background the software counted the number of pixels representing tubules. The total pixel area within the eight fields was then calculated for each well. The average total pixel area and SE were then calculated. There was a significant correlation (r=0.94, p<0.001) between manual and computer analysis of tubule formation.
Tubulin assays: Bovine brain tubulin, prepared as described previously,[16] was used in studies presented here. Assembly IC50 values were determined as described in detail elsewhere.[17] Briefly, 1.0 mg mL−1 (10 μm) tubulin was pre-incubated without GTP with varying compound concentrations for 15 min at 30 °C. Reaction mixtures were placed on ice, and GTP (final concentration, 0.4 mm) was added. The reaction mixtures were transferred to cuvettes, held at 0 °C in a recording spectrophotometer. Baselines were established at 0 °C, and increase in turbidity was followed for 20 min following a rapid (<30 s) jump to 30 °C. Compound concentrations required to decrease the turbidity increase by 50 % were determined. The method for measuring inhibition of the binding of [3H]colchicine to tubulin was described in detail previously.[18] Reaction mixtures contained 0.1 mg mL−1 (1.0 μm) tubulin, 5.0 μm [3H]colchicine, and potential inhibitor at 5.0 μm. Compounds were compared with CA-4, a particularly potent inhibitor of the binding of colchicine to tubulin.[19] Reaction mixtures were incubated for 10 min at 37 °C, a time point at which the binding of colchicine in control reaction mixtures is generally 40–60 % complete. A minimum of two experiments were performed with each compound.
Chemistry
General: All chemicals were either purchased from Aldrich Chemical Co. (Gillingham, UK) or Alfa Aesar (Heysham, UK). Organic solvents of A.R. grade were supplied by Fisher Scientific (Loughborough, UK) and used as supplied. CHCl3, CH2Cl2, DMA, DMF and THF were purchased from Aldrich and stored under a positive pressure of N2 after use. Compounds 13, 18c and 19c were prepared according to literature procedures.[8] Sulfamoyl chloride was prepared by an adaptation of the method of Appel and Berger[20] and was stored in the refrigerator under positive pressure of N2 as a solution in toluene as described by Woo et al.[21] An appropriate volume of this solution was freshly concentrated in vacuo immediately before use. Reactions were carried out at room temperature unless stated otherwise. Thin-layer chromatography (TLC) was performed on pre-coated aluminum plates (Merck, silica gel 60 F254). Product spots were visualised either by UV irradiation at 254 nm or by staining with either alkaline KMnO4 solution or 5 % dodecamolybdophosphoric acid in EtOH, followed by heating. Flash column chromatography was performed using gradient elution (solvents indicated in text) on either pre-packed columns (Isolute) on a Flashmaster II system (Biotage, Uppsala, Sweden) or on a CombiFlash Rf Automated Flash Chromatography System (Teledyne Isco, Lincoln, NE, USA) with RediSep Rf disposable flash columns. 1H and 13C NMR spectra were recorded with either a Delta JMN-GX 270 (Jeol, Peabody, MA, USA) at 270 and 67.5 MHz, respectively, or a Mercury VX 400 NMR spectrometer (Varian, Paolo Alto, CA, USA) at 400 and 100 MHz, respectively. Chemical shifts δ are reported in parts per million (ppm) relative to tetramethylsilane (TMS) as internal standard. Coupling constants J are recorded to the nearest 0.1 Hz. Mass spectra were recorded at the Mass Spectrometry Service Centre, University of Bath, UK. FAB-MS was carried out using m-nitrobenzyl alcohol (NBA) as the matrix. Melting points were determined using a Stuart SMP3 or a Stanford research systems Optimelt MPA100 melting point apparatus (Stanford Research Systems, Sunnyvale, CA, USA) and are uncorrected. All compounds were ≥98 % pure by reversed-phase HPLC run with CH3CN/H2O or MeOH/H2O (Sunfire C18 reversed-phase column, 4.6×150 mm, 3.5 μm pore size).
Crystallographic methods: Single crystals of compound 17 f were analysed using a Nonius Kappa CCD diffractometer using Mo(Kα) radiation. The structure was solved using SHELXS-97[22] and refined using full-matrix least squares in SHELXL-97.[23] The hydrogen atoms attached to N2 were located and refined subject to being a distance of 0.89 Å from the parent atom. CCDC 874341 http://www.ccdc.cam.ac.uk/cgi-bin/catreq.cgicontains the crystallographic data for compound 17 f and can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Computational methods: The Schrödinger software running under Maestro version 9.2.112 was used for all computational work. The crystal structure of 17 f solved in this work was used. One molecule was taken from this structure and run through a brief geometry optimisation procedure. Both dihedral angles were manually and independently adjusted in steps of 30° and the energy of the conformers was calculated. Compounds 16 g and 17 g were built by altering the crystal structure of 17 f to yield the desired structure which was then run through a brief geometry optimisation procedure. Conformers were generated and molecular energies calculated as described above.
5-Hydroxy-6-methoxyindan-1-one (4): 5,6-Dimethoxy-indan-1-one (25.93 g, 135.0 mmol) was placed in a 1 L round-bottom flask. Piperidine (200 mL) and H2O (50 mL) were added and the reaction mixture was stirred at 140 °C for 8 days. The mixture was concentrated in vacuo and aqueous NaOH (2 m, 400 mL) was added. The mixture was extracted with EtOAc (200 mL) and CH2Cl2 (200 mL). The aqueous layer was then acidified with HCl (12 m, 100 mL) and extracted with CH2Cl2 (3×200 mL). This organic layer system was washed with H2O (200 mL), dried (MgSO4), filtered and concentrated in vacuo to give compound 4 as a yellow powder (16.87 g, 70 %). 1H NMR (270 MHz, CDCl3): δ=2.49–2.60 (2 H, m), 2.86–2.98 (2 H, m), 3.83 (3 H, s), 6.86 (1 H, s), 7.09 (1 H, s), 8.03 ppm (1 H, s, br).
5-Benzyloxy-6-methoxyindan-1-one (5): Compound 4 (3.0 g, 16.8 mmol), benzyl bromide (2.1 mL, 17.7 mmol) and potassium carbonate (4.70 g, 34.0 mmol) were suspended in DMF (20 mL) and stirred at RT for 3 days. H2O (50 mL) was added and the mixture was extracted with EtOAc (2×80 mL). The combined organics were washed with H2O and brine, dried (MgSO4) and concentrated in vacuo. Crystallisation from EtOAc/hexane 2:3 afforded compound 5 as a light-yellow powder (4.10 g, 91 %), mp: 139–140 °C. 1H NMR (270 MHz, CDCl3): δ=2.58–2.66 (2 H, m), 2.93–3.01 (2 H, m), 3.89 (3 H, s), 5.22 (2 H, s), 6.88 (1 H, s), 7.18 (1 H, s), 7.27–7.46 ppm (5 H, m).
6-Benzyloxy-7-methoxy-3,4-dihydro-2H-isoquinolin-1-one (6 a): Compound 5 (2.68 g, 10 mmol) was dissolved in CH2Cl2 (10 mL) and methanesulfonic acid (10 mL) and the solution was cooled to 0 °C. Sodium azide (1.32 g, 20 mmol) was added portionwise over 0.5 h. The mixture was allowed to warm to RT and stirred for 16 h. Aqueous NaOH (1.5 m, 50 mL) was added dropwise at 0 °C. The mixture was extracted with EtOAc (3×80 mL). The combined organics were washed with H2O and brine, dried (MgSO4) and concentrated in vacuo. Flash column chromatography (hexane/EtOAc 3:1 to EtOAc) and crystallisation from EtOAc afforded compound 6 a as a white solid (1.45 g, 52 %), mp: 177–178 °C. 1H NMR (270 MHz, CDCl3): δ=2.87 (2 H, t, J=7.6 Hz), 3.53 (2 H, t, J=7.6 Hz), 3.94 (3 H, s), 5.20 (2 H, s), 6.03 (1 H, s, br), 6.70 (1 H, s), 7.29–7.46 (5 H, m), 7.60 ppm (1 H, s); LC–MS (FAB+): m/z 284.44 [M+H]+.
6-Hydroxy-7-methoxy-3,4-dihydro-2H-isoquinolin-1-one (6 b): Compound 6 a (1.418 g, 5.0 mmol) was dissolved in MeOH (50 mL) and filtered. The solution was sucked at 1.0 mL min−1 into the H-cube and treated with full hydrogen over Pd/C (10 %, 140 mg cartridge) at RT. The resulting solution was concentrated in vacuo to afford compound 6 b as a pale-yellow solid (961 mg, 99 %). 1H NMR (270 MHz, CDCl3): δ=2.72 (2 H, t, J=6.7 Hz), 3.27–3.31 (2 H, m), 3.77 (3 H, s), 6.65 (1 H, s), 7.33 (1 H, s), 7.64 (1 H, s, br), 9.65 ppm (1 H, s, br); LC–MS (ES+): m/z 194.2 [M+H]+.
7-Methoxy-(6-triisopropyloxy)-3,4-dihydro-2H-isoquinolin-1-one (6 c): Compound 6 b (3.1 g, 16 mmol), TIPSCl (7.1 mL, 33 mmol) and imidazole (2.4 g, 35 mmol) in DMF (30 mL) was stirred at RT for 16 h. H2O (30 mL) was added and the mixture was extracted with EtOAc (2×80 mL). The combined organic layers were washed with H2O and brine, dried (MgSO4), filtered and concentrated in vacuo. Flash column chromatography (hexane to EtOAc) afforded compound 6 c as a white powder (5.0 g, 93 %), mp: 112–113 °C. 1H NMR (270 MHz, CDCl3): δ=1.07 (18 H, d, J=6.9 Hz), 1.23 (3 H, sept, J=6.9 Hz), 2.85 (2 H, t, J=6.7 Hz), 3.51 (2 H, dt, J=6.7, 2.9 Hz), 3.82 (3 H, s), 5.86 (1 H, s, br), 6.66 (1 H, s), 7.53 ppm (1 H, s); LC–MS (APCI+): m/z 350.50 [M+H]+.
7-Methoxy-2-(2-methoxybenzyl)-6-(2-methoxybenzyloxy)-3,4-dihydroisoquinolin-1(2H)-one (7 a): Compound 6 c (505 mg, 1.44 mmol) was dissolved in anhydrous DMF (5 mL) and cooled to 0 °C. Sodium hydride (60 % in mineral oil, 115 mg, 2.88 mmol) was added portionwise and the suspension was stirred at 0 °C for 0.5 h. 2-Methoxybenzyl chloride (0.24 mL, 1.73 mmol) was added dropwise and the reaction mixture was stirred at RT for 4 days. Ammonium chloride (saturated, 10 mL) was added and the mixture was extracted with EtOAc (80 mL). The organic layer was washed with H2O and brine, dried (MgSO4), filtered and concentrated in vacuo. Flash column chromatography (hexane/EtOAc 10:1 to 2:1) afforded 7 a as a white powder (350 mg, 56 %), mp: 133–134 °C. 1H NMR (270 MHz, CDCl3): δ=2.81 (2 H, t, J=6.7 Hz), 3.50 (2 H, t, J=6.7 Hz), 3.83 (3 H, s), 3.85 (3 H, s), 3.93 (3 H, s), 4.78 (2 H, s), 5.22 (2 H, s), 6.64 (1 H, s), 6.84–6.95 (4 H, m), 7.18–7.32 (3 H, m), 7.44 (1 H, dd, J=7.4, 1.5 Hz), 7.65 ppm (1 H, s); LC–MS (APCI+): m/z 434.56 [M+H]+; HRMS (ES+): m/z found 434.1960; C26H28NO5+ [M+H]+ requires 434.1962.
