

The tumor microenvironment comprises newly formed blood and lymphatic vessels which shape the influx, retention and departure of lymphocytes within the tumor mass. Thus, by influencing the intratumoral composition of lymphocytes, these vessels affect the manner in which the adaptive immune system responds to the tumor, either promoting or impairing effective antitumor immunity. In our study, we utilized a mouse model of carcinogen‐induced fibrosarcoma to examine the composition of tumor‐infiltrating lymphocytes during tumor progression. In particular, we sought to determine whether CD4+Foxp3+ regulatory T cells (Tregs) became enriched during tumor progression thereby contributing to tumor‐driven immunosuppression. This was not the case as the proportion of Tregs and effector CD4+ T cells actually declined within the tumor owing to the unexpected accumulation of naïve T cells. However, we found no evidence for antigen‐driven migration of these T cells or for their participation in an antitumor immune response. Our data support the notion that lymphocytes can enter tumors via aberrantly formed blood and lymphatic vessels. Such findings suggest that targeting both the tumor vasculature and lymphatics will alter the balance of lymphocyte subpopulations that enter the tumor mass. A consideration of this aspect of tumor immunology may be critical to the success of solid cancer immunotherapies.
Keywords: T cells, carcinogen, lymphatics
Short abstract
What's new?
It is well known that a tumors' microenvironment can impair the anti‐tumor immune response. The culprits are usually assumed to be various suppressor cells and cytokines. In this study, however, the authors found that seemingly innocuous, naïve T cells may also play a significant role—simply by accumulating and possibly out‐competing activated effector cells within the tumor. A better understanding of the signals produced by the tumor microenvironment may allow researchers to alter this T‐cell pool, thus enhancing the immune response.
A body of evidence indicates that activation of tumor‐specific T cells is an essential prerequisite for immune‐mediated tumor control (reviewed in Ref. 1). Such evidence for a role of effector T cells in cancer immunity is provided by direct studies in mice, and indirect studies of patients with cancer (reviewed in Ref. 1). However, the immunosuppressive nature of the tumor microenvironment is believed to limit the effector functions of tumor‐specific T cells (reviewed in Ref. 2), preventing them from killing tumor cells and halting tumor progression. Multiple mechanisms may contribute to this immunosuppressive environment, including notably, the suppressive effects of Foxp3+ regulatory T cells (Tregs). Indeed, an increased frequency of Tregs is often associated with tumor progression.3
The type of lymphocyte that enters and accumulates within the tumor mass will be influenced by a variety of factors. These include the type of blood and lymphatic vessels developing around and within the tumor, the chemokines and adhesion molecules expressed by these vessels and within the tumor parenchyma and also on the antigen specificity of the infiltrating T cells. As the combination of receptors for chemokines and adhesion molecules varies between different T‐cell subsets, cues enabling migration and retention of lymphocytes will favor particular T‐cell subsets (reviewed in Ref. 4) and hence dictate the composition of the tumor‐infiltrating T‐cell pool. In our study, we sought to examine the composition of the tumor‐infiltrating CD4+ T cell pool in mouse tumors developing in vivo and how this alters with tumor progression. For this purpose, we used the chemical carcinogen methylcholanthrene (MCA) to induce de novo fibrosarcomas in mice and examined the composition of the tumor‐infiltrating T‐cell pool during tumor progression. The results our study were, however, unexpected as although we observed a pronounced shift in CD4+ T‐cell subsets with tumor progression, this was not owing to an increase in conventional effector T cells or Tregs but rather owing to the accumulation of naïve T cells which eventually dominated the pool of tumor‐infiltrating CD4+ T‐lymphocytes. We then proceeded to examine the mechanisms underpinning the shift toward naïve T‐cell accumulation within tumors focussing on the role of the tumor vasculature and lymphatics.
Material and Methods
Mice
Six‐ to eight‐week‐old female C57BL/6 (Thy1.1) and Foxp3‐GFP (Thy1.2) transgenic mice were used. These mice were housed in specific pathogen‐free conditions and all experiments were conducted in compliance with UK Home Office regulations.
Tumor induction
Mice were anaesthetized and injected subcutaneously (in the hind leg) with 400 µg of 3‐MCA (Sigma Aldrich, Dorset, UK) in 100 µL of olive oil. Tumors occurred between 80 and 150 days after injection. Their development was monitored periodically and mice bearing tumors were culled before the tumors reached 1.5 cm in diameter. Mice showing discomfort or difficulty in walking were culled irrespective of the tumor size.
Lymphocyte isolation
Spleens, tumor‐draining inguinal lymph nodes and the contralateral nondraining lymph nodes were disrupted by mashing through a 40‐µm nylon cell strainer (BD Falcon, Oxford, UK) using a sterile 2‐mL syringe plunger. Tumors were excised and chopped into pieces using scalpel blades. The pieces were then mashed with a syringe plunger and the resulting cell suspension was passed through several 70‐µm nylon cell strainers (BD Falcon). The cell suspensions were centrifuged, and red blood cells were lysed using ammonium–chloride–potassium lysis buffer (Gibco, Paisley, UK).
Flow cytometry and antibodies
Mononuclear cells isolated from spleens, lymph nodes and tumors were first stained with a dead cell marker (LIVE/DEAD Fixable Aqua stain; Invitrogen, Paisley, UK) according to the manufacturer's instructions. Cells were washed and then stained for cell‐surface markers and chemokine receptors. Stained cells were washed, fixed and acquired on a flow cytometer (FACS Canto II, BD). Data analysis was performed using Summit 4.3 software. The following antibodies were used: CD4‐Pacific Blue (Biolegend, San Diego, USA), CD44‐PerCP Cy5.5 (ebioscience, San Diego U) or CD44‐FITC (BD‐Pharrmingen, San Diego, USA), CD62L‐PE‐Cy7 (eBioscience), CCR7‐APC (eBioscience), FoxP3 expression was detected by GFP fluorescence.
Histology
MCA tumors were collected from mice and fixed in neutral buffered formalin and embedded in paraffin. Sections of 5 µm in thickness, were cut and mounted on slides, dewaxed in xylene and hydrated using graded alcohols to tap water. Sections were stained for 3 min in Harris Haematoxylin solution (Thermos scientific, Waltham, USA), washed in tap water for 5 min before blueing in Scotts Tap Water for 1 min. Sections were then washed in tap water and stained in Eosin solution (Sigma‐Aldrich) for 2 min before dehydrating in an ethanol series and mounting in DPX (BDH).
