

The hepatitis delta virus (HDV) ribozyme is a member of the class of small, self-cleaving catalytic RNAs found in a wide range of genomes from HDV to human. Disparate pre- and post-catalysis (precursor and product) crystal structures have been determined, including a most recent trans-acting precursor resembling a previous product structure. Using fluorescence resonance energy transfer (FRET) assays together with a total of 1.8 µsec of molecular dynamics (MD) simulations, the authors show here that the available crystal structures represent intermediates on an overall rugged RNA folding free-energy landscape, where a conformational change accompanies catalysis of the HDV ribozyme.
Keywords: time-resolved FRET, steady-state FRET, small ribozyme, molecular dynamics simulation, conformational change
Abstract
The hepatitis delta virus (HDV) ribozyme is a member of the class of small, self-cleaving catalytic RNAs found in a wide range of genomes from HDV to human. Both pre- and post-catalysis (precursor and product) crystal structures of the cis-acting genomic HDV ribozyme have been determined. These structures, together with extensive solution probing, have suggested that a significant conformational change accompanies catalysis. A recent crystal structure of a trans-acting precursor, obtained at low pH and by molecular replacement from the previous product conformation, conforms to the product, raising the possibility that it represents an activated conformer past the conformational change. Here, using fluorescence resonance energy transfer (FRET), we discovered that cleavage of this ribozyme at physiological pH is accompanied by a structural lengthening in magnitude comparable to previous trans-acting HDV ribozymes. Conformational heterogeneity observed by FRET in solution appears to have been removed upon crystallization. Analysis of a total of 1.8 µsec of molecular dynamics (MD) simulations showed that the crystallographically unresolved cleavage site conformation is likely correctly modeled after the hammerhead ribozyme, but that crystal contacts and the removal of several 2′-oxygens near the scissile phosphate compromise catalytic in-line fitness. A cis-acting version of the ribozyme exhibits a more dynamic active site, while a G-1 residue upstream of the scissile phosphate favors poor fitness, allowing us to rationalize corresponding changes in catalytic activity. Based on these data, we propose that the available crystal structures of the HDV ribozyme represent intermediates on an overall rugged RNA folding free-energy landscape.
INTRODUCTION
The hepatitis delta virus (HDV) ribozyme (Fig. 1) is a small catalytic RNA originally found in the genome and antigenome of the hepatitis delta virus (Lai 1995; Been 2006; Flores et al. 2012; Taylor 2012). It has a minimal length of ∼85 nt and catalyzes the cleavage of its own phosphodiester backbone in cis, resulting in the formation of 5′-OH and 2′,3′-cyclic phosphate products. Self-cleavage of the genomic and antigenomic ribozymes, which share ∼75% sequence identity, is an essential step in the double rolling-circle replication of the HDV (Lai 1995; Been 2006). The overall fold of the HDV ribozyme consists of a double-nested pseudoknot encompassing helices P1, P1.1, P2, P3, and P4 (Fig. 1A,B; Ferre-D'Amare et al. 1998; Pereira et al. 2002; Shih and Been 2002), which leads to a tight-knit structure with a deeply electronegative active site cleft. This cleft attracts metal ions and promotes cleavage by shifting the pKa of an active site cytosine at position 75 (in the genomic ribozyme; C76 in the antigenomic form) toward neutrality (Ferre-D'Amare et al. 1998; Shih and Been 2002). Based on these structural constraints, numerous HDV-like ribozymes were recently discovered in the human and other eukaryotic genomes, suggesting that this RNA motif may have widespread functions in RNA processing and gene regulation (Salehi-Ashtiani et al. 2006; Webb et al. 2009; Webb and Luptak 2011).
FIGURE 1.
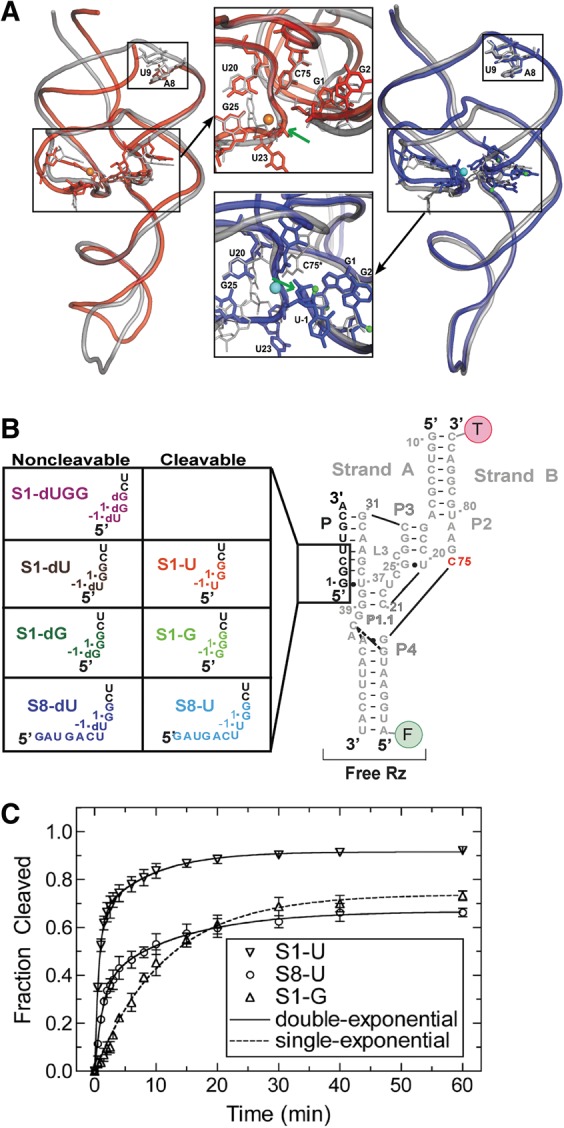
The trans-acting HDV ribozyme studied here. (A) Superpositions of trans-acting (red) (Chen et al. 2010) and cis-acting (blue) (Ke et al. 2004) precursor crystal structures on the cis-acting product structure (silver) (Ferre-D'Amare et al. 1998). Active site magnesium ions are shown in orange and cyan for the trans- and cis-acting ribozymes, respectively. The smaller black boxes on both overlays highlight the region where there is a break in the RNA backbone for the trans-acting ribozyme. The green arrows indicate the cleavage site in both insets. The U-1 nucleotide and scissile phosphate were not resolved in the trans-acting precursor structure. (B) The eight three-stranded precursor and product ribozymes used in our solution experiments. (C) Cleavage assay time courses for S1-U, S8-U, and S1-G. Cleavage assays were conducted at 25°C in 40 mM Tris-HCl, pH 7.5, 25 mM DTT, and 11 mM MgCl2. Data for S1-G were fit with a single-exponential increase function, while the data for S1-U and S8-U were best fit with a double-exponential increase function (raising the R2 as a measure of fit quality of 0.94 for single-exponentials to 0.999 for double-exponentials). Standard deviations from at least three independent assays are shown as error bars.
In addition to numerous studies of self-cleavage by the cis-acting genomic and antigenomic HDV ribozymes (Been and Wickham 1997; Perrotta et al. 1999a,b, 2006; Wadkins et al. 1999; Nakano et al. 2000, 2003; Shih and Been 2002; Harris et al. 2004; Been 2006; Perrotta and Been 2006; Chadalavada et al. 2007; Gong et al. 2007, 2009; Sefcikova et al. 2007a,b; Cerrone-Szakal et al. 2008; Gong et al. 2008; Chen et al. 2009), significant work has also been performed to investigate trans-acting forms of this ribozyme for enzymology and therapeutic purposes (Been 1994; Luptak et al. 2001; Harris et al. 2002; Pereira et al. 2002; Jeong et al. 2003; Tinsley et al. 2003, 2004; Das and Piccirilli 2005; Tinsley and Walter 2007; Walter and Perumal 2009). Generally, trans-acting HDV ribozymes exhibit a 1–2 order-of-magnitude slower catalytic rate constant compared to their cis-acting counterparts, likely due to a less tightly knit structure (Tinsley and Walter 2007). Both cis- and trans-acting ribozymes have been shown to exhibit in solution end-to-end lengthening (defined as between the topmost residues of P2 and the bottommost residues of P4, where our FRET pair is attached as denoted in Fig. 1B) when the catalytic precursor cleaves to form the product, with the cis-acting versions exhibiting more modest changes than the ∼15 Å observed for some trans-acting ribozymes (Pereira et al. 2002; Tanaka et al. 2002; Jeong et al. 2003; Harris et al. 2004).
Since 1998, a number of crystal structures of the cis-acting genomic ribozyme have been solved for both precursor and product forms (Ferre-D'Amare et al. 1998; Ferre-D'Amare and Doudna 2000; Ke et al. 2004; Chen et al. 2010). The emergent crystal structure of the inactivated cis-acting C75U mutant precursor at up to 2.2 Å-resolution (Ke et al. 2004) was probed by independent molecular dynamics (MD) studies in which residue 75 was changed back to C (Krasovska et al. 2005). In combination, these studies led to the discovery of a structural change upon release of the 5′ product and catalytic metal ion, as C75 moves deeper into the active site cleft to form hydrogen bonds with C22's nonbridging oxygen and the 5′ OH leaving group of G1, while the adjacent G25 rotates in conformation from anti to syn (Ke et al. 2004; Krasovska et al. 2005). These changes result in a collapse of the P1 and P3 helices toward the active site and a more compact catalytic core and concomitant overall P2–P4 axial lengthening of the product (Fig. 1A); Ke et al. 2004). These observations are consistent with the results from solution probing using terbium(III) (Pereira et al. 2002; Jeong et al. 2003; Harris et al. 2004), which has been shown to be an effective agent for trapping and accurately probing RNA secondary and tertiary structure (Walter et al. 2000; Todd and Walter 2013).
As is typical, the cis-acting precursor was modified to prevent self-cleavage during crystallization in these studies, which was accomplished by either a C75U mutation, crystallization in the absence of Mg2+, or incorporation of a single 2′-deoxy modification at the cleavage site, although the latter structure was low in resolution (Ke et al. 2004). In 2010, a different team solved a low-pH, 1.9 Å-resolution crystal structure of a trans-acting precursor HDV ribozyme in the presence of Mg2+, inactivated with three 2′-deoxy modifications at the −1, 1, and 2 positions around the cleavage site (Fig. 1A, bottom inset; Chen et al. 2010). It was solved by molecular replacement using the cis-acting product structure and features both active-site and global conformations similar to those of the product (Fig. 1A; Ferre-D'Amare et al. 1998; Chen et al. 2010). For example, G25 is in the syn conformation, and the relative positions of the P1 and P3 helices are more similar to those in the cis-acting product than the precursor ribozyme (Fig. 1A). Such close resemblance to the product suggests that this model may capture an activated conformer past the conformational change needed to activate all previous ribozyme versions. This hypothesis would predict a fast cleavage rate constant; however, the newly crystallized HDV ribozyme cleaves similarly slowly to other trans-acting versions (Chen et al. 2010). Additionally, the substrate strand was sufficiently flexible (and/or nicked) so that no electron density was observed for the scissile phosphate and upstream U-1 nucleotide, further increasing resemblance with the cleavage product. The authors modeled these moieties using the cleavage site of the hammerhead ribozyme (Chen et al. 2010). Consequently, questions remain concerning the relationship of the various proposed and experimentally observed conformations to each other and to the functional activity of the HDV ribozyme.