7-Methoxy-2-(3-methoxybenzyl)-6-(triisopropylsilyloxy)-3,4-dihydroisoquinolin-1(2H)-one (7b1) and 7-Methoxy-2-(3-methoxybenzyl)-6-(3-methoxybenzyloxy)-3,4-dihydroisoquinolin-1(2H)-one (7b2): Method as for 7 a using compound 6 c (505 mg, 1.44 mmol), sodium hydride (60 %, 115 mg, 2.88 mmol) and 3-methoxybenzyl bromide (0.24 mL, 1.73 mmol) in DMF (5 mL) at 0 °C for 0.5 h and at RT for 4 days. Flash column chromatography (hexane/EtOAc 10:1 to 5:1 to 2:1) afforded 7b1 as a colourless oil (120 mg, 18 %) and 7b2 as a white powder (110 mg, 18 %), mp: 85–86 °C. 7b1: 1H NMR (270 MHz, CDCl3): δ=1.07 (18 H, d, J=6.7 Hz), 1.22 (3 H, sept, J=6.7 Hz), 2.79 (2 H, t, J=6.7 Hz), 3.44 (2 H, t, J=6.7 Hz), 3.77 (3 H, s), 3.84 (3 H, s), 4.73 (2 H, s), 6.61 (1 H, s), 6.80 (1 H, ddd, J=8.1, 2.5, 0.8 Hz), 6.85–6.92 (2 H, m), 7.23 (1 H, t, J=7.9 Hz), 7.61 ppm (1 H, s); HRMS (ES+): m/z found 470.2709; C27H40NO4Si+ [M+H]+ requires 470.2721. 7b2: 1H NMR (270 MHz, CDCl3): δ=2.77 (2 H, t, J=6.6 Hz), 3.41 (2 H, t, J=6.6 Hz), 3.75 (3 H, s), 3.77 (3 H, s), 3.92 (3 H, s), 4.72 (2 H, s), 5.14 (2 H, s), 6.61 (1 H, s), 6.76–6.89 (4 H, m), 6.94–7.01 (2 H, m), 7.21 (1 H, t, J=7.9 Hz), 7.25 (1 H, t, J=8.1 Hz), 7.66 ppm (1 H, s); LC–MS (APCI+): m/z 434.56 [M+H]+; HRMS (ES+): m/z found 434.1962; C26H28NO5+ [M+H]+ requires 434.1962.
7-Methoxy-6-(triisopropylsilyloxy)-2-(4-methoxybenzyl)-3,4-dihydroisoquinolin-1(2H)-one (7 c): Method as for 7 a using compound 6 c (524 mg, 1.5 mmol), sodium hydride (60 %, 64 mg, 1.6 mmol) and 4-methoxybenzyl bromide (0.26 mL, 1.8 mmol) in DMF (5 mL) at 0 °C for 0.5 h and at RT for 6 h. Flash column chromatography (hexane/EtOAc 10:1 to 5:1) afforded compound 7 c as a colourless oil (385 mg, 55 %). 1H NMR (270 MHz, CDCl3): δ=1.07 (18 H, d, J=6.9 Hz), 1.22 (3 H, sept, J=6.9 Hz), 2.77 (2 H, t, J=6.7 Hz), 3.42 (2 H, t, J=6.7 Hz), 3.78 (3 H, s), 3.84 (3 H, s), 4.68 (2 H, s), 6.60 (1 H, s), 6.84 (2 H, dt, J=8.6, 2.4 Hz), 7.26 (2 H, dt, J=8.5, 2.3 Hz), 7.60 ppm (1 H, s); LC–MS (APCI+): m/z 470.57 [M+H]+; HRMS (ES+): m/z found 470.2706; C27H40NO4Si+ [M+H]+ requires 470.2721.
6-Benzyloxy-7-methoxy-2-(3,5-dimethoxybenzyl)-3,4-dihydroisoquinolin-1(2H)-one (7 d): Method as for 7 a using compound 6 a (425 mg, 1.5 mmol), sodium hydride (60 %, 120 mg, 3.0 mmol) and 3,5-dimethoxybenzyl bromide (416 mg, 1.8 mmol) in DMF (10 mL) at 0 °C for 0.5 h and at RT for 18 h. Flash column chromatography (hexane/EtOAc 5:1 to 1:1) gave an oil that was stirred in Et2O (50 mL) and hexane (20 mL), filtered and dried in vacuo to afford compound 7 d as a white powder (440 mg, 68 %), mp: 82–83 °C. 1H NMR (270 MHz, CDCl3): δ=2.79 (2 H, t, J=6.7 Hz), 3.43 (2 H, t, J=6.7 Hz), 3.75 (6 H, s), 3.93 (3 H, s), 4.69 (2 H, s), 5.17 (2 H, s), 6.35 (1 H, t, J=2.5 Hz), 6.45 (2 H, d, J=2.5 Hz), 6.62 (1 H, s), 7.27–7.45 (5 H, m), 7.66 ppm (1 H, s); LC–MS (ES+): m/z 456.03 ([M+Na]+, 100 %), 434.05 [M+H]+; HRMS (ES+): m/z found 434.1956; C26H28NO5+ [M+H]+ requires 434.1962.
6-Benzyloxy-7-methoxy-2-(3,4,5-trimethoxybenzyl)-3,4-dihydroisoquinolin-1(2H)-one (7 e): Method as for 7 a using compound 6 a (425 mg, 1.5 mmol), sodium hydride (60 %, 120 mg, 3.0 mmol) and 3,4,5-trimethoxybenzyl chloride (390 mg, 1.8 mmol) in DMF (10 mL) at 0 °C for 0.5 h and at RT for 18 h. The residue was stirred in Et2O, filtered and dried in vacuo to afford compound 7 e as a white powder (570 mg, 82 %), mp: 136–137 °C. 1H NMR (270 MHz, CDCl3): δ=2.80 (2 H, t, J=6.8 Hz), 3.43 (2 H, t, J=6.8 Hz), 3.82 (9 H, s), 3.93 (3 H, s), 4.68 (2 H, s), 5.17 (2 H, s), 6.52 (2 H, s), 6.63 (1 H, s), 7.26–7.43 (5 H, m), 7.66 ppm (1 H, s); LC–MS (ES+): m/z 486.26 ([M+Na]+, 100 %), 464.28 [M+H]+; HRMS (ES+): m/z found 464.2064; C27H30NO6+ [M+H]+ requires 464.2068.
6-Hydroxy-7-methoxy-2-(2-methoxybenzyl)-3,4-dihydroisoquinolin-1(2H)-one (8 a): A mixture of compound 7 a (290 mg, 0.67 mmol) and Pd/C (10 %, 40 mg) in THF (20 mL) and MeOH (20 mL) was stirred under hydrogen at RT for 4 h. After filtration through Celite and evaporation under reduced pressure the residue was stirred in Et2O, filtered and dried in vacuo to afford compound 8 a as a white powder (195 mg, 93 %), mp: 164–165 °C. 1H NMR (270 MHz, CDCl3): δ=2.83 (2 H, t, J=6.7 Hz), 3.51 (2 H, t, J=6.7 Hz), 3.83 (3 H, s), 3.92 (3 H, s), 4.78 (2 H, s), 6.02 (1 H, s), 6.68 (1 H, s), 6.87 (1 H, d, J=7.4 Hz), 6.91 (1 H, dd, J=7.4, 1.0 Hz), 7.23 (1 H, dt, J=7.4, 1.5 Hz), 7.30 (1 H, dd, J=7.9, 1.5 Hz), 7.63 ppm (1 H, s); LC–MS (APCI+): m/z 314.17 [M+H]+; HRMS (ES+): m/z found 314.1384; C18H20NO4+ [M+H]+ requires 314.1387.
6-Hydroxy-7-methoxy-2-(3-methoxybenzyl)-3,4-dihydroisoquinolin-1(2H)-one (8 b): Method as for 8 a using compound 7 b2 (85 mg, 0.196 mmol) and Pd/C (10 %, 20 mg) in THF (10 mL) and MeOH (10 mL) under hydrogen at RT for 2 h. Flash column chromatography (hexane/EtOAc 1:1) afforded compound 8 b as a white powder (52 mg, 76 %), mp: 181–182 °C. 1H NMR (270 MHz, CDCl3): δ=2.82 (2 H, t, J=6.7 Hz), 3.44 (2 H, t, J=6.7 Hz), 3.77 (3 H, s), 3.93 (3 H, s), 4.74 (2 H, s), 6.03 (1 H, s), 6.68 (1 H, s), 6.80 (1 H, dd, J=7.4, 1.7 Hz), 6.84 (1 H, d, J=1.7 Hz), 6.90 (1 H, d, J=7.7 Hz), 7.23 (1 H, t, J=7.9 Hz), 7.64 ppm (1 H, s); LC–MS (APCI+): m/z 314.23 [M+H]+; HRMS (ES+): m/z found 314.1383, C18H20NO4+ [M+H]+ requires 314.1387.
6-Hydroxy-7-methoxy-2-(3-methoxybenzyl)-3,4-dihydroisoquinolin-1(2H)-one (8 b): Compound 7 b1 (110 mg, 0.23 mmol) was dissolved in THF (20 mL). TBAF (1.0 m in THF, 028 mL, 0.28 mmol) was added dropwise and the reaction mixture was stirred at RT for 2 h. H2O was added and the mixture extracted with EtOAc. The organic layer was washed with H2O, brine, dried (MgSO4) and concentrated in vacuo. The resulting yellow solid was stirred in Et2O, filtered and dried in vacuo to afford compound 8 b as a white powder (60 mg, 82 %). Analytical data are identical as shown above.
6-Hydroxy-7-methoxy-2-(4-methoxybenzyl)-3,4-dihydroisoquinolin-1(2H)-one (8 c): Method as for 8 b using compound 7 c (0.24 g, 0.51 mmol) and TBAF (1.0 m in THF, 0.61 mL, 0.61 mmol) in THF (10 mL) at RT for 18 h. Flash column chromatography (hexane/EtOAc 4:1 to 2:1) afforded compound 8 c as a white powder (110 mg, 69 %), mp: 171–172 °C. 1H NMR (270 MHz, CDCl3): δ=2.78 (2 H, t, J=6.7 Hz), 3.41 (2 H, t, J=6.7 Hz), 3.77 (3 H, s), 3.90 (3 H, s), 4.69 (2 H, s), 6.33 (1 H, s, br), 6.66 (1 H, s), 6.84 (2 H, dt, J=8.7, 2.3 Hz), 7.24 (2 H, dt, J=8.9, 2.3 Hz), 7.63 ppm (1 H, s); LC–MS (APCI+): m/z 314.42 [M+H]+; HRMS (ES+): m/z found 314.1381; C18H19NO4+ [M+H]+ requires 314.1387.
6-Hydroxy-7-methoxy-2-(3,5-dimethoxybenzyl)-3,4-dihydroisoquinolin-1(2H)-one (8 d): Method as for 8 a using compound 7 d (330 mg, 0.76 mmol) and Pd/C (10 %, 40 mg) in THF (20 mL) and MeOH (30 mL) under hydrogen at RT for 18 h. The resulting white solid was stirred in Et2O, filtered and dried in vacuo to afford compound 8 d as a white powder (240 mg, 92 %), mp: 166–167 °C. 1H NMR (270 MHz, CDCl3): δ=2.82 (2 H, t, J=6.7 Hz), 3.44 (2 H, t, J=6.7 Hz), 3.75 (6 H, s), 3.93 (3 H, s), 4.70 (2 H, s), 6.04 (1 H, s), 6.35 (1 H, t, J=2.5 Hz), 6.46 (2 H, d, J=2.5 Hz), 6.68 (1 H, s), 7.63 ppm (1 H, s); LC–MS (ES−): m/z 342.09 [M−H]−; HRMS (ES+): m/z found 344.1477; C19H22NO5+ [M+H]+ requires 344.1492.
