

Abstract
Specific language impairment (SLI) is a neurodevelopmental disorder that affects linguistic abilities when development is otherwise normal. We report the results of a genome-wide association study of SLI which included parent-of-origin effects and child genotype effects and used 278 families of language-impaired children. The child genotype effects analysis did not identify significant associations. We found genome-wide significant paternal parent-of-origin effects on chromosome 14q12 (P = 3.74 × 10−8) and suggestive maternal parent-of-origin effects on chromosome 5p13 (P = 1.16 × 10−7). A subsequent targeted association of six single-nucleotide-polymorphisms (SNPs) on chromosome 5 in 313 language-impaired individuals and their mothers from the ALSPAC cohort replicated the maternal effects, albeit in the opposite direction (P = 0.001); as fathers’ genotypes were not available in the ALSPAC study, the replication analysis did not include paternal parent-of-origin effects. The paternally-associated SNP on chromosome 14 yields a non-synonymous coding change within the NOP9 gene. This gene encodes an RNA-binding protein that has been reported to be significantly dysregulated in individuals with schizophrenia. The region of maternal association on chromosome 5 falls between the PTGER4 and DAB2 genes, in a region previously implicated in autism and ADHD. The top SNP in this association locus is a potential expression QTL of ARHGEF19 (also called WGEF) on chromosome 1. Members of this protein family have been implicated in intellectual disability. In summary, this study implicates parent-of-origin effects in language impairment, and adds an interesting new dimension to the emerging picture of shared genetic etiology across various neurodevelopmental disorders.
Keywords: ALSPAC, GWAS, imprinting, neurodevelopmental disorder, specific language impairment
Specific language impairment (SLI) is a complex and heterogeneous neurodevelopmental disorder diagnosed when the child has difficulties with language development despite otherwise showing normal development (Bishop 2006). Specific language impairment affects approximately 7% of preschool children (Tomblin et al. 1997). Familial aggregation and twin studies of SLI indicate a strong genetic component: across several studies, siblings of probands had a 30.3% mean rate of affectedness (higher than in the general population), and a meta-analysis of twin studies showed overall concordances of 83.6% for monozygotic twins and 50.2% for dizygotic twins (Stromswold 1998, 2001). A study of SLI twins which examined quantitative language scores found significant heritability estimates close to 1 for several language scores which account for both expressive and receptive language skills, although these effects were not present when controlling for IQ (Bishop et al. 1995). Further studies also obtained significant heritability estimates, as reviewed by Stromswold (2001). In terms of the prevalence of SLI, males are more frequently affected compared to females (Stromswold 1998), and relatives of males seem to be more frequently affected compared to relatives of females (Conti-Ramsden et al. 2007). However, a twin study that examined same-sex monozygotic twins and opposite-sex dizygotic twins concluded that there were no sex-specific genetic effects on language impairment that can explain the differences in the prevalence between males and females (Viding et al. 2004). Some studies suggest that the sex-bias simply represents a selection bias of males (Stromswold 1998).
Linkage and targeted association studies of SLI have identified several chromosomal regions and genes as candidates for involvement in susceptibility. These include: chromosome 7 (OMIM#602514) (Villanueva et al. 2011), chromosome 13 (OMIM#607134) (Bartlett et al. 2004, 2002), and chromosomes 16 (OMIM#606711) and 19 (OMIM#606712) (Falcaro et al. 2008; Monaco 2007; The SLI Consortium 2002, 2004), CNTNAP2 (OMIM#604569), CMIP (OMIM#610112) and ATP2C2 (OMIM#613082) (Newbury et al. 2009; Vernes et al. 2008)
Previous genome-wide analyses of SLI utilized various linkage methods: parametric or non-parametric, and quantitative or categorical. Certain assumptions may affect the choice of linkage method, including the definition of language impairment itself, which could lead to differences in the loci identified. Linkage may be more efficient than association when different mutations in the same gene contribute to the disorder across families, rather than a specific set of alleles. Association may be more powerful than linkage in detecting variants of small effect sizes, which all contribute to the risk, as expected in complex disorders (Burmeister et al. 2008; Risch & Merikangas 1996; Risch 2000).
Genome-wide association studies of SLI, per se, have yet to be reported but groups have investigated genome-wide effects in related traits. Meaburn et al. (2008) applied a pooled genotyping method across cases and controls with low or high reading abilities from a population cohort, but did not identify any significant associations. In their dyslexia study, Field et al. (2013) reported suggestive association with a region downstream of FGF18. Luciano et al. (2013) investigated quantitative reading and language measures across two population samples and found suggestive association with variants in ABCC13. Eicher et al. (2013) used individuals with low-language and/or reading performance and control individuals in a genome-wide association screen. They reported suggestive association between co-morbid language and reading problems and single-nucleotide-polymorphisms (SNPs) in ZNF385D and COL4A2. Their screen of individuals affected only by oral language deficits highlighted SNPs in NDST4.
When parent-of-origin effects are present, the expression of an allele depends on which parent it was inherited from. Imprinting is an epigenetic phenomenon that results in a parent-of-origin effect. Imprinted genes expressed in the brain are involved in various aspects of neurodevelopmental processes, including: neuronal differentiation, growth and gene regulation, and may even affect the behavior in animal models during their adult lives (Wilkinson et al. 2007). There is some evidence to suggest that epigenetic modifications, including imprinting, may play a role in the etiology of autism (Schanen 2006). Other neurodevelopmental disorders, such as Prader-Willi syndrome and Angelman syndrome, both of which include language deficits, involve imprinted genomic regions, as reviewed by Chamberlain and Lalande (2010). Allele-biased expression, which may result from imprinting, has been reported for genes implicated in autism and schizophrenia (Lin et al. 2012). It has recently been shown that the expression of non-imprinted genes in the mouse may still display parent-of-origin effects through interaction with imprinted loci, and that this phenomenon is a contributing factor in the genetic architecture of complex traits (Mott et al. 2014).
We applied the EMIM tool, which uses family subsets, to test for parent-of-origin effects and child genotype effects. When parent-of-origin effects are present, the EMIM methodology allows for a more accurate model and, consequently, improved detection of associations, compared to that of traditional case–control association methods (Ainsworth et al. 2011).
Materials and methods
Participants
The final (post-quality control) sample included 278 nuclear families (which included 297 affected children) from the SLI Consortium (SLIC) cohort, including samples from five centers across the UK (The Newcomen Centre at Guy’s Hospital, London (now called Evelina Children’s Hospital); the Cambridge Language and Speech Project (CLASP); the Child Life and Health Department at the University of Edinburgh; the Department of Child Health at the University of Aberdeen; and the Manchester Language Study), as described in previous SLI Consortium studies (Falcaro et al. 2008; Monaco 2007; Newbury et al. 2009; The SLI Consortium 2002, 2004). The 278 families included 49 families from the Guy’s Hospital, London cohort which had not been included in previous SLI Consortium studies. All participants had a ‘white British’ background (a principal component analysis was carried out, and samples were excluded based on divergent ancestry, see section on the genotype arrays below). The proband from each family was defined as a case. Any sibling who had an expressive language score or a receptive language score, as obtained with the revised version of the Clinical Evaluation of Language Fundamentals (CELF) (Semel et al. 1992), of 1.5 SD or more below the general population mean for their age was also defined as a case. All cases had a WISC Perceptual Organization Index (a composite score of the non-verbal subtests Picture Completion, Picture Arrangement, Block Design and Object Assembly) of >77.5 (1.5 SD below that expected for their age) and did not have a diagnosis of autism or hearing impairment. Siblings who had both expressive and receptive language scores above the mean were defined as controls. Note that controls did not contribute to the analyses per se but were used in the derivation of expected minor allele frequencies. We did not exclude children on the basis of a diagnosis of ADHD or dyslexia alone, given the high degree of co-occurrence of SLI and ADHD or dyslexia. However, for some of our SLIC samples, data were available for the presence of hyperactivity, coordination and reading problems. From this, we estimate that approximately one third of our SLIC samples showed some evidence of ADHD or developmental coordination disorder, and that approximately one half of our probands had reading problems.