To probe the functional dynamics of the newly crystallized trans-acting HDV ribozyme in solution, here we have employed a combination of experimental and computational approaches. Fluorescence resonance energy transfer (FRET) gel shift assays revealed a heterogeneous population of molecules in solution at physiological pH, suggesting that the crystal structure represents a conformation purified by crystallization. Steady-state and time-resolved FRET measurements consistently detected an ∼8-Å lengthening of the end-to-end distance along the P2–P4 axis accompanying cleavage in solution, an elongation consistent with the range previously found for trans- and cis-acting HDV ribozymes (Pereira et al. 2002; Tanaka et al. 2002; Jeong et al. 2003; Harris et al. 2004). We also performed a total of 1.8 µsec of MD simulations that showed that the hammerhead ribozyme model of the substrate conformation in the active site is compatible with a favorable catalytic in-line fitness (a measure of the poise to undergo catalytic transesterification), as long as C75 is protonated, consistent with previous results (Veeraraghavan et al. 2011). In contrast, intermolecular crystal contacts and 2′-deoxyribose modifications found in the crystal structure near the active site result in unfavorable in-line fitness, offering an explanation for the lack of experimental electron density in this region. A cis-acting version of the crystallized ribozyme inter-converts more frequently between unfavorable and favorable fitness, consistent with a lowered kinetic barrier toward cleavage. Finally, a U-1G mutation results in long-lived states of unfavorable fitness, consistent with its low cleavage activity and the conservation of U-1 in clinical isolates of the virus. Taken together, our data are consistent with a parsimonious model wherein the existing precursor crystal structures, determined for distinct constructs at varying pH, represent alternate RNA conformations on a rugged folding free-energy landscape.
RESULTS
The trans-acting HDV ribozyme preferentially cleaves a short U-1 substrate with biphasic kinetics
The crystallized trans-acting HDV ribozyme (Chen et al. 2010) consists of two strands: the substrate and ribozyme segments. To incorporate site-specific fluorophore labels for solution probing by FRET, we instead synthesized three RNA strands by opening the nonconserved capping loop on helix P4 and labeling the 5′ and 3′-ends of the downstream segment (Strand B) with fluorescein and tetramethylrhodamine, respectively (Fig. 1B), following an established strategy (Pereira et al. 2002). We first tested the catalytic activity of the resulting two-strand ribozyme using radioactive single-turnover cleavage assays at 25°C on three cleavable trans-substrates (Fig. 1B): (1) the short, U-1-containing substrate with the sequence used in the crystal structure (Chen et al. 2010), termed here S1-U; (2) the U-1G mutant substrate S1-G, to check whether this clinically rare HDV ribozyme mutation is as catalytically deleterious as previously observed (Sefcikova et al. 2007b); and (3) the longer, 8-nt substrate S8-U with U-1 and a 5′ sequence selected for its predicted low tendency to form alternate secondary structures. For S1-U and S8-U, the time traces of fraction cleaved were best fit with double-exponential functions (Fig. 1C). The observed biphasic rate constants kobs1 and kobs2 for S1-U were 1.5 min−1 (amplitude: 0.64) and 0.12 min−1 (0.28), respectively. For S8-U, kobs1 and kobs2 were approximately twofold slower, with 0.77 min−1 (0.36) and 0.075 min−1 (0.30), respectively, consistent with a potential of the ribozyme to form competing, weak, alternative secondary structures as predicted by the program mfold (Zuker 2003). A fivefold increase in ribozyme concentration resulted in a modest twofold increase in the kobs values for both S1-U and S8-U, suggesting that the reported values approach the inherent cleavage rate constant (Supplemental Fig. S1). In the case of S1-G, cleavage time traces were best fit with a single-exponential function, yielding a kobs of 0.087 min−1 (Fig. 1C). A fivefold increase in ribozyme concentration did not further increase this low value (Supplemental Fig. S1). The ∼17-fold lower activity of the S1-G compared to the S1-U substrate is consistent with previous observations that G-1 is much disfavored relative to U-1 (Sefcikova et al. 2007b), rationalizing the evolutionary conservation of U-1 in clinical isolates of the virus (Wadkins and Been 2002). The observation of double-exponential cleavage time courses and final fractions cleaved of less than unity for the S1-U and S8-U substrates under single-turnover conditions suggests kinetic heterogeneity of the ribozyme population. Conversely, the fact that the S1-G substrate is cleaved in a single-exponential time course does not rule out such heterogeneity, especially in light of the significantly lower-than-unity cleavage extent (Fig. 1C).
For the crystallized parent ribozyme with S1-U substrate (Chen et al. 2010), a single kobs of ∼6.5 min−1 was reported at 37°C, in 25 mM Tris-HCl, pH 7.0, 10 mM MgCl2 (Chen et al. 2009), slightly higher than our kobs1. We, therefore, asked what impact the higher temperature and slightly different annealing protocol have by repeating our assay under these conditions. We again observed double-exponential cleavage, yielding a kobs1 of 2.4 min−1 (0.63) and a kobs2 of 0.99 min−1 (0.30) (Supplemental Fig. S1). The only small (2.7-fold) difference between our kobs1 at 37°C and the previously reported kobs suggests that our fluorophore-labeled variant is a relevant model system for the crystallized HDV ribozyme (Chen et al. 2010).
The crystallized precursor ribozyme exhibits heterogeneous, catalytically active populations
Fluorescence resonance energy transfer is a powerful tool to determine global conformations of RNA in solution (Walter and Burke 2000; Walter 2001; Walter et al. 2001b). To ask whether the observed heterogeneous cleavage kinetics of the crystallized trans-acting HDV ribozyme (Chen et al. 2010) are reflected in folding heterogeneities, we used electrophoretic mobility shift assays (EMSAs). These assays are sensitive to the distinct charges and hydrodynamic radii of RNA conformers and can be monitored by FRET, as previously described (Pereira et al. 2002; Jeong et al. 2003). In addition to our three cleavable substrates S1-U, S1-G, and S8-U, which are expected to largely convert to product upon ribozyme cleavage during either sample preparation or gel electrophoresis, as well as the product strand P for reference, we generated four noncleavable substrate versions that are expected to remain in the precursor state (Fig. 1B): one version of each substrate with a single 2′-deoxy modification at the cleavage site (i.e., modifying the U-1 or G-1 nucleotide), termed S1-dU, S1-dG, and S8-dU, and one substrate identical to the crystallized one with three 2′-deoxy modifications at the −1, 1, and 2 positions, encompassing the cleavage site (S1-dUGG). (Jeong et al. 2003; Sefcikova et al. 2007b). We then formed substrate-free ribozyme (Strands A and B annealed, or “Free Rz”) (Fig. 1B) and added each one of the cleavable or noncleavable substrates, or the product strand, to compare all complexes with Strand B alone on an EMSA gel (Fig. 2A).
FIGURE 2.
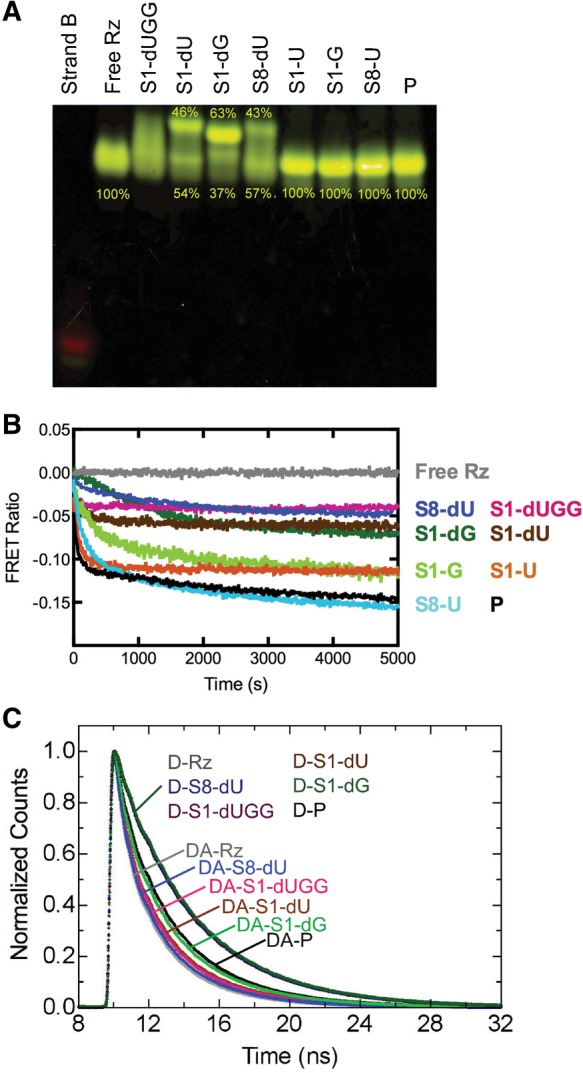
FRET probing of our eight trans-acting precursor and product ribozymes. (A) Fluorescent EMSA of the individual complexes as indicated. Where appropriate, the relative fractions in the main bands of a lane are given. Strand B and free ribozyme (Rz) lanes are shown for reference. (B) Normalized ssFRET time courses upon substrate or product addition, as indicated. A decrease in FRET ratio indicates a lengthening of the fluorophore distance along the longitudinal axis of the ribozyme. Each trace is an average of at least three trials. (C) Representative fluorescein fluorescence decays for free ribozyme (Rz), ribozyme bound to noncleavable substrates, and ribozyme bound to product (P), as indicated. Traces for ribozymes labeled with donor-only (D) and with donor plus acceptor (DA) are plotted. Each trace is one of three replicates but composed of an average of five reads.
The cleavable substrate complexes all comigrate with the single band of the product form of the ribozyme, indicating that they cleave essentially to completeness before or during electrophoresis (Fig. 2A). This product band is only slightly slower migrating than the free ribozyme, suggesting that the addition of the product strand does not significantly increase the hydrodynamic radius of the free ribozyme. In contrast, each of the noncleavable substrates shows more than one distinct band, where one band migrates similarly to the product complex and another is shifted up significantly, with the relative contribution of each band indicated in Figure 2A. The only exception is the crystallized S1-dUGG substrate, whose precursor ribozyme complex runs as a smear spanning the two bands observed for the other noncleavable substrate complexes (Fig. 2A). These observations suggest that each precursor ribozyme complex forms more than one conformation. These conformations either rapidly (compared to the electrophoretic migration time) interconvert or happen to migrate in a continuum of conformations of only slightly different hydrodynamic radius, leading to smearing. Such conformational heterogeneity is consistent with the catalytic heterogeneity observed in our cleavage assays.
The S1-dUGG bound ribozyme appears to exist in solution as a particularly broad set of conformations (Fig. 2A), suggesting that crystallization leads to the relative enrichment (purification) of just one of these conformers (Chen et al. 2010). This observation, together with the fact that the bottom band in all precursor complexes almost comigrates with the product complex, may provide an explanation for why the recent precursor crystal structure resembles the previous product crystal structures more than their precursor counterparts: A more product-like conformation was purified during crystal growth. Consistent with this hypothesis and the notion that the more product-like conformation is catalytically more activated, the noncleavable S1-dG substrate leads to a smaller fractional population of the product-like band (Fig. 2A), as well as to lower catalytic activity (Fig. 1C).