6-Hydroxy-7-methoxy-2-(3,4,5-trimethoxybenzyl)-3,4-dihydroisoquinolin-1(2H)-one (8 e): Method as for 8 a using compound 7 e (375 mg, 0.81 mmol) and Pd/C (10 %, 50 mg) in THF (20 mL) and MeOH (20 mL) under hydrogen at RT for 18 h. The resulting white solid was stirred in Et2O, filtered and dried in vacuo to afford compound 8 e as a white powder (260 mg, 86 %), mp: 168–169 °C. 1H NMR (270 MHz, CDCl3): δ=2.82 (2 H, t, J=6.7 Hz), 3.44 (2 H, t, J=6.7 Hz), 3.82 (9 H, s), 3.92 (3 H, s), 4.69 (2 H, s), 6.08 (1 H, s, br), 6.53 (2 H, s), 6.69 (1 H, s), 7.63 ppm (1 H, s); LC–MS (ES−): m/z 372.07 [M−H]−; HRMS (ES+): m/z found 374.1590; C20H24NO6+ [M+H]+ requires 374.1598.
7-Methoxy-2-(2-methoxybenzyl)-6-sulfamoyloxy-3,4-dihydroisoquinolin-1(2H)-one (9 a): Sulfamoyl chloride (0.6 m in toluene, 1.0 mL, 0.6 mmol) was concentrated in vacuo and dissolved in anhydrous DMA (1.0 mL). Compound 8 a (94 mg, 0.3 mmol) was added as a solid and the reaction mixture was stirred at RT for 18 h. H2O (10 mL) was added and the mixture was extracted with EtOAc (2×50 mL). The combined organic layers were washed with H2O, brine, dried (MgSO4), filtered and concentrated in vacuo. The residue was stirred in Et2O, filtered and dried in vacuo to afford compound 9 a as a white solid (90 mg, 77 %), mp: 142–143 °C. 1H NMR (270 MHz, CDCl3): δ=2.86 (2 H, t, J=6.7 Hz), 3.54 (2 H, t, J=6.7 Hz), 3.84 (3 H, s), 3.93 (3 H, s), 4.77 (2 H, s), 5.16 (2 H, s), 6.87 (1 H, d, J=8.2), 6.91 (1 H, dt, J=7.4, 1.0 Hz), 7.14 (1 H, s), 7.21–7.31 (2 H, m), 7.77 ppm (1 H, s); LC–MS (APCI+): m/z 393.45 [M+H]+; HRMS (ES+): m/z found 393.1118; C18H21N2O6S+ [M+H]+ requires 393.1115.
7-Methoxy-2-(3-methoxybenzyl)-6-sulfamoyloxy-3,4-dihydroisoquinolin-1(2H)-one (9 b): Method as for 9 a using compound 8 b (73 mg, 0.23 mmol) and sulfamoyl chloride (0.46 mmol) in DMA at RT for 18 h. Flash column chromatography (hexane/EtOAc 1:1 to 2:3) afforded compound 9 b as a white solid (60 mg, 67 %), mp: 139–140 °C. 1H NMR (270 MHz, CDCl3): δ=2.86 (2 H, t, J=6.7 Hz), 3.47 (2 H, t, J=6.7 Hz), 3.78 (3 H, s), 3.94 (3 H, s), 4.73 (2 H, s), 5.19 (2 H, s), 6.79–6.89 (3 H, m), 7.15 (1 H, s), 7.24 (1 H, dt, J=7.6, 1.0 Hz), 7.78 ppm (1 H, s); LC–MS (APCI+): m/z 393.64 [M+H]+; HRMS (ES+): m/z found 393.1118; C18H21N2O6S+ [M+H]+ requires 393.1115.
7-Methoxy-2-(4-methoxybenzyl)-6-sulfamoyloxy-3,4-dihydroisoquinolin-1(2H)-one (9 c): Method as for 9 a using compound 8 c (82 mg, 0.26 mmol) and sulfamoyl chloride (0.78 mmol) in DMA (1.0 mL) at RT for 24 h. Flash column chromatography (hexane/EtOAc 3:1 to 1:1) gave a solid that was stirred in Et2O (10 mL), filtered and dried in vacuo to afford compound 9 c as a white solid (75 mg, 73 %), mp: 159–160 °C. 1H NMR (270 MHz, CDCl3/[D4]MeOH 10:1): δ=2.12 (2 H, s), 2.81 (2 H, t, J=6.7 Hz), 3.42 (2 H, t, J=6.7 Hz), 3.76 (3 H, s), 3.90 (3 H, s), 4.67 (2 H, s), 6.83 (2 H, dt, J=8.9, 2.5 Hz), 7.14 (1 H, s), 7.20 (2 H, dt, J=8.9, 2.5 Hz), 7.72 ppm (1 H, s); LC–MS (APCI+): m/z 393.38 [M+H]+; HRMS (ES+): m/z found 393.1117; C18H21N2O6S+ [M+H]+ requires 393.1115.
7-Methoxy-2-(3,5-dimethoxybenzyl)-6-sulfamoyloxy-3,4-dihydroisoquinolin-1(2H)-one (9 d): Method as for 9 a using compound 8 d (100 mg, 0.29 mmol) and sulfamoyl chloride (0.87 mmol) in DMA (1.0 mL) at RT for 24 h. The residue was stirred in Et2O, filtered, washed with Et2O and dried in vacuo to afford compound 9 d as a white powder (95 mg, 77 %), mp: 164–165 °C. 1H NMR (270 MHz, CDCl3): δ=2.90 (2 H, t, J=6.6 Hz), 3.49 (2 H, t, J=6.6 Hz), 3.74 (6 H, s), 3.84 (3 H, s), 4.64 (2 H, s), 6.40–6.45 (3 H, s), 7.26 (1 H, s), 7.61 (1 H, s), 8.08 ppm (2 H, s, br); LC–MS (ES−): m/z 421.13 [M−H]−; HRMS (ES+): m/z found 423.1215; C19H23N2O7S+ [M+H]+ requires 423.1220.
7-Methoxy-6-sulfamoyloxy-2-(3,4,5-trimethoxybenzyl)-3,4-dihydroisoquinolin-1(2H)-one (9 e): Method as for 9 a using compound 8 e (100 mg, 0.27 mmol) and sulfamoyl chloride (0.8 mmol) in DMA (1.0 mL) at RT for 24 h. After addition of H2O (20 mL), EtOAc (100 mL) and THF (50 mL) the organics could not be dissolved. The solvents were evaporated and the resultant solid in the aqueous layer was washed, filtered, washed with H2O, Et2O, EtOAc and dried in vacuo to afford compound 9 e as a white powder (90 mg, 74 %), mp: 201–203 °C. 1H NMR (270 MHz, [D6]DMSO): δ=2.90 (2 H, t, J=6.6 Hz), 3.49 (2 H, t, J=6.6 Hz), 3.63 (3 H, s), 3.74 (6 H, s), 3.84 (3 H, s), 4.64 (2 H, s), 6.61 (2 H, s), 7.26 (1 H, s), 7.61 (1 H, s), 8.08 ppm (2 H, s, br); LC–MS (ES−): m/z 451.18 [M−H]−; HRMS (ES+): m/z found 453.1313; C20H25N2O8S+ [M+H]+ requires 453.1326.
6-Benzyloxy-7-methoxy-2-((2-methoxyphenyl)sulfonyl)-3,4-dihydroisoquinolin-1(2H)-one (10 a): Sodium hydride (60 % in mineral oil, 46 mg, 1.9 mmol) was suspended in anhydrous DMF (5 mL). Compound 6 a (300 mg, 1.0 mmol) was added and the reaction mixture was heated at 50 °C for 0.5 h. The reaction mixture was cooled to RT and 2-methoxybenzenesulfonyl chloride (0.15 mL, 1.0 mmol) was added dropwise. The reaction mixture was stirred for 3.5 h and turned from yellow to almost colourless after addition of the sulfonyl chloride. A further 0.5 equiv (0.07 mL) of the sulfonyl chloride was added and the reaction mixture stirred for a further 2 h. The reaction mixture was poured into sodium bicarbonate (sat., 100 mL) and extracted with CHCl3 (3×50 mL). The combined organic layers were washed with H2O (4×50 mL) and brine (50 mL), dried (MgSO4) and concentrated in vacuo. Flash column chromatography (hexane/EtOAc 2:1) afforded the compound 10 a as a colourless foam (314 mg, 65 %), mp: 202–203 °C. 1H NMR (270 MHz, CDCl3): δ=2.99 (2 H, t, J=6.3 Hz), 3.80 (3 H, s), 3.88 (3 H, s), 4.24 (2 H, t, J=6.3 Hz), 5.19 (2 H, s), 6.66 (1 H, s), 6.96 (1 H, d, J=8.2 Hz), 7.14 (1 H, dt, J=7.6, 1.0 Hz), 7.31–7.44 (6 H, m), 7.52–7.58 (1 H, m), 8.20 ppm (1 H, dd, J=7.9, 1.7 Hz); LC–MS (ES+): m/z 454.46 [M+H]+; HRMS (ES+): m/z found 476.1124; C24H23NO6SNa+ [M+Na]+ requires 476.1144.
6-Benzyloxy-7-methoxy-2-((3-methoxyphenyl)sulfonyl)-3,4-dihydroisoquinolin-1(2H)-one (10 b): Method as for 10 a using compound 6 a (300 mg, 1.1 mmol), sodium hydride (60 % in mineral oil, 64 mg, 1.9 mmol) and 3-methoxybenzenesulfonyl chloride (0.22 mL, 1.6 mmol) in anhydrous DMF (5 mL) at 50 °C for 0.5 h and at RT for 2 h and at 40 °C for 2 h. Flash column chromatography (hexane/EtOAc 2:1) afforded compound 10 b as a colourless foam (369 mg, 77 %), mp: 140–145 °C. 1H NMR (270 MHz, CDCl3): δ=2.99 (2 H, t, J=6.2 Hz), 3.84 (3 H, s), 3.87 (3 H, s), 4.18 (2 H, t, J=6.2 Hz), 5.18 (2 H, s), 6.64 (1 H, s), 7.12 (1 H, ddd, J=8.2, 2.7, 1.0 Hz), 7.31–7.44 (6 H, m), 7.47 (1 H, s), 7.58–7.63 ppm (2 H, m); LC–MS (ES+): m/z 476.50 [M+Na]+; HRMS (ES+): m/z found 476.1116; C24H23NO6SNa+ [M+Na]+ requires 476.1144.
6-Benzyloxy-7-methoxy-2-((4-methoxyphenyl)sulfonyl)-3,4-dihydroisoquinolin-1(2H)-one (10 c): Method as for 10 a using compound 6 a (300 mg, 1.0 mmol), sodium hydride (60 % in mineral oil, 46 mg, 1.9 mmol) and 3-methoxybenzenesulfonyl chloride (0.22 mL, 1.5 mmol) in anhydrous DMF (5 mL) at 50 °C for 0.5 h and at RT for 6 h. Flash column chromatography (hexane/EtOAc 2:1) afforded compound 10 c as a colourless oil (152 mg, 32 %). 1H NMR (270 MHz, CDCl3): δ=2.97 (2 H, t, J=6.3 Hz), 3.82 (3 H, s), 3.87 (3 H, s), 4.16 (2 H, t, J=6.3 Hz), 5.17 (2 H, s), 6.63 (1 H, s), 6.95–6.99 (2 H, m), 7.28–7.41 (5 H, m), 7.46 (1 H, s), 7.98–8.03 ppm (2 H, m); LC–MS (ES+): m/z 454.60 [M+H]+; HRMS (ES+): m/z found 454.1320; C24H24NO6S+ [M+H]+ requires 454.1324.