Ethical agreement for the SLIC study was given by local ethics committees, and all subjects provided informed consent.
Replication cohort
The replication cohort for the maternal parent-of-origin effects analysis comprised 313 language-impaired children and their mothers from the ALSPAC cohort (Boyd et al. 2013; Fraser et al. 2013). We were looking for a replication cohort consisting of children who have been administered language tests and their parents who have additionally been genotyped; the ALSPAC cohort suited that purpose. However, fathers were not genotyped, and therefore paternal parent-of-origin effects were not investigated in this replication cohort. We aimed to select a subset of the ALSPAC cohort that best resembled the SLIC cohort. Preliminary filtering of the cohort therefore involved the exclusion of children who were not of a self-described ‘white’ ethnicity, had hearing problems, had a PIQ score of below 80, or were diagnosed with autism or pervasive developmental disorder. The ALSPAC cohort was not administered the CELF, so we instead selected cases based upon alternative language-related phenotypes: the speech and syntax subscales, from the Children’s Communication Checklist (CCC) (Bishop 1998) to represent expressive language skills, and the comprehension subtest of the Wechsler Objective Language Dimensions (WOLD) (Rust 1996) to represent receptive language skills. Cases were defined as having an expressive (on both speech and syntax subscales) or receptive language score (the WOLD comprehension subtest) at least 1.5 SD below the mean of the entire ALSPAC cohort (after the preliminary filtering). Additionally, all cases had a pragmatic composite score from the CCC of 132 or above, as this cutoff was deemed efficient in discriminating children with SLI from children with pragmatic language impairment, and, potentially, children who might have autism (Bishop & Norbury 2002; Bishop 1998). With regards to the ALSPAC cohort, please note that the study website contains details of all the data that are available through a fully searchable data dictionary (http://www.bris.ac.uk/alspac/researchers/data-access/data-dictionary).
Genotype array and quality control measures
Blood and mouth swab samples were collected and the genomic DNA was extracted by standard protocols. Genomic DNA was quantified using the Quant-iT Pico Green dsDNA Assay Kit (Invitrogen, Grand Island, NY, USA); samples with low amounts of DNA were subject to whole-genome amplification prior to genotyping. Samples were genotyped using the Illumina Human Omni-Express (v12.1) array (Illumina, San Diego, CA, USA). Samples were randomized across plates, with cases and controls being spread evenly across plates, as were different sample types and samples of different origins, while family units were retained with plates. Forty seven samples were duplicated across plates (concordance rate = 0.98968), and checks for inter-plate variances were performed. Quality control measures, as described in Anderson et al. (2010), were performed in two steps: prior to performing the association analyses, SNPs and samples were filtered based on several quality control measures: SNPs and samples with a genotype success rate below 95% and/or heterozygosity rates ±2 SD from the mean were removed, as were all SNPs with a minor allele frequency of less than 1%. Single-nucleotide-polymorphisms with a Gentrain score below 0.5 were removed (Gentrain is a clustering algorithm which produces a score based on the shapes of the genotype clusters of a given SNP and their distances from each other). Single-nucleotide-polymorphisms and samples with an error rate of 1% or higher, as estimated by bad inheritances, were removed. Inheritance data within families were used to exclude SNPs and samples with an error rate of above 1%. Control data (Hapmap release #3) were employed through a principal component analysis to exclude individuals with divergent ancestry, and samples with gross chromosome rearrangements or discordant sex information were removed. In total, 74 individuals from 40 families were removed following the principal component analysis or due to having chromosome rearrangements, extreme heterozygosity rates or discordant sex information. We used PEDSTATS (Wigginton & Abecasis 2005) to make sure no Mendelian errors were present in the final pedigree file. PLINK (Purcell et al. 2007) was used to exclude SNPs based on Hardy-Weinberg equilibrium (HWE) P-values, with a threshold of 0.001. Genotypes were compared with existing SNP genotype data for 700 individuals and 734 SNPs, with an error rate of 0.00967. The total number of SNPs used in the association analyses was 614 937. Following the association analyses, we reexamined the HWE P-values of all SNPs with an association P < 10−4 using PEDSTATS, which selects unrelated samples in a different way from that used by PLINK, resulting in a more powerful test due to increased sample sizes. We removed SNPs that had HWE P < 0.001 if they were not supported by adjacent SNPs (i.e. SNPs with very low P and HWE P-values for which adjacent SNPs did not show any association were removed), and the results were plotted. SNPs that formed association peaks, i.e. a cluster of SNPs in which one SNP has a significant P-value and adjacent SNPs on both sides have slightly higher P-values, were then checked manually for good clustering with Genome Studio.
In the replication analysis, six SNPs that showed association in the SLIC cohort were investigated in the ALSPAC cohort. The ALSPAC samples were genotyped using the Illumina human 660 W-quad array (Illumina, San Diego, CA, USA) (mothers) or the Illumina Human Hap 550-quad array (Illumina, San Diego, CA, USA) (children). ALSPAC quality control measures included the removal of SNPs with more than 5% missing genotype rate, or minor allele frequency of less than 1%. Single-nucleotide-polymorphisms with HWE P-values of less than 10−6 (mothers) or 5 × 10−7 (children) were removed. Samples with a missing genotype rate of more than 5% (mothers) or 3% (children) were removed. Other exclusion criteria included incorrect gender assignments and extreme heterozygosity rates. We found no Mendelian errors for any mother–child duo with the SNPs used in our analysis.
Statistical analyses
We used EMIM (Ainsworth et al. 2011; Howey & Cordell 2012) to perform the association analyses. EMIM estimates the effects of one or more parameters on the increase in the risk of having the disorder. In our analyses of the SLIC cohort, one parameter was estimated in each analysis. These were:
R1: the factor by which the risk is multiplied when the child has a single copy of the risk allele (Ainsworth et al. 2011). In this analysis, the increase in risk when the child has two risk alleles is assumed to be R12 (this is sometimes referred to as a child trend analysis in the EMIM documentation, and is the test which is the most similar to a case–control analysis).
Ip: the factor by which the risk is multiplied when the child receives a risk allele from the father (Ainsworth et al. 2011; Weinberg et al. 1998).
Im: the factor by which the risk is multiplied when the child receives a risk allele from the mother (Ainsworth et al. 2011; Weinberg et al. 1998).