All trans-acting ribozymes undergo a global conformational change accompanying cleavage
To corroborate our EMSA results, we employed a previously developed steady-state FRET (ssFRET) assay (Pereira et al. 2002; Jeong et al. 2003), in which we first form the substrate-free ribozyme, then add a cleavable or noncleavable substrate or the product strand while monitoring in real-time the evolution of the relative FRET ratio. This ratio serves as a measure of changes in distance between the two fluorophores that mark the ends of helixes P4 and P2 (Fig. 1B), thus allowing us to quantify the time dependence of global conformational change. As observed for a majority of similar cases before (Jeong et al. 2003), binding of noncleavable substrate to the free HDV ribozyme leads to a temporal decrease in FRET ratio, reflecting the substrate binding kinetics (Fig. 2B). When adding the product strand P instead, the decrease is more pronounced, suggesting that the P4–P2 end-to-end distance monitored by the FRET pair becomes longer in the product than in any of the precursor structures. The kinetics of the FRET ratio decrease in all cases but the S1-G ribozyme are characterized by a double-exponential decay, providing additional evidence for heterogeneous folding (Fig. 2B). Supporting the notion that formation of product leads to further lengthening of the P4–P2 axis, the combination of binding and subsequent (partial) cleavage upon addition of the cleavable substrates S1-U, S-1G, and S8-U leads to FRET ratio decreases close to that associated with binding of product strand P (Fig. 2B). The fact that, for the S-1U and S-1G substrates, the overall decrease falls between those of binding the corresponding noncleavable strands S1-dU/S1-dG and product strand P is further consistent with the observed incomplete conversion of these substrates into product (Fig. 1C). Taken together, these observations strongly support the result from our EMSAs that the precursor and product forms of the most recently crystallized trans-acting ribozyme version display distinct global conformations. Thus, a global conformational change appears to accompany cleavage, consistent with previous findings on other trans-acting HDV ribozymes (Harris et al. 2002; Pereira et al. 2002; Jeong et al. 2003; Tinsley et al. 2004).
To quantify this conformational change and the resulting P4–P2 end-to-end lengthening, we performed time-resolved FRET (trFRET) measurements as described (Walter et al. 1999, 2001a; Pereira et al. 2002; Jeong et al. 2003; Rueda et al. 2003) on ribozyme free of substrate as well as bound to each one of our noncleavable substrates or the product strand. As expected, the fluorescein-only singly labeled ribozyme complexes display nearly identical fluorescence decay curves, whereas all fluorescein-tetramethylrhodamine doubly labeled ribozymes exhibit distinct, faster decays due to varying FRET efficiencies (Fig. 2C). Analysis of these decay curves yielded the single Gaussian inter-fluorophore distance distributions summarized in Table 1. The free ribozyme exhibits the most compact structure, with a mean inter-fluorophore distance of ∼51 Å. The precursor ribozymes adopt distances of between 53 Å (S8-dU) and 59 Å (S1-dUGG) (Table 1), depending on the noncleavable substrate. The product conformation, with a mean fluorescein-tetramethylrhodamine distance of ∼67 Å, is indeed extended, by ∼8 Å in the P4–P2 end-to-end distance, compared to the crystallized S1-dUGG complex (Table 1). Such an extension is consistent with our ssFRET results as well as previous studies of other trans-acting HDV ribozymes that showed extensions of between 2 and 15 Å upon conversion of the precursor to the product (Pereira et al. 2002; Jeong et al. 2003). Evidence of this extension also underscores the results from our EMSAs, which characterize the product as the fastest-migrating species (Fig. 2A), presumably because their lengthened and thus more oblong conformation is conducive to higher gel mobility.
TABLE 1.
Distance distribution parameters obtained from time-resolved FRET data
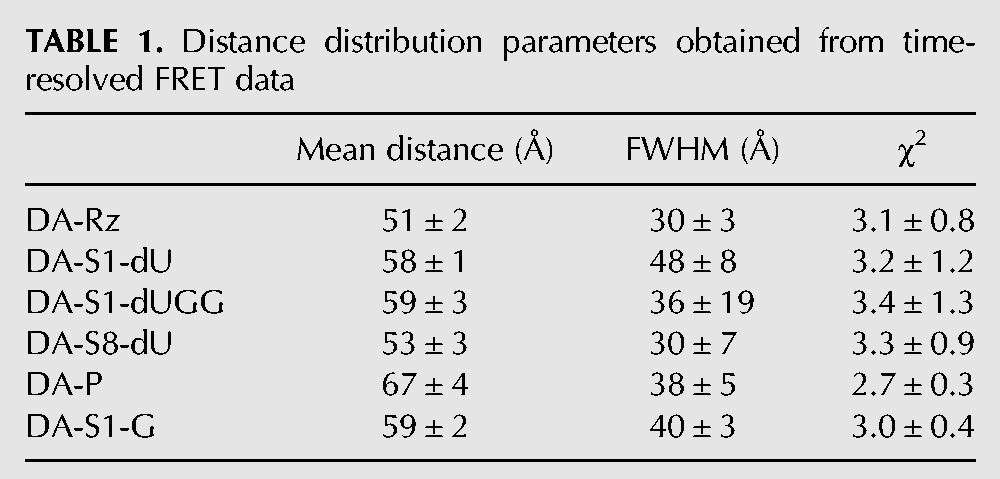
Taken together, our EMSA, ssFRET, and trFRET data consistently show that the newly crystallized trans-acting HDV ribozyme (Chen et al. 2010) in solution undergoes a significant conformational change accompanying cleavage. The ribozyme also displays folding heterogeneity in the precursor form that is removed upon cleavage. The finding that, in our EMSAs, all of our substrates cleave to completion (Fig. 2A) further indicates that all conformational isomers are catalytically active, although to varying degrees as evident from the biphasic kinetics of our cleavage assays (Fig. 1C). That the crystal structure shows a relatively well-defined single global conformation further suggests that crystallization may have purified a more product-like conformer. Consistent with this notion, the P4–P2 end-to-end distance in the crystal is ∼70 Å (Chen et al. 2010), close to that of the product conformer in solution as observed here (∼67 Å).
In MD simulations, the modeled hammerhead ribozyme cleavage site results in favorable in-line fitness, as long as C75 is protonated
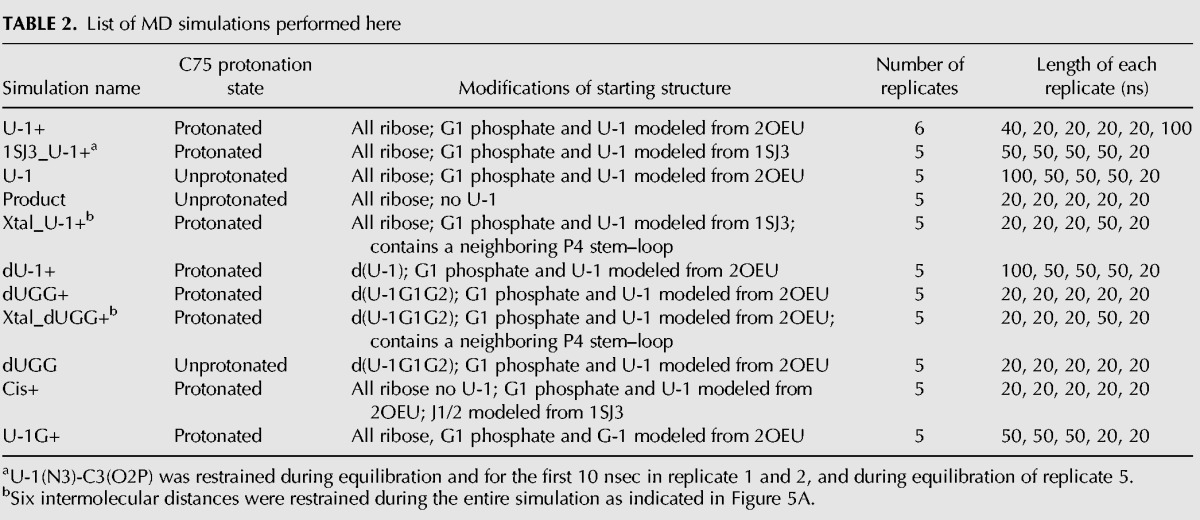
While a single product-like trans-acting HDV ribozyme conformer was crystallized (Chen et al. 2010), our FRET studies show that in solution multiple catalytically active global conformations coexist. This discrepancy raises the question of whether the crystal structure itself may hint at such conformational divergence. Indeed, the U-1 and scissile phosphate were not resolved in the otherwise high-resolution electron density and had to be modeled in (Chen et al. 2010). The two simplest explanations for this lack of electron density are that a significant fraction of molecules degraded during crystal growth or that the crystal contains a conformational ensemble with some flexibility and/or disparity around the active site, a possibility favored by the authors (Chen et al. 2010). To test this notion and provide a window into the rapid (sub-microsecond), small-scale dynamics of the ribozyme, not easily accessible by experimental techniques (Ditzler et al. 2010), we performed MD simulations on the crystallized trans-acting precursor using our established protocols (Krasovska et al. 2005, 2006; Rhodes et al. 2006; McDowell et al. 2007, 2010; Sefcikova et al. 2007a,b; Ditzler et al. 2010). We used the latest parmbsc0χOL3 (Perez et al. 2007; Zgarbova et al. 2011) variant of the Cornell et al. AMBER force field (Cornell et al. 1995), which is essential for stable RNA simulations (Banas et al. 2010). We analyzed 49 simulations with an aggregate 1.8 µsec of simulation time (Table 2). With such an extent of simulations available, we focused our analysis on a parameter that monitors the most critical part of the underlying crystal structure, the modeled cleavage site, and is able to relate its conformation to a functional parameter, catalytic activity. To this end, we used the previously described in-line fitness that combines the distance between the attacking 2′-O nucleophile and the phosphorus atom with the in-line attack angle formed by the 2′-O nucleophile, phosphorus, and 5′-O leaving group (Lee et al. 2011) into a single value reflecting the relative poise to undergo catalysis (Soukup and Breaker 1999). In essence, a “high” fitness value close to 1 indicates a phosphodiester bond geometry more amenable to cleavage than one described by a “low” fitness value closer to 0.
TABLE 2.
List of MD simulations performed here
The cleavage site of the crystallized trans-acting HDV ribozyme (Chen et al. 2010) was proposed to resemble the splayed cleavage site conformation of the crystal structure of the hammerhead ribozyme (PDB ID 2OEU) (Martick and Scott 2006; Chen et al. 2010). Incorporating this model into an all-ribose form of the trans-acting crystal structure with an N3-protonated C75H+, we performed six simulations termed U-1+ (Table 2). We found that half of our simulations predominantly retained high (>0.5) in-line fitness, whereas the other half showed low (<0.5) values with occasional excursions to higher values (Fig. 3A). Alternatively, we modeled the cleavage site using an alternative “U-turn” motif topology that we had identified in the previous cis-acting precursor crystal structure (PDB ID 1SJ3) (Fig. 3B; Sefcikova et al. 2007b). The resulting five 1SJ3_U-1+ simulations (Table 2) exhibited poor in-line fitness, with >99% of the total simulation time resulting in values of <0.5 (Fig. 3A). In addition, when restraining the U-1(N3)…C3(O2P) distance for the first 10 nsec in two simulations (numbers 1 and 2) to that of a hydrogen bond characteristic of the U-turn motif (Sefcikova et al. 2007b), the in-line fitness never increased beyond 0.5 (Fig. 3A). In almost all 1SJ3_U-1+ simulations, C75H+ resided far from G1(O5′), disrupting a hydrogen bond that appears critical for retaining an optimal scissile phosphate geometry (Fig. 3C). Often, the scissile phosphate's two nonbridging oxygens accepted a fluctuating, bifurcated hydrogen bond from the two exocyclic amino protons of A77(N6) (Fig. 3D). In turn, this interaction created a cation-binding pocket different from that observed in the hammerhead model of the cleavage site, with, on average, one ion bound to G1(O1P), and a partially (∼25%) occupied ion bound to A77(N7) and C75H+(O2) (Fig. 3E,F). Such Na+ ion binding is often predictive of Mg2+ binding while avoiding the complications from insufficient sampling and lack of polarization associated with simulating a tightly binding Mg2+ (Krasovska et al. 2006; Ditzler et al. 2010). We conclude that, while the U-turn motif is consistent with the earlier cis-acting precursor crystal structure, it does not lend itself to high in-line fitness in the context of the product-like trans-acting ribozyme. In addition, combining the simulation data, while still far from full sampling, with our experimental findings suggests that the catalytic core of the HDV ribozyme occupies a rugged folding free-energy landscape with multiple low-energy wells (Zhuang et al. 2002; Rueda et al. 2004; Marek et al. 2011).