6-Benzyloxy-7-methoxy-2-((3-chlorophenyl)sulfonyl)-3,4-dihydroisoquinolin-1(2H)-one (10 d): Method as for 10 a using compound 6 a (300 mg, 1.0 mmol), sodium hydride (60 % in mineral oil, 46 mg, 1.9 mmol) and 3-methoxybenzenesulfonyl chloride (0.22 mL, 1.5 mmol) in anhydrous DMF (5 mL) at 50 °C for 0.5 h and at RT for 6 h. Flash column chromatography (hexane/EtOAc 2:1) afforded compound 10 d as a yellow foam (259 mg, 54 %), mp: 155–158 °C. 1H NMR (270 MHz, CDCl3): δ=3.00 (2 H, t, J=6.2 Hz), 3.83 (3 H, s), 4.18 (2 H, t, J=6.2 Hz), 5.18 (2 H, s), 6.65 (1 H, s), 7.27–7.49 (7 H, m), 7.57 (1 H, ddd, J=7.9, 2.0, 1.2 Hz), 7.96–8.01 ppm (2 H, m); LC–MS (ES+): m/z 458.52 [M+H]+; HRMS (ES+): m/z found 480.0621; C23H20ClNO5SNa+ [M+Na]+ requires 480.0643.
Methyl 2-((6-benzyloxy-7-methoxy-1-oxo-3,4-dihydroisoquinolin-2(1H)-yl)sulfonyl)benzoate (10 e): Method as for 10 a using compound 6 a (300 mg, 1.0 mmol), sodium hydride (60 % in mineral oil, 46 mg, 1.9 mmol) and 3-methoxybenzenesulfonyl chloride (0.22 mL, 1.5 mmol) in anhydrous DMF (5 mL) at 50 °C for 0.5 h and at RT for 6 h. Purification by flash column chromatography (hexane/EtOAc 2:1) afforded compound 10 e as a colourless powder (176 mg, 35 %). 1H NMR (270 MHz, CDCl3): δ=3.05 (2 H, t, J=6.2 Hz), 3.83 (3 H, s), 3.92 (3 H, s), 4.19 (2 H, t, J=6.2 Hz), 5.18 (2 H, s), 6.65 (1 H, s), 7.28–7.41 (5 H, m), 7.46 (1 H, s), 7.61–7.71 (3 H, m), 8.55 ppm (1 H, dd, J=6.4, 2.0 Hz); LC–MS (APCI-): m/z 482.29 [M−H]−; HRMS (ES+): m/z found 504.1075; C25H23NO7SNa+ [M+Na]+ requires 504.1087.
6-Hydroxy-7-methoxy-2-((2-methoxyphenyl)sulfonyl)-3,4-dihydroisoquinolin-1(2H)-one (11 a): Method as for 8 a using compound 10 a (316 mg, 0.7 mmol) and Pd/C (10 %, 32 mg) in THF (3 mL) and EtOH (3 mL) under hydrogen at RT for 2 h. The residue was crystallised from dichloromethane/hexane to afford compound 11 a as a colourless powder (145 mg, 57 %), mp: 222–224 °C. 1H NMR (270 MHz, CDCl3): δ=3.02 (2 H, t, J=6.3 Hz), 3.81 (3 H, s), 3.88 (3 H, s), 4.25 (2 H, t, J=6.3 Hz), 6.07 (1 H, s), 6.72 (1 H, s), 6.96 (1 H, d, J=8.4 Hz), 7.12 (1 H, t, J=7.7 Hz), 7.42 (1 H, s), 7.51–7.58 (1 H, m), 8.19 ppm (1 H, dd, J=7.9, 1.7 Hz); LC–MS (ES−): m/z 362.33 [M−H]−; HRMS (ES+): m/z found 386.0651; C17H17NO6SNa+ [M+Na]+ requires 386.0674.
6-Hydroxy-7-methoxy-2-((3-methoxyphenyl)sulfonyl)-3,4-dihydroisoquinolin-1(2H)-one (11 b): Method as for 8 a using compound 10 b (369 mg, 0.81 mmol) and Pd/C (10 %, 37 mg) in THF (5 mL) and EtOH (5 mL) under hydrogen at RT for 2 h. Flash column chromatography (hexane/EtOAc 1:1 to 1:2 to 1:3) afforded compound 11 b as a colourless solid (203 mg, 69 %), mp: 195–197 °C. 1H NMR (270 MHz, CDCl3): δ=3.02 (2 H, t, J=6.3 Hz), 3.84 (3 H, s), 3.86 (3 H, s), 4.19 (2 H, t, J=6.3 Hz), 6.07 (1 H, s), 6.71 (1 H, s), 7.11 (1 H, ddd, J=8.4, 2.6, 1.0 Hz), 7.41 (1 H, t, J=8.1 Hz), 7.45 (1 H, s), 7.57–7.63 ppm (2 H, m); LC–MS (ES−): m/z 362.42 [M−H]−; HRMS (ES+): m/z found 386.0657; C17H17NO6SNa+ [M+Na]+ requires 386.0674.
6-Hydroxy-7-methoxy-2-((4-methoxyphenyl)sulfonyl)-3,4-dihydroisoquinolin-1(2H)-one (11 c): Method as for 8 a using compound 10 c (150 mg, 0.33 mmol) and Pd/C (10 %, 15 mg) in THF (5 mL) and EtOH (5 mL) under hydrogen at RT for 3 h. Flash column chromatography (hexane/EtOAc 2:1) afforded compound 11 c as a colourless powder (74 mg, 62 %), mp: 202–204 °C. 1H NMR (270 MHz, CDCl3): δ=3.00 (2 H, t, J=6.3 Hz), 3.84 (3 H, s), 3.85 (3 H, s), 4.17 (2 H, t, J=6.2 Hz), 6.08 (1 H, s), 6.70 (1 H, s), 6.95–6.99 (2 H, m), 7.45 (1 H, s), 7.99–8.03 ppm (2 H, m); LC–MS (ES−): m/z 362.33 [M−H]−; HRMS (ES+): m/z found 386.0651; C17H17NO6SNa+ [M+Na]+ requires 386.0674.
6-Hydroxy-7-methoxy-2-((3-chlorophenyl)sulfonyl)-3,4-dihydroisoquinolin-1(2H)-one (11 d): Method as for 8 a using compound 10 d (240 mg, 0.53 mmol) and Pd/C (10 %, 24 mg) in THF (5 mL) and EtOH (5 mL) under hydrogen at RT for 1 h. Flash column chromatography (hexane to EtOAc) afforded compound 11 d as a colourless solid (158 mg, 82 %), mp: 178–181 °C. 1H NMR (270 MHz, CDCl3): δ=3.03 (2 H, t, J=6.2 Hz), 3.85 (3 H, s), 4.19 (2 H, t, J=6.3), 6.11 (1 H, s), 6.72 (1 H, s), 7.44 (1 H, s), 7.44–7.50 (1 H, m), 7.54–7.59 (1 H, m), 7.96–8.00 (1 H, m), 8.02 ppm (1 H, t, J=1.5 Hz); LC–MS (ES−): m/z 366.46 [M−H]−; HRMS (ES+): m/z found 390.0160; C16H14ClNO5SNa+ [M+Na]+ requires 390.0173.
Methyl 2-((6-hydroxy-7-methoxy-1-oxo-3,4-dihydroisoquinolin-2(1H)-yl)sulfonyl)benzoate (11 e): Method as for 8 a using compound 10 e (160 mg, 0.33 mmol) and Pd/C (10 %, 90 mg) in THF (5 mL) and EtOH (5 mL) under hydrogen at RT for 3 h. Flash column chromatography (hexane to EtOAc) afforded compound 11 e as a colourless solid (99 mg, 76 %), mp: 236–239 °C. 1H NMR (270 MHz, CDCl3): δ=3.08 (2 H, t, J=6.2 Hz), 3.83 (3 H, s), 3.93 (3 H, s), 4.19 (2 H, t, J=6.2 Hz), 6.08 (1 H, s), 6.71 (1 H, s), 7.44 (1 H, s), 7.60–7.70 (3 H, m), 8.54 ppm (1 H, dd, J=6.2, 1.7 Hz); LC–MS (APCI-): m/z 390.08 [M−H]−; HRMS (ES+): m/z found 414.0609; C18H17NO7SNa+ [M+Na]+ requires 414.0618.
7-Methoxy-2-((2-methoxyphenyl)sulfonyl)-6-(sulfamoyloxy)-3,4-dihydroisoquinolin-1(2H)-one (12 a): Method as for 9 a using compound 11 a (126 mg, 0.35 mmol) and sulfamoyl chloride (0.69 mmol) in anhydrous DMA (1.0 mL) at RT for 22 h. Flash column chromatography (hexane to EtOAc) afforded compound 12 a as a white powder (115 mg, 75 %), mp: 195–197 °C. 1H NMR (270 MHz, [D6]DMSO): δ=3.09 (2 H, t, J=6.2 Hz), 3.78 (3 H, s), 3.89 (3 H, s), 4.18 (2 H, t, J=6.2 Hz), 7.18 (1 H, t, J=7.7 Hz), 7.24 (1 H, d, J=8.4 Hz), 7.37 (1 H, s), 7.44 (1 H, s), 7.68 (1 H, td, J=8.4, 1.6 Hz), 7.79 (1 H, dd, J=7.8, 1.6 Hz), 8.14 ppm (2 H, s, br); LC–MS (ES−): m/z 441.17 [M−H]−; HRMS (ES+): m/z found 465.0394; C17H18N2O8S2Na+ [M+Na]+ requires 465.0402.
7-Methoxy-2-((3-methoxyphenyl)sulfonyl)-6-(sulfamoyloxy)-3,4-dihydroisoquinolin-1(2H)-one (12 b): Method as for 9 a using compound 11 b (200 mg, 0.55 mmol) and sulfamoyl chloride (2.2 mmol) in anhydrous DMA (3.0 mL) at RT for 24 h. Flash column chromatography (hexane to EtOAc) afforded compound 12 b as a colourless powder (118 mg, 49 %), mp: 164–166 °C. 1H NMR (270 MHz, [D6]DMSO): δ=3.12 (2 H, t, J=6.1 Hz), 3.78 (3 H, s), 3.84 (3 H, s), 4.21 (2 H, t, J=6.1 Hz), 7.30 (1 H, dt, J=7.6, 2.1 Hz), 7.36 (1 H, s), 7.47 (2 H, s, br), 7.52–7.61 (2 H, m), 8.15 ppm (2 H, s, br); LC–MS (ES−): m/z 441.38 [M−H]−; HRMS (ES+): m/z found 465.0382; C17H18N2O8S2Na+ [M+Na]+ requires 465.0402.
7-Methoxy-2-((4-methoxyphenyl)sulfonyl)-6-(sulfamoyloxy)-3,4-dihydroisoquinolin-1(2H)-one (12 c): Method as for 9 a using compound 11 c (45 mg, 0.13 mmol) and sulfamoyl chloride (0.5 mmol) in anhydrous DMA (1.0 mL) at RT for 18 h. Flash column chromatography (hexane to EtOAc) gave a colourless solid which was stirred in hexane/CH2Cl2 to afford compound 12 c as a colourless solid (30 mg, 56 %), mp: 177–180 °C. 1H NMR (400 MHz, [D6]DMSO): δ=3.09 (2 H, t, J=6.0 Hz), 3.79 (3 H, s), 3.86 (3 H, s), 4.17 (2 H, t, J=6.2 Hz), 7.12–7.16 (2 H, m), 7.34 (1 H, s), 7.46 (1 H, s), 7.94–7.97 (2 H, m), 8.13 ppm (2 H, s, br); LC–MS (ES−): m/z 441.31 [M−H]−; HRMS (ES+): m/z found 465.0392; C17H18N2O8S2Na+ [M+Na]+ requires 465.0402.