Simulations of similar models which used the above parameters indicated moderate to high power: with 100 case-parents trios, child genotype risk factors of 2 for having one risk allele and 3 for having two risk alleles, and a risk allele frequency of 0.1–0.3, the power was ∼68%, and the type I error rate was 0.058 (Weinberg et al. 1998). Note that our child trend analysis estimated only one risk parameter. When examining parent-of-origin effects with the same sample and a parent-of-origin risk factor of 2.5, the power was higher than 92%, and type I error rate was consistent with the nominal 0.05 (Weinberg et al. 1998). The model employed in our study allowed for the use of additional types of family subsets, which would be expected to increase the power. In addition, our sample included more than 100 case-parents trios on average per SNP, and the parent-of-origin risk factors were estimated to be higher than 2.5 for our top associations.
By default, EMIM treats minor alleles as risk alleles, but this does not mean that the minor allele necessarily increases the risk, and the estimated parameters may have values below 1 if the other allele increases the risk. EMIM estimates the likelihoods of two models, one in which there is no increase in risk resulting from the parameter operating, the null model, and another in which there is an increase in risk (given the parameter being estimated), the alternative model. The baseline risk which the parameters modify is defined as the probability of disease in a child who does not carry the risk allele. To calculate P-values, one may use the value of twice the difference between the maximized log-likelihoods of the two models as a χ2 statistic, with the number of degrees of freedom being the same as the number of parameters tested. EMIM assumes that individuals with missing/unknown affection statuses are controls, which holds true only for rare disorders, and, therefore, control subsets were not used in our analyses.
In the ALSPAC replication sample, in which paternal genotypes were not available, we performed a maternal parent-of-origin analysis only.
Family subsets
Individuals were grouped into case duos and trios with their parents using PREMIM (Howey & Cordell 2012), with the −a option (which includes an estimation of the minor allele frequencies in the sample). Control subsets were not used in our analyses, but they contributed to the estimation of minor allele frequencies. The case and/or parent/s subsets are independent from each other and are constructed per SNP, i.e. if a case and their two parents were used to construct a trio, then they would not be used in any other subset for that SNP. The program built the subsets using our cohort in the following order of preference: case-parents trio, case-mother duo, case-father duo, case, parents of a case, mother of a case, and father of a case. The average numbers of these subsets per SNP in the SLIC sample were: 153 case-parents trios, 54 case-mother duos, 12 case-father duos and 18 cases. In the ALSPAC replication cohort, there were 193 case-mother duos, 75 cases, and 45 mothers of cases on average per SNP. Minor allele frequencies were estimated using all individuals present after the preliminary filtering and prior to the selection of cases.
Manhattan and QQ plots were generated in R with a script written by Stephen Turner and Daniel Capurso (https://raw.github.com/stephenturner/qqman/master/qqman.r). Plots of regions surrounding association peaks were generated with LocusZoom (Pruim et al. 2010).
Results
We found significant evidence for parent-of-origin effects in SLI. We detected significant associations on chromosome 14 with paternal parent-of-origin effects and suggestive associations on chromosome 5 with maternal parent-of-origin effects. The child trend analysis did not detect any significant or suggestive associations. The results of the child trend analysis can be found in Appendix S1 and Fig. 1.
Figure 1.

Results of the child trend association analysis. (a) Manhattan plot for the child trend association analysis. (b) QQ plot for the child trend association analysis with 95% confidence intervals.
We detected paternal parent-of-origin effects on chromosomal band 14q12 (Fig. 2a). All the SNPs which formed the peak passed all quality control measures. The most significant SNP in the peak (rs4280164) reached genome-wide significance with a P-value of 3.74 × 10−8 and deviated from the ‘expected’ line on the QQ plot (Fig. 2b). The peak spans five SNPs (3.74 × 10−8 ≤ P ≤ 1.58 × 10−6), the left-most and right-most of which are at positions 23 839 502 and 23 856 815 (∼17 kb) (hg18), respectively. Levels of linkage disequilibrium (LD) across the associated SNPs reach r2 = 0.8.
Figure 2.

Results of the paternal parent-of-origin effects association analysis. (a) Manhattan plot for the paternal parent-of-origin effects association analysis. (b) QQ plot for the paternal parent-of-origin effects association analysis with 95% confidence intervals.
We detected maternal parent-of-origin effects on chromosomal band 5p13 (Fig. 3a). The highest SNP (rs10447141) in the peak had a P-value of 1.16 × 10−7, and, again, it deviated from the ‘expected’ line on the QQ plot (Fig. 3b). The peak of association spans ∼300 kb (39 784 227–40 086 058, hg18) and encompasses 13 SNPs. The LD levels across the associated SNPs reached r2 = 0.8. Table1 includes the P-values for all SNPs in the peaks on chromosomes 5 and 14, and all associations with P ≤ 10−4 are available in Appendix S1. Figure 4 includes close-up plots of the association peaks.
Figure 3.

Results of the maternal parent-of-origin effects association analysis. (a) Manhattan plot for the maternal parent-of-origin effects association analysis. (b) QQ plot for the maternal parent-of-origin effects association analysis with 95% confidence intervals.
Table 1.
The SNPs which form the peaks on chromosomes 5 and 14
| SNP ID | Chromosome | Position (hg18) | Test | Minor allele/major allele | Increase in risk, Im/Ip relative to risk allele (SLIC) | P (SLIC) | P (ALSPAC) |
|---|---|---|---|---|---|---|---|
| rs1353835 | 5 | 39 784 227 | Maternal parent-of-origin effects | C/A | 2.14 | 2.55 × 10−5 | Not tested |
| rs1994882 | 5 | 39 841 921 | C/A | 2.441 | 1.56 × 10−6 | 0.001 | |
| rs12658486 | 5 | 39 841 974 | G/A | 2.367 | 1.43 × 10−5 | 0.002 | |
| rs980306 | 5 | 39 852 592 | A/G | 3.008 | 3.91 × 10−7 | Not tested | |
| rs10447141 | 5 | 39 852 924 | A/G | 3.08 | 1.16 × 10−7 | Not tested | |
| rs17194068 | 5 | 39 857 074 | G/A | 2.86 | 1.04 × 10−6 | 0.027 | |
| rs6895329 | 5 | 39 861 497 | G/A | 2.926 | 1.02 × 10−6 | Not tested | |
| rs1816088 | 5 | 39 897 583 | A/C | 2.486 | 4.72 × 10−5 | Not tested | |
| rs618051 | 5 | 39 902 670 | C/A | 2.429 | 4.62 × 10−5 | 0.152 | |
| rs542708 | 5 | 39 968 025 | A/G | 2.453 | 8.42 × 10−5 | Not tested | |
| rs17218399 | 5 | 40 061 719 | G/A | 2.4 | 7.54 × 10−5 | Not tested | |
| rs2939378 | 5 | 40 078 002 | G/A | 2.29 | 3.43 × 10−5 | 0.561 | |
| rs1567010 | 5 | 40 086 058 | G/A | 2.628 | 9.93 × 10−6 | 0.329 | |
| rs11158632 | 14 | 23 839 502 | Paternal parent-of-origin effects | C/A | 3.842 | 4.62 × 10−8 | Not tested |
| rs4280164 | 14 | 23 841 124 | A/G | 3.872 | 3.74 × 10−8 | Not tested | |
| rs2144494 | 14 | 23 843 226 | G/A | 3.832 | 4.94 × 10−8 | Not tested | |
| rs2281472 | 14 | 23 845 685 | G/A | 3.421 | 1.12 × 10−7 | Not tested | |
| rs3181384 | 14 | 23 856 815 | A/G | 3.016 | 1.58 × 10−6 | Not tested |
Risk allele.
