

Abstract
Given the complexities of the mammalian CNS, its regeneration is viewed as the holy grail of regenerative medicine. Extraordinary efforts have been made to understand developmental neurogenesis, with the hopes of clinically applying this knowledge. CNS regeneration also involves glia, which comprises at least 50% of the cellular constituency of the brain, and is involved in all forms of injury and disease response, recovery and regeneration. Recent developmental studies have given us unprecedented insight into the processes that regulate the generation of CNS glia. Because restorative processes often parallel those found in development, we will peer through the lens of developmental gliogenesis to gain a clearer understanding of the processes that underlie glial regeneration under pathological conditions. Specifically, this review will focus on key signaling pathways that regulate astrocyte and oligodendrocyte development, and describe how these mechanisms are reutilized in these populations during regeneration and repair after CNS injury.
A central tenet of regenerative biology is that processes controlling tissue generation during development often control its regeneration: Therefore processes regulating developmental gliogenesis in the CNS are likely to provide critical insights into glial repair and its influence on homeostasis in the adult CNS. While injury and pathological states in the adult are comparatively more complex than embryonic and early postnatal development, viewing regeneration through the lens of development lends clarity as well as a starting point for greater insight into the fundamental processes governing glial repair. Therefore, this review will focus on key glial developmental mechanisms that are reused during glial regeneration, how these developmental processes are involved in functional recovery of the CNS, and how they contribute to key neurological disorders. Recently, microglia have been implicated as also playing key roles in CNS regeneration, however because they have different developmental origins from CNS glia, this review will not cover microglia. In discussing some of these fundamental mechanisms and the crucial cellular interactions involved in glial regeneration, we also hope to demonstrate the potential for future interventions based on identifying putative therapeutic targets. Other excellent review articles have recently considered these topics (Burda, 2014), and we will refer to them in providing an overview of the field within the limits of the present article.
The ability of an organism to repair and regenerate its injured nervous system often correlates with the organism’s longevity and complexity. For example, flies and worms have short lifespans and limited capacity to repair their “CNS,” while mice and humans live comparatively much longer and have remarkable ability to repair their damaged CNS. The other side of this equation is that more can go wrong in a CNS with more “moving parts” (cellular elements and complex interactions). Thus, a level of quality control must be in place to ensure that homeostasis is maintained; and the longer you live, the more important this becomes.
In terms of CNS complexity, what key feature separates invertebrates from vertebrates? Besides the existence of many more neuronal subtypes in vertebrates vs. invertebrates, glia are clearly a major differentiating element (Freeman and Rowitch, 2013). Invertebrates have a significantly lower proportion of glial cells, with C. elegans displaying a ratio of 56 glia to 352 neurons (~1:6 ratio), while the glia-to-neuron ratio in vertebrates ranges from 1:1 to 4:1 (depending who you ask!). Coupled with their increasing representation, vertebrates’ glial cells also show escalating diversity and functional complexity. Particularly, oligodendrocytes (OLs) and myelin sheaths, which are not present in smaller invertebrates, are necessary and essential adaptations for rapid nerve conduction in axons of larger vertebrates. And while aspects of astrocyte function are conserved across species (i.e., glutamate transport), their number and morphological complexity largely increase from mice to humans. Indeed, a single hippocampal astrocyte is estimated to make contacts with ~100,000 synapses (Bushsong, et al. 2002). This estimate, coupled with recent reports that transplanting human astrocytes into the mouse brain enhances learning and memory, also suggests additional, undiscovered roles for astrocytes in cognition, further reinforcing their central and increasingly complex role in CNS physiology in higher organisms (Han et al., 2013).
This evolutionary evidence, together with the rapid progress over the last two decades in our understanding of glial biology and physiology, demonstrates an essential role for glia in CNS function. Astrocytes and OLs are able to regenerate in response to CNS injury, and glial regeneration and repair are essential for long-term homeostasis and for complete recovery of integrated functions. Given their unique partnerships with neurons and the extremely limited degree of neurogenesis in the adult CNS, this capacity also functions to preserve neuronal populations post-injury. Furthermore, various types of glial progenitors have the potential to generate neurons under pathological conditions. Thus, glial responses to injury and disease serves two main purposes: 1) repair and preservation of existing cell populations, and 2) regeneration of lost populations, including both neurons and glia.
Astrocyte Development
Generation of astrocytes involves a complex interplay of intrinsic and extrinsic cellular signals that act on neural stem cells (NSCs) and precursor populations to direct their formation. As with the development of any cell lineage, astrocyte differentiation entails a sequential series of events that result in the generation of a mature cell population that actively participates in CNS physiology. Unlike OLs, however, the stages of astrocyte lineage development are poorly defined, lacking stage-specific markers and clearly defined developmental endpoints (Molofsky et al., 2012). Moreover, while astrocytes are a functionally heterogeneous population, the cellular nature of their heterogeneity is just starting to emerge (Chaboub and Deneen, 2012). Many processes associated with astrocyte development have long been shrouded in mystery, but an existing base of knowledge can be used to frame our discussion on reactive astrocytes. The following sections briefly review key aspects of astrocyte development germane to reactive astrocytes and repair (Figure 1).
Figure 1. Development of the Astrocyte Lineage.
Schematic synopsis of the cellular and molecular processes that oversee the specification and differentiation of the astrocyte lineage. Unlike neuronal- or oligodendrocyte- lineage development, the intermediate stages of astrocyte lineage development remain poorly defined, due in part, to the lack of reliable markers and clearly defined functionalendpoints.
Specification and Differentiation
During development, neurogenesis precedes gliogenesis. The transition is often used as a model for identifying factors that regulate specification of astrocytes from NSC populations. This developmental interval – the gliogenic switch – is best characterized in the ventral spinal cord, where several recent studies found that transcription factors Sox9 and NFIA comprise a transcriptional cascade that regulates glial specification (Deneen et al., 2006; Kang et al., 2012; Stolt et al., 2003). Subsequently Sox9/NFIA associate and regulate a set of genes implicated in astrocyte precursor migration and metabolism, key aspects of lineage development (Kang et al., 2012). Interestingly, Shh and Notch signaling have been shown to regulate Sox9 and NFIA, respectively, linking these factors to key developmental pathways active in ventricular zone (VZ) populations (Namihira et al., 2009; Scott et al., 2010). Several studies have implicated both Shh and Notch signaling in astrocyte development and differentiation, though in different regions of the CNS and using different experimental models (Chambers et al., 2001; Garcia et al., 2010).
After specification, astrocyte precursors migrate away from the germinal centers and begin to differentiate. This aspect of astroglial lineage development is defined by the induction of glial fibrillary acidic protein (GFAP), originally identified in reactive astrocytes found in demyelinated multiple sclerosis (MS) plaques (Eng et al., 2000; Gerstl et al., 1970). While GFAP has long been miscast as the definitive marker of astrocytes, identifying the processes that regulate its expression gave crucial insight into the mechanisms regulating astrocyte differentiation. Among key pathways implicated in GFAP regulation, CNTF/LIF/CT-1 cytokine signaling through gp130 and JAK/STAT has been shown in several developmental models to be a key regulator of GFAP and astrocyte differentiation as a whole (Barnabe-Heider et al., 2005; Bonni et al., 1997; Miller and Gauthier, 2007; Sun et al., 2001). Additionally, Notch- and BMP-signaling have been implicated in regulating GFAP-expression and some aspects of astrocyte development, including inhibition of alternative cell fates (Bonaguidi et al., 2005; Grinspan et al., 2000; Namihira et al., 2009; Taylor et al., 2007). Newborn neurons are thought to be the source of these signaling molecules, as separate studies have suggested that CT-1 and Jagged/delta released from neurons promote astrocyte differentiation in vitro (Barnabe-Heider et al., 2005; Namihira et al., 2009). Identifying the in vivo sources of key astrocyte-promoting signaling molecules during development is important for understanding the genesis of reactive astrocytes after CNS injury. Given the complex and heterogeneous cellular response to injury, a knowledge of the normal sources of these key cytokines and signaling molecules can provide insight into paracrine signaling mechanisms that operate after injury (Burda, et al. 2014).
Proliferative Properties
A distinguishing feature of gliogenesis is the proliferation of precursor populations outside the germinal centers. Both OL progenitor cells (OPCs) and astrocyte precursors (ASPs) display robust proliferative potential post-specification (Ge et al, 2012.; Tien et al., 2012; Fruttiger et al., 1999; Richardson et al., 2011). In the early postnatal cortex, up to 55% of GFAP+ astrocytes are generated after migrating away from the germinal centers (Ge et al., 2012). Similarly, in the developing spinal cord, ASP populations retain proliferative potential post-specification, suggesting the existence of an “intermediate astrocyte precursor” (IAP) that governs the expansion of astrocyte numbers throughout lineage development (Tien et al., 2012). The mechanisms governing ASP or IAP proliferation remain poorly defined, but recent studies implicate BRAF-ERK signaling in the spinal cord (Tien et al., 2012). Interestingly, separate studies in the cortex indicate that MEK1/2, which also signals through ERK, regulates gliogenesis through the CNTF-STAT3 axis (Li et al., 2012). Together, these studies suggest a link between the proliferative and differentiative processes that function in ASPs post-specification. Thus, it will be important to identify additional pathways that couple (or uncouple) ASP proliferation and differentiation, and to determine how the existing programs are reused in reactive astrocyte proliferation.
Emerging Heterogeneity
Astrocytes can be broadly classified into two categories: i) protoplasmic astrocytes that occupy the grey matter, and ii) fibrous astrocytes that occupy the white matter (WM). However, an abundant cell type capable of a variety of physiological functions is likely to comprise a vast, reservoir of subtypes and classes within the adult CNS. Indeed, Cajal described the presence of several morphologically distinct types of astrocytes in the cerebellum (Cajal, 1909). Recent studies have begun the complicated task of delineating astrocytes’ cellular heterogeneity, guided by the principles of developmental patterning. A set of recent studies using genetic and lineage-tracing approaches found that molecularly distinct subtypes of astrocytes originate from distinct domains in the developing spinal cord, indicating embryonic patterning as an organizing principle in astrocyte, as well as neuronal, diversity (Hochstim et al., 2008; Tsai et al., 2012). While these patterning principles have yet to be applied to diverse astrocyte populations in the adult brain, a recent study using GFAP- and s100β-reporter mice identified nine morphologically distinct classes of astrocytes distributed in varying constituencies across the adult brain (Emsley and Macklis, 2006). Given the underlying functional heterogeneity of astrocytes and their reactive counterparts, a more comprehensive understanding of their cellular heterogeneity will further resolve their roles after CNS injury.
Astrocytes in CNS injury and repair
With their key role in subserving normal neuronal function by operating as a bridge from neurons to the outside world, a natural extension of astrocyte function after injury would be to reestablish homeostasis in a pathological microenvironment. Reactive astrocytes, the astrocytic response to CNS injury, have been identified in nearly all cases of CNS injury, degeneration, and disease, from multiple sclerosis (MS) and Alzheimer’s disease, to glioma, spinal cord injury (SCI), and stroke (Sofroniew and Vinters, 2010). Despite their ubiquity in injury and disease, the properties, functions, and contributions of reactive astrocytes to CNS repair remain somewhat mysterious. Below, we summarize what is known about reactive astrocytes and their contributions to CNS repair, and how recent studies have changed long-standing views of astrocyte reactivity.
General Properties
Sites of CNS trauma or disease are populated by reactive astrocytes with three general properties: i) expression of GFAP, ii) cellular hypertrophy, and iii) cellular proliferation. Thus, reactive astrocytes can be identified by a combination of molecular markers and cell morphology, coupled with accurate localization of injury. These properties are graded, with increasing proliferation and GFAP expression correlated with more severe injury. Their increased proliferation in severe injury disrupts non-overlapping “tiled” domains of astrocytes, and forms densely packed areas containing extensive overlapping of astrocytic processes.
Astrocytes play diverse and crucial roles in normal CNS physiology, regulating synaptogenesis, neurotransmission, metabolic support, blood-brain-barrier (BBB) formation/maintenance, and other functions not yet characterized (Alvarez et al., 2013; Halassa and Haydon, 2010; Matyash and Kettenmann, 2010). Unsurprisingly, injury-induced proliferation, hypertrophy, and tiling disorganization are likely to disrupt these normal functions; and the introduction of new signaling molecules in an injury milieu raises the question of whether reactive astrocytes after an injury are demonstrating a loss of normal functions or the acquisition of new ones (Burda et al., 2014). It seems likely that normal functions such as synaptic transmission, metabolic support, and BBB formation would aid in repair. Indeed, several studies have linked these functions to reactive astrocytes and repair (Faulkner et al., 2004; Rothstein et al., 1996; Swanson et al., 2004; Voskuhl et al., 2009). But the emergence of new functions, such as managing the immune response to injury, also contributes to the repair process (Dong and Benveniste, 2001; Farina et al., 2007; John et al., 2003). While the exact functions of reactive astrocytes after injury remain poorly defined, one key question remaining is whether different types of injury elicit differential responses from reactive astrocytes and, ultimately, from reactive astrocytes with different repair properties. Recent studies have revealed that stab wounds and ischemia give rise to reactive astrocytes with NSC potential, while degenerative models do not (Shimada et al., 2012; Sirko et al., 2013). These observations hint at complex interplays between type of injury, location, and reactive astrocyte response and function, which are only now being recognized. Decoding these relationships will be essential to understanding how astrocytes respond and contribute to CNS repair.
Good Cop? Bad Cop? Both?
Given the inherent complexity of injury states and the heterogeneous astrocytic response, perhaps a better question at this point is: Are reactive astrocytes detrimental or beneficial to CNS repair? The long-standing view of reactive astrocytes is that they are “bad actors” in the repair process. This view derives from the inhibitory effects of glial scarring on axon regeneration after SCI. The glial scar around trauma sites results from reactive astrocyte proliferation and subsequent accumulation of an astrocytic barrier that physically impedes axon regeneration and releases chondroitin sulfate proteoglycans (CSPGs), which inhibit axon growth (Busch and Silver, 2007; Morgenstern et al., 2002). These observations raise a simple question: If the aftereffects of glial scarring are so bad, why do scars form in the first place? There is general consensus that the reactive astrocyte-glial scar axis functions to “wall off” the damaged area, preventing the spread of inflammatory cells or infectious agents to adjacent healthy tissues; and the axonal inhibitory effects are the cost of this benefit (Rolls et al., 2009). Indeed, ablation or genetic manipulation of reactive astrocytes after SCI increases local inflammation and neuron loss, and generally inhibits recovery, indicating that they are essential for repair (Herrmann et al., 2008; Sofroniew, 2009).
The view that reactive astrocytes inhibit CNS repair is evolving, as numerous studies have found that they enhance recovery in a variety of CNS injury models. While neuronal recovery garners much of the attention, reactive astrocytes also contribute to recovery after white matter injury (WMI), being required for remyelination in several lesion models (Franklin et al., 1991; Talbott et al., 2005). In more complex, inflammatory EAE models of WMI, the loss of reactive astrocytes increases inflammation and neuronal loss, although in some cases it also promotes recovery (Brambilla et al., 2009; Voskuhl et al., 2009). Thus, in cases of neuronal or WM repair, despite emerging evidence that reactive astrocytes facilitate the recovery process, their deleterious effects may impair long-term recovery for the benefit of immediate stability. One key area in gaining an understanding of this duality is the nature of astrocyte heterogeneity. Reactive astrocytes are clearly not homogeneous, and given that normal astrocytes play diverse roles in the normal CNS, it is likely that analogous populations of reactive astrocytes exist after injury and during repair. Future studies should aim at decoding these relationships, as more work in this area is clearly needed.
Linking Astrocyte Development to CNS Injury
The developmental processes regulating how astrocytogenesis contributes to CNS repair remain poorly understood. Unlike the restoration of OLs and neurons, the restoration of astrocyte populations is not a major feature of CNS repair. Thus, the developmental processes that regulate astrocytogenesis are not implemented in a manner paralleling development (see OL sections below). Second, it is possible that astrocytes acquire new properties after injury that are not necessarily associated with their normal functions, but are associated with: i) a new set of regulatory factors, or ii) implementation of the same pathways under vastly different conditions (embryo vs. injured adult). Our basic knowledge of astrocyte development has lagged behind that of OLs and neurons, and this significant gap, combined with the preceding points, widens the chasm between our understandings of normal and injury-associated astrocytes.