2-((3-Chlorophenyl)sulfonyl)-7-methoxy-6-(sulfamoyloxy)-3,4-dihydroisoquinolin-1(2H)-one (12 d): Method as for 9 a using compound 11 d (121 mg, 0.33 mmol) and sulfamoyl chloride (0.66 mmol) in anhydrous DMA (1.0 mL) at RT for 22 h. Flash column chromatography (hexane to EtOAc) afforded compound 12 d as a colourless solid (132 mg, 90 %), mp: 170–173 °C. 1H NMR (270 MHz, [D6]DMSO): δ=3.14 (2 H, t, J=6.2 Hz), 3.79 (3 H, s), 4.24 (2 H, t, J=6.2 Hz), 7.37 (1 H, s), 7.48 (1 H, s), 7.68 (1 H, t, J=8.0 Hz), 7.82–7.85 (1 H, m), 8.00 (1 H, d, J=8.2 Hz), 8.06 (1 H, t, J=1.8 Hz), 8.17 ppm (2 H, s); LC–MS (ES−): m/z 445.30 [M−H]−; HRMS (ES+): m/z found 468.9894; C16H15ClN2O7S2Na+ [M+Na]+ requires 468.9901.
Methyl 2-((7-methoxy-1-oxo-6-(sulfamoyloxy)-3,4-dihydroisoquinolin-2(1H)-yl)sulfonyl)benzoate (12 e): Method as for 9 a using compound 11 e (68 mg, 0.17 mmol) and sulfamoyl chloride (0.52 mmol) in anhydrous DMA (1.0 mL) at RT for 20 h. Flash column chromatography (hexane to EtOAc) afforded compound 12 e as a colourless powder (49 mg, 60 %), mp: 181–186 °C. 1H NMR (270 MHz, [D6]DMSO): δ=3.14 (2 H, t, J=5.8 Hz), 3.80 (3 H, s), 3.88 (3 H, s), 4.13 (2 H, t, J=5.8 Hz), 7.37 (1 H, s), 7.49 (1 H, s), 7.72–7.87 (3 H, m), 8.16 (2 H, s, br), 8.32–8.35 ppm (1 H, m); LC–MS (APCI-): m/z 390.02 [M−SO2NH2]−; HRMS (ES+): m/z found 493.0343; C18H18N2O9S2Na+ [M+Na]+ requires 493.0346.
6-Benzyloxy-2-(2-methoxybenzoyl)-7-methoxy-1,2,3,4-tetrahydroisoquinoline (14 a): Compound 13 (325 mg, 1.2 mmol) was dissolved in CHCl3 (20 mL) and Et3N (1.0 mL, 7.2 mmol). 2-Methoxybenzoyl chloride (239 mg, 1.4 mmol) was added portionwise. The reaction mixture was stirred at RT for 16 h and washed with H2O and brine, dried (MgSO4), filtered and concentrated in vacuo. Flash column chromatography (hexane/EtOAc 3:1 to 2:3) afforded compound 14 a as a white powder (405 mg, 84 %), mp: 92–93 °C. 1H NMR (270 MHz, CDCl3): δ=2.58–2.70 and 2.76–2.86 (2 H, m), 3.38–3.48 and 3.74–3.94 (2 H, m), 3.72, 3.76, 3.81 and 3.88 (6 H, s), 4.21–4.42 and 4.76–4.93 (2 H, m), 5.11 (2 H, s), 6.38 and 6.61 (1 H, s), 6.67 and 6.69 (1 H, s), 6.92 (1 H, d, J=8.2 Hz), 6.99 (1 H, t, J=7.4 Hz), 7.22–7.46 ppm (7 H, m); LC–MS (APCI+): m/z 402.51 (M+-H), m/z 404.46 [M+H]+; HRMS (ES+): m/z found 404.1856; C25H26NO4+ [M+H]+ requires 404.1856.
6-Benzyloxy-2-(3-methoxybenzoyl)-7-methoxy-1,2,3,4-tetrahydroisoquinoline (14 b): Method as for 14 a using compound 13 (404 mg, 1.5 mmol), 3-methoxybenzoyl chloride (0.22 mL, 1.65 mmol) and Et3N (0.42 mL, 3.0 mmol) in CHCl3 (10 mL) at RT for 18 h. The reaction mixture was diluted with EtOAc (80 mL) and washed with H2O and brine, dried (MgSO4), filtered and concentrated in vacuo. Flash column chromatography (hexane/EtOAc 10:1 to 1:1) afforded compound 14 b as a thick colourless oil (500 mg, 83 %). 1H NMR (270 MHz, CDCl3): δ=2.66–2.76 and 2.76–2.88 (2 H, m), 3.52–3.64 and 3.72–3.98 (2 H, m), 3.80 (6 H, s), 4.49 and 4.79 (2 H, s), 5.11 (2 H, s), 6.40 and 6.69 (1 H, br), 6.64 (1 H, br), 6.92–7.05 (2 H, m), 6.97 (1 H, s), 7.26–7.46 ppm (6 H, m); LC–MS (APCI+): m/z 402.45 (M+-H), m/z 404.46 [M+H]+; HRMS (ES+): m/z found 404.1858; C25H26NO4+ [M+H]+ requires 404.1856.
6-Benzyloxy-2-(3-cyanobenzoyl)-7-methoxy-1,2,3,4-tetrahydroisoquinoline (14 c): Method as for 14 a using compound 13 (300 mg, 1.1 mmol), 3-cyanobenzoyl chloride (202 mg, 1.22 mmol) and Et3N (1.0 mL, 7.2 mmol) in CHCl3 (20 mL) at RT for 2 h. Flash column chromatography (hexane/EtOAc 2:1 to 1:2) afforded compound 14 c as a white powder (325 mg, 74 %), mp: 156–157 °C. 1H NMR (270 MHz, CDCl3): δ=2.70–2.87 (2 H, br), 3.52–3.57 and 3.91–3.95 (2 H, br), 3.79 and 3.97 (3 H, s), 4.46 and 4.80 (2 H, br), 5.12 (2 H, s), 6.40, 6.65 and 6.68 (2 H, br), 7.27–7.45 (5 H, m), 7.52–7.58 (1 H, m), 7.66–7.75 ppm (3 H, m); LC–MS (ES+): m/z 421.54 [M+Na]+.
6-Benzyloxy-2-(4-methoxybenzoyl)-7-methoxy-1,2,3,4-tetrahydroisoquinoline (14 d): Method as for 14 a using compound 13 (406 mg, 1.5 mmol), 4-methoxybenzoyl chloride (318 mg, 1.8 mmol) and Et3N (0.42 mL, 3.0 mmol) in CHCl3 (30 mL) at RT for 18 h. The reaction mixture was diluted with EtOAc (80 mL) and washed with H2O and brine, dried (MgSO4), filtered and concentrated in vacuo. Flash column chromatography (hexane/EtOAc 10:1 to 1:1) afforded compound 14 d as a white powder (520 mg, 85 %), mp: 126–127 °C. 1H NMR (270 MHz, CDCl3): δ=2.77 (2 H, br), 3.67 (2 H, br), 3.83 (6 H, s), 4.73 (2 H, br), 5.11 (2 H, s), 6.46 and 6.69 (1 H, br), 6.65 (1 H, s), 6.92 (2 H, dt, J=8.6, 2.3 Hz), 7.26–7.45 ppm (7 H, m); LC–MS (APCI+): m/z 404.53 [M+H]+; HRMS (ES+): m/z found 404.1858; C25H26NO4+ [M+H]+ requires 404.1856.
6-Benzyloxy-2-(3,4-dimethoxybenzoyl)-7-methoxy-1,2,3,4-tetra-hydroisoquinoline (14 e): Method as for 14 a using compound 13 (300 mg, 1.1 mmol), 3,4-dimethoxybenzoyl chloride (240 mg, 1.2 mmol) and Et3N (0.5 mL, 3.6 mmol) in CHCl3 (20 mL) at RT for 18 h. Flash column chromatography (hexane/EtOAc 2:1 to 1:3) afforded compound 14 e as a white powder (370 mg, 77 %), mp: 126–127 °C. 1H NMR (270 MHz, CDCl3): δ=2.77 (2 H, br), 3.83 (2 H, br), 3.88 (3 H, s), 3.89 (3 H, s), 3.90 (3 H, s), 4.61 and 4.74 (2 H, br), 5.11 (2 H, s), 6.65 (1 H, s), 6.86 (1 H, d, J=8.9 Hz), 7.01 (1 H, s), 7.03 (1 H, dd, J=8.9, 2.0 Hz), 7.23–7.46 ppm (6 H, m); LC–MS (APCI+): m/z 432.48 (M+-H), m/z 434.50 [M+H]+; HRMS (ES+): m/z found 434.1966; C26H28NO5+ [M+H]+ requires 434.1962.
6-Benzyloxy-2-(3,5-dimethoxybenzoyl)-7-methoxy-1,2,3,4-tetra-hydroisoquinoline (14 f): Method as for 14 a using compound 13 (404 mg, 1.5 mmol), 3,5-dimethoxybenzoyl chloride (331 mg, 1.65 mmol) and Et3N (0.42 mL, 3.0 mmol) in CHCl3 (20 mL) at RT for 18 h. The reaction mixture was diluted with EtOAc (80 mL) and washed with H2O and brine, dried (MgSO4), filtered and concentrated in vacuo. Flash column chromatography (hexane/EtOAc 10:1 to 1:1) afforded compound 14 f as a white powder (520 mg, 85 %), mp: 171–172 °C. 1H NMR (270 MHz, CDCl3): δ=2.70 and 2.81 (2 H, br), 3.58 and 3.92 (2 H, br), 3.83 (9 H, s), 4.49 and 4.78 (2 H, br), 5.11 (2 H, s), 6.42, 6.63, 6.66 and 6.68 (2 H, br), 6.50 (1 H, t, J=2.2 Hz), 6.55 (2 H, d, J=2.2 Hz), 7.26–7.45 ppm (5 H, m); LC–MS (APCI+): m/z 434.43 [M+H]+; HRMS (ES+): m/z found 434.1962; C26H28NO5+ [M+H]+ requires 434.1962.
6-Benzyloxy-7-methoxy-2-(3,4,5-trimethoxybenzoyl)-1,2,3,4-tetrahydroisoquinoline (14 g): Method as for 14 a using compound 13 (808 mg, 3.0 mmol), 3,4,5-trimethoxybenzoyl chloride (765 mg, 3.3 mmol) and Et3N (2.0 mL, 14.4 mmol) in CHCl3 (20 mL) at RT for 18 h. The reaction mixture was diluted with CHCl3 (80 mL) and washed with H2O and brine, dried (MgSO4), filtered and concentrated in vacuo. Flash column chromatography (hexane/EtOAc 3:1 to 1:2) afforded compound 14 g as a white powder (1.10 g, 79 %), mp: 63–64 °C. 1H NMR (270 MHz, CDCl3): δ=2.80 (2 H, br), 3.60 and 3.80 (2 H, br), 3.85 (6 H, s), 3.86 (6 H, s), 4.50 and 4.77 (2 H, br), 5.12 (2 H, s), 6.44 and 6.65 (4 H, br), 7.25–7.44 ppm (5 H, m); LC–MS (ES+): m/z 496.26 ([M+Na]+, 100 %), 464.28 [M+H]+; HRMS (ES+): m/z found 464.2060; C27H30NO6+ [M+H]+ requires 464.2068.
6-Benzyloxy-2-(2-methoxybenzoyl)-7-methoxy-3,4-dihydroisoquinolin-1(2H)-one (15 a): Compound 14 a (400 mg, 1.0 mmol), KMnO4 (0.79 g, 5.0 mmol) and 18-crown-6 (50 mg, 0.19 mmol) were mixed in dichloromethane (50 mL) and the reaction mixture was stirred at RT for 8 h. The reaction mixture was diluted with CHCl3 and sodium metabisulfite (sat.) was added. The mixture was washed with H2O and brine, dried with MgSO4, filtered and concentrated in vacuo. Flash column chromatography (hexane/EtOAc 3:1) afforded compound 15 a as a white powder (170 mg, 40 %), mp: 163–164 °C. 1H NMR (270 MHz, CDCl3): δ=2.98 (2 H, t, J=6.2 Hz), 3.63 (3 H, s), 3.83 (3 H, s), 4.19 (2 H, t, J=6.2 Hz), 5.22 (2 H, s), 6.72 (1 H, s), 6.86 (1 H, d, J=8.4 Hz), 7.01 (1 H, dt, J=8.4, 0.8 Hz), 7.28–7.46 (7 H, m), 7.56 ppm (1 H, s); LC–MS (ES+): m/z 440.49 [M+Na]+; LC–MS (ES−): m/z 416.44 [M−H]−; HRMS (ES+): m/z found 418.1635; C25H24NO5+ [M+H]+ requires 418.1649.