Association in ALSPAC is in the opposite direction compared to SLIC.
Figure 4.
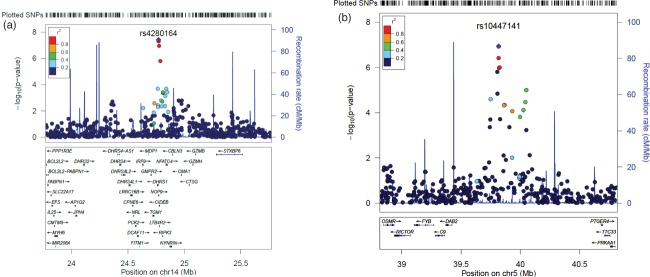
Regional association plots for top associations. (a) SNPs around the association peak on 14q12. (b) SNPs around the association peak on 5p13.
We performed a targeted follow-up analysis in ALSPAC with six SNPs across the SLIC maternal parent-of-origin effects association peak on chromosome 5. This analysis used children with low-language ability and their mothers from the ALSPAC cohort. In this cohort, we found a minimum P-value of 0.001 with SNP rs1994882, and two other SNPs had P ≤ 0.05. The P-values for the replication analyses can be found in Table1. The replicated associations in the ALSPAC cohort were in the opposite direction compared to the associations observed in the SLIC cohort.
To test whether the top association in both analyses were driven mainly by the relevant parent-of-origin effect, we adjusted the null hypothesis to assume parent-of-origin effects of the other type (i.e. when testing for paternal parent-of-origin effects, the null hypothesis will assume maternal parent-of-origin effects are present, and vice versa) at those loci. The P-values for the top SNPs were 3.87 × 10−7 and 1.29 × 10−7 in the paternal and maternal parent-of-origin analyses, respectively, suggesting that even if parental effects of the other type were present, the association is driven mainly by paternal and maternal effects, respectively.
Discussion
In this study, we investigate parent-of-origin effects and child genotype effects in SLI by means of association analyses specifically designed to be used with family data. We detected significant and suggestive associations in the paternal and maternal parent-of-origin effects analyses, respectively. The child trend analysis did not detect significant or suggestive associations. It is possible that the child effects were not strong enough to be detected in this sample, whereas the parent-of-origin effects were.
The maternally-associated peak on 5p13 does not fall within any known gene; it lies ∼863 kbp away from PTGER4 (Prostaglandin E Receptor 4), a member of the G-protein coupled receptor family, and ∼392 kbp away from DAB2 (Disabled Homolog 2, Mitogen-Responsive Phosphoprotein), which is involved in cellular trafficking. Significant downregulation of PTGER4 has been reported in brains of patients with schizophrenia (Schmitt et al. 2011). SNPs found between those two genes have also been associated with Crohn’s disease (Libioulle et al. 2007). Although intergenic, the most highly-associated SNP on 5p13 (rs10447141) is listed in the SCAN database as a potential eQTL of ARHGEF19 (Rho Guanine Nucleotide Exchange Factor 19, also called WGEF) on chromosome 1 (P = 4 × 10−5 using HapMap CEU lymphoblastoid cell line samples) (Gamazon et al. 2010). The WGEF protein belongs to a family of activators of Rho-GTPases, which are involved in a variety of cellular signaling pathways (Van Aelst & D’Souza-Schorey 1997). It has been experimentally shown that WGEF can activate Rho-GTPases and that it induces the rearrangement of cytoskeleton (Wang et al. 2004). WGEF has also been shown to be involved in convergent extension, an important step in embryonic development (Tanegashima et al. 2008). Interestingly, a gene from the same family, ARHGEF6 (Rac/Cdc42 guanine nucleotide exchange factor 6), has been implicated in intellectual disability (Kutsche et al. 2000). It is possible that the mechanism through which these genes are involved in disorders such as intellectual disability relates to their roles in dendritic development and morphology (Newey et al. 2005). Work performed on 3T3-L1 cells revealed that WGEF itself is regulated through methylation, which plays an important role in genomic imprinting (Horii et al. 2009). Interestingly, WGEF lies within the dyslexia susceptibility locus DYX8 (OMIM#608995) on chromosome 1p (Grigorenko et al. 2001, Rabin et al. 1993, Tzenova et al. 2004). This locus, however, was identified through standard linkage analyses.
In a targeted follow-up using six SNPs, we observe consistent association to chromosome 5 in an independent cohort of language-impaired children and their mothers: the most associated SNP has a P-value of 0.001. Two other SNPs also have significant p-values (P ≤ 0.05). Although the most associated SNP in our analysis was not included in the arrays used by ALSPAC, the six SNPs tested are in a region with high LD, and the most associated SNPs in the SLIC and ALSPAC analyses are in high LD with each other (Fig. 4b). The direction of association in the replication analysis was opposite to the one observed in SLIC, but this phenomenon is well-documented in replication studies and could be explained by considering the interactions between the causal variant and the observed variant across populations (Lin et al. 2007). Interestingly, the exact same effect was observed in a previous SLI association study that had used the ALSPAC cohort (Newbury et al. 2009).
The top SNP in the region of paternal association on chromosome 14 (rs4280164) corresponds to a missense variant in the NOP9 gene (also known as C14orf21), yielding an S308N substitution in the encoded protein. The minor allele in our sample, which is the minor allele in the general population as well, encodes the asparagine amino acid and has a frequency of 15.8% in the HapMap CEU population. It is predicted to be ‘possibly damaging’ by PolyPhen (Adzhubei et al. 2010); however, in our analysis it is the major allele which increases the risk when inherited from the father. This locus was not found in the Imprinted Gene and Parent-of-origin Effect Database (Morison et al. 2001). The S308N substitution is found five amino acid positions away from one of the protein’s RNA-binding pumilio domains, and the serine at that position is extremely conserved among mammalian species, with a mean PhyloP score of 1.1. The NOP9 protein has RNA-binding properties according to UniProt (The UniProt Consortium 2012), and the yeast ortholog, Nop9, has been shown to bind RNA and play a role in the nuclear maturation of the ribosomal subunits (Thomson et al. 2007). As reviewed in (Kapeli & Yeo 2012), RNA-binding proteins have been associated with several neurological disorders. This gene in particular has been found to be significantly dysregulated in schizophrenia patients and their unaffected siblings in a study of peripheral blood gene expression (Glatt et al. 2011).