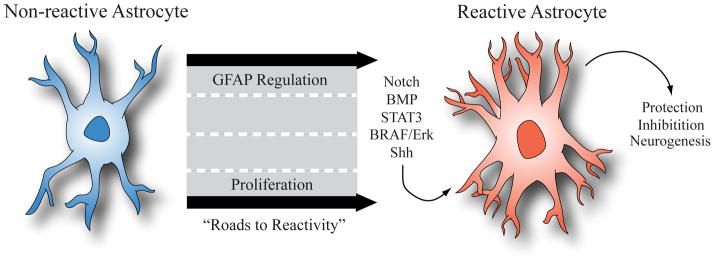
Despite these limitations, fundamental aspects of lineage development that have been molecularly characterized in normal astrocytes and are associated with reactive and regenerative astrocytes may give insight into their injury-associated responses. These features include GFAP expression and cell proliferation. While the molecular processes regulating these facets of astrocyte development are unlikely to be fully conserved after injury, these developmental criteria provide an entryway into the nature of reactive astrocytes after CNS injury (Figure 2).
Figure 2. Astrocyte Roads to Reactivity.

The two key processes associated with astrocyte development that directly contribute to the reactive astrocyte phenotype are, proliferation and GFAP-induction. Molecular regulation of these processes during development is viewed as the road to astrocyte reactivity and understanding how they are recapitulated after injury will serve as a starting point for understanding the nature of reactive astrocytes.
Regulation of GFAP: Gateway to Reactive Astrocyte Signaling
For years, GFAP has been used as the definitive marker of normal astrocyte populations. In some ways this was necessary due to a dearth of reliable astrocyte markers, but in other ways it has hindered the field, as GFAP is not a universal astrocyte marker and relying on it further clouds an already grey area. GFAP has received a lot of bad press lately as the field slowly turns to markers such as Aldh1l1 and Glt1 that are universally expressed in normal astrocyte populations (for complete list, see Molofsky et al., 2012).
Despite these limitations, GFAP upregulation remains a cardinal feature of reactive astrocytes, and logic dictates that the signaling pathways and transcription factors regulating GFAP expression are likely to be associated with reactive astrocytes. This area has received intense investigation, implicating several key signaling pathways and associated transcriptional regulators including BMP, LIF/CNTF, and Notch, in the regulation of astrocyte differentiation and GFAP expression (see above). Each of these pathways has been linked to reactive astrocytes and, given their broad reach, it is likely that they contribute to aspects of reactive astrocyte physiology and function outside of GFAP regulation. Therefore, one logical extension is that GFAP regulation serves as a gateway for identifying factors that influence reactive astrocytes and their biology. Indeed, several recent studies on these core GFAP regulatory pathways in reactive astrocytes have revealed a broader scope of functions after CNS injury, as well as new insights into the biology of reactive astrocytes.
Notch signaling
Notch signaling is a central pathway used by astrocytes during development that regulates both the specification of astrocyte precursors from NSCs and their subsequent maturation. Surprisingly, it has only recently been characterized in reactive astrocytes. Notch receptors are upregulated in reactive astrocytes after CNS injury, and inhibition of the Jagged 1 ligand suppresses GFAP and Endothelin receptor-B (ETB-R) expression in reactive astrocytes found in focally demyelinated lesions (Gadea et al., 2008; Morga et al., 2009; Hammond et al., 2014). Furthermore, γsecretase, a key intracellular regulator of Notch signal transduction, is also upregulated in mouse models of stroke (Arumugam et al., 2006). Interestingly, antisense inhibition of Notch intracellular domain (NICD) expression or treatment with γsecretase inhibitors improved functional recovery after cerebral artery occlusion (Arumugam et al., 2006). The improved recovery correlated with increased immune invasion of the peri-infarct region and a decrease in proliferating astrocytes. Conditional deletion of Notch1 using GFAP-CreER™ similarly resulted in increased immune invasion and decreased astrocyte proliferation, but did not improve recovery, suggesting that the Notch pathway functions in multiple cell systems present in CNS injury (Shimada et al., 2011).
The above studies suggest that suppressing Notch signaling will decrease the reactive astrocyte “load” and thus improve recovery. However, reactive astrocytes also promote CNS recovery, when combined with different injury modalities, in different brain regions, and with different astrocyte constituencies, indicating clearly that the recovery landscape is not a “unified field.” The same principles apply to Notch signaling in reactive astrocytes: A recent landmark study using a photothrombic injury model revealed that Notch signaling is required for subventricular zone (SVZ)-induced astrocytogenesis post-injury (Benner et al., 2013). The authors also demonstrated that Notch-regulated, Thbs4-expressing astrocytes comprise a unique subset of SVZ astrocytes that contribute to microvascular repair. The role of Notch signaling in reactive astrocytes is only now emerging; however, it seems likely to be region- and, perhaps even, injury model-specific. This is underscored by recent studies in an LPS WMI model, in which reactive astrocytes suppress remyelination through ET-1 activation of the Notch ligand Jagged 1 (Hammond et al., 2014). At the cellular level, this suppression is mediated by Jagged 1 activation of Notch-signaling in neighboring OPCs, inhibiting their differentiation. These recent advances in our understanding of Notch signaling in reactive astrocytes highlight the underlying complexity and suggest a nuanced role for this key developmental pathway in reactive astrocytes during CNS recovery.
STAT3
IL-6, CNTF, and CT-1 cytokines have all been linked to GFAP expression through activation of STAT3 in astrocytes (Miller and Gauthier, 2007). Since the discovery that activated STAT3 is a key transcriptional regulator of GFAP expression, STAT3 expression has been chronicled in reactive astrocytes in various injury models across several CNS regions, becoming a hallmark of reactive astrocytes (Choi et al., 2003; Justicia et al., 2000; Xia et al., 2002). Its role in reactive astrocytes is best characterized in glial scarring after SCI, where conditional deletion of STAT3 in NSCs or astrocytes significantly impairs functional recovery (Herrmann et al., 2008; Okada et al., 2006). In both these studies, glial scar formation was compromised by decreased proliferation and migration of STAT3−/− astrocytes, resulting in greater inflammation and lesion volume. Mechanistic studies suggest that STAT3 mediates scar formation and recovery by regulating responses to oxidative stress and sequestration of inflammatory cells (Sarafian et al., 2010).
Generation of reactive astrocytes after injury occurs in a cellular milieu comprising several cell types, including immune cells and OLs. Given that reactive astrocytes are generated in a complex pathological cellular environment, it is no surprise that the cytokines regulating STAT3 expression are also found in inflammatory cells in several injury models (Choi et al., 2003; Okada et al., 2004; Sriram et al., 2004). Equally interesting is the cellular interplay of OLs, microglia, and reactive astrocytes orchestrated by STAT3. In a recent study, deletion of STAT3 in astrocytes suppressed remyelination after WMI. Mechanistically, STAT3−/− astrocytes promoted TGFβ1 expression in microglia, which subsequently suppressed OPC differentiation after injury (Nobuta et al., 2012). This study highlights not only new roles for STAT3 in reactive astrocytes, but also the complexities of paracrine signaling between cellular compartments present in injury. Undoubtedly, future studies will investigate whether similar paracrine mechanisms underlie STAT3 function in reactive astrocytes in other CNS injury and regeneration models, as injury conditions or location may dictate the STAT3-response in reactive astrocytes themselves.
BMP signaling
The convergence of cellular constituency and signaling complexity after CNS injury - and its influence on regeneration and repair processes - are perhaps best illustrated by the BMP pathway. BMP signaling plays a key role in astrocyte differentiation and is also activated in reactive astrocytes after various CNS injuries, including SCI, ischemia, hypoxia, and WMI (Lewen et al., 1997; Martinez et al., 2001; See and Grinspan, 2009; Setoguchi et al., 2001). Several groups have manipulated BMP signaling in a variety of injury models by administering noggin or BMP ligands, each having a different effect on repair. For example, adding BMP ligand after ischemic injury promoted functional recovery, while several studies indicate that BMP secreted from reactive astrocytes suppresses remyelination in WMI models (Chang et al., 2003; Martinez et al., 2001; Sabo et al., 2011; Wang et al., 2011). The latter findings are supported by several developmental studies, indicating that BMP signaling suppresses normal OPC differentiation (Gomes et al., 2003; See et al., 2004). Thus, BMP signaling in reactive astrocytes during injury response is both detrimental and beneficial, depending on the type of injury and the cell population being repaired. This duality is best shown in SCI models, where conditional deletions of BMPR1a or BMPR1b in astrocytes have opposing effects on glial scar formation (Sahni et al., 2010). BMPR1a-cKO demonstrate reduced gliosis, increased lesion volume, and impaired motor recovery, while the BMPR1b-cKO display increased GFAP expression and decreased lesion volume in acute injury, accompanied by reduced scarring in chronic states. The vastly different effects of these receptor subunits on reactive astrocytes in SCI add another layer of regulatory complexity to BMP signaling in CNS injury. Such complexities are likely to be present in other signaling systems that operate in reactive astrocytes or other cell types associated with injury, but have yet to be discovered.
Astrocyte Proliferation: A path to regeneration?
The mechanisms regulating astrocyte proliferation during development are poorly understood, partly because a clearly defined post-mitotic stage has not been identified during astrocyte maturation. However, the presence of GFAP-expressing astrocytes with proliferative potential in the postnatal cortex parallels the transient amplifying “astrocyte” population in the stem cell niche of the SVZ, allowing us to infer that normal astrocytes harbor latent NSC potential associated with their proliferative response to injury (Kriegstein and Alvarez-Buylla, 2009). Reactive astrocytes demonstrate enhanced production of multipotent NSCs in vitro, suggesting that under proper conditions proliferating reactive astrocytes can acquire NSC-like properties (Robel et al., 2011), and raising the question of whether the mechanisms controlling post-injury astrocyte proliferation similarly regulate de-differentiation to NSCs and possible neuronal regeneration. Several recent studies are unraveling the processes regulating astrocyte proliferation and their role, if any, in the astrocyte-NSC axis.
BRAF/Mek/ERK signaling axis
Recently Mek and BRAF have been implicated in astrocytogenesis and precursor proliferation during development, respectively (Li et al., 2012; Tien et al., 2012). Given that both Mek and BRAF function through the MAPK-ERK signaling axis, these findings implicate a central signaling pathway in astrocyte generation. While specific expression of activated Mek and BRAF in reactive astrocytes remains limited, activated ERK and MAPK have been detected in reactive astrocytes in various injury models (Carbonell and Mandell, 2003; Mandell et al., 2001; Mandell and VandenBerg, 1999). In ischemia, pharmacological manipulation of p38 MAPK or ERK promotes recovery, though its effect on reactive astrocyte proliferation was never examined (Gadea et al., 2008; Namura et al., 2001). In WMI injury models, activated ERK is present in reactive astrocytes and mediates ET-1-dependent astrocyte proliferation in lesions (Gadea et al., 2008). ERK has also been implicated recently in S1P signaling in reactive astrocytes associated with human MS (Fischer et al., 2011). However, the precise cellular site of action and its influence on the role of MAP-ERK signaling repair remain poorly defined.
More broadly, delineating the upstream and downstream biology surrounding possible BRAF-Mek-ERK signaling in reactive astrocytes is extremely complex. This pathway mediates a myriad of cellular responses in various forms of CNS injury. However, in the context of the astrocyte-NSC axis, EGF signaling plays a major role in NSC formation and functions through the MEK-ERK pathway, which is also implicated in NSC formation (Burrows et al., 1997). This fact, coupled with observed expression of EGFR in reactive astrocytes and EGF in sites of injury, leads to speculation that EGF signaling through the BRAF-Mek-ERK axis in proliferating reactive astrocytes provokes conversion to a NSC-like identity, ultimately stimulating neurogenesis (Planas et al., 1998). Such endogenous repair mechanisms are intriguing and somewhat advantageous, but newborn neurons derived from reactive astrocytes after injury have not been documented in vivo, and cortical transplantation of NSCs derived from reactive astrocytes from ischemia models did not support neurogenesis (Shimada et al., 2012).
Sonic Hedgehog Signaling
The role of Shh in normal astrocytes is complex, as it is required for patterning and therefore indirectly contributes to generating patterned astrocyte subpopulations (Ericson et al., 1997; Hochstim et al., 2008). Consistent with a role in astrocyte development, deletion of smoothened (Smo) results in gliosis in subsets of astrocytes in the cortex and spinal cord, suggesting that Shh signaling suppresses their proliferation and implying stage-specific roles for Shh in astrocyte patterning and differentiation (Garcia et al., 2010; Yu et al., 2013). However, in a stab-wound/freeze CNS injury model, Shh is expressed in reactive astrocytes and pharmacological inhibition via cyclopamine impairs their proliferation (Amankulor et al., 2009). Further evidence of a role for Shh is the expression of the Shh target, Olig2, in reactive astrocytes associated with varied CNS injuries. Analogous to Shh inhibition, conditional deletion of Olig2 in astrocytes results in decreased reactive astrocyte proliferation after injury (Chen et al., 2008). Thus, the role of Shh appears to vary significantly between normal and reactive astrocytes and points again to the differing cellular milieus in development and in injury.
Given the link between astrocyte proliferation and NSC-like states, and the effect of Shh as a potent inducer of NSCs and reactive astrocyte proliferation, it seems natural that Shh may regulate aspects of the astrocyte-NSC axis associated with injury. Indeed, these observations were beautifully integrated in a recent study characterizing Shh responses in reactive astrocytes across a spectrum of CNS injury models (Sirko et al., 2013). The authors demonstrate that reactive astrocytes acquire NSC-like properties in stab-wound, but not degenerative CNS injury. Shh expression correlates with reactive astrocytes having NSC-like properties; and conditional knockout of Smo in reactive astrocytes decreases their proliferative response and NSC-forming capacity. These observations reinforce the broader point that not all reactive astrocytes are “created equal” and this heterogeneity may be induced by injury states. Alternatively, the role of Shh signaling in patterning and astrocyte diversity may also indicate that subsets of astrocytes are differentially responsive to signaling and act accordingly. Indeed, recent studies in human ESCs found that astrocytes derived from Olig2-expressing neural progenitor cells promote recovery after ischemia, suggesting that Shh signaling may be differentially used in reactive astrocyte populations after injury (Jiang et al., 2013).
While provocative and promising, these findings must be interpreted with tempered optimism, as characterizing the NSC nature of these reactive astrocytes relies on in vitro analysis. In vitro culture is well known to change the properties of CNS-derived cells, and the Shh-Olig2 axis is especially prone to such deregulation in NSC culture (Gabay et al., 2003). Definitive proof that reactive astrocytes acquire NSC-like properties with neurogenic potential will require complementary conditional lineage tracing analysis, to distinguish the “actual” from the “possible” (Anderson, 2001).
Oligodendrocyte Development
Our knowledge of OL development derives from an extensive range of genetic and biochemical tools – particularly cellular markers and cell-specific gene promoters – that have enabled detailed characterization of OPCs and lineages able to generate OLs during embryonic and postnatal development (Emery, 2010a; Kessaris et al., 2006; Richardson et al., 2006). These tools have also been used to analyze OL turnover and regeneration in adult brain (Fancy et al., 2011; Franklin and ffrench-Constant, 2008; McTigue and Tripathi, 2008; Miller and Mi, 2007). The opportunity to identify and fate-map developing OLs in diverse brain regions has revealed cellular factors and intrinsic molecular pathways functionally involved in OL development and regeneration. This analysis has also illuminated and defined cellular interactions between OLs and other neural cells with critical roles in development and pathology. Damaged OLs can be replaced by new cells derived from OPCs that proliferate and differentiate after injury or in disease. Thus, studies elucidating altered development in a perturbed cellular environment will reveal how interactions between intrinsic OPC properties and the injury milieu determine the cells’ fate. As many excellent articles have recently reviewed OL development in embryonic and postnatal brain (Emery, 2010b; Liu and Casaccia, 2010; Meijer et al., 2012; Miller, 2002; Mitew et al., 2013; Nishiyama et al., 2009; Richardson et al., 2006), we will focus on some salient aspects of this topic.