Evans Blue
In all, 80 µL of Evans Blue Dye (1% w/v, Sigma‐Aldrich) was injected into the base of the leg and approximately 60 min later tumor, inguinal and popliteal lymph nodes were removed and then photographed and either prepared for immunohistochemistry or dye was extracted using formamide and measured by spectrophotometry.
Fluorescein isothiocyanate–dextran
In brief, 50–100 µL of fluorescein isothiocyanate (FITC)–dextran (5,000 kDa; 10 mg/mL; Sigma‐Aldrich) was injected into the base of the leg and approximately 60 min later tumor, inguinal and popliteal lymph nodes were removed and prepared for immunohistochemistry.
Immunohistochemistry
Frozen sections. MCA tumors and lymph nodes from mice injected with Evans Blue Dye or FITC Dextran were embedded in optimum cutting temperature (OCT) compound (RA Lamb) and snap frozen. Sections, 5 µm in thickness, were fixed for 10 min in ice‐cold acetone and left to dry at room temperature. Slides were washed in phosphate‐buffered saline (PBS) and blocked with 2.5% of normal horse serum (Vector Labs, Petersborough, UK) in PBS for 30 min. Sections were stained overnight at 4°C with rabbit polyclonal anti‐LYVE‐15 (Dr. David Jackson; Oxford University, Oxford, United Kingdom), rat anti‐CD31‐FITC (clone 390; eBioscience) and biotinylated rat anti‐mouse CD169 (clone MOMA‐1; Abcam, Cambridge, UK). Primary antibodies were detected with Alexa Fluor 568 donkey anti‐rabbit, Alexa Fluor 488 goat anti‐rat, Alexa Fluor streptavidin 555, Alexa Fluor 488 goat anti‐rabbit (Invitrogen Life Sciences). Sections were sometimes counter stained with TOTO‐3 (Invitrogen Life Sciences) to detect nuclei. Sections were mounted in Vectashield, containing 4,6‐diamidino‐2‐phenylindole (DAPI) (Vector Labs). Images were collected with a Zeiss LSM5 Pascal confocal microscope. Images were assembled in Adobe Photoshop software.
Paraffin sections. MCA tumors were collected from mice and fixed, embedded, cut and dewaxed as mentioned above. After performing antigen retrieval, by microwaving in 10 mM of Tris, 1 mM of ethylenediaminetetraacetic acid buffer, pH 9, for 8 min, sections were cooled for 30 min and then equilibrated in PBS. Nonspecific antibody binding was blocked by incubating sections with 2.5% of normal horse serum (Vector Labs) in PBS for 30 min. Sections were stained overnight at 4°C with goat polyclonal anti‐CCL21 antibody (R+D Systems, Abingdon, UK) rabbit polyclonal anti‐LYVE‐15 (Dr. David Jackson Oxford University, Oxford, United Kingdom), and rat anti‐CD31 (clone SZ31; Dianova, Hamburg, Germany). Primary antibodies were detected with Alexa Fluor 555 donkey anti‐goat, Alexa Fluor 488 donkey anti‐rabbit, Alexa Fluor 568 goat anti‐rat and Alexa Fluor 488 donkey anti‐rat (Invitrogen Life Sciences). Sections were counter stained with TOTO‐3 (Invitrogen Life Sciences) to detect nuclei. Sections were mounted in Vectashield, containing DAPI (Vector Labs). Images were collected with a Zeiss LSM5 Pascal confocal microscope. Images were assembled in Adobe Photoshop software.
T‐cell transfer
Spleen cells purified from naïve wild‐type (WT) or CCR7−/− animals were loaded with the fluorescent dye label PKH26 (Sigma‐Aldrich) or CFSE, respectively, and 3 × 107 cells were injected (100 µL) into the base of the hind leg. Single‐cell suspensions were prepared from the inguinal lymph nodes and tumors 24 hr later, stained with antibodies against CD4. Stained cells were analyzed by flow cytometry as described above.
T‐cell receptor repertoire analyses
For cell sorting, cells were stained with anti‐CD4 Pacific Blue (BD), anti‐CD25 PE (eBioscience), anti‐CD44 APC (BD) and anti‐CD62L PE‐Cy7 (eBioscience). Cell sorting was performed using a customized 20‐parameter FACSAria II flow cytometer (BD). CD4+GFP+ lymphocytes were sorted as Tregs and CD4+GFP− lymphocytes were sorted as conventional T cells (Tconv). Tconv were further separated into antigen‐experienced (CD44hiCD62Llo) and naïve T cells. Post‐sort purity was >98% in all cases. RNA was extracted from sorted cells using a magnetic column‐based RNA extraction kit (Miltenyi, Surrey, UK). cDNA was reverse transcribed from 30 to 200 ng of total RNA using Superscript III (Invitrogen) with random hexamers. For the analysis of the total T‐cell receptor (TCR) repertoire, an unbiased template‐switch anchored RT‐PCR was used as described previously.6 The PCR products were then transformed into chemically competent Escherichia coli (TOP10; Invitrogen) and grown on selective LB plates (100 µg/mL ampicillin with IPTG and X‐gal for blue/white screening). Up to 96 colonies per sort were picked and DNA sequenced (Beckman Coulter, Danvers, USA). The TRB gene usage and CDR3 amino acid composition was established using IMGT/V‐QUEST software.
RNA extraction and gene expression analysis
Tumor and LN samples were cut in 10‐µM sections from frozen OCT‐embedded tissue and RNA was extracted using TRIzol reagent (Invitrogen). Quantity and quality of RNA were determined by the Agilent 2100 bioanalyzer (Agilent Technologies, Wokingham, UK), and only high‐quality samples with an RIN (RNA intergrity number) of ≥8 were used for gene expression analysis. Gene expression profiling was carried out using MouseRef‐8v2.0 whole genome expression bead chip (Illumina) as recommended by the manufacturer. Probe intensity values were corrected by background subtraction in GenomeStudio software and subsequently log‐2 and baseline (median) transformed using Genespring software (Agilent Technologies) before the analsyis of genes.