6-Benzyloxy-7-methoxy-2-(3-methoxybenzoyl)-3,4-dihydroisoquinolin-1(2H)-one (15 b): Method as for 15 a using compound 14 b (560 mg, 1.38 mmol), KMnO4 (1.1 g, 6.9 mmol) and 18-crown-6 (70 mg, 0.26 mmol) in dichloromethane (50 mL) at RT for 16 h. Flash column chromatography (hexane/EtOAc 3:1) afforded compound 15 b as a white powder (235 mg, 41 %), mp: 141–143 °C. 1H NMR (270 MHz, CDCl3): δ=3.03 (2 H, t, J=6.2 Hz), 3.81 (3 H, s), 3.85 (3 H, s), 4.08 (2 H, t, J=6.2 Hz), 5.23 (2 H, s), 6.73 (1 H, s), 7.01 (1 H, ddd, J=8.2, 2.7, 1.2 Hz), 7.11–7.15 (2 H, m), 7.26–7.45 (6 H, m), 7.56 ppm (1 H, s); LC–MS (ES+): m/z 440.43 [M+Na]+; LC–MS (ES−): m/z 416.38 [M−H]−; HRMS (ES+): m/z found 418.1636; C25H24NO5+ [M+H]+ requires 418.1649.
6-Benzyloxy-2-(3-cyanobenzoyl)-7-methoxy-3,4-dihydroisoquinolin-1(2H)-one (15 c): Method as for 15 a using compound 14 c (315 mg, 0.79 mmol), KMnO4 (0.62 g, 3.8 mmol) and 18-crown-6 (40 mg, 0.15 mmol) in dichloromethane (40 mL) at RT for 16 h. Flash column chromatography (hexane/EtOAc 3:2) afforded compound 15 c as a white powder (100 mg, 31 %), mp: 195–196 °C. 1H NMR (270 MHz, CDCl3): δ=3.05 (2 H, t, J=6.2 Hz), 3.82 (3 H, s), 3.86 (2 H, t, J=6.2 Hz), 5.24 (2 H, s), 6.74 (1 H, s, 7.30–7.45 (5 H, m), 7.49–7.55 (2 H, m), 7.74 (1 H, ddd, J=7.9, 1.7, 1.5 Hz), 7.79 (1 H, ddd, J=7.9, 1.7, 1.2 Hz), 7.82 ppm (1 H, d, J=1.5, 1.2 Hz); LC–MS (ES+): m/z 435.58 [M+Na]+; HRMS (ES+): m/z found 413.1496; C25H21N2O4+ [M+H]+ requires 413.1496.
6-Benzyloxy-2-(4-methoxybenzoyl)-7-methoxy-3,4-dihydroisoquinolin-1(2H)-one (15 d): Method as for 15 a using compound 14 d (403 mg, 1.0 mmol), KMnO4 (0.79 g, 5.0 mmol) and 18-crown-6 (53 mg, 0.2 mmol) in dichloromethane (50 mL) at RT for 16 h. Flash column chromatography (hexane/EtOAc 4:1 to 1:1 to EtOAc) afforded compound 15 d as a white powder (188 mg, 45 %), mp: 151–152 °C. 1H NMR (270 MHz, CDCl3): δ=3.02 (2 H, t, J=6.1 Hz), 3.83 (3 H, s), 3.86 (3 H, s), 4.04 (2 H, t, J=6.1 Hz), 5.23 (2 H, s), 6.73 (1 H, s), 6.88 (2 H, d, J=8.7 Hz), 7.30–7.46 (5 H, m), 7.58 (1 H, s), 7.62 ppm (2 H, d, J=8.7 Hz); HRMS (ES+): m/z found 418.1651; C25H24NO5+ [M+H]+ requires 418.1649.
6-Benzyloxy-2-(3,4-dimethoxybenzoyl)-7-methoxy-3,4-dihydroisoquinolin-1(2H)-one (15 e): Method as for 15 a using compound 14 e (450 mg, 1.04 mmol), KMnO4 (0.82 g, 5.2 mmol) and 18-crown-6 (50 mg, 0.19 mmol) in dichloromethane (50 mL) at RT for 8 h. Flash column chromatography (hexane/EtOAc 3:1) afforded compound 15 e as a white powder (170 mg, 37 %), mp: 200–201 °C. 1H NMR (270 MHz, CDCl3): δ=3.04 (2 H, t, J=6.2 Hz), 3.87 (3 H, s), 3.90 (6 H, s), 4.04 (2 H, t, J=6.2 Hz), 5.23 (2 H, s), 6.74 (1 H, s), 6.81 (1 H, d, J=8.4 Hz), 7.20 (1 H, dd, J=8.4, 2.0 Hz), 7.26 (1 H, d, J=2.0 Hz), 7.28–7.46 (5 H, m), 7.58 ppm (1 H, s); LC–MS (ES+): m/z 470.52 [M+Na]+; LC–MS (ES−): m/z 446.47 [M−H]−.
6-Benzyloxy-2-(3,5-dimethoxybenzoyl)-7-methoxy-3,4-dihydroisoquinolin-1(2H)-one (15 f): Method as for 15 a using compound 14 f (433 mg, 1.0 mmol), KMnO4 (0.79 g, 5.0 mmol) and 18-crown-6 (53 mg, 0.2 mmol) in dichloromethane (50 mL) at RT for 16 h. Flash column chromatography (hexane/EtOAc 10:1 to 4:1) afforded compound 15 f as a white powder (193 mg, 43 %), mp: 180–181 °C. 1H NMR (270 MHz, CDCl3): δ=3.02 (2 H, t, J=6.2 Hz), 3.78 (6 H, s), 3.85 (3 H, s), 4.06 (2 H, t, J=6.2 Hz), 5.23 (2 H, s), 6.56 (1 H, t, J=2.2 Hz), 6.70 (2 H, d, J=2.2 Hz), 6.73 (1 H, s), 7.29–7.46 (5 H, m), 7.56 ppm (1 H, s); LC–MS (APCI+): m/z 448.54 [M+H]+; HRMS (ES+): m/z found 448.1756; C26H26NO6+ [M+H]+ requires 448.1755.
6-Benzyloxy-7-methoxy-2-(3,4,5-trimethoxybenzoyl)-3,4-dihydroisoquinolin-1(2H)-one (15 g): Method as for 15 a using compound 14 g (0.85 g, 1.83 mmol) and 18-crown-6 (48 mg, 0.18 mmol) in CHCl3 (30 mL) at 0 °C for 2 h then at RT 16 h. The reaction mixture was diluted with CHCl3 and sodium bisulfite (sat.) and HCl (2 m, 1.0 mL, 2.0 mmol) were added. The mixture was washed with H2O and brine, dried with MgSO4, filtered and concentrated in vacuo. Flash column chromatography (hexane/EtOAc 3:1 to 1:1) afforded compound 15 g as a white powder (220 mg, 25 %), mp: 195–196 °C. 1H NMR (270 MHz, CDCl3): δ=3.05 (2 H, t, J=6.2 Hz), 3.83 (6 H, s), 3.87 (3 H, s), 3.87 (3 H, s), 4.05 (2 H, t, J=6.2 Hz), 5.23 (2 H, s), 6.75 (1 H, s), 6.84 (2 H, s), 7.29–7.46 (5 H, m), 7.56 ppm (1 H, s); LC–MS (ES+): m/z 500.18 ([M+Na]+, 100 %), 478.20 [M+H]+; HRMS (ES+): m/z found 478.1844; C27H28NO7+ [M+H]+ requires 478.1860.
6-Hydroxy-2-(2-methoxybenzoyl)-7-methoxy-3,4-dihydroisoquinolin-1(2H)-one (16 a): Method as for 8 a using compound 15 a (145 mg, 0.35 mmol) and Pd/C (10 %, 20 mg) in THF (10 mL) and MeOH (10 mL) under hydrogen at RT for 2 h. The residue was stirred in EtOAc (5 mL), filtered and dried in vacuo to afford compound 16 a as a white powder (105 mg, 92 %), mp: 199–200 °C. 1H NMR (270 MHz, [D6]DMSO): δ=2.94 (2 H, t, J=6.2 Hz), 3.57 (3 H, s), 3.74 (3 H, s), 4.05 (2 H, t, J=6.2 Hz), 6.76 (1 H, s), 6.93–7.00 (2 H, m), 7.27 (1 H, dd, J=7.4, 1.7 Hz), 7.32 (1 H, s), 7.34–7.41 (1 H, m), 10.21 ppm (1 H, s); LC–MS (ES−): m/z 326.51 [M−H]−; HRMS (ES+): m/z found 328.1167; C18H18NO5+ [M+H]+ requires 328.1179.
6-Hydroxy-2-(3-methoxybenzoyl)-7-methoxy-3,4-dihydroisoquinolin-1(2H)-one (16 b): Method as for 8 a using compound 15 b (200 mg, 0.48 mmol) and Pd/C (10 %, 25 mg) in THF (10 mL) and MeOH (10 mL) under hydrogen at RT for 1 h. The residue was stirred in EtOAc (5 mL), filtered and dried in vacuo to afford compound 16 b as a white powder (125 mg, 80 %), mp: 198–199 °C. 1H NMR (270 MHz, [D6]DMSO): δ=3.03 (2 H, t, J=5.9 Hz), 3.75 (3 H, s), 3.76 (3 H, s), 3.95 (2 H, t, J=5.9 Hz), 6.77 (1 H, s), 7.05–7.10 (3 H, m), 7.32 (1 H, dt, J=6.7, 1.0 Hz), 7.34 (1 H, s), 10.21 ppm (1 H, s); LC–MS (ES−): m/z 326.58 [M−H]−; HRMS (ES+): m/z found 328.1168; C18H18NO5+ [M+H]+ requires 328.1179.
2-(3-Cyanobenzoyl)-6-hydroxy-7-methoxy-3,4-dihydroisoquinolin-1(2H)-one (16 c): Method as for 8 a using compound 15 c (155 mg, 0.38 mmol) and Pd/C (10 %, 20 mg) in THF (10 mL) and MeOH (10 mL) under hydrogen at RT for 1 h. The residue was dissolved in hot EtOAc (5 mL), filtered and concentrated then stirred in Et2O (20 mL), filtered and dried in vacuo to afford compound 16 c as a white powder (71 mg, 59 %), mp: 197–198 °C. 1H NMR (270 MHz, [D6]acetone): δ=3.14 (2 H, t, J=6.2 Hz), 3.85 (3 H, s), 4.08 (2 H, t, J=6.2 Hz), 6.83 (1 H, s), 7.42 (1 H, s), 7.65 (1 H, dd, J=8.2, 7.6 Hz), 7.86–7.91 (2 H, m), 8.00–8.06 (1 H, m), 8.71 ppm (1 H, s, br); LC–MS (ES−): m/z 321.47 (M+-H); HRMS (ES+): m/z found 323.1012; C18H15N2O4+ [M+H]+ requires 323.1026.