Embryonic Development
In the embryo, OLs originate from multiple progenitor cell pools, which populate distinct regions of the developing spinal cord and brain, and mature during different time periods. Although a specialized OL domain (pMN domain) was originally thought to exist in the ventral ventricular zone (VZ) of the spinal cord and perhaps in the forebrain (reviewed in Rowitch, 2004), recent investigation has demonstrated dorsal – as well as ventral – origins of OLs in both spinal cord and brain (reviewed in Richardson et al., 2006). This conclusion is based on analysis of Nkx6.1 and Nkx6.2 double-mutant mice, in which pMN-derived OPCs are absent from the spinal cord due to impaired Olig2 activation by Nkx6. In these mice, OPCs that express all the proper markers – including the paired box gene Pax7 – are still produced in the dorsal spinal cord (Cai et al., 2005; Vallstedt et al., 2005). Further Cre-lox fate-mapping analysis using Dbx1 or Msx gene promoter elements to control Cre recombinase expression showed that dorsally derived OPCs arise from different domains and generate 10–50% of dorsal OLs, depending on their origin (Fogarty et al., 2005). Dorsal OPCs are also generated later in development than pMN-derived OPCs (Cai et al., 2005; Fogarty et al., 2005; Vallstedt et al., 2005). Shh signaling activates Olig2 in the pMN domain (Lu et al., 2002), whereas dorsally derived BMPs and Wnts support generation of astrocytes and prevent OL development at earlier embryonic stages (Mekki-Dauriac et al., 2002; Shimizu et al., 2005; Vallstedt et al., 2005). It has also been suggested that FGF signaling is required to specify OLs in the dorsal spinal cord (Vallstedt et al., 2005). In any case, distinct and restricted OPC domains exist in developing spinal cord and are responsible for multiple waves of OPC production influenced by different cellular factors.
In the forebrain, fate-mapping analysis also demonstrated distinct temporal waves of OPC production from separate domains (Kessaris et al., 2006). The main separate domains identified were the medial ganglionic eminence (Nkx2.1-derived ventral precursors from about E12.5), the lateral ganglionic eminence (Gsh2-derived; a few days later), and the cortex Emx1-derived precursors that produce OPCs predominantly after birth. All precursors migrate vigorously from the VZ to populate distant brain regions, and the population of most ventrally derived precursors disappears after birth (Kessaris et al., 2006).
Postnatal Development
In the postnatal brain, the SVZ is the major source of OPCs and OLs (Aguirre et al., 2007; Menn et al., 2006; Nait Oumesmar et al., 1999). Located around the tips of the lateral ventricles, this region is mostly derived from the embryonic lateral ganglionic eminence and lateral cortex. Pioneering work using retroviral labeling of dividing cells in the SVZ showed that cells in this brain region are able to generate both OLs and astrocytes (Levison and Goldman, 1993); and more recent analysis of postnatal and adult brains demonstrated that proliferative OPCs in the SVZ express NG2 and Olig2, as well as other OPC and progenitor markers including PDGFRα and Mash1 (Aguirre et al., 2004; Aguirre et al., 2007; Menn et al., 2006). In the adult SVZ, OPCs express a type C cell phenotype and derive from type B stem cells (Aguirre et al., 2004). During postnatal development, OPCs migrate from the SVZ into WM regions, where they stop dividing and differentiate into mature myelinating OLs (Aguirre et al., 2007; Gonzalex-Perez et al., 2009; Levison and Goldman, 1993; Menn et al., 2006). The accessibility of the postnatal SVZ and its ease of manipulation have facilitated the analysis and identification of cellular factors and molecular pathways regulating OPC proliferation, migration, and differentiation. This highly gliogenic region of the postnatal/adult brain has been intensively studied in animal models of injury and pathology to define pathways that could be used for OL regeneration.
Oligodendrocyte Regeneration in the Developing Brain
Animal Models of Developmental Injury and Pathology
This section will focus on examples of pathological insults in the immature brain, to illustrate the differential responses of distinct OPC populations and to relate these responses to regeneration and repair. We limit our discussion to models of injury and pathology that have contributed to defining cellular and molecular pathways involved in OL and myelin regeneration and repair during development. OPCs’ ability to react to injury may be greater in the young adult brain brain (Ruckh et al., 2012) and these early mechanisms are potentially applicable to the less favorable environment of the pathological adult brain (van Wijngaarden and Franklin, 2013). Furthermore, interventions targeting specific progenitor cells and molecular pathways may be more successful during a period of ongoing developmental programs that would permit a higher degree of neural plasticity. Identification of signalling systems activated after brain injury or during the course of neurological disorders can guide strategies that lead to more robust amplification of the OPC pool and to enhanced repopulation, regeneration, and functional recovery of the injured brain.
Perinatal hypoxia and ischemia
Animal models of neurodevelopmental disorders and neonatal brain injury attempt to reproduce different aspects of the pathologies found in humans. However, these animal models should satisfy the following criteria: i) the model must accurately reflect the histopathological spectrum of a specific injury to the developing brain; ii) the model should display a phenotype that correlates with developmental changes observed in humans, and iii) the model should display abnormal functional outcomes also observed in human pathology.
Cerebral WMI and brain gray matter atrophy are the most frequent brain insults in preterm infants and major causes of disability in survivors. WM abnormalities in preterm infants persist throughout adolescence and young adulthood (Hack et al., 2002). Chronic periventricular white matter injuries (PWMIs) comprise a broad spectrum, including the most severe form – focal cystic necrotic lesions (periventricular leukomalacia; PVL) – or diffuse hypomyelination (diffuse white matter injury; DWMI). PWMI is directly associated with adverse outcomes such as cerebral palsy, epilepsy, cognitive delay, and learning disabilities (Hack et al., 2002). Although other animal models have been extensively utilized (Back and Rosenberg, 2014; Salmaso et al., 2014), the two primary animal models of premature brain injury represent hypoxia/ischemia (HI) (Rice et al., 1981) and chronic perinatal hypoxia (HX) (reviewed in Back et al., 2006; Scafidi et al., 2009; Vaccarino and Ment, 2004). Both models result in neuropathological changes resembling those seen in premature human infants, including severe hypomyelination and decreased gray matter volume (Salmasio et al., 2014).
Glial cell types susceptible to insult in chronic PWMI have been identified by their expression of cell-specific markers. OLs are particularly vulnerable to PWMI, but microglial activation and astrogliosis are also observed, along with axonal injury. In chronic PWMI, the maturation of late OPCs/pre-OLs is believed to arrest at a pre-myelinating stage, contributing to myelination failure in PWMI (Back et al., 2002). Indeed, maximal PWMI susceptibility in preterm infants and perinatal rodents corresponds to developmental stages characterized by a sizeable and susceptible population of late OPCs (Back, 2006). In perinatal HX, analysis of different cell populations in the OL lineage also indicated occurrence of delayed OL maturation, along with specific abnormalities in myelin ultrastructure, even when overall myelination levels appeared normal (Jablonska et al., 2012; Scafidi et al., 2014). Consistent with this finding, WM changes showed recovery in some prematurely born adolescents or young adults, but physiological and behavioral abnormalities of WM function still occurred (Nagy et al., 2003; Fjell et al., 2011). Thus, early intervention is critical for OL regeneration after PWMI and requires identification of unique specific cell populations in the brains of premature infants to promote WM recovery after neonatal injury.
Sources of OPCs
Paralleling the effects on OPC differentiation and delayed OL maturation described above, HI- or HX-induced WMI also produced regenerative responses. Several studies demonstrated enhanced SVZ progenitor cell proliferation and migration after insult to the perinatal brain – an event resulting in significant expansion of neuronal and glial progenitor pools. Increased SVZ cell proliferation was observed within 2 days after HI and within 6 days of chronic perinatal HX, and persisted for several weeks (Fagel et al., 2006; Felling et al., 2006). HX-induced proliferation of NG2-/Olig2-expressing OPC occurs in the SVZ within the first few days after insult and depends on activation of the Cdk4 pathway (Jablonska et al., 2012). Hypoxic rearing doubled the number of Blbp+Glast+GFAP+ cells in mouse SVZ during the first week of recovery (Fagel et al., 2006), and a similar proliferative response occurred after perinatal HI (Ong et al., 2005; Plane et al., 2004). After HI, SVZ precursor cells display multipotency in vitro and generate more neurons and OLs in vivo (Yang and Levison, 2006; Zaidi et al., 2004). These cellular dynamics suggest that the early postnatal SVZ is a potential source of OPCs as well as other progenitor cell populations for repair. Studies of the SVZ have shown that OPC proliferation is key to repair and that identifying regions with this potential is essential for targeting specific progenitor cell populations and signaling pathways involved in OL regeneration.
Another major source of OPCs is the parenchyma, including the WM itself. OPCs in the parenchyma express the same cellular markers (NG2, PDGFRα, Olig2) as their SVZ counterpart (Aguirre et al., 2007; Jablonska et al., 2012; Scafidi et al., 2014). Fate-mapping analysis showed PDGFRα-expressing OPCs to be a major source of newly generated OLs in the WM following HX (Scafidi et al., 2014
OPC Proliferation Drives Oligodendrocyte Regeneration
The studies discussed above indicate that proliferation of immature progenitor cell populations is necessary for glial regeneration. Therefore, we will briefly describe some molecular mechanisms that regulate OPC proliferation after neonatal brain injury.
Significant OL death in the brain occurs within the first 2 weeks after perinatal HX, resulting in transient hypomyelination (Jablonska et al., 2012). OPCs in the parenchyma – particularly in subcortical WM – actively divide and regenerate OLs after HX (Jablonska et al., 2012; Scafidi et al., 2014). Immunocytochemical and fate-mapping analysis demonstrated that NG2+PDGFRα+ OPCs proliferate within a week after HX and generate new OLs for the following 3–4 weeks (Jablonska et al., 2012; Scafidi et al., 2014). Unlike the SVZ, OPC proliferation in WM depends on Cdk2 activation and formation of the Cdk2/CyclinE complex, a major regulator of G1-S transition in the OPC cell cycle in both developing and adult brains (Jablonska et al., 2012; Belachew et al., 2002). The proliferative response of endogenous OPCs to HX is crucial for expanding this progenitor pool and for regenerating a normal number of OLs, restoring normal myelin protein levels (Jablonska et al., 2012; Scafidi et al., 2014). Conversely, blockage of signaling pathways (e.g., EGF/EGF-receptors) that promote OPC expansion after HX impairs OL regeneration and functional recovery (Scafidi, et al., 2014).
What molecular mechanisms delay or prevent timely OL maturation from OPCs after HX? Do these mechanisms involve intrinsic molecular pathways that regulate OL proliferation under normal physiological conditions? At least two pathways of functional importance in OL development play essential roles in OL regeneration: p27Kip1 and Wnt signaling. Importantly, alterations in molecular elements of these pathways have been noted in animal models of premature brain injury, as well as in postmortem brain tissue of infants who died of hypoxic/ ischemic encephalopathy (Jablonska et al., 2012; Scafidi et al., 2014).
Many studies have contributed to identifying p27Kip1 as an important regulator of OL differentiation (Casaccia-Bonnefil et al., 1997): p27Kip1 knockout mice display fewer OLs, and signals that prevent OL maturation also increase p27Kip1 levels in OPCs (Casaccia-Bonnefil et al., 1997; Ghiani et al., 1999). p27Kip1 levels are significantly reduced in the OL lineage of mice exposed to HX and in human postmortem tissue, and the effects of HX on OL development are enhanced in p27Kip1-null mice (Jablonska et al., 2012). HX regulates p27Kip1 via at least two mechanisms: i) by reducing expression of FoxO transcription factors, which are required to maintain normal p27Kip1 levels, and ii) by enhancing activity of the p27Kip1-degrading enzyme Skp2, which causes ubiquitination of the cyclin kinase inhibitor (Jablonska et al., 2012).
Wnt-β-catenin plays an important role in OL development, as downregulation of this signaling pathway is required for OL maturation (Fancy et al, 2009). Transcription factors (e.g., Sox17) and cellular signals that promote OPC differentiation and myelination suppress Wnt-β-catenin signaling (Chew et al., 2011). Axin2, one of the negative regulators of Wnt-β-catenin, is expressed at higher levels in OPCs than in OLs and induces β-catenin degradation (Fancy et al., 2011a). HX destabilizes Axin2, but treatment with a small molecule inhibitor of the enzyme tankyrase prevents this effect and promotes new OL generation and timely myelination (Fancy et al., 2011a). The findings above on p27Kip1 and on Wnt-β-catenin not only demonstrate clear links between OL development and regeneration/repair, but also show that manipulation of molecular pathways regulating OL maturation under normal physiological conditions may hold important therapeutic implications for promoting regeneration of these cells and preventing hypomyelination after neonatal brain injury.
Growth and Trophic Factors: Therapeutic Insights
A multitude of growth and trophic factors regulate OL development, especially OPC proliferation, migration, and differentiation (Bansal, 2002; Baron et al., 2005; Miller, 2002). Similarly, OL survival and myelin maintenance depend on growth and trophic factors (Casaccia-Bonnefil, 2000; Nave and Trapp, 2008). An important question, which may have significant therapeutic implications, is whether these factors’ availability is altered in the pathological immature brain after neonatal injury. If changes in the overall levels of specific growth factors induced by injury mimic those seen during normal development that would indicate reactivation of endogenous developmental programs aimed at OL recovery. Several studies have harnessed our developmental knowledge of these factors for therapeutic interventions aimed at OL regeneration.
Fibroblast growth factor 2 (FGF2), epidermal growth factor (EGF), and leukemia inhibitory factor (LIF) are upregulated in the SVZ after hypoxic insult (Covey and Levison, 2007; Ganat et al., 2002; Scafidi et al., 2014). These factors promote OL generation from OPCs, as well as OL maturation and survival during normal development. Experimental analysis indicates that these factors cause: i) expansion of the SVZ progenitor pools observed after HX, and ii) generation of new OLs from these progenitors (Covey and Levison, 2007; Ganat et al., 2002; Scafidi et al., 2014). Similarly, insulin-like growth factor-1 (IGF-1) plays a crucial role in promoting oligodendrogenesis and myelination in early brain development (Carson et al., 1993; Ye et al., 2002). IGF-1 treatment after neonatal brain injury recapitulates some developmental effects of this growth factor and promotes OL regeneration. IGF-1 administration prevented immature OL death, enhanced myelination after HI and protected OPCs in the SVZ and WM regions (Lin et al., 2005; Zhong et al., 2009).
We recently observed upregulation of endogenous EGF in WM and the SVZ after perinatal HX and showed that EGFR overexpression in the OL lineage enhances OL regeneration and promotes functional recovery in WM (Scafidi et al., 2014). Consistent with these findings and with EGF’s effects on OL during normal development, noninvasive intranasal EGF delivery during a critical developmental time window immediately after HX causes significant expansion of the OPC pool in WM, and promotes OL regeneration and functional/behavioral recovery (Scafidi et al., 2014). Although EGF appears to act via various mechanisms, a major effect is to prevent HX-induced Notch activation in OL lineage cells, inhibiting OPC differentiation and contributing to the delay in OL maturation observed after HX (Scafidi et al., 2014). This EGF-Notch interaction occurs in the SVZ during normal development and regulates the size of the OPC pool in this neurogenic region (Scafidi et al., 2014). To summarize: EGF prevents the delay in OL maturation caused by HX by generating a significant number of new OLs and inhibiting signaling pathways that arrest OPC maturation.
A similar therapeutic intervention developed in an animal model of intraventricular hemorrhage (IVH) mimics a major cause of WMI in preterm infants. The intervention was based on the well-established idea that thyroid hormone (TH) signaling – involving TH receptor α (TRα and TRβ – strongly promotes OL maturation and myelination. It was hypothesized that IVH would decrease TH levels and reduce TH signaling. Interestingly, in both preterm brain tissue and rabbits with IVH, levels of TH-activating enzyme deiodinase-2 were reduced, whereas TH-deactivating enzyme deiodinase-3 increased (Vose et al., 2013). Treating IVH pups with TH accelerated OPC proliferation and maturation into OLs, enhanced myelination, and restored neurological function (Vose et al., 2013).
Together, the EGF and the TH treatment paradigms demonstrate that targeting specific factors required during normal development for OPC proliferation, survival, and differentiation might represent a viable therapeutic strategy in pathologies associated with premature brain injury. The studies also uncover the existence of rather specific time windows of intervention that closely follow the injury and likely permit enhancing plasticity processes to occur in the developing WM. Importantly, a study in human neonatal tissue demonstrated that neurogenesis ceases after 18 months, but OPCs are detected in the SVZ after this age (Sanai et al., 2011), strongly indicating that gliogenesis continues and highlighting the potential for regeneration at later developmental stages of the postnatal brain.