FIGURE 3.

Testing the hammerhead ribozyme model for the cleavage site. (A) In-line fitness of our U-1+ and 1SJ3_U-1+ simulations (Table 2). (B) Overlay of 1SJ3_U-1+ and U-1+ scissile phosphates. The 1SJ3_U-1+ structure is shown in color, while the U-1+ structure is shown in transparent cartoon with relevant residues shown as thickened lines. The U-1(N3)C3(O2P) heavy atom distance restrained in some simulations (Table 2) is denoted with a black dashed line, while the destabilized C75(N3H3)G1(O5′) distance is denoted with a light blue dashed line. For reference, the stable C75(N3H3)G1(O5′) distance is denoted in the gray U-1+ structure with a black dashed line. In all stick representations of 1SJ3_U-1+ structures, carbons are colored dark red, nitrogens are blue, oxygens are red, and hydrogens are white. (C) Comparison of C75H+(N3H3)…G1(O5′) hydrogen bonding occupancies of U-1+ and 1SJ3_U-1+ simulations. Traces were computed using the Simulaid program and in-house scripts, with cutoffs of ≥120° and ≤3.5 Å to indicate the presence of a hydrogen bond. (D) Monitoring of alternate A77(N6)…G1(O1/O2P) hydrogen bonding interactions for U-1+ and 1SJ3_U-1+ simulations. (E) The alternative ion cage observed in 1SJ3_U-1+ simulations. Overlays of 1SJ3_U-1+ and U-1 are shown in the left-hand portion of the figure. As in panel B, the 1SJ3_U-1+ structure is shown in color, while the U-1+ structure is in transparent gray cartoon for reference. Relevant residues are shown in sticks and thickened lines, respectively. Red dashed lines indicate hydrogen bonds/ionic interactions that form and are stable in 1SJ3_U-1+ trajectories, while blue dashed lines indicate hydrogen bonds in U-1+ simulations that are destabilized due to the new scissile phosphate conformation of the 1SJ3_U-1+ structure. A sodium ion is shown as a large navy sphere. The cyan lines in panel A denote from which replicate and at what times the snapshots were taken. (F) Coordination number of sodium ions at a given distance from each of the functional groups specified. The plotted curves are weighted averages from all available simulations of a given type.
In our simulations so far, we used an N3-protonated C75H+, which we found leads to a C75H+(N3)…G1(5′O) hydrogen bond critical for maintaining a favorable in-line fitness. Previous MD studies have identified two protonated cytosines as structurally important: C41, that upon protonation forms a rare stabilizing quadruple interaction just below the active site (Krasovska et al. 2005; Veeraraghavan et al. 2010), and C75, that upon protonation promotes a stable active site in the context of the trans-acting precursor (Veeraraghavan et al. 2011) as well as cis-acting product structure (Krasovska et al. 2005). Protonation in the context of the new, trans-acting ribozyme is justified since the crystal structure was obtained at a pH of 5.0 (Chen et al. 2010); however, since the pKa of C75 of the HDV ribozyme was measured as ∼6.4 (Nakano et al. 2000; Gong et al. 2007), at the more physiological pH of 7.5 used in our solution, probing a protonated C75H+ will represent only a minor (as low as 10%) species. To ask what impact an unprotonated C75 has on in-line fitness, we performed five U-1 simulations without protonating C75 in the starting structure (Table 2). Indeed, very few excursions to high (>0.5) in-line fitness occurred (Fig. 4A), and without the C75H+(N3)…G1(5′O) hydrogen bond, this distance increased to typically 5–10 Å (Fig. 4B). Such distances are not typically seen in the U-1+ simulations (Fig. 4B). On average, a single Na+ ion occupied a position close to the unprotonated C75(N3), as well as C75(O2), essentially adopting the position of the H(C75N3) proton in the U-1+ simulations, occupying a pocket of the ribozyme rendered more negative in electrostatic surface potential due to deprotonation of C75H+ (Fig. 4C). The fractional residency of this ion was 101 ± 29% for C75(N3) (indicating that occasionally more than one Na+ binds in the pocket) and 78 ± 21% for C75(O2) (Fig. 4D). These observations are consistent with previous experimental studies that suggested anti-cooperativity between protonation of C75 and relatively weak binding of an active-site magnesium ion (Nakano et al. 2001; Krasovska et al. 2006). Taken together, our data strongly suggest that protonation of C75 to form a C75H+(N3)…G1(5′O) hydrogen bond is critical for the trans-acting HDV ribozyme precursor to adopt a favorable in-line fitness value, positioning its scissile phosphate for catalysis.
FIGURE 4.

A C75(N3)…G1(O5′) hydrogen bond supports a product-like active site. (A) In-line fitness of U-1 trajectories. (B) Comparison of C75(N3)…G1(O5′) heavy atom distances in U-1+ and U-1 trajectories. (C) Structural overlay of U-1 and U-1+ active sites. The U-1 structure is in color, while the U-1+ structure is in transparent gray. Relevant residues are shown as sticks and thickened lines, respectively. Red dashed lines indicate stable coordination of the orange sodium ion. Light blue dashed lines indicate the C75H+(N3H3+)…G1(O5′) hydrogen bond lost in the U-1 structures. Colored atoms in stick representations are colored as indicated in Figure 3. The cyan lines in panel B denote from which replicate and at what times the snapshots were taken. (D) Running coordination numbers of U-1+ and U-1 ion-binding residues. The plotted curves indicate the number of sodium ions coordinated by a given residue at a range of distances and are weighted averages from all available simulations of a given set. (E) Time traces for the reverse G1(OH5′)…C75(N3) hydrogen bond in product trajectories.
Further supporting the notion that the C75H+(N3)…G1(5′O) hydrogen bond is a critical anchoring point for a product-like structure, we found that five simulations of a product form of the trans-acting crystal structure (with U-1 removed, G1(5′O) protonated, and C75 left unprotonated) (termed “Product” in Table 2) showed only a subtle difference within the active site relative to our U-1+ simulations. In this case of a less crowded active site without U-1, an even more stable G1(OH5′)…C75(N3) hydrogen bond forms. This hydrogen bond is present in all five Product simulations, with occupancies ranging from 30% to 80% (Fig. 4E).
Intermolecular crystal contacts and 2′-deoxy modifications decrease in-line fitness and stacking of U-1 on U23
Upon inspection of the crystal structure, we noticed an intermolecular contact between the closing GAAA tetraloop of P4 of one molecule in the unit cell and the active site in another (Fig. 5A; Chen et al. 2010). While the several hydrogen bonds from this contact may have fortuitously aided in stabilizing crystals, we sought to understand whether they may have an impact on the cleavage site. To this end, we prepared a starting structure of a ribozyme together with the adjacent P4 stem–loop from a second ribozyme in the unit cell, using six distance restraints to keep the latter P4 stem–loop from changing its relative positioning (Fig. 5A). The resulting five simulations termed Xtal_U-1+ (Table 2) exhibited considerably poorer fitness than our U-1+ simulations (Fig. 5B). A closer look revealed that the loop's formation of hydrogen bonds with U23 and C22 resulted in increased distances of U-1 from U23 and C22 (Fig. 5A). These increased distances hinder formation of a U-1(O2′)…U23(O1P) hydrogen bond, which, in turn, destabilizes a specific U-1 conformation wherein it stacks on U23, a stacking interaction that occurred frequently in our U-1+ simulations (Fig. 5A). The crystal packing contact thus may contribute to the lack of U-1 electron density (Chen et al. 2010) since it confers a less well-defined positioning of U-1.
FIGURE 5.

Effects of crystallization parameters on active site geometry. (A) Whole-molecule view of our crystal structure model, with length of long-range restraints and participating residues indicated. (Top inset) Short-range restraints and participating residues in our Xtal simulations. (Bottom inset) A 100-psec-averaged snapshot from a Xtal_U-1+ simulation (gold) is overlaid with a 100-psec-averaged snapshot from a U-1+ simulation (navy). The adjacent P4 loop interacts with U23 and C22 resulting in increased distances of these residues from U-1. As a result, interactions important for stabilizing favorable fitness cannot easily form. (B) Comparison of in-line fitness values in Xtal_dU-1+ and dU-1+ simulations. (C) Comparison of in-line fitness “equivalent” values (see the Results section for definition) in dUGG+, Xtal_dUGG+, and dUGG simulations. (D) Comparison of C75(N3)…G1(O5′) heavy atom distances in dUGG+ and dUGG simulations.
Another unusual feature of the crystal structure (Chen et al. 2010) is the presence of three 2′-deoxy modifications in U-1, G1, and G2 flanking the cleavage site (Fig. 1A). While a single such modification of the N-1 nucleotide is often used to render a ribozyme substrate noncleavable by removing the 2′-OH nucleophile that serves as a nucleophile during cleavage, multiple 2′-deoxy modifications are less common. To ask what effect these extended modifications may have on the local structural dynamics of the cleavage site, we first performed five dU-1+ simulations with a single 2′-deoxy modification at U-1 (Table 2) and compared them to our all-ribose U-1+ simulations. Because there is no U-1(2′O) in the dU-1+ simulations, an in-line fitness “equivalent” was calculated by using U-1(2′H) in place of U-1(2′O). In two of our dU-1+ simulations, in-line fitness started off at favorable values but later sampled unfavorable values as well (Fig. 5B). In two other dU-1+ simulations, fitness started with and remained at unfavorable values (Fig. 5B). This behavior resembles that of the all-ribose U-1+ simulations (Fig. 3A).
We then performed five dUGG+ simulations that carried all three 2′-deoxy modifications of the crystal structure (Table 2). In this case, less frequent adoption of favorable in-line fitness was observed and, if adopted, high in-line fitness values were often rapidly lost (Fig. 5C). These observations suggest that, as with the intermolecular crystal contacts, 2′-deoxy modifications near the cleavage site may reduce catalytic poise. Specifically, the absence of a G1(2′OH)…U-1(5′O) interaction seems to affect this difference, whereas hydrogen bonding by G2(2′OH) does not appear to have distinct effects. Interestingly, the dynamic exchange between high and low fitness values appears to be accelerated for the dUGG+ simulations (Fig. 5C), consistent with the large number of closely related (i.e., almost comigrating) conformations we observed in our EMSAs for S1-dUGG ribozymes (Fig. 2A). Such an enhanced flexibility may further have contributed to the lack of experimental electron density for U-1 and the cleavage site in the 2′-deoxy-modified crystal structure (Chen et al. 2010). When we performed simulations with the crystal contacts and all three 2′-deoxy modifications at the active site (“Xtal_dUGG+”) (Table 2), we observed a greater dominance of in-line fitness values <0.4 in each replicate compared to Xtal_U-1+ or dUGG+ (Fig. 5C). Finally, when performing five dUGG simulations with an unprotonated C75, we observed behavior similar to that of the C75 unprotonated dU simulations with particularly low in-line fitness. We also observed generally unfavorable C75(N3)…G1(5′O) heavy atom distances in both the dUGG+ and dUGG simulations (Fig. 5D), indicating that the incorporation of all three 2′-deoxy modifications contributes to further active site destabilization beyond simply unfavorable in-line fitness.