Results
Composition of the tumor‐infiltrating CD4+ T‐cell pool
Fibrosarcomas were induced in mice approximately 80–150 days after injection of the carcinogen, MCA. The relative proportions of CD4+ Foxp3− and CD4+ Foxp3+ T cells among tumor‐infiltrating lymphocytes (TILs) were assessed and (Fig. 1a) there was a large range in Treg representation within the tumor‐infiltrating T‐cell pool (7–42%). The ratio of Foxp3− cells and Foxp3+ cells among CD4+ lymphocytes from disaggregated tumors (average, 4:1) was different from that observed in blood (average, 19:1, data not shown and Ref. 7), implying that these lymphocytes are tumor‐infiltrating cells and not simply blood‐borne contaminants. To further delineate the phenotype of these CD4+ T‐cell populations, a detailed phenotypic analysis of CD4+ TILs was performed. Distinct subpopulations of CD4+ T cells within the tumor were observed as defined by the expression of CD62L and CCR7. Although the majority of CD4+Foxp3+ T cells (Tregs) were CD62LloCCR7− and hence of an effector memory (Tem) phenotype (Figs. 1b and 1d), we found that on average less than half of the CD4+Foxp3− T cells were Tem cells (Figs. 1c and 1e). Notably, within the CD4+Foxp3− population, a significant number of cells expressed both CD62L and CCR7, markers compatible with a naïve or central memory T‐cell phenotype.8 Costaining with CD44 revealed low CD44 expression on the majority of the CD62Lhi TILs, supporting the premise that these cells are of a naïve phenotype (Fig. 1f and Supporting Information Fig. 1). This phenomenon contrasted with CD4+ T cells recovered from the lungs of influenza‐infected mice, where almost all were CD44hi, that is they have an activated phenotype. As expected, the majority of CD62LhiCD4+ T cells in the thymus have low CD44 expression indicative of a naïve phenotype (Supporting Information Fig. 1).
Figure 1.
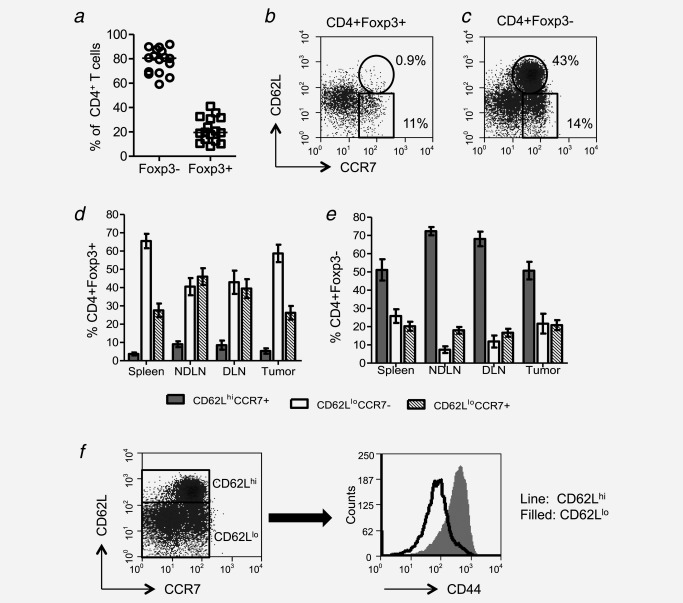
A significant proportion of tumor‐infiltrating CD4+ T Cells are CD62LhiCCR7+CD44lo. Tumor‐cell suspensions were stained with CD4‐, Foxp3‐, CD62L‐ and CCR7‐specific monoclonal antibodies (mAbs) and analyzed by flow cytometry. Proportion of CD4+ cells that are Foxp3− or Foxp3+ are shown (a). Examples of CD62L and CCR7 staining on gated CD4+Foxp3+ and CD4+Foxp3− cells (b and c). Proportions of tumor‐infitrating CD4+Foxp3+and Foxp3− cells from spleens, tumors, draining and nondraining lymph nodes, represented as CD62LhiCCR7+, CD62LloCCR7+ and CD62LloCCR7− (d and e) (n = 7). Expression of CD44 by CD62Lhi and CD62Llo cells (f).
Tumor‐infiltrating CD44loCD62LhiCCR7+ lymphocytes are bona fide naïve CD4+ T cells
As CD44loCD62LhiCCR7+ T cells represented a significant population within the tumor‐infiltrating CD4+Foxp3− pool, we sought to determine whether they were indeed naïve T cells. These cells expressed high levels of CD27 (Fig. 2a), compatible with a naïve phenotype.9 Short‐term functional assays were conducted whereby interferon (IFN)‐γ was measured after a brief period of stimulation with PMA/ionomycin. Although IFN‐γ expression was observed among the CD44hi cells, the CD44lo cells did not express IFN‐γ (Fig. 2b), confirming that these CD44lo cells are also functionally naïve. As it has been reported that exhausted T cells, which express PD‐1 and Tim‐3, can downregulate CD44, expression of these markers was evaluated.10 Many TILs did indeed express both markers but expression was confined to CD44hi cells, indicating that the CD44lo cells were not exhausted cells masquerading as naïve cells (Figs. 2c and 2d). Thus, in conclusion, these data demonstrate that a significant number of naïve T cells had indeed migrated to the tumor mass.
Figure 2.