A different approach was undertaken in rodent models of HI to target iron, an essential trophic factor for OL development and survival. Iron plays a crucial role in oxygen consumption and ATP production in all cells, but OLs are the major brain cells that stain for iron, indicating higher levels (Todorich et al., 2009). Iron deficiency causes hypomyelination and neurological abnormalities, which can be reversed by intracranial injection of apotransferrin (aTf) at early developmental stages (Badaracco et al., 2008). The use of aTf to restore physiological levels of iron transport has been explored in neonatal WMI after HI, as HI increases iron release in WM regions (Guardia Clausi et al., 2012). Intranasal administration of aTf immediately after HI promoted neuroprotection of the neonatal mouse brain by reducing WM damage, astrogliosis, and OL death. The treatment also enhanced OPC proliferation in the SVZ and corpus callosum and promoted OPC differentiation. Overall, this non-invasive treatment acting on a signal with a crucial role in normal development also promoted functional recovery. The results obtained with aTf also suggest that future studies could investigate the use of combined treatments (e.g., EGF+aTf) delivered at different times after neonatal injury.
Transcription Factors and Epigenetic Regulation
Despite extensive characterization of transcriptional regulation of OPC development/ differentiation, the reuse of these processes after neonatal HX or HI remains poorly defined. Myt1 and NFIA, two transcription factors with important roles in OL development, were recently studied in animal models of neonatal HX and in PVL tissue. Myelin transcription factor 1 (Myt1) is a zinc finger DNA-binding protein expressed at higher levels in OPCs than in mature OLs (Armstrong et al., 1995). Based on its expression pattern, regulation, and functional analysis, Myt1 is thought to modulate OPC transition from a proliferative to a terminally differentiated state that requires high levels of myelin gene transcription (Armstrong et al., 1995; Nielsen et al., 2004). Interestingly, in normally developing human brains, many Myt1-expressing glial cells are found between gestational weeks 19 and 26 but decrease by 1 year of age (Hirayama et al., 2003). In PVL brains, a large increase in Myt1-expressing cells is observed around necrotic foci and in regions displaying higher levels of myelin protein immunoreactivity (Hirayama et al., 2003), strongly suggesting their role in human OL pathology/regeneration.
A similar pattern of developmental regulation was observed for NFIA in OLs. During mouse embryonic development, NFIA is expressed in OPCs but not in differentiated OLs; its overexpression prevents OPC differentiation by directly repressing myelin gene expression (Fancy et al., 2012). Notably, in HIE lesions, NFIA is expressed in OPCs, but not in PLP-expressing cells (Fancy et al., 2012), indicating that NFIA dysregulation may contribute to the delayed OPC maturation and hypomyelination found in HIE, by repressing myelin gene expression. Indeed, misexpression of NFIA suppressed remyelination in an adult LPS model, suggesting a conserved function after WMI.
Epigenetic mechanisms, including DNA and histone methylation and acetylation, represent a different level of developmental regulation of the OPC-OL transition. In normal brain, timely OPC differentiation and myelination require histone deacetylation (HDAC) during a critical developmental time window (Shen et al., 2005), and HDAC activity regulates the regenerative potential of OPCs during development (Shen et al., 2008). Some hypothesize that epigenetic mechanisms – including acetylation/deacetylation and methylation – are involved in neurodegenerative diseases and stroke as regulators of the extent of injury (López-Otín et al., 2013; Schweizer et al., 2013). Altered gene expression caused by ischemia is an important contributor to neuronal or glial damage, and HDAC inhibitors have attracted significant attention as potential neuroprotective agents (Kumral et al., 2009 and 2013). Although the role of HDAC and HDAC inhibitors has been specifically investigated only in regeneration and remyelination of adult OL (see below), particular alterations in HIF-mediated gene transcription have been observed in neonatal HI (Kumral et al., 2009; Singh et al., 2012). Thus, it will be important to define changes in DNA methylation and acetylation following HX or HI, as these changes will strongly influence the expression of genes involved in OL regeneration and WM recovery in the neonatal brain. Finally, as significant sex differences have been observed in the extent of premature brain injury – males being more susceptible than females – sex differences in histone modifications (Tsai et al., 2009) might account at least partially for differential vulnerability seen in WM of male and female infants.
Future studies will further investigate the links between specific growth factor signaling pathways and differing levels of transcriptional regulators after developmental injury and during regeneration. Understanding these signaling axes will be crucial to generating new therapeutic approaches based on molecular mechanisms regulating normal glial development.
Oligodendrocyte Regeneration and Repair in the Adult Brain
The hope for OL replacement in various myelin disorders and in adult WMI rests on the established finding that the adult brain contains populations of undifferentiated OPCs able to divide and regenerate myelinating OLs. Recent reviews have extensively discussed the cellular and molecular properties of these “activated” OPCs and their developmental potential in the context of demyelination/remyelination of adult brain (Gallo and Franklin, 2014; Zuo and Nishiyama, 2013; Fancy et al., 2011b). Our discussion will focus on the signaling pathways shared between development and remyelination, and whether these represent true recapitulation of developmental programs. Many developmental signals and associated molecular pathways have been identified as either negative or positive regulators of OL regeneration, and studies have investigated how those mechanisms interact with cellular changes that occur in the pathological microenvironment of the adult brain. This section will focus on OL regeneration and remyelination in animal models of demyelination, particularly models of MS.
Animal Models of Injury and Pathology of the Adult White Matter
Recent reviews have comprehensively and elegantly discussed all available animal models of MS and demyelinating diseases (Franklin & Ffrench Constant, 2008; Ransohoff, 2012). These animal models (summarized in Table 1) have been used extensively for revealing the endogenous remyelination potential of the central nervous system as a possible therapeutic approach to functional recovery. For example, the presence of OPCs in MS lesions is well established (Chang et al., 2002; Wolswijk et al., 1998), so understanding their origin and response to demyelinating insults is fundamentally important to developing new cell-based strategies for remyelination. OPC activation (i.e., changes in morphology, proliferation, migration, and expression of specific genes) and recruitment occur in all animal models of adult brain demyelination, including experimental autoimmune encephalomyelitis (EAE), and cuprizone- and lysolecithin (LPC)-induced demyelination (Aguirre et al., 2007; Cate et al., 2010; Messersmith et al., 2000; Nait-Oumesmar et al., 1999; Picard-Riera et al., 2002). These changes also occur in “activated” OPCs in MS tissue, probably induced by signals present in lesions (Nait-Oumesmar et al., 2007).
Table 1.
Models of Adult Demyelination/Remyelination
| Model | Type | Mechanism/Description |
|---|---|---|
| Cuprizone | Toxin |
|
| Lysolecithin | Toxin |
|
| Ethidium Bromide | Toxin |
|
| Experimental Autoimmune Encephalomyelitis (EAE) | Immune Mediated |
|
| Theiler’s Murine Encephalomyelitis (TME) | Immune Mediated |
|
| Murine Hepatitis | Immune Mediated |
|
| Diphtheria Toxin (DT) Receptor Targeting | Genetic |
|
Enhancers of oligodendrocyte development and regeneration
Several intrinsic and extrinsic regulators of OL development have been identified as enhancers of regeneration (Figure 3), opening up new opportunities to design molecularly targeted approaches aimed at enhancing remyelination.
Figure 3. Positive and negative regulators of oligodendrocyte development and regeneration.

Schematic synopsis of the major developmental phases of oligodendrocyte maturation - i.e. proliferation, cell cycle exit/differentiation and myelination – after demyelination of the adult brain. Green arrows refer to enhancers of these processes under pathological conditions, whereas red arrows refer to inhibitory pathways. Preventing or suppressing the inhibitory effects of specific pathways, as well as leveraging on enhancers promotes oligodendrocyte maturation and myelination, and might define future cell-based therapeutic approaches to demyelinating diseases.
Growth Factors
During normal development, OPC proliferation and cell cycle progression are positively regulated by two potent mitogenic factors – platelet-derived growth factor (PDGF) and fibroblast growth factor 2 (FGF2) – that act either individually or cooperatively through PDGFRα and FGFR1 to generate mature OLs (Calver et al., 1998; Fortin et al., 2005; McKinnon et al., 1990; Miller, 2002). Expression of the ligand PDGF-A is upregulated in reactive astrocytes of demyelinated lesions (Armstrong et al., 2002: Messersmith et al., 2000), and both FGF2 and FGFRs are upregulated in areas of acute or chronic cuprizone-induced demyelination (Messersmith et al., 2000). PDGF and FGF2 control differential responses in the OL lineage after demyelination (Murtie et al., 2005). Consistent with a role of PDGF in proliferation, OPC amplification and subsequent repopulation of demyelinated lesions are impaired in a heterozygote PDGFRα+/− or in a GFAP-driven PDGF-A-null mouse (Murtie e et al., 2005). Conversely, FGF2 acts to inhibit remyelination through FGFR1 (see below).
Stimulation of epidermal growth factor (EGF) signaling is an example of successfully leveraging a developmental mechanism to promote regeneration in the adult brain. EGF exerts multiple effects on the OL lineage, both OPCs and mature OLs (Aguirre et al., 2007; Scafidi et al., 2014). Selective EGFR overexpression in OL lineage cells promotes OPC proliferation and migration, enhancing the extent of functional remyelination of the corpus callosum after LPC-induced demyelination (Aguirre et al., 2007). Consistent with these findings, EGF infusion into the lateral ventricle (Gonzalez-Perez et al., 2009) or intranasal HB-EGF administration (Cantarella et al., 2008) promoted OPC recruitment from SVZ to demyelinated lesions. Furthermore, viral-mediated EGFR overexpression in WM OPCs increased proliferation and maintained an undifferentiated phenotype (Ivkovic et al., 2008). Other growth factors are also involved in regulating OPC number under physiological or pathological conditions: for example, analysis of null mouse strains demonstrated that brain-derived neurotrophic factor (BDNF) is required to regulate the number of OPCs during normal development and during remyelination (VonDran et al., 2010 and 2011). Together, these studies demonstrate that, similar to the perinatal brain, specific GFs activate endogenous pools of OPCs that can be redirected from proliferative areas to injury sites to promote recovery in adults. The OPC response to demyelination and to cellular factors present in a lesion is integrated by numerous cell cycle regulatory proteins, including Cdk2 and p27Kip1, which regulate the number of OPCs recruited to a lesion (Caillava et al., 2011; Crockett et al., 2005).
Developmental studies and analyses in animal models of demyelination have also identified factors that positively regulate OPC differentiation and maturation of myelinating OLs under normal physiological and pathological conditions. During normal development, OL maturation is enhanced by ciliary neurotrophic factor (CNTF) (Stankoff et al., 2002), which also exhibits a strong pro-myelinating effect after demyelination (Albrecht PJ et al., 2003). Growth factor BDNF also promotes OPC differentiation and myelin protein expression after cuprizone-induced demyelination, demonstrating that this factor modulates diverse phases of the OL lineage (VonDran et al., 2011). Similarly, the neurotrophin NT-3 produced a broad range of developmental and regenerative effects in OLs – promoting OPC proliferation and supporting survival of mature OLs through TrkC receptors and MAPK phosphorylation (Cohen et al., 1996). Notably, direct NT-3 injection into the corpus callosum promoted oligodendrogenesis and remyelination in a model of LPC-induced demyelination (Jean et al., 2003).
Insulin-like growth factor 1 (IGF-1) was identified early on as a necessary factor for OL differentiation and myelination in vitro and in vivo (Mozell and McMorris, 1991; Werther et al., 1998). In vivo studies demonstrated that IGF-1 ablation causes reduced brain size and hypomyelination (Beck et al., 1995; Ye et al., 2002). The IGF-1R (IGF type 1 receptor) is continuously expressed throughout the entire OL lineage, and IGF-1 appears to act at multiple developmental stages, promoting OPC proliferation, OL survival and myelin synthesis (Barres et al., 1993; Ye and D’Ercole, 1999; Zeger et al., 2007). Consistent with its multiple developmental roles, IGF-1 significantly attenuates the damage induced by a demyelinating insult. In a transgenic mouse strain expressing higher IGF-1 levels in the brain, the cellular effects of cuprizone-induced demyelination on OL death were significantly reduced, although significant demyelinaton was seen. Furthermore, the remyelination rate was much higher than in wt controls (Mason JL et al., 2000). Consistent with these findings, IGF-1 decreased lesion severity and clinical abnormalities in the EAE mouse model (Yao et al., 1995). These results indicate that IGF-1 may improve recovery from a demyelinating insult by selectively enhancing OPC differentiation and supporting survival of the more mature OL population. However, the experimental paradigm of enhanced IGF-1 expression might be crucial, as increasing local IGF-1 mRNA levels by adenoviral vector failed to promote remyelination (O’Leary et al., 2002).
How does IGF-1 promote OL survival and differentiation? Analysis of signaling in OLs demonstrated IGF-1 to be a potent activator of the PI3K/Akt and mTOR pathway (Min et al., 2012). Exposure to IGF-1 maintains Akt signaling in differentiating OPCs (Ness et al., 2002), consistent with the idea that IGF-1 signaling through PI3K/Akt and mTOR is functionally linked to OPC differentiation into mature OLs. Recent research identified mTOR as a critical signaling kinase in OL differentiation and myelination and showed its effects occurring through the mTORC1 and mTORC2 signaling complexes (Tyler et al., 2009).
The preceding discussion of growth factors in demyelination and remyelination points to the need for in vivo studies in various animal models of pathology, each model designed to define specific cellular and developmental effects of individual growth factors on OL regeneration. The discussion also suggests that sustained expression of various factors during distinct phases of injury and recovery from a myelin insult might be used to coordinate and integrate multiple cellular mechanisms that lead to successful functional remyelination. Furthermore, analysis of glial regeneration and repair should be extended to other cellular factors that modulate glial development. For example, hepatocyte growth factor (HGF) has been identified as a positive regulator of oligodendrocyte proliferation and migration, and an inhibitor of differentiation (Yan and Rivlkees, 2002: Ohya et al., 2007; Mela and Goldman, 2013). The role of HGF in remyelination has not been explored, but it might promote early phases of OPC expansion and repopulation of demyelinated lesions.
Hormones
Several lines of evidence indicate that specific hormonal states regulate OL numbers, developmental myelination, and myelin repair. Sexual dimorphism exists in rodent subcortical WM, where females display a higher proportion of unmyelinated axons (Kim and Juraska, 1997) and higher OL turnover (Cerghet et al., 2006). MS occurs more frequently in females and undergoes remission during pregnancy (Confraveux et al., 1998), although the hormonal causes are poorly understood. Interestingly, pregnant mice demonstrate an increase in newly generated OLs and in the number of myelinated axons in corpus callosum (Gregg et al., 2007); and pregnant mice show enhanced ability to remyelinate after LPC-induced focal demyelination (Gregg et al., 2007). And prolactin has been identified as a major regulator of OL plasticity and WM repair during pregnancy, inducing OPC proliferation and enhancing OL regeneration in vivo (Gregg et al., 2007).
Thyroid hormone (triiodothyronine; T3) is the best-studied hormone for its effects on OL development. T3 regulates the timing of OPC differentiation (Barres et al., 1994) and promotes morphological maturation via a metabolic switch in OPCs that supports myelin synthesis in mature OLs (Baas et al., 1997; Younes-Rapozo et al., 2006). Specific cellular changes induced by T3 have been extensively analyzed in purified cultured OPCs or in reaggregating cultures (Baas et al., 1997; Jones et al., 2003; Matthieu et al., 1992). In vivo, hypothyroidism causes delayed and poor deposition of myelin during development (Ferreira et al., 2004; Ibarrola N and Rodríguez-Peña, 1997), while hyperthyroidism accelerates myelination (Marta et al., 1998). Prolonged neonatal hypothyroidism decreases the number of myelinated axons in adult rats, although most of the myelinated axons display normal thickness of the myelin sheath. Two animal models of myelin pathology demonstrated the beneficial effects of T3 on OL regeneration and remyelination. In the cuprizone model, systemic T3 injection in rats significantly enhanced remyelination by increasing the ratio of mature OLs to OPCs (Franco GP et al., 2008). In the EAE model, T3 treatment yielded myelin sheath and axonal protection and enhanced nerve conduction (Del’Acqua et al., 2012). These cellular and physiological studies support previous molecular analyses demonstrating that T3 thyroid receptors can directly activate MBP and PLP gene promoters in mature OLs (Farsetti et al., 1991; Bogazzi et al., 1994).
The analysis of hormones in vivo reveals that both prolactin and T3 promote WM plasticity during development and in regeneration, implicating these hormones as potential therapeutic agents for targeting axons and OLs in demyelinating diseases.