A cis-acting precursor model exhibits greater fluctuations in fitness, whereas multimodal stacking of a U-1G mutation with U23 results in decreased fitness
A wealth of experimental evidence has indicated that cis-acting HDV ribozymes generally cleave ∼10–100 times faster than their trans-acting counterparts (Pereira et al. 2002; Shih and Been 2002; Sefcikova et al. 2007a; Tinsley and Walter 2007; Chen et al. 2009). It has been hypothesized that the loss in catalytic activity of trans-acting ribozymes is due to a less poised or stable cleavage site architecture as a result of the distal discontinuity of the RNA backbone (Shih and Been 2002; Tinsley and Walter 2007). To test this hypothesis in the context of the trans-acting crystal structure, we modeled into it two J1/2 joiner nucleotides (A8 and U9) from the previous cis-acting product crystal structure (Ferre-D'Amare et al. 1998) to arrive at a corresponding, C75(N3)-protonated (or C75H+) cis-acting precursor model (Cis+) (see Table 2 and Fig. 6A). While generally similar in the probability distribution of in-line fitness values (Supplemental Fig. S2), we found this model to exhibit greater dynamics in fitness than our U-1+ simulations, as all five of our Cis+ simulations converted multiple times between high and low in-line fitness values (Fig. 6B). This finding contrasts with our U-1+ simulations that show either mostly high or mostly poor in-line fitness over entire 20 nsec simulations (Fig. 3A). These more rapid fluctuations of Cis+ in-line fitness compared to that of the U-1+ simulations are consistent with a lower barrier to transitioning between inactive and active local conformations once C75 is protonated and may rationalize the generally higher catalytic activity of cis-acting HDV ribozymes.
FIGURE 6.

Simulating a cis-acting precursor model and a U-1G mutant. (A) Global structure of our Cis+ simulations. Modeled-in residues are denoted in purple. (B) In-line fitness of our Cis+ simulations. (C) In-line fitness (top plot), stacking behavior (middle plot), and representative stacking snapshots (bottom images) of our U-1G+ simulations. Stacking distances were calculated as the distance between the center of mass of U23 heavy ring atoms, and the center of mass of G-1 atoms C4 and C5. Both snapshots overlay a snapshot from the U-1+ simulation to contrast the stacking behavior. The red double-headed arrow in the left snapshot indicates the ability of G-1 to slide. The curved arrow in the right snapshot depicts the “flipping up” movement of G-1 as it unstacks. The U-1G+ snapshots are in color, while the U-1+ snapshot is in transparent gray. Relevant residues are shown in sticks and thickened lines. Atom colors are as described in Figure 3. The cyan lines in the middle plot denote from which replicate and at what times the snapshots were taken.
Conversely, previous studies have shown that a G-1 upstream of the scissile phosphate results in a decrease in catalytic rate relative to the U-1 found conserved in clinical isolates of HDV (Wadkins and Been 2002; Sefcikova et al. 2007b). Our bulk FRET assays also indicated that this mutation results in kinetic behavior distinct from that of the native U-1. To ask whether the trans-acting ribozyme structure may offer an explanation for both of these effects, we analyzed five simulations with a G-1 in place of the U-1, in the presence of an N3-protonated C75H+ (U-1G+) (Table 2). We found 98.5% of the total frames of these trajectories to adopt in-line fitness <0.5 (Fig. 6C). This trend was mainly determined by unfavorable in-line attack angles, which, in turn, appeared to be due to variable stacking of G-1 with U23; in addition to a sliding motion of U23 on the larger G-1, significant unstacking of the two bases was observed in three out of five trajectories (Fig. 6C). These observations rationalize the decrease in catalytic activity upon U-1G mutation of trans-acting HDV ribozymes (Sefcikova et al. 2007b) and further support the functional relevance of our MD simulations.
DISCUSSION
Disparate crystal structure models of the cleavage precursor of the HDV ribozyme have raised questions about the validity of the particular modifications needed for each specific construct to crystallize. In the case of the cis-acting precursor, either the catalytic C75 was mutated to U or divalent metal ions were omitted (Ke et al. 2004), both of which may have led to trapping of an off-pathway conformation. In the case of a more recent trans-acting precursor structure (Chen et al. 2010), three 2′-deoxy modifications surrounding the cleavage site and the establishment of intermolecular contacts during crystal growth may have led to the poorly defined cleavage site, which lacks electron density for the U-1 nucleotide and scissile phosphate. In addition, this structure was grown at pH 5.0, protonating C75 into an ionized species that would be relatively rare at physiological pH given C75's pKa of ∼6.4 and the competition of protonation with Mg2+ binding (Nakano et al. 2000; Gong et al. 2007); this protonation, in turn, likely trapped a rare, more product-like conformation that would be slow to react, as cleavage is disfavored at low pH due to the scarcity of the required general base (Perrotta et al. 1999b; Nakano et al. 2000).
Our data support the unifying minimal model summarized in Figure 7 that resolves these questions. We find that the crystallized trans-acting ribozyme shows multiple distinct conformations in solution at physiological pH as evidenced by biphasic cleavage kinetics (Fig. 1C) and a smear upon native gel electrophoresis (EMSA) (Fig. 2A). The fact that the EMSA smear is converted into a single product band upon cleavage in the gel (Fig. 2A) suggests that all of these conformations eventually cleave and, hence, are catalytically active even if with varying rate constants. Accompanying cleavage in solution at physiological pH, the crystallized trans-acting HDV ribozyme undergoes a significant conformational change, as observed by ssFRET and trFRET in the form of a significant lengthening along the longitudinal axis (Fig. 2B,C). This finding is similar to observations for all previous trans- and cis-acting ribozymes (Pereira et al. 2002; Tanaka et al. 2002; Jeong et al. 2003; Harris et al. 2004), suggesting that it represents an immutable, common feature of the HDV ribozyme in general. Taken together, these observations suggest that occasional, stochastic protonation of C75 at physiologic pH leads to the transient adoption of an ionized state that moves C75H+ closer to the cleavage site so that its N3 donates a hydrogen bond to the G1(O5′) leaving group (while its exocyclic amine donates a hydrogen bond to one of C22's nonbridging oxygens; it remains unclear whether the adjacent G25 rotates from anti to syn before, during, or after this event). This conformational adjustment sets the stage for the adoption of favorable in-line fitness. If, concomitantly, a Mg2+ ion bound to the adjacent electronegative cleft carries a deprotonated water molecule in its inner-coordination sphere, cleavage can proceed with C75H+ and Mg(OH)+ serving as the general acid and base, respectively (Fig. 7). Since the opposing protonation states of C75H+ and Mg(OH)+ are rare and transient, cleavage is slow compared to protein-based ribonucleases (Bevilacqua 2003; Bevilacqua et al. 2004; Walter 2007). According to this model, the conformational change accompanying catalysis (Ke et al. 2004) is induced by the—at physiological pH—rare protonation of C75. The resulting transient adoption of an active site conformation similar to that modeled into the trans-acting ribozyme structure was enriched by crystallization at low pH. Conversely, our solution probing at physiological pH detects a dominant conformation more similar to the cis-acting precursor crystal structure, prior to the conformational change, which represents a legitimate alternate conformation and on-pathway catalytic intermediate. (We note that there must also be other, more globally distinct conformations present in solution to explain the broad smear and multiple bands of the various trans-acting ribozyme observed during EMSA, as well as the biphasic cleavage kinetics, which points to deeply trapped conformations [Zhuang et al. 2002; Rueda et al. 2004; Marek et al. 2011].)
FIGURE 7.
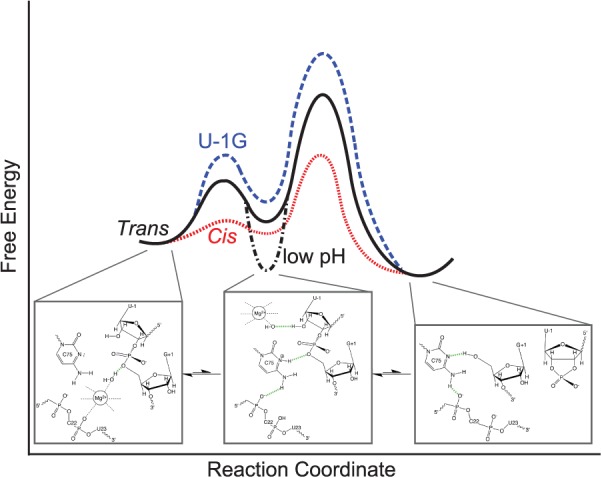
A parsimonious, unifying, minimal model of the HDV ribozyme folding free-energy landscape focused on the catalytic reaction coordinate, indicating the divergence of the trans-acting, cis-acting, and U-1G mutant ribozymes. Below each of the main on-pathway intermediates, structural models as derived from the available crystal structures are depicted. The cis-acting precursor crystal structure (Ke et al. 2004) resembles most closely an early intermediate dominant at physiological pH (left). This intermediate still needs to undergo a conformational change that positions the C75H+ for general acid catalysis and was trapped at low pH in the trans-acting precursor structure (Chen et al. 2010) (middle). If a magnesium hydroxide is bound to the active site simultaneously, cleavage into the product leads to loss of the 5′-precursor sequence and magnesium ion (right).
Energetically, we have evidence that the cis-acting HDV ribozyme has a lower barrier toward catalysis than the trans-acting version (Fig. 7). First, all cis-acting ribozymes are 1–2 orders of magnitude faster in cleavage (Tinsley and Walter 2007). Second, in our MD simulations, a cis-acting precursor model appears to exhibit more rapid fluctuations between conformations of high and low fitness (Fig. 6B), suggesting that the barrier between the pre- and post-conformational change states and, as a consequence, also the overall energetic barrier of catalysis, are lowered (Fig. 7). However, at physiological pH, the preconformational change state is still thermodynamically favored, leading to the observed preconformational cis-acting crystal structure. Conversely, a U-1G mutation leads to generally low in-line fitness, and thus a higher catalytic barrier, by disfavoring the conformational change needed to adopt a suitable active site conformation (Fig. 7), at least partially due to trapping of G-1 in a shifted stacking interaction with U23 (Fig. 6C). Our data and resulting parsimonious model thus offer rationalizations for a number of independent experimental observations. In addition, they further advance a widely held view of RNA folding free-energy landscapes as particularly rugged (compared to, for example, those of proteins) (Woodson 2010; Marek et al. 2011).
Our MD simulations provide a glimpse of how subtle some of the conformational rearrangements may be that control a catalytically critical parameter such as the in-line fitness, which needs to reach a value of >0.5 for RNA cleavage to become significant (Soukup and Breaker 1999). Efficient ribozymes are likely to stabilize favorable in-line fitness, as well as position general acid and base catalysts. The cis-acting HDV ribozyme is one of the most efficient self-cleaving ribozymes known, yet is still many orders of magnitude slower than RNase A (Walter 2007; Nakano et al. 2000). One reason may be the electrostatic competition between C75 protonation and Mg2+ binding to the active site (Nakano et al. 2001). Furthermore, while C75H+ is a rare protonated species needed for catalysis, the hydrated Mg2+ ion with its high pKa of ∼12 needs to adopt a rare deprotonated state to act as the general base, contrasting events that will have a low probability to coincide. Protonation of C75, in turn, leads to a C75H+(N3)…G1(O5′) hydrogen bond that aids in adopting a favorable in-line fitness. It appears that it is the required temporal coincidence of these partially competing, dynamic low-probability protonation and conformational change events that slows RNA self-cleavage to, at best, some 10s to 100s min−1 (Sefcikova et al. 2007b; McDowell et al. 2010). Future studies will likely reveal more such examples for the complex relationship between the folding dynamics and function of RNA. Linking results from experimental structural and functional probing with those from computational MD simulations, as accomplished here through the use of in-line fitness as a parameter transcending the gap in accessible timescales, is likely to pave the way for more advances in this direction.