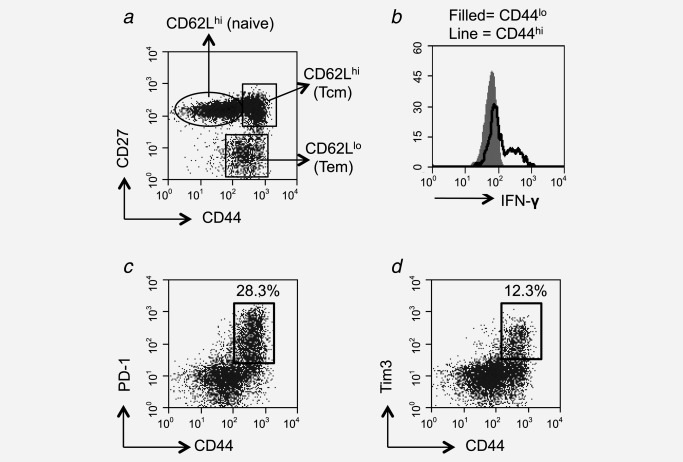
Tumor‐infiltrating CD4+CD62LhiCCR7+CD44lo T cells are bona fide naïve T cells. Expression of CD27 and CD44 by CD4+ T cells derived from the tumor. Individual populations were gated (a) and analyzed for the expression of CD62L. Relative expression levels of CD62L are indicated on the plot (a). Tumor cell suspensions were stained with CD4‐ and CD44‐specific mAbs and subsequently stimulated for 4 hr with PMA/ionomycin before intracellular staining for IFN‐γ. IFN‐γ staining was examined on lymphocytes gated as CD4+CD44lo (filled) and CD4+CD44hi (black line) (b). Tumor cell suspensions were also stained with PD‐1‐ and Tim3‐specific mAbs and representative plots are shown of either PD‐1 and CD44 or Tim3 and CD44 on CD4‐gated cells cells (c and d).
Route of entry of naïve CD4+ T cells
Under nonpathological conditions, naïve lymphocytes mainly recirculate between lymph nodes; however, there is evidence that naïve T cells do enter nonlymphoid organs as part of their normal migratory pathway (Ref. 11 and reviewed in Ref. 12). As naïve T cells are significantly enriched in the tumors described above compared to the previous reports of other tissues,11 it is likely that mechanisms other than those that allow migration of naïve T cells into nonlymphoid organs, under homeostatic conditions, are at play. It has been reported that naïve T cells can access sites of chronic inflammation and autoimmunity through neogenesis of high endothelial venules (HEVs) within ectopic lymphoid structures. HEVs are specialized blood vessels, which, under nonpathological conditions, are found only in secondary lymphoid organs where they allow naïve T‐cell entry (reviewed in Ref. 13). As we have reported previously, however, HEVs are not found in unmanipulated MCA‐induced fibrosarcomas and thus cannot account for the enrichment of naïve T cells in these tumors.14
Tumor angiogenesis is a process through which new blood vessels are formed to supply oxygen and nutrients required to sustain tumor growth. This is a characteristically chaotic process in tumors, leading to the development of irregular and often leaky blood vessels (reviewed in Ref. 15). With this fact in mind, it is possible that T cells, including naïve T cells, could enter the tumor parenchyma randomly by egress through these blood vessels. Upon examination of H&E‐stained fibrosarcoma sections, red blood cells were clearly observed outside blood vessels and moreover, areas of significant haemorrhage were readily detected within the tumor mass (Fig. 3). These data clearly indicate that aberrantly formed hyperpermeable blood vessels could serve as entry points through which cells, including naïve T cells, could enter the tumor mass. Leaky blood vessels may not completely account for the presence of naïve T cells within the tumors as the relative proportions of different T‐cell subsets in blood and tumors were, as mentioned above, distinct.
Figure 3.
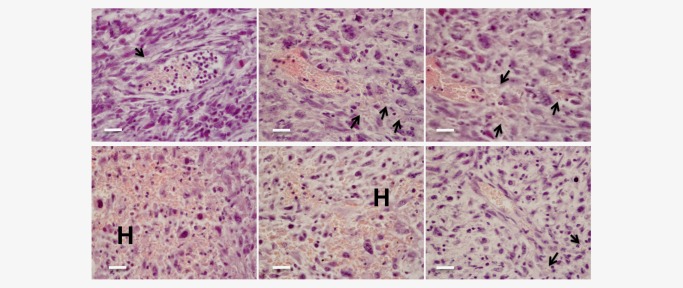
Blood vessels are leaky in MCA‐induced tumors. Sections prepared from paraffin‐embedded tumors were H&E stained and examined for red blood cells. Arrows point to red blood cells identified outside of blood vessels and H indicates areas of extensive haemorrhage. Scale bars: 100 µm.
Tumor development is also associated with lymphatic hyperplasia and the sprouting of new lymphatic vessels, processes that facilitate dissemination of cells from the primary tumor via peritumoral lymphatics, and metastasis to tumor draining lymph nodes (reviewed in Ref. 16). Here, we considered the novel possibility that the development of an expanded network of lymphatics in and around fibrosarcomas might facilitate entry of naïve T cells from neighboring tissues via afferent lymph. In support of this hypothesis, naïve T cells have been reported in afferent lymph, albeit in lower numbers than memory cells.17 Uptake of lymph into fibrosarcomas was assessed by monitoring the accumulation of Evans Blue Dye,18 after injection of the dye into the base of the leg of a tumor‐bearing mouse (Supporting Information Fig. 2). After approximately 1 hr, the primary tumor, as well as the draining ipsilateral and contralateral inguinal lymph nodes, was removed for examination (Supporting Information Fig. 2). Upon macroscopic inspection, tumors, as well as the draining lymph nodes, were visibly blue (Supporting Information Fig. 2). To confirm dye uptake by the tumor, Evans Blue Dye was extracted and measured by spectrometry (Supporting Information Fig. 2). Finally, as Evans Blue Dye is autofluorescent, sections were prepared from tumors and inguinal lymph nodes for examination by confocal microscopy. Uptake of dye in both tumors and draining lymph nodes was clearly observed (Figs. 4a and 4b). To further examine the tumors for the presence of a lymphatic network, tumors from Evans Blue‐injected mice were costained with LYVE‐1‐, CD31‐ and MOMA‐1‐specific antibodies. Colocalization of LYVE‐1 with both CD31 and MOMA‐1 was observed, indicating the development of lymphatic vessels and a subcapsular‐sinus‐like structure within the tumor. To further confirm that the lymphatic vessels can deliver lymph constituents to the tumor, we injected dextran–FITC into the base of the leg of a tumor‐bearing mouse and stained recovered tumors with LYVE‐1‐specific antibodies. As shown in Figure 4c, we observed colocalization of dextran–FITC and LYVE‐1 within both draining lymph nodes and tumors. Moreover, dextran–FITC could also be observed intratumorally, indicating that lymph constituents can enter the tumor body (Fig. 4c).
Figure 4.