Inflammatory Cytokines and Chemokines
Among the influences on OL development and regeneration, cytokines and chemokines have been studied as factors predominantly upregulated after demyelination. Tumor necrosis factor-α (TNFα) is an inflammatory cytokine able to induce a broad range of cellular responses, including cell proliferation and cell death, mediated by the activation of two distinct receptors – TNFR1 (p55) and TNFR2 (p75) (Dopp et al., 1997; Raine et al., 1998; Tchelingerian et al., 1995). In the brain, TNFα is synthesized and released by astrocytes and microglia (Liu et al., 1998). This cytokine was shown to negatively regulate OPC number and differentiation, as well as OL survival in developing cultured cells (Cammer and Zhang, 1999). In keeping with these findings, studies in the EAE model showed that genetic ablation or blockade of TNFα signaling attenuated myelin pathology (Hövelmeyer et al., 2005). However, analysis of TNFα and TNFR1, and TNFR2 mutant mice in a different model of demyelination also revealed a role for this cytokine in OL regeneration and remyelination. Cuprizone-induced demyelination substantially increased TNFα and TNFR2 in lesions, and genetic ablation of the cytokine caused significant reduction in OL apoptosis, delayed demyelination, and ultimately enhanced remyelination (Arnett et al., 2001). TNFR2 was identified as the primary receptor mediating the influence of TNFα on remyelination, which was attributed to potent effects on OPC proliferation and to generation of new OLs during demyelination/remyelination (Arnett et al., 2001).
The mechanism of OL regeneration and its modulation by TNFR2 appear complex. More recent analysis demonstrated that TNFR2 also regulates expression of the chemokine CXCL12, which acts on the CXCR4 receptor to modulate remyelination. Interestingly, genetic ablation of TNFR2 causes significant downregulation of CXCL12 expression in cuprizone-induced lesions and decreases the number of CXCR4-expressing OPCs (Patel et al., 2012). Cellular analysis, combined with rescue experiments, demonstrated that TNFR2 activation in astrocytes induces CXCL12 expression in demyelinated corpus callosum, in turn promoting OPC proliferation and differentiation. Consistent with this hypothesis, loss of CXCR4 signaling decreased OPC maturation and prevented remyelination after cuprizone-induced demyelination (Patel et al., 2010). As CXCL12 also regulates normal OL development – in particular OPC proliferation, migration and differentiation (Dziembowska et al., 2005; Patel JR et al., 2012) – the findings described above indicate that reactivation of CXCL12 signaling has similar effects on OL regeneration and remyelination.
Another cellular mechanism of action was attributed to the pro-regenerative effects of yet another cytokine – Interleukin-1β(IL-1β). This cytokine was shown to promote OL differentiation after acute demyelination, as IL-1β−/− mice failed to properly remyelinate despite an OL depletion rate similar to that of wt mice (Mason JL et al., 2001). The cytokine’s effects were indirect and were attributed to delayed OPC differentiation due to lack of IGF-1 produced by astrocytes and microglia (Mason et al., 2001). These findings show cytokines acting at multiple levels – both directly and indirectly – to regulate OL regeneration and remyelination; and cellular interactions between OPCs, astrocytes, and microglia modulate all phases of remyelination. These mechanisms are likely important for cell-based therapies, as recent studies of NSCs engrafted after demyelination revealed a requirement for CXCL12 and CXCR4 signaling in NSC migration and proliferation giving rise to OLs (Carbajal et al., 2010).
Transcription Factors
Converting OPCs to a regenerative phenotype is thought to involve enhanced expression and activity of several transcription factors playing important roles in OL development. A number of studies indicate that multiple transcription factor networks probably regulate OPCs’ response to demyelination, modulating OL regeneration. During development, Olig1, Olig2, and Nkx2.2 are crucial in OL specification and maturation (Lu QR, 2002; Soula C et al., 2001), and expression of transcription factors that regulate OL fates – including Olig2 and Nkx2.2 – is enhanced in adult OPCs after demyelination (Fancy et al., 2004). This upregulation is generally observed during acute phases of demyelination and during early remyelination, indicating that OPC transition to a regenerative phenotype probably involves recapitulation of developmental programs. Although some transcription factors may play roles in both development and repair of glia, others – like Olig1 – appear to be primarily involved in repair. Given that remyelination is less efficient in Olig1−/− mice, Olig1 is regarded as crucial in myelin repair in the adult brain, although this factor is redundant in OL development (Arnett et al., 2004). Indeed, Olig1 displays nuclear localization in early remyelinating lesions in animal models of demyelination and in MS tissue, but is mostly cytosolic in normal WM (Ligon et al., 2006). A similar expression pattern is observed for transcription factor Myt1, which appears to modulate OPC proliferation and contribute to the timing of differentiation (Nielsen et al., 2004; Vana et al., 2007).
Two other transcription factors – Ascl1/Mash1 and Sox17– belong to distinct families and play differing roles in OL development and regeneration. The proneural gene Ascl1/Mash1 was newly identified as a regulator of embryonic and postnatal oligodendrogenesis (Parras et al. 2004 and 2007). In the embryonic CNS, Ascl1 is expressed in ventral forebrain and cooperates with Olig2 to specify OPCs (Parras et al., 2007). In the postnatal SVZ, Ascl1 is expressed in progenitors of both OL and neuronal lineages (Parras et al., 2004; Nakatani et al., 2013) and positively regulates OPC specification as well as the balance between proliferation and differentiation.
Another group of transcription factors are induced by injury, inhibit OPC proliferation, and increase cell differentiation, but have complex effects on various neural cell lineages. In the OL lineage, expression of Sox17 RNA and protein peaks at P15 and precedes myelin gene expression (Sohn et al., 2006). Unlike Ascl1, Sox17 does not appear to be involved in OPC specification but rather in cell cycle exit and initiation of differentiation (Sohn et al., 2006). Developmental studies in cultured cells and transgenic mice in vivo demonstrated that Sox17 promotes OPC differentiation by inhibiting the Wnt/β-catenin pathway (Chew et al., 2011; Ming et al., 2013). Extensive analysis of Sox17 expression and regulation in animal models of demyelination demonstrated that, during remyelination, Sox17 expression is highest in newly generated OLs, suggesting a role for this transcription factor in OL regeneration (Moll et al., 2013). In MS tissue, Sox17 is highly expressed in chronic active lesions (Moll et al., 2013). Finally, selective overexpression of Sox17 in the OL lineage strongly attenuated the effects of LPC-induced demyelination by promoting OPC differentiation and OL survival (Ming et al., 2013), indicating that sustained activation of Sox17 promotes OL regeneration and myelin repair.
Retinoic acid receptor signaling strongly induces neuronal differentiation during embryonic development and in adult NSCs (Bain et al., 1996; Goncalves et al., 2005) also, retinoid X receptor gamma (RXR-gamma) (Huang et al., 2011) positively regulates OL differentiation during myelination and remyelination. Nuclear translocation of RXR-gamma was noted during OPC differentiation and OL regeneration and, importantly, predominantly nuclear RXR-gamma was detected in OPCs in MS tissue within active lesions, periplaque WM, and remyelinated plaques. Thus, its presence in the nucleus was consistent with cellular activity and repair capacity in MS. The finding that systemic administration of 9-cis retinoic acid after demyelination promoted axonal remyelination (Huang et al., 2011) demonstrates that activating this pathway is a potentially effective treatment for demyelinating lesions.
The findings described above indicate that several intrinsic regulators of OL development are important in regeneration and myelin repair. While direct targeting of transcription factors may represent a challenge in regenerative therapies, elucidating the signaling pathways regulated by these factors will open new avenues for molecularly targeted approaches to myelin repair. Furthermore, identifying the transcription factors that promote an OL phenotype under physiological and pathological conditions will enhance the efficiency of OL generation in cell-based therapies involving NSCs or induced pluripotent stem cells (iPSCs).
Other signaling pathways
The morphogen Sonic Hedgehog (Shh) is another example of reactivation of a developmental signal during OL regeneration (Ferent et al., 2013). Shh is required for OPC specification during embryonic development (Orentas et al., 1999; Spassky et al., 2001) and regulates OL production in adult cortex and corpus callosum (Yung et al., 2002). In LPC-induced focal demyelination, Shh transcripts and protein were upregulated in OL lineage cells within the lesion in a time-dependent fashion; furthermore, increased transcription of Shh target genes showed significant reactivation of the Shh pathway. Gain- and loss-of-function analyses demonstrated that Shh signaling plays a positive role in OL regeneration and myelin repair, enhancing OPC proliferation and differentiation as well as OL survival. Yet, activation of Shh signaling decreased astrogliosis and macrophage infiltration (Ferent et al., 2013), indicating that some of these beneficial effects may be indirect.
Reactivation of signaling pathways that stimulate OPC migration also occurs after demyelination. Netrin-1 positively regulates OPC migration in the postnatal optic nerve (Spassky et al., 2002), and a recent study demonstrated involvement of Netrin-1 in promoting migration of OPCs out of the adult SVZ after LPC-induced demyelination (Cayre et al., 2013). Netrin-1 was strongly upregulated in the SVZ after demyelination, and was identified as the signal involved in local angiogenesis and progenitor cell migration, likely by activation of deleted in colorectal cancer (DCC) and neogenin receptors (Cayre et al., 2013). Demyelination induced vascular remodeling in the SVZ, and VEGF-mediated progenitor cell recruitment occurred with specific association between reactive blood vessels and OPCs (Cayre et al., 2013).
Metalloproteinase inhibitors may also play positive roles in OL regeneration in response to injury. Recent analysis demonstrated that myelin injury upregulates tissue inhibitor of metalloproteinase (TIMP-1) in reactive astrocytes, regulating secretion of matrix metalloproteinases from these cells (Nygardas and Hinkkanen, 2002). Analysis in TIMP-1-null mice showed that oligodendrogenesis and compact myelin formation are significantly impaired, at least partly due to a reduced number of OPCs (Moore CS, 2011). In EAE, TIMP-1 expression is associated with astrocytes in demyelinated lesions, and remyelination is impaired in TIMP-1-null mice (Crocker, S.J., 2006). And in clinical studies, robust TIMP-1 expression was found in acute demyelinating diseases that display some degree of remyelination (Ichiyama, T., 2006), while cerebrospinal TIMP-1 levels are not modified in MS (Avolio, C., 2003).
Inhibitors of OL development and regeneration
Mechanisms underlying the failure of remyelination in WM lesions include depletion of the endogenous OPC pool, limited migration of endogenous OPCs in a pathological environment, loss of axonal signals that support OPC-derived remyelination, and presence of inhibitors (likely produced by both astrocytes and infiltrating microglia) preventing their complete differentiation (Chong and Chan, 2010; Mason et al., 2004; Miron et al., 2013). Many of these inhibitors also negatively regulate normal OPC development (Figure 3).
A fundamental phase in OL development consists of OPC proliferation and the generation of sufficient OLs. Proteins that regulate cell cycle progression and cell cycle exit in OPCs – i.e., ultimately, the number of OPCs – have been intensely analyzed for their roles in development and repair. During normal development, type 2 cyclin-dependent kinase (Cdk2) is necessary for OPC proliferation but does not affect differentiation (Belachew et al., 2002). However, after demyelination, loss of Cdk2 accelerates CNS remyelination by altering OPC cycle exit and enhancing differentiation, indicating Cdk2 to be an inhibitor of OL maturation in pathology (Caillava et al., 2011). Recruitment of progenitors to a demyelinated lesion in spinal cord is negatively regulated by Cdk2 inhibitor p27Kip1, as more OPCs were found in the lesion of p27Kip1-null mice than in wt controls, and these cells differentiated into mature OLs (Crockett et al., 2005). The other Cdk inhibitor, p57Kip2, appears to have a different role, as it prevents OL differentiation and remyelination in the EAE model (Kremer et al., 2009). This is consistent with the finding that suppressing p57Kip2 expression promotes oligodendrogenesis from NSCs (Jadasz et al., 2012). Together, these findings indicate that failure to remyelinate may be due, at least partly, to cell cycle regulators that inhibit OL maturation from OPCs.
Studies of cellular factors identified inhibitors that should, ideally, be eliminated from a demyelinated lesion to promote regeneration of mature OLs. These inhibitors – BMP, FGF2, Notch, and Wnt signaling – act similarly in preventing oligodendrogenesis during normal development. During development, BMP signaling suppresses oligodendrogenesis, promoting neurogenesis (Colak et al., 2008) and astrogliogenesis (Gomes et al., 2003; Samanta et al., 2007; See et al., 2007; See et al., 2009) BMPs negatively control OPC specification and lineage commitment, as well as OPC maturation into myelinating OLs (See et al., 2007). These effects likely involve induction of Id (Inhibitor of differentiation) expression and inhibition of Olig activity (Samanta and Kessler, 2004; Chen et al., 2007). BMP expression is enhanced in several animal models of CNS injury (Ara et al., 2008; Chen et al., 2005; Hampton et al., 2007; Matsuura et al., 2008) and - consistent with the complex developmental functions of BMPs - this can exert opposite effects on neural cell regeneration after injury. In animal models of demyelination, the expression of BMP-4, -6, and -7 is upregulated (Ara et al., 2008; Fuller et al., 2007; Sabo et al., 2011) and BMPs likely released by neurons and astrocytes inhibit remyelination and impair recovery (See et al., 2009). Infusing the BMP antagonist Noggin into demyelinated lesions increased the density of mature OLs during remyelination of the CC (Sabo et al., 2011). The BMP antagonist chordin was identified as a SVZ signal that promotes oligodendrogenesis from GAD65- and Dcx-expressing neuroblasts of the adult SVZ (Figure 4), indicating that niche-dependent mechanisms regulate lineage plasticity of progenitors in the adult brain (Jablonska et al., 2010). In contrast to the role of FGF2 (or FGFR1) in normal development, their genetic ablation under pathological conditions dramatically enhanced OL regeneration by promoting OPC differentiation (Armstrong et al., 2002; Zhou et al., 2012), indicating that this growth factor actually inhibits OPC maturation in a demyelinated lesion. Thus, manipulation of BMP and FGF in a demyelinated lesion environment illustrates how the extent of remyelination from endogenous OPCs can be enhanced by differentially modulating activity of specific developmental signals.
Figure 4. Oligodendrocyte regeneration through lineage plasticity of GAD-expressing neuroblasts.

Demyelination of the adult white matter (corpus callosum) causes fate plasticity in GAD-expressing (GAD+) neuroblasts of the subventricular zone (SVZ). Under normal physiological conditions, Olig2 expression is repressed by BMP signaling in GAD+ neuroblasts. Demyelination causes upregulation in the levels of the BMP antagonist chordin in the SVZ, which prevents BMP signaling and results in induction of Olig2 expression in GAD+ cells. These cells migrate out of the SVZ into the corpus callosum, where they generate mature, myelinating oligodendrocytes (see Jablonska et al., 2010).
Notch signaling was also identified as a developmentally regulated inhibitor of OPC maturation, as downregulation of this pathway is required for OL differentiation and myelination (Hu et al., 2003; Wang et al., 1998). Suppressors of Notch signaling enhance OPC maturation during normal development (Aguirre et al., 2010; Hu et al., 2003; Wang et al., 1998). Interestingly, Notch signaling in adult brain is reactivated after demyelination of the CC (Hammond et al., 2014; Zhang et al., 2009), and Jagged 1 upregulation found in MS chronic lesions (John et al., 2002) suggests that activated Notch contributes to preventing endogenous OPC maturation and remyelination. The question is: Which endogenous signals activate Notch after demyelination, and how is Notch activated in OPCs? Our recent study using a transgenic Notch-reporter mouse showed that the peptide Endothelin-1 (ET-1) is a major regulator of Notch after focal LPC-induced demyelination (Hammond et al., 2014). ET-1 is upregulated in mouse and MS reactive astrocytes and induces Jagged 1 expression in these cells, which in turn activates Notch and delays OL maturation after demyelination. These findings suggest that ET-1 is one of many signals that regulate Notch in a demyelinated lesion and in MS, and that various cellular factors in pathological WM might converge on the inhibitory Notch “hub” in developing and adult brain.
OL differentiation and activation of the myelination program are both essential in timely myelination. Like Notch, LINGO-1 – another inhibitor of OL differentiation and myelination – is a modulator of cell-cell interactions that precisely regulate the timing of myelination (Mi et al., 2005). LINGO-1 is a transmembrane protein selectively expressed in the CNS, specifically in axons and OPCs (Mi et al., 2005). LINGO-1 is a component of the Nogo receptor complex and its expression is developmentally regulated (Mi et al., 2004). Studies in cultured cells and knockout mice demonstrated that downregulation of LINGO-1 promotes OL differentiation and myelination during development (Mi et al., 2005). Importantly, LINGO-1 is upregulated in a variety of animal models of CNS pathology, including the EAE model of MS in which functional blockage of LINGO-1 enhances OL regeneration and remyelination and promotes functional recovery (Mi et al., 2007). Two other molecules expressed in neurons directly regulate OPC development, myelination, and remyelination: Semaphorin 3A and 3F (Sema 3A and 3F) exert opposite effects – chemorepulsive and chemoattractive, respectively – on OPC migration (Okada et al., 2007; Syed et al., 2011; Xiang et al., 2012). Both Sema 3A and 3F are upregulated in demyelinated lesions and in MS but appear to play different functional roles in OPC-derived remyelination. Loss- and gain-of-function analysis demonstrated that Sema 3A is needed for OPC recruitment and repopulation of the demyelinated lesion, whereas Sema 3F promotes both OPC recruitment and remyelination (Syed et al., 2011). Together, these findings indicate that population of the adult WM, as well as OL differentiation during myelination and remyelination, are finely regulated by multiple interactions between OPCs and neurons.