MATERIALS AND METHODS
Preparation of RNAs
All sequences used here were based on the recent trans-acting HDV ribozyme crystal structure (Chen et al. 2010). The GAAA loop of the P4 stem was replaced with an extension of the P4 helix to generate a three-stranded version of the ribozyme (Fig. 1B), as done before (Pereira et al. 2002). Additionally, U55 in the P4 helix was omitted to prevent potential misfolding. All RNA oligonucleotides were purchased from the Keck Biotechnology Resource Laboratory at the Yale University School of Medicine (http://keck.med.yale.edu/oligo). Some strands were ordered already 2′-de-protected and cartridge-(Glen-Pak)purified, while others were 2′-de-protected in-house according to the manufacturer's instructions and then purified as described (Walter 2002) by denaturing, 8 M urea, 20% polyacrylamide (19:1 acrylamide:bisacrylamide) gel electrophoresis. The bands of interest were cut out, diffusion-eluted into either 1 mM EDTA or “Crush ‘n Soak” buffer (0.5 M NH4OAc, 0.1 mM EDTA), ethanol-precipitated, and resuspended in autoclaved double-distilled water, similar to previous protocols (Walter 2002). Oligonucleotides that were cartridge-purified by Keck were analyzed by denaturing, 8 M urea, 20% polyacrylamide gel electrophoresis to ensure high purity. RNA concentrations were calculated from absorption at 260 nm.
Strand B was purchased already labeled with a 5′-fluorescein and a 3′-C7-amino modifier in preparation for further in-house labeling with 5-carboxytetramethylrhodamine (TMR) succinimidyl ester (Invitrogen) according to the manufacturer's protocol, using overnight incubation at room temperature (22°C). The doubly labeled RNA was then purified by denaturing, 8 M urea, 20% polyacrylamide gel electrophoresis. The bands of interest were cut and diffusion-eluted either into 1 mM EDTA or Crush ‘n Soak buffer. The RNA was then ethanol-precipitated and resuspended using autoclaved, double-distilled water, similar to previous protocols (Walter 2002). The concentration of the doubly labeled Strand B was determined from the absorption spectrum and estimated to contain ∼35% of singly labeled starting material.
Radioactive cleavage assays
Radioactive cleavage assays to determine ribozyme activity were performed similar to those previously described (Pereira et al. 2002). Cleavable substrate strands were 5′-end-labeled using T4 polynucleotide kinase (NEB) and [γ-32P] ATP. All cleavage assays were carried out under single-turnover conditions, where the free ribozyme concentration was ∼125-fold above the substrate concentration, unless otherwise noted. Standard assay conditions were a buffer of 40 mM Tris-HCl, pH 7.5, 25 mM DTT, and 11 mM MgCl2 at 25°C, unless otherwise stated. Ribozymes were preannealed by mixing Strand A and twice the concentration of Strand B (labeled with fluorescein and TMR) under standard assay conditions without magnesium, heating to 90°C for 2 min, and then cooling to 25°C for 10 min using a water bath. MgCl2 was added to a final concentration of 11 mM. The free ribozyme was incubated at 50°C for 10 min, followed by cooling to 25°C for 10 min. The final ribozyme concentration, based on the concentration of Strand A, was 750 nM. Cleavage assays were initiated by adding a trace amount (<6 nM) of 5′-32P-labeled substrate. Alternatively, the conditions of a previous study (Chen et al. 2009) were followed by mixing a 250-fold excess of ribozyme over the 32P-labeled substrate in 25 mM Tris-HCl, pH 7.0, heating at 90°C for 2 min, and cooling to room temperature for 10 min. The mixture was then incubated at 37°C for 2 min. Magnesium chloride was added to initiate the assay to be carried out at 37°C. In either case, aliquots of 5 µL were taken at appropriate time intervals and quenched with 10 µL of stop solution (75% [v/v] formamide, 150 mM EDTA, and 0.1% [w/v] each of bromophenol blue and xylene cyanol dyes). The 5′ cleavage product was separated from the uncleaved substrate by gel electrophoresis using denaturing, 8 M urea, 20% polyacrylamide gel electrophoresis at a limiting power of 25 W for 1.5 to 2 h at room temperature. The gel was then exposed to a phosphorimager screen for 18–22 h and scanned using a Typhoon 9410 Variable Mode Imager. Bands were quantified using ImageJ (Schneider et al. 2012), and the calculated fraction cleaved at each time point was normalized to the sum of the substrate and product bands. Error bars were derived from 3–5 independent assays. Time traces of product formation were fitted with either a single-exponential (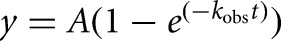 ) or double-exponential increase function (
) or double-exponential increase function (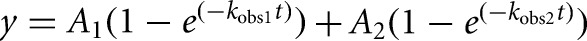 ), utilizing the Marquardt-Levenberg nonlinear least-squares regression method (GraphPad Prism version 4.0b for Mac, GraphPad Software), where A is the pre-exponential factor and kobs is the observed rate constant for the reaction.
), utilizing the Marquardt-Levenberg nonlinear least-squares regression method (GraphPad Prism version 4.0b for Mac, GraphPad Software), where A is the pre-exponential factor and kobs is the observed rate constant for the reaction.
Gel electrophoretic mobility shift assays (EMSAs)
Similar to previous protocols (Pereira et al. 2002), 50 pmol of substrate or product strand were incubated with 30 pmol of Strand A and 8 pmol of Strand B in standard buffer without magnesium (final concentration of 40 mM Tris-HCl, pH 7.5, 25 mM DTT), supplemented with 10% (v/v) glycerol. The samples were then incubated using the protocol outlined for cleavage assays, with two minor changes: The initial 90°C incubation lasted 1 instead of 2 min, and the two 10-min incubations were conducted at room temperature on the bench top instead of in a 25°C water bath. MgCl2 was added to a final concentration of 11 mM and the solution incubated for at least 10 min at 50°C. The electrophoresis unit had been allowed to pre-equilibrate at 4°C for at least 2 h. After incubation, the samples were loaded onto a nondenaturing 10% polyacrylamide (19:1 acrylamide:bisacrylamide) gel containing 11 mM Mg(OAc)2 and 40 mM Tris(OAc), pH 7.5. An electric field of ∼10 V/cm was applied to the samples, which were electrophoresed for 12–14 h at 4°C. The gel was then imaged using a Typhoon 9410 Variable Mode Imager as described (Pereira et al. 2002). A dual-color image was generated by the scanner software and opened in ImageQuant. Relative band intensities were determined by analysis using ImageJ (Schneider et al. 2012); relative band percentages were obtained by summing the intensities from the fluorescein and TMR scans and normalizing them to their sum for each lane.
Steady-state FRET measurements
Solutions of 43–49 nM Strand B (final concentration after full assembly) in standard buffer were annealed as specified in the EMSA protocol. One hundred forty microliters of such a solution were added to a Starna quartz fluorescence cuvette, its fluorescein label excited at 470 nm (bandwidth 8 nm) and emissions detected at 520 nm (bandwidth 16 nm) and 585 nm (bandwidth 16 nm) for fluorescein and TMR, respectively. After Strand B signal proved stable, 5 µL Strand A were added in a 3.25-fold excess over Strand B for S8-U, S8-dU, S1-U, S1-dU, and S1-dUGG and P runs, and in a 3.8-fold excess over Strand B for S1-G and S1-dG runs. The resulting change in FRET was recorded for 30 min. Once this signal was stable, traces were further followed upon the addition of a substrate or product strand in a 5.42-fold excess over Strand B for S8-U, S8-dU, S1-U, S1-dU, S1-dUGG, and P runs, and in a 5.8-fold excess for the S1-G and S1-dG runs. All substrate and product strands were added in 5-µL volumes, for a total final volume of 150 µL. The resultant signal change was measured in all cases for at least 5000 sec. In the case of the free Rz runs, 5 μL of standard buffer were added instead of the third strand, so that the final reaction volume was consistent. The ratio of acceptor to donor signal was calculated and normalized to the starting baseline. Each trace was then smoothed using a running average with a window of 10 and further normalized to control runs. Average curves of three runs for substrates and control, and four runs for the product, are reported here. Results were plotted as the relative FRET ratio decrease using Prism (GraphPad Prism version 4.0b for Mac, GraphPad Software).
Time-resolved FRET measurements
Preparations of 53 nM of Strand B (six singly labeled with fluorescein and six doubly labeled with fluorescein and TMR) were incubated in standard buffer without magnesium for 1 min at 90°C, followed by 10 min at 25°C. MgCl2 was added to a final concentration of 11 mM, and the solution was incubated for 10 min at 50°C, followed by 10 min at 25°C. Strand A was then added so that its final concentration would be threefold that of Strand B. Strands A and B were incubated at 25°C for 30 min. The third strand, i.e., one of the noncleavable substrate strands or the product strand, or buffer in the case of the free ribozyme sample, was then added to a final concentration that was 50-fold that of Strand B. Each of the six pairs of samples was incubated at 25°C for 2 h and then kept at 4°C when not being used to measure decay lifetimes.
An ISS Alba confocal fluctuation microscope (based on an Olympus IX-81 frame) was used to collect fluorescein decay lifetimes. Forty microliters of sample were loaded onto a coverslip, which was placed on a 60 × 1.2 numerical aperture water immersion objective. A blue-enhanced supercontinuum laser (Fianium SC 400-6-PP, operated at 6 W) excited fluorescein at 488 nm with 100-psec pulse width and 20-MHz pulse picking. Decays were collected at 531 nm using a photomultiplier tube (Hamamatsu H7422P-40) feeding into a time-correlated single-photon counting acquisition card SPCM-830 (Becker & Hickl), with 4096 bins at 12.20 psec/bin. An instrument response function was obtained by measuring the back reflection off a coverslip without sample. For each sample, decays of donor (fluorescein) only and donor with acceptor (TMR) present were collected. Decays were collected so that the maximum raw peak count was between 40,000 and 60,000 counts. Five consecutive measurement reads were collected for a single sample, and there were three inter-day replicates for each sample. Table 1 summarizes the distance distribution parameters averaged from two or three inter-day replicates.
The donor-only traces with fluorescence intensity ID were fit with a sum of exponentials 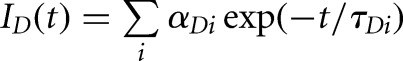 to extract three intrinsic donor lifetimes (τDi is the intrinsic donor lifetime, and αDi is the fractional contribution of each lifetime). Using these donor lifetimes, the doubly labeled samples’ decays with fluorescence intensity IDA were fit with the following model for a distance distribution:
to extract three intrinsic donor lifetimes (τDi is the intrinsic donor lifetime, and αDi is the fractional contribution of each lifetime). Using these donor lifetimes, the doubly labeled samples’ decays with fluorescence intensity IDA were fit with the following model for a distance distribution:
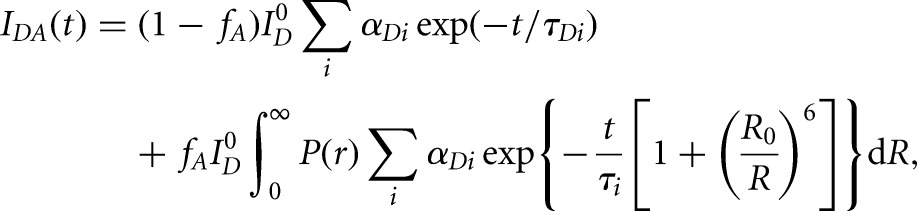 |
where 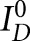 is the donor intensity at time 0, fA is the fraction of molecules labeled with acceptor (allowing to correct for any singly labeled RNA), R is the distance between donor and acceptor, R0 is the Förster distance (55 Å for the fluorescein and TMR pair), and P(r) is the distance distribution. Distance distributions assumed a three-dimensional Gaussian function model based on the equation:
is the donor intensity at time 0, fA is the fraction of molecules labeled with acceptor (allowing to correct for any singly labeled RNA), R is the distance between donor and acceptor, R0 is the Förster distance (55 Å for the fluorescein and TMR pair), and P(r) is the distance distribution. Distance distributions assumed a three-dimensional Gaussian function model based on the equation:
where a and b are parameters that describe the shape of the distribution and c is the normalization constant.