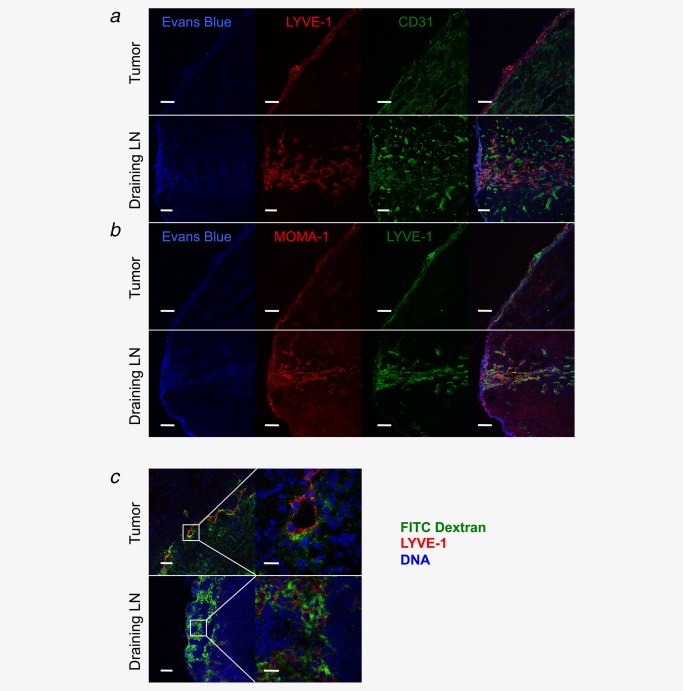
Afferent lymphatic vessel function detected in MCA‐induced tumors. Evans Blue was injected into the base of the leg and approximately 60 min later the tumor and draining lymph nodes were removed. Frozen sections were prepared and examined for Evans Blue autofluorescence and also stained for LYVE‐1 and CD31 (a) and MOMA‐1 and LYVE‐1 (b). FITC–dextran (5,000 kDa) was injected into the base of the leg and approximately 60 min later the tumor and draining lymph nodes were removed. Frozen sections were prepared and stained for LYVE‐1 and were also counter stained with TOTO‐3 to detect DNA. Low‐power images are shown on the left and high‐power images of the area in the white box are shown on the right (c). Scale bars a, b, and c left: 100 µm. Scale bars c right: 20 µm.
MCA‐induced fibrosarcomas express the lymphoid chemokine, CCL21
The majority of naïve T cells lack receptors for inflammatory chemokine‐mediated migration, the expression of which is linked to T‐cell activation. Naïve T cells express CCR7, a chemokine receptor that binds to the homeostatic lymphoid chemokines CCL19 and CCL21, which help direct and retain naïve T cells in secondary lymph nodes. CCL21 is expressed on lymphatic vessels where its synthesis is significantly increased in response to inflammation.19 It is reportedly expressed by many tumors and there is evidence that exogenous expression of CCL21 promotes accumulation of CCR7+ cells and the development of stromal zones within tumors resembling lymph node stroma.20 As conventional CD4+ T cells, but not Tregs, downregulate CCR7 upon activation, it has been proposed that CCL21 expression in tumors serves to preferentially attract Tregs thereby contributing to immunosuppression within the tumor microenvironment.20 As naïve CD4+ T cells express CCR7 and many of these as well as CCR7+ Tregs are observed in the fibrosarcomas described here (Supporting Information Fig. 3), it was clearly of relevance to determine whether these tumors express CCL21.
Gene expression analysis was performed using mRNA isolated from individual tumors. As shown in Supporting Information Figure 3, CCL21‐ser (predominantly found in lymphoid organs) and CCL21‐leu (predominantly found in lymphatics) were detected in tumors albeit at lower levels than in lymph nodes. In contrast, we observed no expression of the lymphoid chemokine, CCL19. To confirm CCL21 expression in tumors, paraffin‐embedded tumor sections were prepared and stained with CCL21‐specific antibodies recognizing both CCL21 isoforms. CCL21 was observed in the fibrosarcomas and costaining of sections with LYVE‐1‐ or CD31‐specific antibodies, indicating colocalization of CCL21 with lymphatic endothelial cells (Figs 5a–5c).
Figure 5.
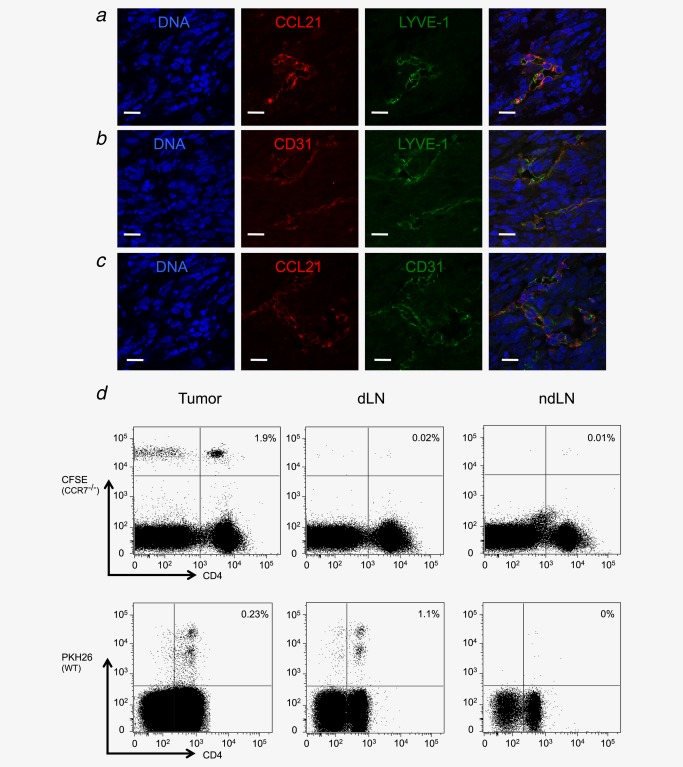
Afferent lymphatic vessels are CD31+ and contain CCL21 in MCA‐induced tumors. Frozen tumor sections were prepared and stained for LYVE‐1 and CCL21 (a), CD31 and LYVE‐1 (b) and CCL21 and CD31 (c). All sections were also counter stained with TOTO‐3 to detect DNA. PKH26‐labeled WT and CFSE‐labeled CCR7−/− spleen cells were subcutaneously injected into the base of the leg of a tumor‐bearing mouse. Around 18 hr later, the tumors, draining and nondraining lymph nodes were harvested and single‐cell suspensions were stained with CD4‐specific antibodies. Lymphocytes were gated using a live/dead marker and proportions of dye‐labeled CD4+ cells in each of the three compartments are shown (d). Scale bars a–c: 20 µm.