Genome-wide screening has been extensively used to identify novel regulators of OPC development, and a screen performed in remyelinating rodent lesions found approximately 50 transcription factors regulated during regeneration (Fancy et al., 2009). These included the Wnt pathway mediator Tcf4, indicating that activation of this pathway may be involved in delaying myelin recovery. Subsequent gain- and loss-of-function analysis demonstrated that the canonical Wnt pathway negatively regulates OL differentiation during developmental myelination and remyelination (Fancy et al., 2009). The effects of Wnt are mediated by BMPs (Feigensen, 2011). But how do OPCs and OLs eventually overcome this pathway’s inhibitory effects when remyelination does occur? Recent analysis identified transcription factor Sox17 as a suppressor of Wnt/β-catenin, consistent with its role as a positive regulator of OL maturation and regeneration during normal development (Chew et al., 2011). Sox17 was first identified as a transcription factor that promotes OPC cycle exit and induces differentiation. After demyelination, Sox17 levels are highest in newly generated OLs and active MS lesions (Moll et al., 2013), and its selective overexpression in the OL lineage promotes remyelination (Ming et al., 2013). These findings, along with enhanced remyelination induced by Axin 2 targeting (Fancy et al., 2011), indicate the Wnt/β-catenin pathway as a strong therapeutic target in adult myelin regeneration.
Another type of inhibitor of OL maturation is directly linked to a specific role in OL survival. Death receptor 6 (DR6), a member of the TNF receptor (TNFR) superfamily, is expressed at high levels in immature OLs but downregulated in myelinating cells (Mi et al., 2011). Enhanced DR6 expression causes caspase 3 (casp3) activation and cell death, whereas attenuation of DR6 function promotes OL maturation, myelination, and casp3 downregulation (Mi et al., 2011). In two different animal models of demyelination, genetic ablation of DR6 enhanced remyelination, and treatment with a DR6 antagonist antibody had the same effect (Mi et al., 2011). These findings reveal not only a crucial role for DR6 in OL development, regeneration, and in remyelination, but also a cellular mechanism that could be targeted in MS to promote remyelination and inhibit the autoimmune component of the disease.
Epigenetic Regulation
Epigenetic mechanisms also regulate OL regeneration and remyelination in adult WM. After demyelination of young brains, HDACs are efficiently recruited to promoter regions of differentiation inhibitors to promote remyelination; however this process is impaired in old animals, whose accumulated transcriptional inhibitors prevent myelin gene expression and efficient remyelination (Shen et al., 2008). Treating young animals with HDAC-inhibitor valproic acid reproduces the defect observed in adults (Shen et al., 2008), indicating that the age-dependent decrease in remyelination efficiency is – at least in part – due to epigenetic modulation of OL differentiation potential. Importantly, the OL-specific transcription factor TCF7L2/TCF4 – a bipartite co-effector of β-cateinin – was also shown to mediate cross talk between HDAC1/2 and the Wnt signaling pathway to regulate OL differentiation (Ye et al., 2009). Similar to mice lacking HADC1 and 2, TCF7L2-null mutants displayed impaired remyelination (Ye et al., 2009). Together, these results indicate that HDACs represent important molecular targets for promoting remyelination under pathological conditions.
Conclusions and Future Directions
The rapid and significant progress in understanding astrocyte and OL development and pathology continues to raise important questions about the role of these cells in human neurological disorders and brain injury. These glial cells clearly play crucial roles in virtually all brain pathologies, and defining their role in brain disease will also provide information about the function of astrocytes and OLs in normal brain development. Continuing to define the cellular, molecular, and physiological properties of glial populations is essential to gain a true understanding of relationships between neural development and regeneration.
A first critical issue in future studies on the roles of astrocyte and OL progenitors in disorders of the developing and adult brain is the lack of specific molecular tools that enable the heterogeneity of these cell populations to be resolved. Once glial populations of different brain regions are identified and isolated, advanced molecular and genetic techniques can be applied to discover crucial genes that regulate their functional properties under normal and pathological conditions. Lineage reporters combined with gene expression profiling, and ChIP-on-Chip, ChIP-Seq, and RNA-Seq assays can be also used to isolate genes preferentially expressed in glial cell subtypes, and to define sets of developmentally regulated glial genes. Genome-wide screening in specific identified glial progenitor cell subpopulations will help identify spatiotemporally restricted expression patterns of transcription factors, as well as other molecular regulators of glial development (Fu et al., 2009) and regeneration (Fancy et al., 2009). Such studies are essential entry points for the isolation and subsequent characterization of these factors and regulators in the developing and injured CNS. These approaches will also help to determine how genetic defects affect glial lineages and the impact of altered glial development on normal brain function. Protein products of genes involved in neurodevelopmental disorders (Pacey et al., 2007) or in adult neurological diseases (Rempe et al., 2010) may be expressed in glial progenitor cells and impair specific signaling pathways. Future research will reveal how specific glial defects contribute to these and other brain disorders, and to brain injury.
A second critical issue for future research is glial progenitor fate plasticity, particularly the identification in vivo of microenvironmental factors that favor or restrict fate plasticity in glia within the intact CNS. Understanding this phenomenon and its underlying mechanisms is important for manipulating not only the fate of endogenous glial progenitor cells but also the fate of progenitors grafted into the pathological brain. Recent studies have identified new roles for the vasculature and endothelial cells’ response to injury in modulating glial progenitor cell proliferation, migration and differentiation (Cayre et al., 2013; Goldman and Chen, 2011). Therefore, signaling pathways that regulate glial progenitor-vasculature interactions might also represent attractive targets for enhancing glial regeneration under pathological conditions.
Another topic for future study is the progressive changes in cellular and molecular properties experienced by glial progenitor cells during CNS development and aging. For example, the remyelinating potential of OPCs decreases from early postnatal development to adulthood (Sim et al., 2002) and astrocytes outside the classic neurogenic areas undergo progressive lineage restriction and lose their neurogenic potential (Berninger, 2010; Bi et al., 2011; Layell et al., 2000). It is crucial to continue characterizing the molecular and cellular mechanisms that regulate timely generation and lineage progression of different glial types in the developing CNS, to define: i) the determinants of developmental time windows for neurogenesis, gliogenesis, and proper functional maturation of the brain, and ii) the potential for extending these time windows for regenerative purposes.
Finally, advances in iPS and ES cell technology hold great promise for cell-replacement therapies across a spectrum of tissues and associated pathologies. Several groups have established protocols to derive astrocytes and OLs, providing a foundation for translational applications. The likely application of this technology in glial repair is in WMI and OL replacement, as myelin sheath restoration is the clear focus of these therapeutic interventions. For astrocytes, the goal is less clear, as their complex roles in injury (protective vs. inhibitory), injury conditions, and cellular heterogeneity make modeling of these conditions very difficult. Future in vivo studies aimed at understanding how different forms of injury differentially harness the reactive astrocyte response are essential for developing iPS or ES approaches to generating protective and regenerative astrocytes for cell-based therapies.
Resolving the issues in brain pathology discussed above will be critical to further elucidate relationships between glial cell populations in rodent models and their human counterparts, providing a better perspective on how to harness knowledge of glial development for therapeutic purposes. In addition, new techniques applied in parallel to human and animal brains – in particular noninvasive neuroimaging (e.g., fMRI, DTI, in vivo spectroscopy), as well as molecular analysis of postmortem tissue or surgical specimens – will determine how findings in animal models of disease relate to human conditions. The generation of more sophisticated animal models that incorporate the genetic heterogeneity seen in humans (Niu et al., 2014) will also promote better translation of animal research to clinical conditions. Thus, studies of glial cells and their responses to injury and pathology present a unique opportunity to interface studies in animal models of disease with neurodevelopmental disorders and diseases of the adult human brain.
Acknowledgments
This work was supported by the NIH (R01NS045702, R01NS056427 and P01NS062686 to V.G., and R01NS071153 to B.D.) and the National Multiple Sclerosis Society RG4706 (V.G.) and RG4623 (B.D.). This work was also supported by Eunice Kennedy Shriver Intellectual and Developmental Disabilities Research Centers P30HD40677 (Children’s National MedicalCenter) and P30HD024064 (Baylor College of Medicine). We thank Jonathan Ritter for figures and Li-Jin Chew for comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguirre A, Dupree JL, Mangin JM, Gallo V. A functional role for EGFR signaling in myelination and remyelination. Nat Neurosci. 2007;10:990–1002. doi: 10.1038/nn1938. [DOI] [PubMed] [Google Scholar]
- Aguirre A, Rubio ME, Gallo V. Notch and EGFR pathway interaction regulates neural stem cell number and self-renewal. Nature. 2010;467:323–327. doi: 10.1038/nature09347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre AA, Chittajallu R, Belachew S, Gallo V. NG2-expressing cells in the subventricular zone are type C-like cells and contribute to interneuron generation in the postnatal hippocampus. J Cell Biol. 2004;165:575–589. doi: 10.1083/jcb.200311141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht PJ, Murtie JC, Ness JK, Redwine JM, Enterline JR, Armstrong RC, Levison SW. Astrocytes produce CNTF during the remyelination phase of viral-induced spinal cord demyelination to stimulate FGF-2 production. Neurobiol Dis. 2003;13:89–101. doi: 10.1016/s0969-9961(03)00019-6. [DOI] [PubMed] [Google Scholar]
- Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia. 2013;61:1939–1958. doi: 10.1002/glia.22575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amankulor NM, Hambardzumyan D, Pyonteck SM, Becher OJ, Joyce JA, Holland EC. Sonic hedgehog pathway activation is induced by acute brain injury and regulated by injury-related inflammation. J Neurosci. 2009;29:10299–10308. doi: 10.1523/JNEUROSCI.2500-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DJ. Stem cells and pattern formation in the nervous system: the possible versus the actual. Neuron. 2001;30:19–35. doi: 10.1016/s0896-6273(01)00260-4. [DOI] [PubMed] [Google Scholar]
- Ara J, See J, Mamontov P, Hahn A, Bannerman P, Pleasure D, Grinspan JB. Bone morphogenetic proteins 4, 6, and 7 are up-regulated in mouse spinal cord during experimental autoimmune encephalomyelitis. J Neurosci Res. 2008;86:125–135. doi: 10.1002/jnr.21462. [DOI] [PubMed] [Google Scholar]
- Armstrong RC, Dorn HH, Kufta CV, Friedman E, Dubois-Dalcq ME. Pre-oligodendrocytes from adult human CNS. J Neurosci. 1992;12:1538–1547. doi: 10.1523/JNEUROSCI.12-04-01538.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RC, Kim JG, Hudson LD. Expression of myelin transcription factor I (MyTI), a “zinc-finger” DNA-binding protein, in developing oligodendrocytes. Glia. 1995;14:303–321. doi: 10.1002/glia.440140407. [DOI] [PubMed] [Google Scholar]
- Armstrong RC, Le TQ, Frost EE, Borke RC, Vana AC. Absence of fibroblast growth factor 2 promotes oligodendroglial repopulation of demyelinated white matter. J Neurosci. 2002;22:8574–8585. doi: 10.1523/JNEUROSCI.22-19-08574.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett HA, Fancy SP, Alberta JA, Zhao C, Plant SR, Kaing S, Raine CS, Rowitch DH, Franklin RJ, Stiles CD. bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science. 2004;306:2111–2115. doi: 10.1126/science.1103709. [DOI] [PubMed] [Google Scholar]
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4:1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- Arumugam TV, Chan SL, Jo DG, Yilmaz G, Tang SC, Cheng A, Gleichmann M, Okun E, Dixit VD, Chigurupati S, et al. Gamma secretase-mediated Notch signaling worsens brain damage and functional outcome in ischemic stroke. Nat Med. 2006;12:621–623. doi: 10.1038/nm1403. [DOI] [PubMed] [Google Scholar]
- Avolio C, Ruggieri M, Giuliani F, Liuzzi GM, Leante R, Riccio P, Livrea P, Trojano M. Serum MMP-2 and MMP-9 are elevated in different multiple sclerosis subtypes. J Neuroimm. 2003;136:46–53. doi: 10.1016/s0165-5728(03)00006-7. [DOI] [PubMed] [Google Scholar]
- Baas D, Bourbeau D, Sarlieve LL, Ittel ME, Dussault JH, Puymirat J. Oligodendrocyte maturation and progenitor cell proliferation are independently regulated by thyroid hormone. Glia. 1997;19:324–332. doi: 10.1002/(sici)1098-1136(199704)19:4<324::aid-glia5>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Back SA. Perinatal white matter injury: the changing spectrum of pathology and emerging insights into pathogenetic mechanisms. Ment Ret Dev Dis Res Rev. 2006;12:129–140. doi: 10.1002/mrdd.20107. [DOI] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Rosenberg PA. Pathophysiology of glia in perinatal white matter injury. Glia. 2014 Mar 31; doi: 10.1002/glia.22658. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badaracco ME, Ortiz EH, Soto EF, Connor J, Pasquini JM. Effect of transferrin on hypomyelination induced by iron deficiency. J Neurosci Res. 2008;86:2663–2673. doi: 10.1002/jnr.21709. [DOI] [PubMed] [Google Scholar]
- Bain G, Ray WJ, Yao M, Gottlieb DI. Retinoic acid promotes neural and represses mesodermal gene expression in mouse embryonic stem cells in culture. Biochem Biophys Res Comm. 1996;223:691–694. doi: 10.1006/bbrc.1996.0957. [DOI] [PubMed] [Google Scholar]
- Bansal R. Fibroblast growth factors and their receptors in oligodendrocyte development: implications for demyelination and remyelination. Dev Neurosci. 2002;24:35–46. doi: 10.1159/000064944. [DOI] [PubMed] [Google Scholar]
- Barnabe-Heider F, Wasylnka JA, Fernandes KJ, Porsche C, Sendtner M, Kaplan DR, Miller FD. Evidence that embryonic neurons regulate the onset of cortical gliogenesis via cardiotrophin-1. Neuron. 2005;48:253–265. doi: 10.1016/j.neuron.2005.08.037. [DOI] [PubMed] [Google Scholar]
- Baron W, Colognato H, ffrench-Constant C. Integrin-growth factor interactions as regulators of oligodendroglial development and function. Glia. 2005;49:467–479. doi: 10.1002/glia.20132. [DOI] [PubMed] [Google Scholar]
- Barres BA, Lazar MA, Raff MC. A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development. 1994;120:1097–1108. doi: 10.1242/dev.120.5.1097. [DOI] [PubMed] [Google Scholar]