6-Hydroxy-2-(4-methoxybenzoyl)-7-methoxy-3,4-dihydroisoquinolin-1(2H)-one (16 d): Method as for 8 a using compound 15 d (96 mg, 0.23 mmol) and Pd/C (10 %, 20 mg) in THF (10 mL) and EtOH (10 mL) under hydrogen at RT for 0.5 h. The residue was stirred in EtOAc (10 mL) and hexane (2 mL), filtered and dried in vacuo to afford compound 16 d as a white powder (72 mg, 96 %), mp: 224–225 °C. 1H NMR (270 MHz, [D6]DMSO): δ=3.01 (2 H, t, J=5.7 Hz), 3.77 (3 H, s), 3.82 (3 H, s), 3.90 (2 H, t, J=5.7 Hz), 6.77 (1 H, s), 6.95 (2 H, d, J=8.6 Hz), 7.36 (1 H, s), 7.55 (2 H, d, J=8.6 Hz), 10.19 ppm (1 H, s, br); LC–MS (APCI+): m/z 328.46 [M+H]+; HRMS (ES+): m/z found 328.1181; C18H18NO5+ [M+H]+ requires 328.1179.
6-Hydroxy-2-(3,4-dimethoxybenzoyl)-7-methoxy-3,4-dihydroisoquinolin-1(2H)-one (16 e): Method as for 8 a using compound 15 e (165 mg, 0.37 mmol) and Pd/C (10 %, 20 mg) in THF (150 mL) under hydrogen at RT for 2 h. Crystallisation from EtOAc (5 mL) afforded compound 16 e as a white powder (115 mg, 87 %), mp: 199–200 °C. 1H NMR (400 MHz, [D6]DMSO): δ=3.02 (2 H, t, J=5.9 Hz), 3.75 (3 H, s), 3.76 (3 H, s), 3.81 (3 H, s), 3.90 (2 H, t, J=5.9 Hz), 6.77 (1 H, s), 6.96 (1 H, d, J=9.2 Hz), 7.15–7.18 (2 H, m), 7.36 (1 H, s), 10.20 ppm (1 H, s, br); LC–MS (ES−): m/z 356.60 [M−H]−; HRMS (ES+): m/z found 358.1273; C19H20NO6+ [M+H]+ requires 358.1285.
6-Hydroxy-2-(3,5-dimethoxybenzoyl)-7-methoxy-3,4-dihydroisoquinolin-1(2H)-one (16 f): Method as for 8 a using compound 15 f (200 mg, 0.45 mmol) and Pd/C (10 %, 30 mg) in THF (15 mL) and MeOH (15 mL) under hydrogen at RT for 3 h. The residue was stirred in Et2O, filtered and dried in vacuo to afford compound 16 f as a white powder (130 mg, 81 %), mp: 198–199 °C. 1H NMR (270 MHz, CDCl3): δ=3.05 (2 H, t, J=6.2 Hz), 3.78 (6 H, s), 3.86 (3 H, s), 4.07 (2 H, t, J=6.2 Hz), 6.16 (1 H, s), 6.56 (1 H, t, J=2.2 Hz), 6.71 (2 H, d, J=2.2 Hz), 6.79 (1 H, s), 7.54 ppm (1 H, s); LC–MS (APCI+): m/z 358.25 [M+H]+; HRMS (ES+): m/z found 358.1285; C19H20NO6+ [M+H]+ requires 358.1285.
6-Hydroxy-7-methoxy-2-(3,4,5-trimethoxybenzoyl)-3,4-dihydroisoquinolin-1(2H)-one (16 g): Method as for 8 a using compound 15 g (180 mg, 0.38 mmol) and Pd/C (10 %, 40 mg) in THF (20 mL) and MeOH (20 mL) under hydrogen at RT for 24 h. The residue was stirred in Et2O, filtered, washed with Et2O and dried in vacuo to afford compound 16 g as a white powder (140 mg, 96 %), mp: 175–176 °C. 1H NMR (270 MHz, CDCl3): δ=3.07 (2 H, t, J=6.4 Hz), 3.83 (6 H, s), 3.88 (3 H, s), 3.89 (3 H, s), 4.06 (2 H, t, J=6.4 Hz), 6.18 (1 H, s), 6.80 (1 H, s), 6.85 (2 H, s), 7.55 ppm (1 H, s); HRMS (ES+): m/z found 388.1380; C20H22NO7+ [M+H]+ requires 388.1391; LC–MS (ES−): m/z 386.24 [M−H]−.
2-(2-Methoxybenzoyl)-7-methoxy-6-sulfamoyloxy-3,4-dihydroisoquinolin-1(2H)-one (17 a): Method as for 9 a using compound 16 a (70 mg, 0.21 mmol) and sulfamoyl chloride (0.43 mmol) in DMA (1.0 mL) at RT for 16 h. The residue was stirred in Et2O (30 mL), filtered and dried in vacuo to afford compound 17 a as a white powder (65 mg, 75 %), mp: 165–166 °C. 1H NMR (400 MHz, [D6]DMSO): δ=3.05 (2 H, t, J=6.2 Hz), 3.60 (3 H, s), 3.80 (3 H, s), 4.11 (2 H, t, J=6.2 Hz), 6.96–7.03 (2 H, m), 7.31 (1 H, dd, J=7.7, 1.7 Hz), 7.39 (1 H, s), 7.41 (1 H, dt, J=8.4, 1.7 Hz), 7.53 (1 H, s), 8.18 ppm (2 H, s, br); LC–MS (ES−): m/z 405.50 [M−H]−; HRMS (ES+): m/z found 407.0898; C18H19N2O7S+ [M+H]+ requires 407.0907.
2-(3-Methoxybenzoyl)-7-methoxy-6-sulfamoyloxy-3,4-dihydroisoquinolin-1(2H)-one (17 b): Method as for 9 a using compound 16 b (80 mg, 0.24 mmol) and sulfamoyl chloride (0.48 mmol) in DMA (1.0 mL) at RT for 16 h. The residue was stirred in Et2O (30 mL), filtered and dried in vacuo to afford compound 17 b as a white powder (75 mg, 77 %), mp: 173–174 °C. 1H NMR (400 MHz, [D6]DMSO): δ=3.15 (2 H, t, J=5.9 Hz), 3.77 (3 H, s), 3.81 (3 H, s), 4.01 (2 H, t, J=5.9 Hz), 7.08–7.18 (3 H, m), 7.31–7.36 (1 H, m), 7.41 (1 H, s), 7.57 (1 H, s), 8.18 ppm (2 H, s, br); LC–MS (ES−): m/z 405.43 [M−H]−; HRMS (ES+): m/z found 407.0895; C18H19N2O7S+ [M+H]+ requires 407.0907.
2-(3-Cyanobenzoyl)-7-methoxy-6-sulfamoyloxy-3,4-dihydroisoquinolin-1(2H)-one (17 c): Method as for 9 a using compound 16 c (50 mg, 0.16 mmol) and sulfamoyl chloride (0.40 mmol) in DMA (0.5 mL) at RT for 16 h. Crystallisation from EtOAc afforded compound 17 c as a white powder (45 mg, 70 %), mp: 171–172 °C. 1H NMR (270 MHz, [D6]acetone): δ=3.23 (2 H, t, J=6.2 Hz), 3.88 (3 H, s), 4.14 (2 H, t, J=6.2 Hz), 7.24 (2 H, s, br), 7.40 (1 H, s), 7.58 (1 H, s), 7.69 (1 H, dt, J=7.9, 0.8 Hz), 7.91–7.98 (2 H, m), 8.06 ppm (1 H, dt, J=1.8, 0.8 Hz); LC–MS (ES−): m/z 400.53 [M−H]−; HRMS (ES+): m/z found 402.0739; C18H16N3O6S+ [M+H]+ requires 402.0754.
2-(4-Methoxybenzoyl)-7-methoxy-6-sulfamoyloxy-3,4-dihydroisoquinolin-1(2H)-one (17 d): Method as for 9 a using compound 16 d (90 mg, 0.28 mmol) and sulfamoyl chloride (0.55 mmol) in DMA (1.0 mL) at RT for 18 h. Flash column chromatography (hexane/EtOAc 10:1 to 4:1) afforded compound 17 d as a white powder (80 mg, 71 %), mp: 171–172 °C. 1H NMR (270 MHz, CDCl3/[D4]MeOH 5:1): δ=3.08 (2 H, s, br), 3.10 (2 H, t, J=6.4 Hz), 3.83 (3 H, s), 3.85 (3 H, s), 4.03 (2 H, t, J=6.4 Hz), 6.88 (2 H, dt, J=8.9, 2.5 Hz), 7.31 (1 H, s), 7.61 (2 H, dt, J=8.9, 2.5 Hz), 7.67 ppm (1 H, s); LC–MS (APCI+): m/z 407.42 [M+H]+; HRMS (ES+): m/z found 407.0904; C18H19N2O7S+ [M+H]+ requires 407.0907.
2-(3,4-Dimethoxybenzoyl)-7-methoxy-6-sulfamoyloxy-3,4-dihydroisoquinolin-1(2H)-one (17 e): Method as for 9 a using compound 16 e (71 mg, 0.20 mmol) and sulfamoyl chloride (0.40 mmol) in DMA (1.0 mL) at RT for 16 h. After addition of H2O (5 mL) the reaction mixture was extracted into EtOAc (2×50 mL), the organic layers washed with H2O and brine, then dried (MgSO4) and evaporated. The residue was stirred in EtOAc (5 mL), filtered and dried in vacuo to afford compound 17 e as a white powder (55 mg, 77 %), mp: 180–181 °C. 1H NMR (270 MHz, ([D6]acetone): δ=3.19 (2 H, t, J=6.2 Hz), 3.81 (3 H, s), 3.87 (3 H, s), 3.88 (3 H, s), 4.03 (2 H, t, J=6.2 Hz), 6.96 (1 H, d, J=8.9 Hz), 7.21 (2 H, s, br), 7.28 (1 H, s), 7.30 (1 H, d, J=8.9 Hz), 7.39 (1 H, s), 7.62 ppm (1 H, s); LC–MS (ES−): m/z 435.44 [M−H]−; HRMS (ES+): m/z found 437.1011; C19H21N2O8S+ [M+H]+ requires 437.1013.
2-(3,5-Dimethoxybenzoyl)-7-methoxy-6-sulfamoyloxy-3,4-dihydroisoquinolin-1(2H)-one (17 f): Method as for 9 a using compound 16 f (80 mg, 0.22 mmol) and sulfamoyl chloride (0.45 mmol) in DMA (1.0 mL) at RT for 18 h. The residue was stirred in Et2O, filtered and dried in vacuo to afford compound 17 f as a white powder (76 mg, 78 %), mp: 169–170 °C. 1H NMR (270 MHz, (CDCl3/[D4]MeOH 10:1): δ=2.46 (2 H, s, br), 3.08 (2 H, t, J=6.2 Hz), 3.75 (6 H, s), 3.82 (3 H, s), 4.05 (2 H, t, J=6.2 Hz), 6.56 (1 H, t, J=2.3 Hz), 6.68 (2 H, d, J=2.3 Hz), 7.29 (1 H, s), 7.65 ppm (1 H, s); LC–MS (APCI+): m/z 437.39 [M+H]+; HRMS (ES+) m/z found 437.1011; C19H21N2O8S+ [M+H]+ requires 437.1013.
7-Methoxy-2-(3,4,5-trimethoxybenzoyl)-6-sulfamoyloxy-3,4-dihydroisoquinolin-1(2H)-one (17 g): Method as for 9 a using compound 16 g (80 mg, 0.21 mmol) and sulfamoyl chloride (0.6 mmol) in DMA (1.0 mL) at RT for 24 h. H2O (20 mL) was added and the mixture extracted with EtOAc (50 mL) and THF (50 mL). The organic layer was washed with H2O and brine, dried (MgSO4), filtered and concentrated in vacuo. The residue was stirred in Et2O (5 mL) and EtOAc (5 mL), filtered and dried to afford compound 17 g as a white powder (80 mg, 82 %), mp: 196–197 °C. 1H NMR (270 MHz, [D6]DMSO): δ=3.16 (2 H, t, J=5.7 Hz), 3.72 (3 H, s), 3.75 (6 H, s), 3.81 (3 H, s), 3.98 (2 H, t, J=5.7 Hz), 6.90 (2 H, s), 7.40 (1 H, s), 7.57 (1 H, s), 8.18 ppm (2 H, s, br); LC–MS (ES−): m/z 465.22 [M−H]−; HRMS (ES+): m/z found 467.1109; C20H23N2O9S+ [M+H]+ requires 467.1119.