To determine whether the CCL21–CCR7 axis is important for mediating entry of lymphocytes via the lymphatics, we loaded donor CCR7−/− and WT spleen cells with CFSE and PKH26, respectively, prior to subcutaneous injection into recipient tumor‐bearing mice. After 18 hr, T cells were recovered from the tumor and draining (inguinal) and nondraining (contralateral inguinal) lymph nodes (Fig. 5d). At this time point, we found virtually no transferred cells in the spleens and contralateral lymph nodes of the recipient mice consistent with migration via the lymph. In accordance with the previous reports, we could recover WT but not CCR7−/− cells from the draining inguinal lymph nodes supporting the previous studies, indicating that CCR7 regulates T‐cell exit from peripheral tissues.21 To our surprise, however, both WT and CCR7−/− T cells could be recovered from tumors. These results suggest that T cells can access fibrosarcomas via lymphatics by a mechanism that does not involve the CCL21/CCR7 axis.
Proportions of naïve CD4+ T cells increase with tumor progression but fail to become activated
When we examined TIL composition in small versus large tumors, the results were striking, revealing that the proportion of CD4+CD44loCD62LhiCCR7+ T cells increases significantly as tumors progress (Fig. 6a). This was owing to an increase in the proportion of CD4+Foxp3− CD44loCD62LhiCCR7+ T cells (Figs. 6b and 6c) and not owing to an increase in CD4+Foxp3+ T cells which are likely to prevent their intratumoral activation (Fig. 6d). Furthermore, the proportion of CD4+Foxp3− TILs with an activated effector phenotype decreased with tumor progression (Fig. 6e). These data indicate that naïve conventional CD4+ T cells accumulate with tumor progression, eventually outnumbering both effector/memory T cells and Tregs in larger tumors.
Figure 6.
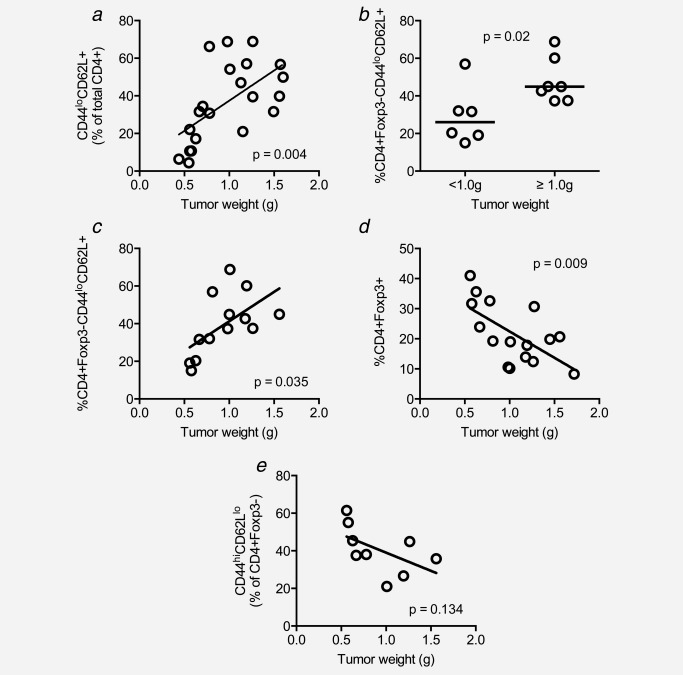
The proportion of intratumoral naïve T cells increases significantly with tumor progression. Tumor cell suspensions were stained with CD4‐, CD44‐, CCR7‐ and CD62L‐specific mAbs and analyzed by flow cytometry. Proportions of CD4+CD44loCD62L+ cells were analyzed with respect to tumor weight at the time of sample preparation (a). Frequency of naïve CD4+ TILs in small versus large tumor (b). In (c), Foxp3+ cells were excluded from the analysis to more accurately demonstrate the relationship between naïve CD4+ cells and tumor weight. The relationship between CD4+Foxp3+ cells and tumor weight (d). The relationship between activated effector CD4+Foxp3− TILs and tumor weight (e).
To determine whether the intratumoral naïve T cells become activated within the tumor, we performed TCR clonotyping where we compared the repertoires of naïve (CD44loCD62Lhi) and activated T cells (CD44hiCD62Llo) within the intratumoral and draining lymph node CD4+Foxp3− T‐cell populations.6 We found that although there was a pronounced narrowing of the TCR repertoire and expansion of certain clonotypes within the intratumoral activated T‐cell population, a similar pattern was not observed among the CD44loCD62Lhi population (Supporting Information Fig. 4). These data support the premise that tumor‐specific T cells are activated in the draining lymph node before migrating to the tumor, where they expand in an antigen‐driven fashion and exert their antitumor effects.
Discussion
Through a detailed examination of tumor‐infiltrating T cells, our study has revealed that naïve T cells can accumulate within tumors. The key questions that arise from this finding relate to the route through which T cells may access a tumor mass and the implications of this event for tumor progression. It has been reported previously that a measureable number of naïve T cells can enter nonlymphoid organs under normal homeostatic conditions (reviewed in Refs. 11). In these previous studies, where naïve T cells have been recovered from nonlymphoid organs, the numbers were reportedly low constituting but a very small proportion of T cells migrating through the tissues.11 We surmized that mechanisms allowing the influx of naïve T cells into tumors must therefore be distinct from those defining the normal migratory pathway of naïve T cells as proportions of naïve T cells, at least within MCA‐induced fibrosarcomas are very high and significantly, increase during tumor progression. This observation points to alterations in the tumor architecture that allow influx of naïve T cells. Our investigations produced clear evidence for blood vessel leakage within the fibrosarcomas, indicating that this was at least one point through which naïve T cells could enter the tumor mass. We also examined whether T‐cell ingress might occur via lymphatics. Injection of dextran–FITC at the base of the tumor‐bearing leg resulted in ingress of dye into the tumor, suggesting that lymph can indeed access the tumor. Moreover, both CCR7−/− and CCR7+/+ T cells injected into the skin adjacent to primary fibrosarcomas could be recovered from the tumor tissue within 18 hr. Although the mechanism by which this occurs remains unclear, the implication is that naïve T cells, reported to be present in the skin, can enter tumors via lymphatics. These may be co‐opted skin lymphatics that enter the tumor or newly formed lymphatics induced by the tumor. As a result of the perturbed environment created by the tumor, the nature of these lymphatic vessels may allow entry of T cells via CCR7‐independent mechanisms.