- Barres BA, Schmid R, Sendnter M, Raff MC. Multiple extracellular signals are required for long-term oligodendrocyte survival. Development. 1993;118:283–295. doi: 10.1242/dev.118.1.283. [DOI] [PubMed] [Google Scholar]
- Beck KD, Powell-Braxton L, Widmer HR, Valverde J, Hefti F. Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron. 1995;14:717–730. doi: 10.1016/0896-6273(95)90216-3. [DOI] [PubMed] [Google Scholar]
- Belachew S, Aguirre AA, Wang H, Vautier F, Yuan X, Anderson S, Kirby M, Gallo V. Cyclin-dependent kinase-2 controls oligodendrocyte progenitor cell cycle progression and is downregulated in adult oligodendrocyte progenitors. J Neurosci. 2002;22:8553–8562. doi: 10.1523/JNEUROSCI.22-19-08553.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner EJ, Luciano D, Jo R, Abdi K, Paez-Gonzalez P, Sheng H, Warner DS, Liu C, Eroglu C, Kuo CT. Protective astrogenesis from the SVZ niche after injury is controlled by Notch modulator Thbs4. Nature. 2013;497:369–373. doi: 10.1038/nature12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berninger B. Making neurons from mature glia: a far-fetched dream? Neuropharmacol. 2010;58:894–902. doi: 10.1016/j.neuropharm.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Bi B, Salmaso N, Komitova M, Simonini MV, Silbereis J, Cheng E, Kim J, Luft S, Ment LR, Horvath TL, et al. Cortical glial fibrillary acidic protein-positive cells generate neurons after perinatal hypoxic injury. J Neurosci. 2011;31:9205–9221. doi: 10.1523/JNEUROSCI.0518-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogazzi F, Hudson LD, Nikodem VM. A novel heterodimerization partner for thyroid hormone receptor. Peroxisome proliferator-activated receptor. J Biol Chem. 1994;269:11683–11686. [PubMed] [Google Scholar]
- Bonaguidi MA, McGuire T, Hu M, Kan L, Samanta J, Kessler JA. LIF and BMP signaling generate separate and discrete types of GFAP-expressing cells. Development. 2005;132:5503–5514. doi: 10.1242/dev.02166. [DOI] [PubMed] [Google Scholar]
- Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997;278:477–483. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Persaud T, Hu X, Karmally S, Shestopalov VI, Dvoriantchikova G, Ivanov D, Nathanson L, Barnum SR, Bethea JR. Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J Immunol. 2009;182:2628–2640. doi: 10.4049/jimmunol.0802954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229–248. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows RC, Wancio D, Levitt P, Lillien L. Response diversity and the timing of progenitor cell maturation are regulated by developmental changes in EGFR expression in the cortex. Neuron. 1997;19:251–267. doi: 10.1016/s0896-6273(00)80937-x. [DOI] [PubMed] [Google Scholar]
- Busch SA, Silver J. The role of extracellular matrix in CNS regeneration. Current opinion in neurobiology. 2007;17:120–127. doi: 10.1016/j.conb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Qi Y, Hu X, Tan M, Liu Z, Zhang J, Li Q, Sander M, Qiu M. Generation of oligodendrocyte precursor cells from mouse dorsal spinal cord independent of Nkx6 regulation and Shh signaling. Neuron. 2005;45:41–53. doi: 10.1016/j.neuron.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Caillava C, Vandenbosch R, Jablonska B, Deboux C, Spigoni G, Gallo V, Malgrange B, Baron-Van Evercooren A. Cdk2 loss accelerates precursor differentiation and remyelination in the adult central nervous system. J Cell Biol. 2011;193:397–407. doi: 10.1083/jcb.201004146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajal SR. Histologie du systeme nerveux de l’homme et des vertebres. Vol. 1. Paris: Maloine; 1909. [Google Scholar]
- Calver AR, Hall AC, Yu WP, Walsh FS, Heath JK, Betsholtz C, Richardson WD. Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron. 1998;20:869–882. doi: 10.1016/s0896-6273(00)80469-9. [DOI] [PubMed] [Google Scholar]
- Cammer W, Zhang H. Maturation of oligodendrocytes is more sensitive to TNF alpha than is survival of precursors and immature oligodendrocytes. J Neuroimmunol. 1999;97:37–42. doi: 10.1016/s0165-5728(99)00045-4. [DOI] [PubMed] [Google Scholar]
- Cantarella C, Cayre M, Magalon K, Durbec P. Intranasal HB-EGF administration favors adult SVZ cell mobilization to demyelinated lesions in mouse corpus callosum. Dev Neurobiol. 2008;68:223–236. doi: 10.1002/dneu.20588. [DOI] [PubMed] [Google Scholar]
- Carbajal KS, Schaumburg C, Strieter R, Kane J, Lane TE. Migration of engrafted neural stem cells is mediated by CXCL12 signaling through CXCR4 in a viral model of multiple sclerosis. Proc Natl Acad Sci (USA) 2010;107:11068–11073. doi: 10.1073/pnas.1006375107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonell WS, Mandell JW. Transient neuronal but persistent astroglial activation of ERK/MAP kinase after focal brain injury in mice. J Neurotrauma. 2003;20:327–336. doi: 10.1089/089771503765172282. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Behringer RR, Brinster RL, McMorris FA. Insulin-like growth factor I increases brain growth and central nervous system myelination in transgenic mice. Neuron. 1993;10:729–740. doi: 10.1016/0896-6273(93)90173-o. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P. Cell death in the oligodendrocyte lineage: a molecular perspective of life/death decisions in development and disease. Glia. 2000;29:124–135. doi: 10.1002/(sici)1098-1136(20000115)29:2<124::aid-glia5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Tikoo R, Kiyokawa H, Friedrich V, Jr, Chao MV, Koff A. Oligodendrocyte precursor differentiation is perturbed in the absence of the cyclin-dependent kinase inhibitor p27Kip1. Genes & Dev. 1997;11:2335–2346. doi: 10.1101/gad.11.18.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cate HS, Sabo JK, Merlo D, Kemper D, Aumann TD, Robinson J, Merson TD, Emery B, Perreau VM, Kilpatrick TJ. Modulation of bone morphogenic protein signalling alters numbers of astrocytes and oligodendroglia in the subventricular zone during cuprizone-induced demyelination. J Neurochem. 2010;115:11–22. doi: 10.1111/j.1471-4159.2010.06660.x. [DOI] [PubMed] [Google Scholar]
- Cayre M, Courtes S, Martineau F, Giordano M, Arnaud K, Zamaron A, Durbec P. Netrin 1 contributes to vascular remodeling in the subventricular zone and promotes progenitor emigration after demyelination. Development. 2013;140:3107–3117. doi: 10.1242/dev.092999. [DOI] [PubMed] [Google Scholar]
- Cerghet M, Skoff RP, Bessert D, Zhang Z, Mullins C, Ghandour MS. Proliferation and death of oligodendrocytes and myelin proteins are differentially regulated in male and female rodents. J Neurosci. 2006;26:1439–1447. doi: 10.1523/JNEUROSCI.2219-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaboub LS, Deneen B. Developmental origins of astrocyte heterogeneity: the final frontier of CNS development. Dev Neurosci. 2012;34:379–388. doi: 10.1159/000343723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers CB, Peng Y, Nguyen H, Gaiano N, Fishell G, Nye JS. Spatiotemporal selectivity of response to Notch1 signals in mammalian forebrain precursors. Development. 2001;128:689–702. doi: 10.1242/dev.128.5.689. [DOI] [PubMed] [Google Scholar]
- Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. New Engl J Med. 2002;346:165–173. doi: 10.1056/NEJMoa010994. [DOI] [PubMed] [Google Scholar]
- Chang CF, Lin SZ, Chiang YH, Morales M, Chou J, Lein P, Chen HL, Hoffer BJ, Wang Y. Intravenous administration of bone morphogenetic protein-7 after ischemia improves motor function in stroke rats. Stroke. 2003;34:558–564. doi: 10.1161/01.str.0000051507.64423.00. [DOI] [PubMed] [Google Scholar]
- Chen HL, Panchision DM. Concise review: bone morphogenetic protein pleiotropism in neural stem cells and their derivatives--alternative pathways, convergent signals. Stem Cells. 2007;25:63–68. doi: 10.1634/stemcells.2006-0339. [DOI] [PubMed] [Google Scholar]
- Chen J, Leong SY, Schachner M. Differential expression of cell fate determinants in neurons and glial cells of adult mouse spinal cord after compression injury. Eur J Neurosci. 2005;22:1895–1906. doi: 10.1111/j.1460-9568.2005.04348.x. [DOI] [PubMed] [Google Scholar]
- Chen Y, Miles DK, Hoang T, Shi J, Hurlock E, Kernie SG, Lu QR. The basic helix-loop-helix transcription factor olig2 is critical for reactive astrocyte proliferation after cortical injury. J Neurosci. 2008;28:10983–10989. doi: 10.1523/JNEUROSCI.3545-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew LJ, Shen W, Ming X, Senatorov VV, Jr, Chen HL, Cheng Y, Hong E, Knoblach S, Gallo V. SRY-box containing gene 17 regulates the Wnt/beta-catenin signaling pathway in oligodendrocyte progenitor cells. J Neurosci. 2011;31:13921–13935. doi: 10.1523/JNEUROSCI.3343-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JS, Kim SY, Cha JH, Choi YS, Sung KW, Oh ST, Kim ON, Chung JW, Chun MH, Lee SB, Lee MY. Upregulation of gp130 and STAT3 activation in the rat hippocampus following transient forebrain ischemia. Glia. 2003;41:237–246. doi: 10.1002/glia.10186. [DOI] [PubMed] [Google Scholar]
- Chong SY, Chan JR. Tapping into the glial reservoir: cells committed to remaining uncommitted. J Cell Biol. 2010;188:305–312. doi: 10.1083/jcb.200905111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen RI, Marmur R, Norton WT, Mehler MF, Kessler JA. Nerve growth factor and neurotrophin-3 differentially regulate the proliferation and survival of developing rat brain oligodendrocytes. J Neurosci. 1996;16:6433–6442. doi: 10.1523/JNEUROSCI.16-20-06433.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak D, Mori T, Brill MS, Pfeifer A, Falk S, Deng C, Monteiro R, Mummery C, Sommer L, Gotz M. Adult neurogenesis requires Smad4-mediated bone morphogenic protein signaling in stem cells. J Neurosci. 2008;28:434–446. doi: 10.1523/JNEUROSCI.4374-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Confavreux C, Hutchinson M, Hours MM, Cortinovis-Tourniaire P, Moreau T. Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group. New Engl J Med. 1998;339:285–291. doi: 10.1056/NEJM199807303390501. [DOI] [PubMed] [Google Scholar]
- Covey MV, Levison SW. Leukemia inhibitory factor participates in the expansion of neural stem/progenitors after perinatal hypoxia/ischemia. Neuroscience. 2007;148:501–509. doi: 10.1016/J.NEUROSCIENCE.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker SJ, Whitmire JK, Frausto RF, Chertboonmuang P, Soloway PD, Whitton JL, Campbell IL. Persistent macrophage/microglial activation and myelin disruption after experimental autoimmune encephalomyelitis in tissue inhibitor of metalloproteinase-1-deficient mice. Am J Pathol. 2006;169:2104–2116. doi: 10.2353/ajpath.2006.060626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crockett DP, Burshteyn M, Garcia C, Muggironi M, Casaccia-Bonnefil P. Number of oligodendrocyte progenitors recruited to the lesioned spinal cord is modulated by the levels of the cell cycle regulatory protein p27Kip-1. Glia. 2005;49:301–308. doi: 10.1002/glia.20111. [DOI] [PubMed] [Google Scholar]
- Dell’Acqua ML, Lorenzini L, D’Intino G, Sivilia S, Pasqualetti P, Panetta V, Paradisi M, Filippi MM, Baiguera C, Pizzi M, et al. Functional and molecular evidence of myelin- and neuroprotection by thyroid hormone administration in experimental allergic encephalomyelitis. Neuropathol Appl Neurobiol. 2012;38:454–470. doi: 10.1111/j.1365-2990.2011.01228.x. [DOI] [PubMed] [Google Scholar]
- Deneen B, Ho R, Lukaszewicz A, Hochstim CJ, Gronostajski RM, Anderson DJ. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron. 2006;52:953–968. doi: 10.1016/j.neuron.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Dopp JM, Mackenzie-Graham A, Otero GC, Merrill JE. Differential expression, cytokine modulation, and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J Neuroimmunol. 1997;75:104–112. doi: 10.1016/s0165-5728(97)00009-x. [DOI] [PubMed] [Google Scholar]
- Dziembowska M, Tham TN, Lau P, Vitry S, Lazarini F, Dubois-Dalcq M. A role for CXCR4 signaling in survival and migration of neural and oligodendrocyte precursors. Glia. 2005;50:258–269. doi: 10.1002/glia.20170. [DOI] [PubMed] [Google Scholar]
- Emery B. Transcriptional and post-transcriptional control of CNS myelination. Curr Op Neurobiol. 2010a;20:601–607. doi: 10.1016/j.conb.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Emery B. Regulation of oligodendrocyte differentiation and myelination. Science. 2010b;330:779–782. doi: 10.1126/science.1190927. [DOI] [PubMed] [Google Scholar]
- Emsley JG, Macklis JD. Astroglial heterogeneity closely reflects the neuronal-defined anatomy of the adult murine CNS. Neuron Glia Biol. 2006;2:175–186. doi: 10.1017/S1740925X06000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000) Neurochem Res. 2000;25:1439–1451. doi: 10.1023/a:1007677003387. [DOI] [PubMed] [Google Scholar]
- Ericson J, Briscoe J, Rashbass P, van Heyningen V, Jessell TM. Graded sonic hedgehog signaling and the specification of cell fate in the ventral neural tube. Cold Spring Harb Symp Quant Biol. 1997;62:451–466. [PubMed] [Google Scholar]
- Fagel DM, Ganat Y, Silbereis J, Ebbitt T, Stewart W, Zhang H, Ment LR, Vaccarino FM. Cortical neurogenesis enhanced by chronic perinatal hypoxia. Exp Neurol. 2006;199:77–91. doi: 10.1016/j.expneurol.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Fancy SP, Baranzini SE, Zhao C, Yuk DI, Irvine KA, Kaing S, Sanai N, Franklin RJ, Rowitch DH. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes & Dev. 2009;23:1571–1585. doi: 10.1101/gad.1806309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Chan JR, Baranzini SE, Franklin RJ, Rowitch DH. Myelin regeneration: a recapitulation of development? Ann Rev Neurosci. 2011b;34:21–43. doi: 10.1146/annurev-neuro-061010-113629. [DOI] [PubMed] [Google Scholar]
- Fancy SP, Glasgow SM, Finley M, Rowitch DH, Deneen B. Evidence that nuclear factor IA inhibits repair after white matter injury. Annals Neurol. 2012;72:224–233. doi: 10.1002/ana.23590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, Bruce CC, Otero JJ, Huang EJ, Nusse R, et al. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat Neurosci. 2011a;14:1009–1016. doi: 10.1038/nn.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Zhao C, Franklin RJ. Increased expression of Nkx2.2 and Olig2 identifies reactive oligodendrocyte progenitor cells responding to demyelination in the adult CNS. Mol Cell Neurosci. 2004;27:247–254. doi: 10.1016/j.mcn.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Farsetti A, Mitsuhashi T, Desvergne B, Robbins J, Nikodem VM. Molecular basis of thyroid hormone regulation of myelin basic protein gene expression in rodent brain. The J Biol Chem. 1991;266:23226–23232. [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felling RJ, Snyder MJ, Romanko MJ, Rothstein RP, Ziegler AN, Yang Z, Givogri MI, Bongarzone ER, Levison SW. Neural stem/progenitor cells participate in the regenerative response to perinatal hypoxia/ischemia. J Neurosci. 2006;26:4359–4369. doi: 10.1523/JNEUROSCI.1898-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferent J, Zimmer C, Durbec P, Ruat M, Traiffort E. Sonic Hedgehog signaling is a positive oligodendrocyte regulator during demyelination. J Neurosci. 2013;33:1759–1772. doi: 10.1523/JNEUROSCI.3334-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira AA, Nazario JC, Pereira MJ, Azevedo NL, Barradas PC. Effects of experimental hypothyroidism on myelin sheath structural organization. J Neurocytol. 2004;33:225–231. doi: 10.1023/b:neur.0000030697.78488.63. [DOI] [PubMed] [Google Scholar]