Both association peaks are found in regions that have been previously implicated in various neurodevelopmental disorders. The peak on chromosomal band 14q12 falls within a region implicated in a homozygosity mapping study of intellectual disability (OMIM#611095) (Abou Jamra et al. 2011). The maternally-associated peak on 5p13 falls within a region implicated in both autism and attention deficit hyperactivity disorder (ADHD), ADHD4 (OMIM#608906). In a genome-wide linkage study of ADHD, a region of strong linkage was found on chromosomal band 5p13 (Ogdie et al. 2003), with the closest marker being D5S418 (chr5:40051524–40051931, hg18), which is ∼200 kbp proximal to our top SNP (rs10447141). A subsequent fine mapping of the region confirmed the linkage (Ogdie et al. 2004). A pooled analysis which used the cohort from the above studies and an independent cohort found that the only common risk locus was on 5p13 (Ogdie et al. 2006). A genome-wide screen for autism susceptibility loci obtained the highest linkage peak (under the broad-phenotype scheme used in the study) on 5p13 at marker D5S2494 (chr5:40253968–40254209), which is ∼400 kbp proximal to our top SNP (Liu et al. 2001). Moreover, the majority of the contribution to the IBD sharing (the linkage method used was affected-sib pair analysis) was from the maternal side, which is in line with the maternal parent-of-origin effects in our analysis. A more recent genome-wide screen of parent-of-origin effects in autism found a maternally-linked region with a peak on 5p13.1 (Fradin et al. 2010), which is the location of the top SNP in our association peak. Duplications on 5p13 have also been associated with developmental delay and intellectual disability (Yan et al. 2009), but these do not overlap with our associations. It is not clear whether imprinting plays a role in the etiology of these disorders, or whether the region on 5p13 is imprinted regardless of the disease context, which means that only the maternal allele, in this case, will have an effect. As discussed in the Introduction section, even when a locus is not imprinted, it may interact with imprinted loci and thereby show parent-of-origin effects. With regard to the studies discussed above, which used linkage methods, it should be noted that linkage and association methods test for different things; while linkage methods find regions that may harbor genes involved in a disease, association methods test for the statistical correlation between genetic variants and a disease.
The association peak on chromosomal band 14q12 reached genome-wide significance in terms of the widely-used threshold of 5 × 10−8 as proposed by Risch and Merikangas (1996). Both associations (the top SNPs on 5p13 and 14q12) are supported by association trends in adjacent SNPs. The peak on chromosomal band 5p13 did not reach the 5 × 10−8 threshold; this threshold, however, was based on an adjustment for 106 independent tests, while we had 614937 SNPs, many of which are in LD with each other. Furthermore, the 5 × 10−8 threshold may not be appropriate for some study designs (Hoggart et al. 2008). In particular, the methods we used are different from case–control association methods, to which this threshold is traditionally applied. Even then, some studies use different thresholds, e.g. 5 × 10−7 (The Wellcome Trust Case Control Consortium 2007). Given the association trends of the adjacent SNPs, the fact that the associations are more significant than expected (Fig. 3b), the good clustering for all the SNPs that form the peak and the replicated association effects, we believe that the association on chromosomal band 5p13 is not a false positive. We have performed several analyses which were all mutually exclusive in terms of the parameters that were estimated while performing them. For this reason, we present thresholds corrected for the number of SNPs, but not the number of analyses. In other words, we did not have a global null hypothesis (Krawczak 2006). We note, however, that if the P-values of our top associations were corrected for the total number of analyses in a Bonferroni manner, they would not remain genome-wide significant.
The data presented here provide compelling and significant evidence for parent-of-origin effects in SLI on chromosomes 5 and 14, which may overlap with other disorders, namely, ADHD, autism and intellectual disability. The relationship between SLI, autism and ADHD is not yet completely understood. While it is evident that language is likely to be impaired in both autism (Kjelgaard & Tager-Flusberg 2001) and ADHD (Baird et al. 2000), the exact nature of the language impairment may not be the same in all three (Bishop 2008; Bishop & Baird 2001). Nonetheless, at least some children with SLI manifest language problems which are common in autism, and vice versa (Bishop 2003). Moreover, several genetic overlaps between SLI, autism and ADHD have been observed, the most prominent of which is the implication of the CNTNAP2 gene in all three disorders (Alarcon et al. 2008; Elia et al. 2010; Vernes et al. 2008). Bishop proposed a model which can account for the genetic overlaps and the phenotypic similarities and dissimilarities between these disorders (Bishop 2010). This model incorporates gene–gene interactions and does not rely solely on additive effects. In line with this model, it is plausible that the causal variants underlying the detected associations play a role in genetic pathways that are shared between the three disorders. While the aforementioned genetic studies mention parent-of-origin effects only in the context of autism, there have been reports of parent-of-origin effects (namely, paternal over-transmission of risk alleles) in studies of ADHD candidate genes (Hawi et al. 2010, 2005). However, other studies failed to confirm overall parent-of-origin effects in ADHD (Anney et al. 2008; Kim et al. 2007). A recent GWAS of parent-of-origin effects in ADHD identified several loci that showed such effects, but none were on chromosome 5 (Wang et al. 2012).
Further support for parent-of-origin effects in SLI and other neurodevelopmental disorders will require additional replication cohorts of individuals with neurodevelopmental disorders for whom parental genetic data are also available; we replicated the association on chromosome 5, but in order to attempt to replicate the association on chromosome 14, paternal genotypes need to be available. Moreover, it should be noted that the SLIC cohort is not a large one, which affects the power of the analyses to detect true associations. This is of particular importance in the context of the child trend analysis discussed in this article.
In conclusion, this article presents novel evidence for parent-of-origin effects in SLI. Furthermore, the loci identified in this study overlapped with regions in other neurodevelopmental disorders, supporting the notion of shared and imprinted genetic pathways across several neurodevelopmental disorders.
Acknowledgments
We would like to thank all the families, professionals and individuals who participated in this research. In particular, we would like to thank Simon Fiddy for his assistance with data transformation. Dianne Newbury is an MRC Career Development Fellow and a Junior Research Fellow at St John’s College, University of Oxford. The work of the Newbury lab is funded by the Medical Research Council [G1000569/1 and MR/J003719/1]. Ron Nudel is funded by a University of Oxford Nuffield Department of Medicine Prize Studentship. The genotyping of samples was funded by the Max Planck Society. Silvia Paracchini is a Royal Society University Research Fellow. The analyses of the ALSPAC cohort were supported by a grant from the Medical Research Council [G0800523/86473]. The collection of the SLIC samples was supported by the Wellcome Trust (060774 and 076566). Patrick Bolton is supported by a National Institute of Health Research (UK) Senior Investigator award and the Biomedical Research Centre in Mental Health at the South London & Maudsley NHS Trust Hospital, London. The work of the Wellcome Trust Centre in Oxford is supported by the Wellcome Trust [090532/Z/09/Z]. The authors declare no conflicts of interest.
We are very grateful to the other members of the SLI Consortium for their contributions to this work: V. Slonims (Newcomen Centre, Evelina Children’s Hospital, London, UK), A. Clark, J. Watson (Speech and Hearing Sciences, Queen Margaret University, Edinburgh, UK), E. Simonoff, A. Pickles (King’s College London, Institute of Psychiatry); A. Everitt (University Child Health and DMDE, University of Aberdeen); J. Seckl (Molecular Medicine Centre, University of Edinburgh); H. Cowie (Department of Speech and Language Therapy, Royal Hospital for Sick Children, Edinburgh); W. Cohen (Psychological Sciences and Health, University of Strathclyde); J. Nasir (Division of Biomedical Sciences, St George’s University of London); D.V.M. Bishop (Department of Experimental Psychology, University of Oxford); Z. Simkin (School of Psychological Sciences, University of Manchester).