Collectively, the data described above point to two routes whereby naïve T cells might access the tumor parenchyma in an antigen nonspecific manner. The fate of these naïve T cells is, however, unclear. Other studies using largely transgenic T cells indicate that T‐cell priming can occur within a tumor mass. Through transfecting tumor cells with LIGHT, a cytokine capable of driving development of HEV in tumors, it has been shown that naïve transgenic T cells entering via these HEV can become primed within the tumor and contribute significantly to limiting tumor progression.25 Similarly, delivery of lymphotoxin‐α fused to tumor‐specific antibodies induces HEV development in B16 melanoma, resulting in infiltration and priming of effective antitumor T cells.26 In another study, adoptively transferred naïve transgenic T cells were shown to infiltrate tumors where they became activated and proliferated in response to antigens presented by cells within the tumor parenchyma.27 These reports are interesting as they demonstrate the potential for T‐cell priming in tumors and the capacity for this event to contribute to control of tumor growth. Our data, however, argue that in the case where physiological precursor frequencies of T cells capable of recognizing tumor antigens are present within the naïve T‐cell pool, and when antigen‐driven signals are not required for guiding entry of the cells into tumors, these naïve T cells probably make little, if any contribution to the antitumor immune response.
Although the T cells that enter via the routes, described above, may play little role in shaping the progression of the tumor, our study has uncovered a hitherto unreported event namely the capacity of tumor‐associated lymphatics to deliver lymph to the tumor. Normally such vessels drain interstitial fluids as well as antigens and various other soluble factors for filtration to downstream lymph nodes, in response to the raised interstitial pressure within the tumor body.28 It is possible that delivery of lymph represents an important step in promoting the acquisition of lymphoid‐like features in tumors. It is known that activated cells and not naïve T cells home to sites of inflammation, whereas effector cells tend to be excluded from lymphoid organs29 and thus acquisition of lymphoid‐like features by tumors is very likely to shape the nature of the immune infiltrate present within the tumor microenvironment. Our understanding of how such events contribute to induction of tolerance versus immunity remains, however, extremely poor. Shields et al.20 recently reported that overexpression of CCL21 in tumors can promote the development of lymphoid structures, inducing local immunological tolerance through facilitating migration and retention of Tregs. On the other hand, in a study of more than 300 breast tumors, it was reported that HEV, specialized blood vessels normally found only in secondary lymphoid organs, could be identified in tumors. In this case, the presence of CCL21 was associated with HEV and infiltration of large numbers of effector T cells, the presence of which was indicative of a good prognosis.30 The impact of CCL21 expression may not be the same in all tumors. Although Shields et al.20 reported Treg accumulation in rapidly progressing orthotopic tumors expressing CCL21, we observed an accumulation of naïve T cells in carcinogen‐induced tumors that develop in vivo over a period of months. Our experiments indicate no role, however, for CCL21 in guiding entry of T cells into tumors at least not via the lymphatics. What role might CCL21 therefore play? Previous experiments tracking the migration and accumulation of rat lymphocytes have revealed that although naïve T cell numbers increase in lymph nodes over time, their numbers in nonlymphoid organs remain constant or decrease over time (reviewed in Ref. 23). Perhaps CCL21 in the tumor serves to position and/or retain naïve T cells within the tumor mass, thereby facilitating their accumulation and allowing them to become more abundant as tumors progress. Further experiments are required to test this hypothesis.
It is noteworthy that experiments carried out previously have demonstrated that afferent lymph is required to maintain HEV in lymph nodes.18 In these experiments, shutdown of afferent lymphatics was shown to significantly reduce HEV in lymph nodes with concomitant reduction in the degree of lymphocytic infiltrate. Although no HEV could be detected in the MCA‐induced tumors described here, it has been previously shown by our group that HEV can develop in these tumors when Tregs are depleted.14 It is thus possible that delivery of lymph, which may provide a source of key factors such as chemokines and cytokines, is essential for this process.32 It is also tempting to speculate that the development of afferent‐like lymphatics may be an essential prerequisite to HEV development in tumors, thereby forming an important part of facilitating an effective antitumor immune response.
Outgrowth of tumors may, at least in part, be owing to a failure of the immune response (reviewed in Ref. 2). This failure has been attributed, among other things, to loss of immunogenicity, anergy, suppression by Tregs and myeloid‐derived suppressor cells. It is becoming increasingly clear that the tumor microenvironment, specifically blood vessels and lymphatics feeding the tumor, dictate the immunological composition of tumor‐infiltrating leukocytes. In case of some tumors, this may be through the development of a lymphoid‐like stroma that promotes accumulation of Tregs. However, in the case of the tumors described here, where CCL21 is expressed on lymphatic endothelial cells and lymph is delivered to the tumors, the result is the accumulation of antigen nonspecific naïve T cells and a complete failure to drive an antitumor immune response. It is possible that these naïve T cells impinge on the antitumor immune response by competing with effector T cells for important nutrients and resources. Understanding the signals that dictate the nature of the tumor microenvironment should enable manipulation of the composition of the tumor‐infiltrating T‐cell pool to tip the balance away from tolerance or nonresponsiveness and toward the development and maintenance of a productive response capable of sustained elimination of the tumor cells.
Supplementary Material
Additional Supporting Information may be found in the online version of this article.
Supplementary Information Figures
Acknowledgements
This study was supported by a project grant from the Medical Research Council (G0801190) and University Award (Awen Gallimore) from the Wellcome Trust (086983/Z/08/Z). The authors are very grateful to Maria Ulvmar, Igor Novitsky‐Basso and Antal Rot for the provision of CCR7−/− mice. The authors are also grateful to the staff at Cardiff University JBIOS for their continued support.