2-(3,4-Dimethoxybenzoyl)-6-hydroxy-7-methoxy-1,2,3,4-tetrahydroisoquinoline (18 a): Method as for 8 a using 14 e (403 mg, 0.93 mmol) and Pd/C (10 %, 40 mg) in THF (15 mL) and MeOH (15 mL) under hydrogen at RT for 1.5 h. The resulting solid was stirred in Et2O, filtered and dried in vacuo to afford compound 18 a as a white solid (300 mg, 94 %), mp: 132–133 °C. 1H NMR (270 MHz, CDCl3): δ=2.80 (2 H, br), 3.83 (2 H, br), 3.88 (3 H, s), 3.90 (6 H, s), 4.61 and 4.72 (2 H, br), 5.62 (1 H, s), 6.60 (1 H, br), 6.69 (1 H, br), 6.86 (1 H, d, J=8.9 Hz), 7.02 (1 H, s), 7.03 (1 H, d, J=8.9 Hz), 8.84 ppm (1 H, s); LC–MS (APCI+): m/z 344.27 [M+H]+; HRMS (ES+): m/z found 344.1493; C19H22NO5+ [M+H]+ requires 344.1492.
2-(3,5-Dimethoxybenzoyl)-6-hydroxy-7-methoxy-1,2,3,4-tetrahydroisoquinoline (18 b): Method as for 8 a using 14 f (390 mg, 0.90 mmol) and Pd/C (10 %, 40 mg) in THF (15 mL) and MeOH (15 mL) under hydrogen at RT for 2 h. The resulting solid was stirred in Et2O, filtered and dried in vacuo to afford compound 18 b as a white solid (290 mg, 94 %), mp: 136–137 °C. 1H NMR (270 MHz, CDCl3): δ=2.73 and 2.84 (2 H, br), 3.59 and 3.93 (2 H, br), 3.79 (9 H, s), 4.48 and 4.77 (2 H, br), 5.58 (1 H, s), 6.38 and 6.64 (1 H, br), 6.50 (1 H, t, J=2.2 Hz), 6.55 (2 H, d, J=2.2 Hz), 6.68 ppm (1 H, br); LC–MS (APCI+): m/z 344.33 [M+H]+; HRMS (ES+): m/z found 344.1493; C19H22NO5+ [M+H]+ requires 344.1492.
2-(3,4-Dimethoxybenzoyl)-7-methoxy-6-sulfamoyloxy-1,2,3,4-tetrahydroisoquinoline (19 a): Method as for 9 a using 18 a (75 mg, 0.24 mmol) and sulfamoyl chloride (0.48 mmol) in anhydrous DMA (1.0 mL) at RT for 24 h. The residue was stirred in Et2O, filtered and dried in vacuo to afford compound 19 a as a white powder (80 mg, 85 %), mp: 155–156 °C. 1H NMR (270 MHz, CDCl3/[D4]MeOH 5:1): δ=2.73 (2 H, br), 3.59 and 3.72 (2 H, br), 3.77 (3 H, s), 3.79 (6 H, s), 4.52 and 4.65 (2 H, br), 6.61 and 6.70 (1 H, br), 6.80 (1 H, d, J=8.2 Hz), 6.87 (1 H, d, J=2.0 Hz), 6.90 (1 H, dd, J=8.2, 2.0 Hz), 7.03 ppm (1 H, s); LC–MS (APCI+): m/z 423.42 [M+H]+; HRMS (ES+): m/z found 423.1220; C19H23N2O7S+ [M+H]+ requires 423.1220.
2-(3,5-Dimethoxybenzoyl)-7-methoxy-6-sulfamoyloxy-1,2,3,4-tetrahydroisoquinoline (19 b): Method as for 9 a using 18 b (75 mg, 0.24 mmol) and sulfamoyl chloride (0.48 mmol) in anhydrous DMA (1.0 mL) at RT for 24 h. The residue was stirred in Et2O, filtered and dried in vacuo to afford compound 19 b as a white powder (80 mg, 85 %), mp: 170–171 °C. 1H NMR (270 MHz, CDCl3/[D4]MeOH 5:1): δ=2.70 and 2.80 (2 H, t, J=5.3 Hz), 3.55 and 3.82 (2 H, t, J=5.3 Hz), 3.63 (2 H, s), 3.71 (9 H, s), 4.46 and 4.72 (2 H, br), 6.44 (2 H, s), 6.46 and 6.74 (1 H, br), 7.05 ppm (1 H, br); LC–MS (APCI+): m/z 423.35 [M+H]+; HRMS (ES+): m/z found 423.1224; C19H23N2O7S+ [M+H]+ requires 423.1220.
Acknowledgments
We thank Ms. Alison Smith for technical support and the NCI DTP for providing in vitro screening resources. This work was supported by Sterix Ltd., a member of the IPSEN Group, and in part by the Wellcome Trust (Program Grant 082837 to B.V.L.P.). B.V.L.P. is a Wellcome Trust Senior Investigator (Grant 101010).
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Bubert C, Leese MP, Mahon MF, Ferrandis E, Regis-Lydi S, Kasprzyk PG, Newman SP, Ho YT, Purohit A, Reed MJ, Potter BVL. J. Med. Chem. 2007;50:4431–4443. doi: 10.1021/jm070405v. [DOI] [PubMed] [Google Scholar]
- 2.Jourdan FL, Leese MP, Dohle W, Ferrandis E, Newman SP, Chander S, Purohit A, Potter BVL. J. Med. Chem. 2011;54:4863–4879. doi: 10.1021/jm200483x. [DOI] [PubMed] [Google Scholar]
- 3.Jourdan FL, Leese MP, Dohle W, Hamel E, Ferrandis E, Newman SP, Purohit A, Reed MJ, Potter BVL. J. Med. Chem. 2010;53:2942–2951. doi: 10.1021/jm9018806. [DOI] [PubMed] [Google Scholar]
- 4.Leese MP, Hejaz HAM, Mahon MF, Newman SP, Purohit A, Reed MJ, Potter BVL. J. Med. Chem. 2005;48:5243–5256. doi: 10.1021/jm050066a. [DOI] [PubMed] [Google Scholar]
- 5.Leese MP, Jourdan FL, Gaukroger K, Mahon MF, Newman SP, Foster PA, Stengel C, Regis-Lydi S, Ferrandis E, Di Fiore A, De Simone G, Supuran CT, Purohit A, Reed MJ, Potter BVL. J. Med. Chem. 2008;51:1295–1308. doi: 10.1021/jm701319c. [DOI] [PubMed] [Google Scholar]
- 6.Leese MP, Leblond B, Smith A, Newman SP, Di Fiore A, De Simone G, Supuran CT, Purohit A, Reed MJ, Potter BVL. J. Med. Chem. 2006;49:7683–7696. doi: 10.1021/jm060705x. [DOI] [PubMed] [Google Scholar]
- 7.Leese MP, Jourdan FL, Dohle W, Kimberley MR, Thomas MP, Bai R, Hamel E, Ferrandis E, Potter BVL. ACS Med. Chem. Lett. 2012;3:5–9. doi: 10.1021/ml200232c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leese MP, Jourdan FL, Major MR, Dohle W, Hamel E, Ferrandis E, Fiore A, Kasprzyk PG, Potter BVL. ChemMedChem. 2014;9:85–108. doi: 10.1002/cmdc.201300261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dohle W, Leese MP, Jourdan F, Major MR, Bai R, Hamel E, Ferrandis E, Kasprzyk PG, Fiore A, Newman SP, Purohit A, Potter BVL. ChemMedChem. 2014;9:350–370. doi: 10.1002/cmdc.201300412. [DOI] [PubMed] [Google Scholar]
- 10.Leese MP, Jourdan F, Kimberley MR, Cozier GE, Thiyagarajan N, Stengel C, Regis-Lydi S, Foster PA, Newman SP, Acharya KR, Ferrandis E, Purohit A, Reed MJ, Potter BVL. Chem. Commun. 2010;46:2907–2909. doi: 10.1039/c002558e. [DOI] [PubMed] [Google Scholar]
- 11.Dohle W, Jourdan F, Chapman CJ, Leese MP, Hamel E, Ferrandis E, Potter BVL. ChemMedChem. 2014 doi: 10.1002/cmdc.201402025. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okada M, Iwashita S, Koizumi N. Tetrahedron Lett. 2000;41:7047–7051. [Google Scholar]
- 13a.Ho YT, Purohit A, Vicker N, Newman SP, Robinson JJ, Leese MP, Ganeshapillai D, Woo LWL, Potter BVL, Reed MJ. Biochem. Biophys. Res. Commun. 2003;305:909–914. doi: 10.1016/s0006-291x(03)00865-9. [DOI] [PubMed] [Google Scholar]
- 13b.Newman SP, Ireson CR, Tutill HJ, Day JM, Parsons MFC, Leese MP, Potter BVL, Reed MJ, Purohit A. Cancer Res. 2006;66:324–330. doi: 10.1158/0008-5472.CAN-05-2391. [DOI] [PubMed] [Google Scholar]
- 13c.Ireson CR, Chander SK, Purohit A, Perera S, Newman SP, Parish D, Leese MP, Smith AC, Potter BVL, Reed MJ. Br. J. Cancer. 2004;90:932–937. doi: 10.1038/sj.bjc.6601591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newman SP, Leese MP, Purohit A, James DRC, Rennie CE, Potter BVL, Reed MJ. Int. J. Cancer. 2004;109:533–540. doi: 10.1002/ijc.20045. [DOI] [PubMed] [Google Scholar]
- 15. Crystal Data for 17 f: C19H20N2O8S, Mr=436.43 Da, λ =0.71073 Å, monoclinic, space group P21c, a =9.8720(3), b=12.2900(4),c =15.9220(6) Å, β =97.321(1)°, U =1916.02(11) Å3, Z =4, ρcalcd =1.513 g cm−3, μ =0.222 mm−1, F (000)=912, crystal size 0.30×0.07×0.07 mm, unique reflections=4376 [Rint =0.0994], observed I>2σI =2503, data/restraints/parameters=4376/2/283, obsd. data R1=0.0558 wR2=0.1238; all data R1=0.1226 wR2=0.1458; max peak/hole 0.381 and −0.508 e Å−3. CCDC 874341 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
- 16.Hamel E, Lin CM. Biochemistry. 1984;23:4173–4184. doi: 10.1021/bi00313a026. [DOI] [PubMed] [Google Scholar]
- 17.Hamel E. Cell Biochem. Biophys. 2003;38:1–22. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 18.Verdier-Pinard P, Lai JY, Yoo HD, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Mol. Pharmacol. 1998;53:62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 19.Lin CM, Ho HH, Pettit GR, Hamel E. Biochemistry. 1989;28:6984–6991. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- 20.Appel R, Berger G. Chem. Ber. 1958;91:1339–1341. [Google Scholar]
- 21.Woo LWL, Lightowler M, Purohit A, Reed MJ, Potter BVL. J. Steroid Biochem. Mol. Biol. 1996;57:79–88. doi: 10.1016/0960-0760(95)00244-8. [DOI] [PubMed] [Google Scholar]
- 22.Sheldrick GM. Acta Crystallogr. Sect. A. 1990;46:467–473. [Google Scholar]
- 23. G. M. Sheldrick, SHELXL-97, a computer program for crystal structure refinement, University of Göttingen (Germany), 1997.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information