We are extremely grateful to all the families who took part in this study, the midwives for their help in recruiting them, and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses. The UK Medical Research Council and the Wellcome Trust (Grant ref: 092731) and the University of Bristol provide core support for ALSPAC.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher’s web-site
Annotated summary for top associations.
References
- Abou Jamra R, Wohlfart S, Zweier M, Uebe S, Priebe L, Ekici A, Giesebrecht S, Abboud A, Al Khateeb MA, Fakher M, Hamdan S, Ismael A, Muhammad S, Nothen MM, Schumacher J. Reis A. Homozygosity mapping in 64 Syrian consanguineous families with non-specific intellectual disability reveals 11 novel loci and high heterogeneity. Eur J Hum Genet. 2011;19:1161–1166. doi: 10.1038/ejhg.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS. Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainsworth HF, Unwin J, Jamison DL. Cordell HJ. Investigation of maternal effects, maternal-fetal interactions and parent-of-origin effects (imprinting), using mothers and their offspring. Genet Epidemiol. 2011;35:19–45. doi: 10.1002/gepi.20547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, Sebat J, Wigler M, Martin CL, Ledbetter DH, Nelson SF, Cantor RM. Geschwind DH. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008;82:150–159. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CA, Pettersson FH, Clarke GM, Cardon LR, Morris AP. Zondervan KT. Data quality control in genetic case–control association studies. Nat Protoc. 2010;5:1564–1573. doi: 10.1038/nprot.2010.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anney RJ, Hawi Z, Sheehan K, et al. Parent of origin effects in attention/deficit hyperactivity disorder (ADHD): analysis of data from the international multicenter ADHD genetics (IMAGE) program. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1495–1500. doi: 10.1002/ajmg.b.30659. [DOI] [PubMed] [Google Scholar]
- Baird J, Stevenson JC. Williams DC. The evolution of ADHD: a disorder of communication? Q Rev Biol. 2000;75:17–35. doi: 10.1086/393256. [DOI] [PubMed] [Google Scholar]
- Bartlett CW, Flax JF, Logue MW, Vieland VJ, Bassett AS, Tallal P. Brzustowicz LM. A major susceptibility locus for specific language impairment is located on 13q21. Am J Hum Genet. 2002;71:45–55. doi: 10.1086/341095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett CW, Flax JF, Logue MW, Smith BJ, Vieland VJ, Tallal P. Brzustowicz LM. Examination of potential overlap in autism and language loci on chromosomes 2, 7, and 13 in two independent samples ascertained for specific language impairment. Hum Hered. 2004;57:10–20. doi: 10.1159/000077385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop DVM. Development of the Children’s Communication Checklist (CCC): a method for assessing qualitative aspects of communicative impairment in children. J Child Psychol Psychiatry. 1998;39:879–891. [PubMed] [Google Scholar]
- Bishop DVM. Autism and specific language impairment: categorical distinction or continuum? Novartis Found Symp. 2003;251:213–226. discussion 226–234, 281–297. [PubMed] [Google Scholar]
- Bishop DVM. What causes specific language impairment in children? Curr Dir Psychol Sci. 2006;15:217–221. doi: 10.1111/j.1467-8721.2006.00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop DVM, Bishop DVM, Norbury CF, Tomblin JB. Specific language impairment, dyslexia, and autism: Using genetics to unravel their relationship Understanding developmental language disorders: from theory to practice. New York, NY: Psychology press; 2008. pp. 67–78. [Google Scholar]
- Bishop DVM. Overlaps between autism and language impairment: phenomimicry or shared etiology? Behav Genet. 2010;40:618–629. doi: 10.1007/s10519-010-9381-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop DVM. Baird G. Parent and teacher report of pragmatic aspects of communication: use of the children’s communication checklist in a clinical setting. Dev Med Child Neurol. 2001;43:809–818. doi: 10.1017/s0012162201001475. [DOI] [PubMed] [Google Scholar]
- Bishop DV. Norbury CF. Exploring the borderlands of autistic disorder and specific language impairment: a study using standardised diagnostic instruments. J Child Psychol Psychiatry. 2002;43:917–929. doi: 10.1111/1469-7610.00114. [DOI] [PubMed] [Google Scholar]
- Bishop DVM, North T. Donlan C. Genetic basis of specific language impairment: evidence from a twin study. Dev Med Child Neurol. 1995;37:56–71. doi: 10.1111/j.1469-8749.1995.tb11932.x. [DOI] [PubMed] [Google Scholar]
- Boyd A, Golding J, Macleod J, Lawlor DA, Fraser A, Henderson J, Molloy L, Ness A, Ring S. Davey Smith G. Cohort profile: the ‘children of the 90s’—the index offspring of the Avon Longitudinal Study of Parents and Children. Int J Epidemiol. 2013;42:111–127. doi: 10.1093/ije/dys064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister M, McInnis MG. Zollner S. Psychiatric genetics: progress amid controversy. Nat Rev Genet. 2008;9:527–540. doi: 10.1038/nrg2381. [DOI] [PubMed] [Google Scholar]
- Chamberlain SJ. Lalande M. Neurodevelopmental disorders involving genomic imprinting at human chromosome 15q11-q13. Neurobiol Dis. 2010;39:13–20. doi: 10.1016/j.nbd.2010.03.011. [DOI] [PubMed] [Google Scholar]
- Conti-Ramsden G, Falcaro M, Simkin Z. Pickles A. Familial loading in specific language impairment: patterns of differences across proband characteristics, gender and relative type. Genes Brain Behav. 2007;6:216–228. doi: 10.1111/j.1601-183X.2006.00250.x. [DOI] [PubMed] [Google Scholar]
- Eicher JD, Powers NR, Miller LL, et al. Genome-wide association study of shared components of reading disability and language impairment. Genes Brain Behav. 2013;12:792–801. doi: 10.1111/gbb.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia J, Gai X, Xie HM, et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry. 2010;15:637–646. doi: 10.1038/mp.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcaro M, Pickles A, Newbury DF, Addis L, Banfield E, Fisher SE, Monaco AP, Simkin Z. Conti-Ramsden G. Genetic and phenotypic effects of phonological short-term memory and grammatical morphology in specific language impairment. Genes Brain Behav. 2008;7:393–402. doi: 10.1111/j.1601-183X.2007.00364.x. [DOI] [PubMed] [Google Scholar]
- Field LL, Shumansky K, Ryan J, Truong D, Swiergala E. Kaplan BJ. Dense-map genome scan for dyslexia supports loci at 4q13, 16p12, 17q22; suggests novel locus at 7q36. Genes Brain Behav. 2013;12:56–69. doi: 10.1111/gbb.12003. [DOI] [PubMed] [Google Scholar]