References
- 1.Vesely MD, Kershaw MH, Schreiber RD, et al Natural innate and adaptive immunity to cancer. Annu Rev Immunol 2011;29:235–71 [DOI] [PubMed] [Google Scholar]
- 2.Kerkar SP, Restifo NP. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res 2012;72:3125–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gallimore A, Godkin A. Regulatory T cells and tumour immunity—observations in mice and men. Immunology 2008;123:157–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu H, Wang A, Mauro C, et al T lymphocyte trafficking: molecules and mechanisms. Front Biosci 2013;18:422–40 [DOI] [PubMed] [Google Scholar]
- 5.Banerji S, Ni J, Wang SX, et al LYVE‐1, a new homologue of the CD44 glycoprotein, is a lymph‐specific receptor for hyaluronan. J Cell Biol 1999;144:789–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Price DA, Brenchley JM, Ruff LE, et al Avidity for antigen shapes clonal dominance in CD8+ T cell populations specific for persistent DNA viruses. J Exp Med 2005;202:1349–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Betts G, Twohig J, Van den Broek M, et al The impact of regulatory T cells on carcinogen‐induced sarcogenesis. Br J Cancer 2007;96:1849–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sallusto F, Lenig D, Forster R, et al Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999;401:708–12 [DOI] [PubMed] [Google Scholar]
- 9.Kurosawa K, Kobata T, Tachibana K, et al Differential regulation of CD27 expression on subsets of CD4 T cells. Cell Immunol 1994;158:365–75 [DOI] [PubMed] [Google Scholar]
- 10.Wherry EJ, Ha SJ, Kaech SM, et al Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007;27:670–84 [DOI] [PubMed] [Google Scholar]
- 11.Cose S, Brammer C, Khanna KM, et al Evidence that a significant number of naive T cells enter non‐lymphoid organs as part of a normal migratory pathway. Eur J Immunol 2006;36:1423–33 [DOI] [PubMed] [Google Scholar]
- 12.Lewis M, Tarlton JF, Cose S. Memory versus naive T‐cell migration. Immunol Cell Biol 2008;86:226–31 [DOI] [PubMed] [Google Scholar]
- 13.Drayton DL, Liao S, Mounzer RH, et al Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol 2006;7:344–53 [DOI] [PubMed] [Google Scholar]
- 14.Hindley JP, Jones E, Smart K, et al T‐cell trafficking facilitated by high endothelial venules is required for tumor control after regulatory T‐cell depletion. Cancer Res 2012;72:5473–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011;473:298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alitalo K. The lymphatic vasculature in disease. Nat Med 2011;17:1371–80 [DOI] [PubMed] [Google Scholar]
- 17.Braun A, Worbs T, Moschovakis GL, et al Afferent lymph‐derived T cells and DCs use different chemokine receptor CCR7‐dependent routes for entry into the lymph node and intranodal migration. Nat Immunol 2011;12:879–87 [DOI] [PubMed] [Google Scholar]
- 18.Liao S, Ruddle NH. Synchrony of high endothelial venules and lymphatic vessels revealed by immunization. J Immunol 2006;177:3369–79 [DOI] [PubMed] [Google Scholar]
- 19.Johnson LA, Jackson DG. Inflammation‐induced secretion of CCL21 in lymphatic endothelium is a key regulator of integrin‐mediated dendritic cell transmigration. Int Immunol 2010;22:839–49 [DOI] [PubMed] [Google Scholar]
- 20.Shields JD, Kourtis IC, Tomei AA, et al Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science 2010;328:749–52 [DOI] [PubMed] [Google Scholar]
- 21.Debes GF, Arnold CN, Young AJ, et al Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat Immunol 2005;6:889–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hindley JP, Ferreira C, Jones E, et al Analysis of the T‐cell receptor repertoires of tumor‐infiltrating conventional and regulatory T cells reveals no evidence for conversion in carcinogen‐induced tumors. Cancer Res 2011;71:736–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Westermann J, Ehlers EM, Exton MS, et al Migration of naive, effector and memory T cells: implications for the regulation of immune responses. Immunol Rev 2001;184:20–37 [DOI] [PubMed] [Google Scholar]
- 24.Westermann J, Pabst R. How organ‐specific is the migration of ‘naive’ and ‘memory’ T cells? Immunol Today 1996;17:278–82 [DOI] [PubMed] [Google Scholar]
- 25.Yu P, Lee Y, Liu W, et al Priming of naive T cells inside tumors leads to eradication of established tumors. Nat Immunol 2004;5:141–9 [DOI] [PubMed] [Google Scholar]
- 26.Schrama D, thor Straten P, Fischer WH, et al Targeting of lymphotoxin‐alpha to the tumor elicits an efficient immune response associated with induction of peripheral lymphoid‐like tissue. Immunity 2001;14:111–21 [DOI] [PubMed] [Google Scholar]
- 27.Thompson ED, Enriquez HL, Fu YX, et al. Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J Exp Med 2010;207:1791–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swartz MA, Lund AW. Lymphatic and interstitial flow in the tumour microenvironment: linking mechanobiology with immunity. Nat Rev Cancer 2012;12:210–9 [DOI] [PubMed] [Google Scholar]
- 29.Weninger W, Crowley MA, Manjunath N, et al Migratory properties of naive, effector, and memory CD8(+) T cells. J Exp Med 2001;194:953–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinet L, Garrido I, Filleron T, et al Human solid tumors contain high endothelial venules: association with T‐ and B‐lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res 2011;71:5678–87 [DOI] [PubMed] [Google Scholar]
- 31.Hendriks HR, Eestermans IL. Disappearance and reappearance of high endothelial venules and immigrating lymphocytes in lymph nodes deprived of afferent lymphatic vessels: a possible regulatory role of macrophages in lymphocyte migration. Eur J Immunol 1983;13:663–9 [DOI] [PubMed] [Google Scholar]
- 32.Mebius RE, Streeter PR, Breve J, et al The influence of afferent lymphatic vessel interruption on vascular addressin expression. J Cell Biol 1991;115:85–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gretz JE, Norbury CC, Anderson AO, et al Lymph‐borne chemokines and other low molecular weight molecules reach high endothelial venules via specialized conduits while a functional barrier limits access to the lymphocyte microenvironments in lymph node cortex. J Exp Med 2000;192:1425–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article.
Supplementary Information Figures