Molecular dynamics simulations
All MD trajectories were generated using the AMBER10/11 suite of programs (Case et al. 2008, 2010) and the ff99 force field with recent modifications to χ (essential for RNA) and α/γ torsions (ff99bsc0χOL3 version, current AMBER default) (Perez et al. 2007; Zgarbova et al. 2011). Structures were net-neutralized with Na+ ions placed at locations of optimal electrostatics using the LEaP module of AMBER10/11 and solvated using a rectangular periodic TIP3P water box model (Jorgensen et al. 1983) extended to a distance of ≥10 Å from any ribozyme atom or Na+ ion. The ABC protocol (Lavery et al. 2010; Beveridge et al. 2012) was used to minimize and equilibrate structures prior to production runs. The minimization and equilibration protocol as well as the production runs were performed using the PMEMD module of the AMBER suite of programs. For the production runs, the particle mesh Ewald (PME) method was employed with a grid spacing of 1.0 Å, cubic-spline interpolation, and a heuristic pair list update with a Lennard-Jones cutoff of 9.0 Å. Production runs were carried out at 300 K with constant-pressure boundary conditions and the Berendsen temperature coupling algorithm (Berendsen et al. 1984) with a time constant of 1.0 psec. The SHAKE procedure (Ryckaert et al. 1977) was applied to constrain bonds involving hydrogen. Parameters for the protonated cytosine were derived using the Cornell et al. procedure (Cornell et al. 1995; Banas et al. 2012). Partial atomic charges were calculated for a protonated N1-methylcytosine using restrained electrostatic potential (RESP) fitting (Cornell et al. 1993) and the HF/6-31G(d) method. Ab initio calculations were done using the program Gaussian03 (Frisch et al. 2004). These charges were then used for the protonated cytosine; phosphate and sugar charges were kept the same as in unprotonated cytidine phosphate. Residual charge was added to C1′ to ensure a net charge of zero for the entire nucleotide. The U-1+ starting structure was prepared by globally aligning the hammerhead ribozyme 2OEU and trans-acting HDV ribozyme 3NKB (Chen et al. 2010) structures to copy the active site residues from the former to the latter. The 1SJ3_U-1+ structure was prepared by globally aligning the cis-acting 1SJ3 (Ke et al. 2004) and trans-acting 3NKB (Chen et al. 2010) structures. The coordinates for U-1, the scissile phosphate, and G1 were then copied from 1SJ3 (Ke et al. 2004) into 3NKB (Chen et al. 2010). The resulting structures were minimized and equilibrated using a protocol that involved first restraining the newly incorporated nucleotides, and subsequently removing those restraints. After these stages, the structure was equilibrated using the ABC protocol. Following minimization and equilibration, the structure was carefully checked for the presence of unfavorable geometry before carrying out production runs. For the U-1G mutation, the U-1 residue was mutated to G-1 by first deleting the atoms unique to uracil that do not overlay with guanine. The AMBER LEaP module was then used to fill in the missing atoms for guanine. The cis-acting model of the trans-acting ribozyme was created similarly to that of the 1SJ3_U-1+ structure, by modeling in the J1/2 joiner nucleotides from the cis-acting product crystal structure (PDB ID 1SJ3) (Ferre-D'Amare et al. 1998). First, the product and trans-acting structures were globally aligned, then the J1/2 joiner region was cut and pasted into the trans-acting structure using PyMol. The newly created cis-acting structure was minimized and equilibrated using a protocol that involved first restraining the newly incorporated J1/2 joiner region and subsequently removing those restraints. Following minimization and equilibration, the structure was carefully checked for the presence of unfavorable geometry before carrying out production runs. The Sander module was used to set distance restraints for appropriate simulations (Table 2).
MD trajectories were analyzed using the AMBER ptraj module and the program Simulaid (Mezei 2010). The ptraj module was used to calculate heavy-atom distances, angles, and radial distribution functions. All snapshots in the figures were generated from 100-psec-averaged structures calculated using ptraj. Simulaid was used to determine hydrogen bonding time traces using parameters of ≥120° donor-hydrogen-acceptor angle and ≤3.5 Å donor-acceptor distance. In-line fitness and other time traces were plotted using Matlab (2010b) (The MathWorks Inc.). In-line fitness (F) was calculated using the following equation (Soukup and Breaker 1999):
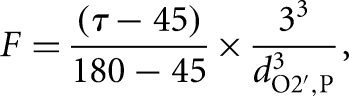 |
where τ is the angle between the 2′ oxygen nucleophile (O2′), the phosphorus electrophile (P), and the 5′ oxygen of the leaving group (O5′), and d is the distance between the 2′ oxygen nucleophile and the phosphorus electrophile.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Tristan Tabouillot, Kaushik Gunrunathan, and May Daher for protocols, training, and assistance with the time-resolved FRET experiments. We also thank Nad'a Špačková and Kamila Réblová for protocols and assistance with MD simulations. We acknowledge funding from NIH grant R01 GM062357 to N.G.W., grant P208/12/1878 from the Grant Agency of the Czech Republic to J.S. and M.O., Operational Program Research and Development for Innovations—European Regional Development Fund (project CZ.1.05/2.1.00/03.0058) and the Operational Program Education for Competitiveness—European Social Fund (CZ.1.07/2.3.00/20.0017 and CZ.1.07/2.3.00/20.0058) of the Ministry of Education, Youth and Sports of the Czech Republic to M.O. and P.B., “CEITEC—Central European Institute of Technology” (CZ.1.05/1.1.00/02.0068) from European Regional Development Fund to J.S., and a Canadian NSERC Postgraduate Scholarship to W.W.T. We also thank the Single Molecule Analysis in Real-Time (SMART) Center of the University of Michigan, seeded by NSF MRI-R2-ID award DBI-0959823 to N.G.W., for use of the ISS Alba confocal fluctuation microscope.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.044982.114.
REFERENCES
- Banas P, Hollas D, Zgarbova M, Jurecka P, Orozco M, Cheatham TE III, Sponer J, Otyepka M 2010. Performance of molecular mechanics force fields for RNA simulations: stability of UUCG and GNRA hairpins. J Chem Theory Comput 6: 3836–3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banas P, Sklenovsky P, Wedekind JE, Sponer J, Otyepka M 2012. Molecular mechanism of preQ1 riboswitch action: a molecular dynamics study. J Phys Chem B 116: 12721–12734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Been MD 1994. Cis- and trans-acting ribozymes from a human pathogen, hepatitis δ virus. Trends Biochem Sci 19: 251–256 [DOI] [PubMed] [Google Scholar]
- Been MD 2006. HDV ribozymes. Curr Top Microbiol Immunol 307: 47–65 [DOI] [PubMed] [Google Scholar]
- Been MD, Wickham GS 1997. Self-cleaving ribozymes of hepatitis δ virus RNA. Eur J Biochem 247: 741–753 [DOI] [PubMed] [Google Scholar]
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR 1984. Molecular dynamics with coupling to an external bath. J Chem Phys 81: 3684–3690 [Google Scholar]
- Beveridge DL, Cheatham TE III, Mezei M 2012. The ABCs of molecular dynamics simulations on B-DNA, circa 2012. J Biosci 37: 379–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua PC 2003. Mechanistic considerations for general acid-base catalysis by RNA: revisiting the mechanism of the hairpin ribozyme. Biochemistry 42: 2259–2265 [DOI] [PubMed] [Google Scholar]
- Bevilacqua PC, Brown TS, Nakano S, Yajima R 2004. Catalytic roles for proton transfer and protonation in ribozymes. Biopolymers 73: 90–109 [DOI] [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheatham TE III, Simmerling CL, Wang J, Duke RE, Luo R, Crowley M, Walker RC, Zhang W, et al. 2008. AMBER 10. University of California, San Francisco, CA [Google Scholar]
- Case DA, Darden TA, Cheatham TE III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, et al. 2010. AMBER 11. University of California, San Francisco, CA [Google Scholar]
- Cerrone-Szakal AL, Siegfried NA, Bevilacqua PC 2008. Mechanistic characterization of the HDV genomic ribozyme: Solvent isotope effects and proton inventories in the absence of divalent metal ions support C75 as the general acid. J Am Chem Soc 130: 14504–14520 [DOI] [PubMed] [Google Scholar]
- Chadalavada DM, Cerrone-Szakal AL, Bevilacqua PC 2007. Wild-type is the optimal sequence of the HDV ribozyme under cotranscriptional conditions. RNA 13: 2189–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JH, Gong B, Bevilacqua PC, Carey PR, Golden BL 2009. A catalytic metal ion interacts with the cleavage site G⋅U wobble in the HDV ribozyme. Biochemistry 48: 1498–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JH, Yajima R, Chadalavada DM, Chase E, Bevilacqua PC, Golden BL 2010. A 1.9 Å crystal structure of the HDV ribozyme precleavage suggests both Lewis acid and general acid mechanisms contribute to phosphodiester cleavage. Biochemistry 49: 6508–6518 [DOI] [PubMed] [Google Scholar]
- Cornell WD, Cieplak P, Bayly CI, Kollman PA 1993. Application of RESP charges to calculate conformational energies, hydrogen bond energies and free energies of solvation. J Am Chem Soc 115: 9620–9631 [Google Scholar]
- Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KMJ, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc 117: 5179–5197 [Google Scholar]
- Das SR, Piccirilli JA 2005. General acid catalysis by the hepatitis δ virus ribozyme. Nat Chem Biol 1: 45–52 [DOI] [PubMed] [Google Scholar]
- Ditzler MA, Otyepka M, Sponer J, Walter NG 2010. Molecular dynamics and quantum mechanics of RNA: conformational and chemical change we can believe in. Acc Chem Res 43: 40–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre-D'Amare AR, Doudna JA 2000. Crystallization and structure determination of a hepatitis δ virus ribozyme: use of the RNA-binding protein U1A as a crystallization module. J Mol Biol 295: 541–556 [DOI] [PubMed] [Google Scholar]
- Ferre-D'Amare AR, Zhou K, Doudna JA 1998. Crystal structure of a hepatitis δ virus ribozyme. Nature 395: 567–574 [DOI] [PubMed] [Google Scholar]
- Flores R, Ruiz-Ruiz S, Serra P 2012. Viroids and hepatitis δ virus. Semin Liver Dis 32: 201–210 [DOI] [PubMed] [Google Scholar]
- Frisch MJT, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, et al. 2004. Gaussian 03. Gaussian, Inc, Wallingford, CT [Google Scholar]
- Gong B, Chen JH, Chase E, Chadalavada DM, Yajima R, Golden BL, Bevilacqua PC, Carey PR 2007. Direct measurement of a pKa near neutrality for the catalytic cytosine in the genomic HDV ribozyme using Raman crystallography. J Am Chem Soc 129: 13335–13342 [DOI] [PubMed] [Google Scholar]
- Gong B, Chen Y, Christian EL, Chen JH, Chase E, Chadalavada DM, Yajima R, Golden BL, Bevilacqua PC, Carey PR 2008. Detection of innersphere interactions between magnesium hydrate and the phosphate backbone of the HDV ribozyme using Raman crystallography. J Am Chem Soc 130: 9670–9672 [DOI] [PubMed] [Google Scholar]
- Gong B, Chen JH, Bevilacqua PC, Golden BL, Carey PR 2009. Competition between Co(NH3)63+ and inner sphere Mg2+ ions in the HDV ribozyme. Biochemistry 48: 11961–11970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris DA, Rueda D, Walter NG 2002. Local conformational changes in the catalytic core of the trans-acting hepatitis δ virus ribozyme accompany catalysis. Biochemistry 41: 12051–12061 [DOI] [PubMed] [Google Scholar]