- Fradin D, Cheslack-Postava K, Ladd-Acosta C, Newschaffer C, Chakravarti A, Arking DE, Feinberg A. Fallin MD. Parent-of-origin effects in autism identified through genome-wide linkage analysis of 16,000 SNPs. PLoS One. 2010;5:e12513. doi: 10.1371/journal.pone.0012513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser A, Macdonald-Wallis C, Tilling K, Boyd A, Golding J, Davey Smith G, Henderson J, Macleod J, Molloy L, Ness A, Ring S, Nelson SM. Lawlor DA. Cohort profile: the avon longitudinal study of parents and children: ALSPAC mothers cohort. Int J Epidemiol. 2013;42:97–110. doi: 10.1093/ije/dys066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon ER, Zhang W, Konkashbaev A, Duan S, Kistner EO, Nicolae DL, Dolan ME. Cox NJ. SCAN: SNP and copy number annotation. Bioinformatics. 2010;26:259–262. doi: 10.1093/bioinformatics/btp644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatt SJ, Stone WS, Nossova N, Liew CC, Seidman LJ. Tsuang MT. Similarities and differences in peripheral blood gene-expression signatures of individuals with schizophrenia and their first-degree biological relatives. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:869–887. doi: 10.1002/ajmg.b.31239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorenko EL, Wood FB, Meyer MS, Pauls JE, Hart LA. Pauls DL. Linkage studies suggest a possible locus for developmental dyslexia on chromosome 1p. Am J Med Genet. 2001;105:120–129. [PubMed] [Google Scholar]
- Hawi Z, Segurado R, Conroy J, Sheehan K, Lowe N, Kirley A, Shields D, Fitzgerald M, Gallagher L. Gill M. Preferential transmission of paternal alleles at risk genes in attention-deficit/hyperactivity disorder. Am J Hum Genet. 2005;77:958–965. doi: 10.1086/498174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawi Z, Kent L, Hill M, Anney RJ, Brookes KJ, Barry E, Franke B, Banaschewski T, Buitelaar J, Ebstein R, Miranda A, Oades RD, Roeyers H, Rothenberger A, Sergeant J, Sonuga-Barke E, Steinhausen HC, Faraone SV, Asherson P. Gill M. ADHD and DAT1: further evidence of paternal over-transmission of risk alleles and haplotype. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:97–102. doi: 10.1002/ajmg.b.30960. [DOI] [PubMed] [Google Scholar]
- Hoggart CJ, Clark TG, De Iorio M, Whittaker JC. Balding DJ. Genome-wide significance for dense SNP and resequencing data. Genet Epidemiol. 2008;32:179–185. doi: 10.1002/gepi.20292. [DOI] [PubMed] [Google Scholar]
- Horii T, Morita S, Kimura M. Hatada I. Epigenetic regulation of adipocyte differentiation by a Rho guanine nucleotide exchange factor, WGEF. PLoS One. 2009;4:e5809. doi: 10.1371/journal.pone.0005809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howey R. Cordell HJ. PREMIM and EMIM: tools for estimation of maternal, imprinting and interaction effects using multinomial modelling. BMC Bioinformatics. 2012;13:149. doi: 10.1186/1471-2105-13-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapeli K. Yeo GW. Genome-wide approaches to dissect the roles of RNA binding proteins in translational control: implications for neurological diseases. Front Neurosci. 2012;6:144. doi: 10.3389/fnins.2012.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Waldman ID, Faraone SV, Biederman J, Doyle AE, Purcell S, Arbeitman L, Fagerness J, Sklar P. Smoller JW. Investigation of parent-of-origin effects in ADHD candidate genes. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:776–780. doi: 10.1002/ajmg.b.30519. [DOI] [PubMed] [Google Scholar]
- Kjelgaard MM. Tager-Flusberg H. An investigation of language impairment in autism: implications for genetic subgroups. Lang Cogn Process. 2001;16:287–308. doi: 10.1080/01690960042000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczak M. Multiple Significance Testing in Genetic Epidemiology. eLS. Chichester: John Wiley & Sons, Ltd; 2006. [Google Scholar]
- Kutsche K, Yntema H, Brandt A, Jantke I, Nothwang HG, Orth U, Boavida MG, David D, Chelly J, Fryns JP, Moraine C, Ropers HH, Hamel BC, van Bokhoven H. Gal A. Mutations in ARHGEF6, encoding a guanine nucleotide exchange factor for Rho GTPases, in patients with X-linked mental retardation. Nat Genet. 2000;26:247–250. doi: 10.1038/80002. [DOI] [PubMed] [Google Scholar]
- Libioulle C, Louis E, Hansoul S, et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 2007;3:e58. doi: 10.1371/journal.pgen.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PI, Vance JM, Pericak-Vance MA. Martin ER. No gene is an island: the flip-flop phenomenon. Am J Hum Genet. 2007;80:531–538. doi: 10.1086/512133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Hrabovsky A, Pedrosa E, Wang T, Zheng D. Lachman HM. Allele-biased expression in differentiating human neurons: implications for neuropsychiatric disorders. PLoS One. 2012;7:e44017. doi: 10.1371/journal.pone.0044017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Nyholt DR, Magnussen P, Parano E, Pavone P, Geschwind D, Lord C, Iversen P, Hoh J, Ott J. Gilliam TC. A genomewide screen for autism susceptibility loci. Am J Hum Genet. 2001;69:327–340. doi: 10.1086/321980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano M, Evans DM, Hansell NK, Medland SE, Montgomery GW, Martin NG, Wright MJ. Bates TC. A genome-wide association study for reading and language abilities in two population cohorts. Genes Brain Behav. 2013;12:645–652. doi: 10.1111/gbb.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaburn EL, Harlaar N, Craig IW, Schalkwyk LC. Plomin R. Quantitative trait locus association scan of early reading disability and ability using pooled DNA and 100K SNP microarrays in a sample of 5760 children. Mol Psychiatry. 2008;13:729–740. doi: 10.1038/sj.mp.4002063. [DOI] [PubMed] [Google Scholar]
- Monaco AP. Multivariate linkage analysis of specific language impairment (SLI) Ann Hum Genet. 2007;71:660–673. doi: 10.1111/j.1469-1809.2007.00361.x. [DOI] [PubMed] [Google Scholar]
- Morison IM, Paton CJ. Cleverley SD. The imprinted gene and parent-of-origin effect database. Nucleic Acids Res. 2001;29:275–276. doi: 10.1093/nar/29.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott R, Yuan W, Kaisaki P, Gan X, Cleak J, Edwards A, Baud A. Flint J. The architecture of parent-of-origin effects in mice. Cell. 2014;156:332–342. doi: 10.1016/j.cell.2013.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbury DF, Winchester L, Addis L, et al. CMIP and ATP2C2 modulate phonological short-term memory in language impairment. Am J Hum Genet. 2009;85:264–272. doi: 10.1016/j.ajhg.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newey SE, Velamoor V, Govek EE. Van Aelst L. Rho GTPases, dendritic structure, and mental retardation. J Neurobiol. 2005;64:58–74. doi: 10.1002/neu.20153. [DOI] [PubMed] [Google Scholar]
- Ogdie MN, Macphie IL, Minassian SL, Yang M, Fisher SE, Francks C, Cantor RM, McCracken JT, McGough JJ, Nelson SF, Monaco AP. Smalley SL. A genomewide scan for attention-deficit/hyperactivity disorder in an extended sample: suggestive linkage on 17p11. Am J Hum Genet. 2003;72:1268–1279. doi: 10.1086/375139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogdie MN, Fisher SE, Yang M, Ishii J, Francks C, Loo SK, Cantor RM, McCracken JT, McGough JJ, Smalley SL. Nelson SF. Attention deficit hyperactivity disorder: fine mapping supports linkage to 5p13, 6q12, 16p13, and 17p11. Am J Hum Genet. 2004;75:661–668. doi: 10.1086/424387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogdie MN, Bakker SC, Fisher SE, Francks C, Yang MH, Cantor RM, Loo SK, van der Meulen E, Pearson P, Buitelaar J, Monaco A, Nelson SF, Sinke RJ. Smalley SL. Pooled genome-wide linkage data on 424 ADHD ASPs suggests genetic heterogeneity and a common risk locus at 5p13. Mol Psychiatry. 2006;11:5–8. doi: 10.1038/sj.mp.4001760. [DOI] [PubMed] [Google Scholar]
- Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR. Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ. Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabin M, Wen XL, Hepburn M, Lubs HA, Feldman E. Duara R. Suggestive linkage of developmental dyslexia to chromosome 1p34-p36. Lancet. 1993;342:178. doi: 10.1016/0140-6736(93)91384-x. [DOI] [PubMed] [Google Scholar]
- Risch NJ. Searching for genetic determinants in the new millennium. Nature. 2000;405:847–856. doi: 10.1038/35015718. [DOI] [PubMed] [Google Scholar]
- Risch N. Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Rust J. WOLD Wechsler Objective Language Dimensions Manual. London: The Psychological Corporation; 1996. [Google Scholar]
- Schanen NC. Epigenetics of autism spectrum disorders. Hum Mol Genet. 2006;15(Spec 2):R138–R150. doi: 10.1093/hmg/ddl213. [DOI] [PubMed] [Google Scholar]
- Schmitt A, Leonardi-Essmann F, Durrenberger PF, Parlapani E, Schneider-Axmann T, Spanagel R, Arzberger T, Kretzschmar H, Herrera-Marschitz M, Gruber O, Reynolds R, Falkai P. Gebicke-Haerter PJ. Regulation of immune-modulatory genes in left superior temporal cortex of schizophrenia patients: a genome-wide microarray study. World J Biol Psychiatry. 2011;12:201–215. doi: 10.3109/15622975.2010.530690. [DOI] [PubMed] [Google Scholar]
- Semel EM, Wiig EH. Secord W. Clinical Evaluation of Language Fundamentals – Revised. San Antonio, TX: Phychological Corporation; 1992. [Google Scholar]
- Stromswold K. Genetics of spoken language disorders. Hum Biol. 1998;70:297–324. [PubMed] [Google Scholar]
- Stromswold K. The heritability of language: a review and metaanalysis of twin, adoption, and linkage studies. Language. 2001;77:647–723. [Google Scholar]
- Tanegashima K, Zhao H. Dawid IB. WGEF activates Rho in the Wnt-PCP pathway and controls convergent extension in Xenopus gastrulation. EMBO J. 2008;27:606–617. doi: 10.1038/emboj.2008.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The SLI Consortium. A genomewide scan identifies two novel loci involved in specific language impairment. Am J Hum Genet. 2002;70:384–398. doi: 10.1086/338649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The SLI Consortium. Highly significant linkage to the SLI1 locus in an expanded sample of individuals affected by specific language impairment. Am J Hum Genet. 2004;74:1225–1238. doi: 10.1086/421529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson E, Rappsilber J. Tollervey D. Nop9 is an RNA binding protein present in pre-40S ribosomes and required for 18S rRNA synthesis in yeast. RNA. 2007;13:2165–2174. doi: 10.1261/rna.747607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomblin JB, Records NL, Buckwalter P, Zhang X, Smith E. O’Brien M. Prevalence of specific language impairment in kindergarten children. J Speech Lang Hear Res. 1997;40:1245–1260. doi: 10.1044/jslhr.4006.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzenova J, Kaplan BJ, Petryshen TL. Field LL. Confirmation of a dyslexia susceptibility locus on chromosome 1p34-p36 in a set of 100 Canadian families. Am J Med Genet B Neuropsychiatr Genet. 2004;127:117–124. doi: 10.1002/ajmg.b.20139. [DOI] [PubMed] [Google Scholar]
- Van Aelst L. D’Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- Vernes SC, Newbury DF, Abrahams BS, Winchester L, Nicod J, Groszer M, Alarcon M, Oliver PL, Davies KE, Geschwind DH, Monaco AP. Fisher SE. A functional genetic link between distinct developmental language disorders. N Engl J Med. 2008;359:2337–2345. doi: 10.1056/NEJMoa0802828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viding E, Spinath FM, Price TS, Bishop DVM, Dale PS. Plomin R. Genetic and environmental influence on language impairment in 4-year-old same-sex and opposite-sex twins. J Child Psychol Psychiatry. 2004;45:315–325. doi: 10.1111/j.1469-7610.2004.00223.x. [DOI] [PubMed] [Google Scholar]
- Villanueva P, Newbury DF, Jara L, De Barbieri Z, Mirza G, Palomino HM, Fernandez MA, Cazier JB, Monaco AP. Palomino H. Genome-wide analysis of genetic susceptibility to language impairment in an isolated Chilean population. Eur J Hum Genet. 2011;19:687–695. doi: 10.1038/ejhg.2010.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Suzuki H, Yokoo T, Tada-Iida K, Kihara R, Miura M, Watanabe K, Sone H, Shimano H, Toyoshima H. Yamada N. WGEF is a novel RhoGEF expressed in intestine, liver, heart, and kidney. Biochem Biophys Res Commun. 2004;324:1053–1058. doi: 10.1016/j.bbrc.2004.09.153. [DOI] [PubMed] [Google Scholar]
- Wang KS, Liu X, Zhang Q, Aragam N. Pan Y. Parent-of-origin effects of FAS and PDLIM1 in attention-deficit/hyperactivity disorder. J Psychiatry Neurosci. 2012;37:46–52. doi: 10.1503/jpn.100173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg CR, Wilcox AJ. Lie RT. A log-linear approach to case-parent-triad data: assessing effects of disease genes that act either directly or through maternal effects and that may be subject to parental imprinting. Am J Hum Genet. 1998;62:969–978. doi: 10.1086/301802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigginton JE. Abecasis GR. PEDSTATS: descriptive statistics, graphics and quality assessment for gene mapping data. Bioinformatics. 2005;21:3445–3447. doi: 10.1093/bioinformatics/bti529. [DOI] [PubMed] [Google Scholar]
- Wilkinson LS, Davies W. Isles AR. Genomic imprinting effects on brain development and function. Nat Rev Neurosci. 2007;8:832–843. doi: 10.1038/nrn2235. [DOI] [PubMed] [Google Scholar]
- Yan J, Zhang F, Brundage E, Scheuerle A, Lanpher B, Erickson RP, Powis Z, Robinson HB, Trapane PL, Stachiw-Hietpas D, Keppler-Noreuil KM, Lalani SR, Sahoo T, Chinault AC, Patel A, Cheung SW. Lupski JR. Genomic duplication resulting in increased copy number of genes encoding the sister chromatid cohesion complex conveys clinical consequences distinct from Cornelia de Lange. J Med Genet. 2009;46:626–634. doi: 10.1136/jmg.2008.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Annotated summary for top associations.