- Harris DA, Tinsley RA, Walter NG 2004. Terbium-mediated footprinting probes a catalytic conformational switch in the antigenomic hepatitis δ virus ribozyme. J Mol Biol 341: 389–403 [DOI] [PubMed] [Google Scholar]
- Jeong S, Sefcikova J, Tinsley RA, Rueda D, Walter NG 2003. Trans-acting hepatitis δ virus ribozyme: Catalytic core and global structure are dependent on the 5′ substrate sequence. Biochemistry 42: 7727–7740 [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML 1983. Comparison of simple potential functions for simulating liquid water. J Chem Phys 79: 926–935 [Google Scholar]
- Ke A, Zhou K, Ding F, Cate JH, Doudna JA 2004. A conformational switch controls hepatitis δ virus ribozyme catalysis. Nature 429: 201–205 [DOI] [PubMed] [Google Scholar]
- Krasovska MV, Sefcikova J, Spackova N, Sponer J, Walter NG 2005. Structural dynamics of precursor and product of the RNA enzyme from the hepatitis δ virus as revealed by molecular dynamics simulations. J Mol Biol 351: 731–748 [DOI] [PubMed] [Google Scholar]
- Krasovska MV, Sefcikova J, Reblova K, Schneider B, Walter NG, Sponer J 2006. Cations and hydration in catalytic RNA: molecular dynamics of the hepatitis δ virus ribozyme. Biophys J 91: 626–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai MM 1995. The molecular biology of hepatitis δ virus. Annu Rev Biochem 64: 259–286 [DOI] [PubMed] [Google Scholar]
- Lavery R, Zakrzewska K, Beveridge D, Bishop TC, Case DA, Cheatham T III, Dixit S, Jayaram B, Lankas F, Laughton C, et al. 2010. A systematic molecular dynamics study of nearest-neighbor effects on base pair and base pair step conformations and fluctuations in B-DNA. Nucleic Acids Res 38: 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, Giambasu G, Harris ME, York DM 2011. Characterization of the structure and dynamics of the HDV ribozyme at different stages along the reaction path. J Phys Chem Lett 2: 2538–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luptak A, Ferre-D'Amare AR, Zhou K, Zilm KW, Doudna JA 2001. Direct pKa measurement of the active-site cytosine in a genomic hepatitis δ virus ribozyme. J Am Chem Soc 123: 8447–8452 [DOI] [PubMed] [Google Scholar]
- Marek MS, Johnson-Buck A, Walter NG 2011. The shape-shifting quasispecies of RNA: one sequence, many functional folds. Phys Chem Chem Phys 13: 11524–11537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martick M, Scott WG 2006. Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell 126: 309–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell SE, Spackova N, Sponer J, Walter NG 2007. Molecular dynamics simulations of RNA: an in silico single molecule approach. Biopolymers 85: 169–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell SE, Jun JM, Walter NG 2010. Long-range tertiary interactions in single hammerhead ribozymes bias motional sampling toward catalytically active conformations. RNA 16: 2414–2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezei M 2010. Simulaid: a simulation facilitator and analysis program. J Comput Chem 31: 2658–2668 [DOI] [PubMed] [Google Scholar]
- Nakano S, Chadalavada DM, Bevilacqua PC 2000. General acid-base catalysis in the mechanism of a hepatitis δ virus ribozyme. Science 287: 1493–1497 [DOI] [PubMed] [Google Scholar]
- Nakano S, Proctor DJ, Bevilacqua PC 2001. Mechanistic characterization of the HDV genomic ribozyme: assessing the catalytic and structural contributions of divalent metal ions within a multichannel reaction mechanism. Biochemistry 40: 12022–12038 [DOI] [PubMed] [Google Scholar]
- Nakano S, Cerrone AL, Bevilacqua PC 2003. Mechanistic characterization of the HDV genomic ribozyme: classifying the catalytic and structural metal ion sites within a multichannel reaction mechanism. Biochemistry 42: 2982–2994 [DOI] [PubMed] [Google Scholar]
- Pereira MJ, Harris DA, Rueda D, Walter NG 2002. Reaction pathway of the trans-acting hepatitis δ virus ribozyme: A conformational change accompanies catalysis. Biochemistry 41: 730–740 [DOI] [PubMed] [Google Scholar]
- Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE III, Laughton CA, Orozco M 2007. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys J 92: 3817–3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotta AT, Been MD 2006. HDV ribozyme activity in monovalent cations. Biochemistry 45: 11357–11365 [DOI] [PubMed] [Google Scholar]
- Perrotta AT, Nikiforova O, Been MD 1999a. A conserved bulged adenosine in a peripheral duplex of the antigenomic HDV self-cleaving RNA reduces kinetic trapping of inactive conformations. Nucleic Acids Res 27: 795–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotta AT, Shih I, Been MD 1999b. Imidazole rescue of a cytosine mutation in a self-cleaving ribozyme. Science 286: 123–126 [DOI] [PubMed] [Google Scholar]
- Perrotta AT, Wadkins TS, Been MD 2006. Chemical rescue, multiple ionizable groups, and general acid-base catalysis in the HDV genomic ribozyme. RNA 12: 1282–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes MM, Reblova K, Sponer J, Walter NG 2006. Trapped water molecules are essential to structural dynamics and function of a ribozyme. Proc Natl Acad Sci 103: 13380–13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda D, Wick K, McDowell SE, Walter NG 2003. Diffusely bound Mg2+ ions slightly reorient stems I and II of the hammerhead ribozyme to increase the probability of formation of the catalytic core. Biochemistry 42: 9924–9936 [DOI] [PubMed] [Google Scholar]
- Rueda D, Bokinsky G, Rhodes MM, Rust MJ, Zhuang X, Walter NG 2004. Single-molecule enzymology of RNA: Essential functional groups impact catalysis from a distance. Proc Natl Acad Sci 101: 10066–10071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert J-P, Ciccotti G, Berendsen HJC 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23: 327–341 [Google Scholar]
- Salehi-Ashtiani K, Luptak A, Litovchick A, Szostak JW 2006. A genomewide search for ribozymes reveals an HDV-like sequence in the human CPEB3 gene. Science 313: 1788–1792 [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sefcikova J, Krasovska MV, Spackova N, Sponer J, Walter NG 2007a. Impact of an extruded nucleotide on cleavage activity and dynamic catalytic core conformation of the hepatitis δ virus ribozyme. Biopolymers 85: 392–406 [DOI] [PubMed] [Google Scholar]
- Sefcikova J, Krasovska MV, Sponer J, Walter NG 2007b. The genomic HDV ribozyme utilizes a previously unnoticed U-turn motif to accomplish fast site-specific catalysis. Nucleic Acids Res 35: 1933–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih IH, Been MD 2002. Catalytic strategies of the hepatitis δ virus ribozymes. Annu Rev Biochem 71: 887–917 [DOI] [PubMed] [Google Scholar]
- Soukup GA, Breaker RR 1999. Relationship between internucleotide linkage geometry and the stability of RNA. RNA 5: 1308–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Tagaya M, Hori T, Sakamoto T, Kurihara Y, Katahira M, Uesugi S 2002. Cleavage reaction of HDV ribozymes in the presence of Mg2+ is accompanied by a conformational change. Genes Cells 7: 567–579 [DOI] [PubMed] [Google Scholar]
- Taylor JM 2012. Virology of hepatitis D virus. Semin Liver Dis 32: 195–200 [DOI] [PubMed] [Google Scholar]
- Tinsley RA, Walter NG 2007. Long-range impact of peripheral joining elements on structure and function of the hepatitis δ virus ribozyme. Biol Chem 388: 705–715 [DOI] [PubMed] [Google Scholar]
- Tinsley RA, Harris DA, Walter NG 2003. Significant kinetic solvent isotope effects in folding of the catalytic RNA from the hepatitis δ virus. J Am Chem Soc 125: 13972–13973 [DOI] [PubMed] [Google Scholar]
- Tinsley RA, Harris DA, Walter NG 2004. Magnesium dependence of the amplified conformational switch in the trans-acting hepatitis δ virus ribozyme. Biochemistry 43: 8935–8945 [DOI] [PubMed] [Google Scholar]
- Todd GC, Walter NG 2013. Secondary structure of bacteriophage T4 gene 60 mRNA: implications for translational bypassing. RNA 19: 685–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraraghavan N, Bevilacqua PC, Hammes-Schiffer S 2010. Long-distance communication in the HDV ribozyme: insights from molecular dynamics and experiments. J Mol Biol 402: 278–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraraghavan N, Ganguly A, Golden BL, Bevilacqua PC, Hammes-Schiffer S 2011. Mechanistic strategies in the HDV ribozyme: chelated and diffuse metal ion interactions and active site protonation. J Phys Chem B 115: 8346–8357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadkins TS, Been MD 2002. Ribozyme activity in the genomic and antigenomic RNA strands of hepatitis δ virus. Cell Mol Life Sci 59: 112–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadkins TS, Perrotta AT, Ferre-D'Amare AR, Doudna JA, Been MD 1999. A nested double pseudoknot is required for self-cleavage activity of both the genomic and antigenomic hepatitis δ virus ribozymes. RNA 5: 720–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter NG 2001. Structural dynamics of catalytic RNA highlighted by fluorescence resonance energy transfer. Methods 25: 19–30 [DOI] [PubMed] [Google Scholar]
- Walter NG 2002. Probing RNA structural dynamics and function by fluorescence resonance energy transfer (FRET). Curr Protoc Nucleic Acid Chem 11: 11.10.11–11.10.23 [DOI] [PubMed] [Google Scholar]
- Walter NG 2007. Ribozyme catalysis revisited: Is water involved? Mol Cell 28: 923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter NG, Burke JM 2000. Fluorescence assays to study structure, dynamics, and function of RNA and RNA-ligand complexes. Methods Enzymol 317: 409–440 [DOI] [PubMed] [Google Scholar]
- Walter NG, Perumal S 2009. The small ribozymes: common and diverse features observed through the FRET lens. Springer Ser Biophys 13: 103–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter NG, Burke JM, Millar DP 1999. Stability of hairpin ribozyme tertiary structure is governed by the interdomain junction. Nat Struct Biol 6: 544–549 [DOI] [PubMed] [Google Scholar]
- Walter NG, Yang N, Burke JM 2000. Probing non-selective cation binding in the hairpin ribozyme with Tb(III). J Mol Biol 298: 539–555 [DOI] [PubMed] [Google Scholar]
- Walter NG, Chan PA, Hampel KJ, Millar DP, Burke JM 2001a. A base change in the catalytic core of the hairpin ribozyme perturbs function but not domain docking. Biochemistry 40: 2580–2587 [DOI] [PubMed] [Google Scholar]
- Walter NG, Harris DA, Pereira MJ, Rueda D 2001b. In the fluorescent spotlight: global and local conformational changes of small catalytic RNAs. Biopolymers 61: 224–242 [DOI] [PubMed] [Google Scholar]
- Webb CH, Luptak A 2011. HDV-like self-cleaving ribozymes. RNA Biol 8: 719–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb CH, Riccitelli NJ, Ruminski DJ, Luptak A 2009. Widespread occurrence of self-cleaving ribozymes. Science 326: 953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodson SA 2010. Taming free energy landscapes with RNA chaperones. RNA Biol 7: 677–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgarbova M, Otyepka M, Sponer J, Mladek A, Banas P, Cheatham TE III, Jurecka P 2011. Refinement of the Cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J Chem Theory Comput 7: 2886–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X, Kim H, Pereira MJ, Babcock HP, Walter NG, Chu S 2002. Correlating structural dynamics and function in single ribozyme molecules. Science 296: 1473–1476 [DOI] [PubMed] [Google Scholar]
- Zuker M 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]