- Fischer I, Alliod C, Martinier N, Newcombe J, Brana C, Pouly S. Sphingosine kinase 1 and sphingosine 1-phosphate receptor 3 are functionally upregulated on astrocytes under pro-inflammatory conditions. PloS One. 2011;6:e23905. doi: 10.1371/journal.pone.0023905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Westlye LT, Amlien IK, Walhovd KB. Reduced white matter integrity is related to cognitive instability. J Neurosci. 2011;31:18060–18072. doi: 10.1523/JNEUROSCI.4735-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty M, Richardson WD, Kessaris N. A subset of oligodendrocytes generated from radial glia in the dorsal spinal cord. Development. 2005;132:1951–1959. doi: 10.1242/dev.01777. [DOI] [PubMed] [Google Scholar]
- Fortin D, Rom E, Sun H, Yayon A, Bansal R. Distinct fibroblast growth factor (FGF)/FGF receptor signaling pairs initiate diverse cellular responses in the oligodendrocyte lineage. J Neurosci. 2005;25:7470–7479. doi: 10.1523/JNEUROSCI.2120-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco PG, Silvestroff L, Soto EF, Pasquini JM. Thyroid hormones promote differentiation of oligodendrocyte progenitor cells and improve remyelination after cuprizone-induced demyelination. Exp Neurol. 2008;212:458–467. doi: 10.1016/j.expneurol.2008.04.039. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Crang AJ, Blakemore WF. Transplanted type-1 astrocytes facilitate repair of demyelinating lesions by host oligodendrocytes in adult rat spinal cord. J Neurocytol. 1991;20:420–430. doi: 10.1007/BF01355538. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nature Rev Neurosci. 2008;9:839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Gallo V. The translational biology of remyelination: Past, present, and future. Glia. 2014 Jan 20; doi: 10.1002/glia.22622. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Freeman MR, Rowitch DH. Evolving concepts of gliogenesis: a look way back and ahead to the next 25 years. Neuron. 2013;80:613–623. doi: 10.1016/j.neuron.2013.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MR, Rowitch DH. Evolving concepts of gliogenesis: a look way back and ahead to the next 25 years. Neuron. 2013;80:613–623. doi: 10.1016/j.neuron.2013.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruttiger M, Karlsson L, Hall AC, Abramsson A, Calver AR, Bostrom H, Willetts K, Bertold CH, Heath JK, Betsholtz C, Richardson WD. Defective oligodendrocyte development and severe hypomyelination in PDGF-A knockout mice. Development. 1999;126:457–467. doi: 10.1242/dev.126.3.457. [DOI] [PubMed] [Google Scholar]
- Fu H, Cai J, Clevers H, Fast E, Gray S, Greenberg R, Jain MK, Ma Q, Qiu M, Rowitch DH, et al. A genome-wide screen for spatially restricted expression patterns identifies transcription factors that regulate glial development. J Neurosci. 2009;29:11399–11408. doi: 10.1523/JNEUROSCI.0160-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller ML, DeChant AK, Rothstein B, Caprariello A, Wang R, Hall AK, Miller RH. Bone morphogenetic proteins promote gliosis in demyelinating spinal cord lesions. Annals Neurol. 2007;62:288–300. doi: 10.1002/ana.21179. [DOI] [PubMed] [Google Scholar]
- Gabay L, Lowell S, Rubin LL, Anderson DJ. Deregulation of dorsoventral patterning by FGF confers trilineage differentiation capacity on CNS stem cells in vitro. Neuron. 2003;40:485–499. doi: 10.1016/s0896-6273(03)00637-8. [DOI] [PubMed] [Google Scholar]
- Gadea A, Schinelli S, Gallo V. Endothelin-1 regulates astrocyte proliferation and reactive gliosis via a JNK/c-Jun signaling pathway. J Neurosci. 2008;28:2394–2408. doi: 10.1523/JNEUROSCI.5652-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganat Y, Soni S, Chacon M, Schwartz ML, Vaccarino FM. Chronic hypoxia up-regulates fibroblast growth factor ligands in the perinatal brain and induces fibroblast growth factor-responsive radial glial cells in the sub-ependymal zone. Neuroscience. 2002;112:977–991. doi: 10.1016/s0306-4522(02)00060-x. [DOI] [PubMed] [Google Scholar]
- Garcia AD, Petrova R, Eng L, Joyner AL. Sonic hedgehog regulates discrete populations of astrocytes in the adult mouse forebrain. J Neurosci. 2010;30:13597–13608. doi: 10.1523/JNEUROSCI.0830-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge WP, Miyawaki A, Gage FH, Jan YN, Jan LY. Local generation of glia is a major astrocyte source in postnatal cortex. Nature. 2012;484:376–380. doi: 10.1038/nature10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstl B, Eng LF, Tavaststjerna M, Smith JK, Kruse SL. Lipids and proteins in multiple sclerosis white matter. J Neurochem. 1970;17:677–689. doi: 10.1111/j.1471-4159.1970.tb00547.x. [DOI] [PubMed] [Google Scholar]
- Ghiani CA, Eisen AM, Yuan X, DePinho RA, McBain CJ, Gallo V. Neurotransmitter receptor activation triggers p27(Kip1)and p21(CIP1) accumulation and G1 cell cycle arrest in oligodendrocyte progenitors. Development. 1999;126:1077–1090. doi: 10.1242/dev.126.5.1077. [DOI] [PubMed] [Google Scholar]
- Goldman SA, Chen Z. Perivascular instruction of cell genesis and fate in the adult brain. Nat Neurosci. 2011;14:1382–1389. doi: 10.1038/nn.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes WA, Mehler MF, Kessler JA. Transgenic overexpression of BMP4 increases astroglial and decreases oligodendroglial lineage commitment. Dev Biol. 2003;255:164–177. doi: 10.1016/s0012-1606(02)00037-4. [DOI] [PubMed] [Google Scholar]
- Gomes WA, Mehler MF, Kessler JA. Transgenic overexpression of BMP4 increases astroglial and decreases oligodendroglial lineage commitment. Dev Biol. 2003;255:164–177. doi: 10.1016/s0012-1606(02)00037-4. [DOI] [PubMed] [Google Scholar]
- Goncalves MB, Boyle J, Webber DJ, Hall S, Minger SL, Corcoran JP. Timing of the retinoid-signalling pathway determines the expression of neuronal markers in neural progenitor cells. Dev Biol. 2005;278:60–70. doi: 10.1016/j.ydbio.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Perez O, Romero-Rodriguez R, Soriano-Navarro M, Garcia-Verdugo JM, Alvarez-Buylla A. Epidermal growth factor induces the progeny of subventricular zone type B cells to migrate and differentiate into oligodendrocytes. Stem Cells. 2009;27:2032–2043. doi: 10.1002/stem.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg C, Shikar V, Larsen P, Mak G, Chojnacki A, Yong VW, Weiss S. White matter plasticity and enhanced remyelination in the maternal CNS. J Neurosci. 2007;27:1812–1823. doi: 10.1523/JNEUROSCI.4441-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinspan JB, Edell E, Carpio DF, Beesley JS, Lavy L, Pleasure D, Golden JA. Stage-specific effects of bone morphogenetic proteins on the oligodendrocyte lineage. J Neurobiol. 2000;43:1–17. [PubMed] [Google Scholar]
- Guardia Clausi M, Paez PM, Campagnoni AT, Pasquini LA, Pasquini JM. Intranasal administration of aTf protects and repairs the neonatal white matter after a cerebral hypoxic-ischemic event. Glia. 2012;60:1540–1554. doi: 10.1002/glia.22374. [DOI] [PubMed] [Google Scholar]
- Hack M, Flannery DJ, Schluchter M, Cartar L, Borawski E, Klein N. Outcomes in young adulthood for very-low-birth-weight infants. New Engl J Med. 2002;346:149–157. doi: 10.1056/NEJMoa010856. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Ann Rev Physiol. 2010;72:335–355. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond TR, Gadea A, Dupree J, Kerninon C, Nait-Oumesmar B, Aguirre A, Gallo V. Astrocyte-derived endothelin-1 inhibits remyelination through notch activation. Neuron. 2014;81:588–602. doi: 10.1016/j.neuron.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton DW, Asher RA, Kondo T, Steeves JD, Ramer MS, Fawcett JW. A potential role for bone morphogenetic protein signalling in glial cell fate determination following adult central nervous system injury in vivo. Eur J Neurosci. 2007;26:3024–3035. doi: 10.1111/j.1460-9568.2007.05940.x. [DOI] [PubMed] [Google Scholar]
- Han X, Chen M, Wang F, Windrem M, Wang S, Shanz S, Xu Q, Oberheim NA, Bekar L, Betstadt S, et al. Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell. 2013;12:342–353. doi: 10.1016/j.stem.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama A, Oka A, Ito M, Tanaka F, Okoshi Y, Takashima S. Myelin transcription factor 1 (MyT1) immunoreactivity in infants with periventricular leukomalacia. Dev Brain Res. 2003;140:85–92. doi: 10.1016/s0165-3806(02)00585-0. [DOI] [PubMed] [Google Scholar]
- Hochstim C, Deneen B, Lukaszewicz A, Zhou Q, Anderson DJ. Identification of positionally distinct astrocyte subtypes whose identities are specified by a homeodomain code. Cell. 2008;133:510–522. doi: 10.1016/j.cell.2008.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovelmeyer N, Hao Z, Kranidioti K, Kassiotis G, Buch T, Frommer F, von Hoch L, Kramer D, Minichiello L, Kollias G, et al. Apoptosis of oligodendrocytes via Fas and TNF-R1 is a key event in the induction of experimental autoimmune encephalomyelitis. J Immunol. 2005;175:5875–5884. doi: 10.4049/jimmunol.175.9.5875. [DOI] [PubMed] [Google Scholar]
- Hu QD, Ang BT, Karsak M, Hu WP, Cui XY, Duka T, Takeda Y, Chia W, Sankar N, Ng YK, et al. F3/contactin acts as a functional ligand for Notch during oligodendrocyte maturation. Cell. 2003;115:163–175. doi: 10.1016/s0092-8674(03)00810-9. [DOI] [PubMed] [Google Scholar]
- Huang JK, Jarjour AA, Nait Oumesmar B, Kerninon C, Williams A, Krezel W, Kagechika H, Bauer J, Zhao C, Baron-Van Evercooren A, et al. Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat Neurosci. 2011;14:45–53. doi: 10.1038/nn.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarrola N, Rodriguez-Pena A. Hypothyroidism coordinately and transiently affects myelin protein gene expression in most rat brain regions during postnatal development. Brain Res. 1997;752:285–293. doi: 10.1016/s0006-8993(96)01480-1. [DOI] [PubMed] [Google Scholar]
- Ichiyama T, Kajimoto M, Suenaga N, Maeba S, Matsubara T, Furukawa S. Serum levels of matrix metalloproteinase-9 and its tissue inhibitor (TIMP-1) in acute disseminated encephalomyelitis. J Neuroimmunol. 2006;172:182–186. doi: 10.1016/j.jneuroim.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Ivkovic S, Canoll P, Goldman JE. Constitutive EGFR signaling in oligodendrocyte progenitors leads to diffuse hyperplasia in postnatal white matter. J Neurosci. 2008;28:914–922. doi: 10.1523/JNEUROSCI.4327-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonska B, Aguirre A, Raymond M, Szabo G, Kitabatake Y, Sailor KA, Ming GL, Song H, Gallo V. Chordin-induced lineage plasticity of adult SVZ neuroblasts after demyelination. Nat Neurosci. 2010;13:541–550. doi: 10.1038/nn.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonska B, Scafidi J, Aguirre A, Vaccarino F, Nguyen V, Borok E, Horvath TL, Rowitch DH, Gallo V. Oligodendrocyte regeneration after neonatal hypoxia requires FoxO1-mediated p27Kip1 expression. J Neurosci. 2012;32:14775–14793. doi: 10.1523/JNEUROSCI.2060-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadasz JJ, Rivera FJ, Taubert A, Kandasamy M, Sandner B, Weidner N, Aktas O, Hartung HP, Aigner L, Kury P. p57kip2 regulates glial fate decision in adult neural stem cells. Development. 2012;139:3306–3315. doi: 10.1242/dev.074518. [DOI] [PubMed] [Google Scholar]
- Jean I, Lavialle C, Barthelaix-Pouplard A, Fressinaud C. Neurotrophin-3 specifically increases mature oligodendrocyte population and enhances remyelination after chemical demyelination of adult rat CNS. Brain Res. 2003;972:110–118. doi: 10.1016/s0006-8993(03)02510-1. [DOI] [PubMed] [Google Scholar]
- Jiang P, Chen C, Wang R, Chechneva OV, Chung SH, Rao MS, Pleasure DE, Liu Y, Zhang Q, Deng W. hESC-derived Olig2+ progenitors generate a subtype of astroglia with protective effects against ischaemic brain injury. Nat Commun. 2013;4:2196. doi: 10.1038/ncomms3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John GR, Lee SC, Brosnan CF. Cytokines: powerful regulators of glial cell activation. Neuroscientist. 2003;9:10–22. doi: 10.1177/1073858402239587. [DOI] [PubMed] [Google Scholar]
- John GR, Shankar SL, Shafit-Zagardo B, Massimi A, Lee SC, Raine CS, Brosnan CF. Multiple sclerosis: re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat Med. 2002;8:1115–1121. doi: 10.1038/nm781. [DOI] [PubMed] [Google Scholar]
- Jones SA, Jolson DM, Cuta KK, Mariash CN, Anderson GW. Triiodothyronine is a survival factor for developing oligodendrocytes. Mol Cell End. 2003;199:49–60. doi: 10.1016/s0303-7207(02)00296-4. [DOI] [PubMed] [Google Scholar]
- Justicia C, Gabriel C, Planas AM. Activation of the JAK/STAT pathway following transient focal cerebral ischemia: signaling through Jak1 and Stat3 in astrocytes. Glia. 2000;30:253–270. doi: 10.1002/(sici)1098-1136(200005)30:3<253::aid-glia5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Kang P, Lee HK, Glasgow SM, Finley M, Donti T, Gaber ZB, Graham BH, Foster AE, Novitch BG, Gronostajski RM, Deneen B. Sox9 and NFIA coordinate a transcriptional regulatory cascade during the initiation of gliogenesis. Neuron. 2012;74:79–94. doi: 10.1016/j.neuron.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. 2006;9:173–179. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Juraska JM. Sex differences in the development of axon number in the splenium of the rat corpus callosum from postnatal day 15 through 60. Dev Brain Res. 1997;102:77–85. doi: 10.1016/s0165-3806(97)00080-1. [DOI] [PubMed] [Google Scholar]
- Kremer D, Heinen A, Jadasz J, Gottle P, Zimmermann K, Zickler P, Jander S, Hartung HP, Kury P. p57kip2 is dynamically regulated in experimental autoimmune encephalomyelitis and interferes with oligodendroglial maturation. Proc Natl Acad Sci (USA) 2009;106:9087–9092. doi: 10.1073/pnas.0900204106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Ann Rev Neurosci. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumral A, Tuzun F, Tugyan K, Ozbal S, Yilmaz O, Yesilirmak CD, Duman N, Ozkan H. Role of epigenetic regulatory mechanisms in neonatal hypoxic-ischemic brain injury. Early Hum Dev. 2013;89:165–173. doi: 10.1016/j.earlhumdev.2012.09.016. [DOI] [PubMed] [Google Scholar]
- Kumral A, Tuzun F, Yesilirmak D, Duman N, Ozkan H. Role of epigenetic regulatory mechanisms in neonatal hypoxic-ischemic brain injury. Med Hyp. 2009;72:692–693. doi: 10.1016/j.mehy.2008.10.032. [DOI] [PubMed] [Google Scholar]
- Laywell ED, Rakic P, Kukekov VG, Holland EC, Steindler DA. Identification of a multipotent astrocytic stem cell in the immature and adult mouse brain. Proc Natl Acad Sci (USA) 2000;97:13883–13888. doi: 10.1073/pnas.250471697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levison SW, Goldman JE. Both oligodendrocytes and astrocytes develop from progenitors in the subventricular zone of postnatal rat forebrain. Neuron. 1993;10:201–212. doi: 10.1016/0896-6273(93)90311-e. [DOI] [PubMed] [Google Scholar]
- Lewen A, Soderstrom S, Hillered L, Ebendal T. Expression of serine/threonine kinase receptors in traumatic brain injury. Neuroreport. 1997;8:475–479. doi: 10.1097/00001756-199701200-00020. [DOI] [PubMed] [Google Scholar]
- Li X, Newbern JM, Wu Y, Morgan-Smith M, Zhong J, Charron J, Snider WD. MEK Is a Key Regulator of Gliogenesis in the Developing Brain. Neuron. 2012;75:1035–1050. doi: 10.1016/j.neuron.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon KL, Fancy SP, Franklin RJ, Rowitch DH. Olig gene function in CNS development and disease. Glia. 2006;54:1–10. doi: 10.1002/glia.20273. [DOI] [PubMed] [Google Scholar]
- Lin S, Fan LW, Pang Y, Rhodes PG, Mitchell HJ, Cai Z. IGF-1 protects oligodendrocyte progenitor cells and improves neurological functions following cerebral hypoxia-ischemia in the neonatal rat. Brain Res. 2005;1063:15–26. doi: 10.1016/j.brainres.2005.09.042. [DOI] [PubMed] [Google Scholar]
- Liu J, Casaccia P. Epigenetic regulation of oligodendrocyte identity. Trends Neurosci. 2010;33:193–201. doi: 10.1016/j.tins.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Marino MW, Wong G, Grail D, Dunn A, Bettadapura J, Slavin AJ, Old L, Bernard CC. TNF is a potent anti-inflammatory cytokine in autoimmune-mediated demyelination. Nat Med. 1998;4:78–83. doi: 10.1038/nm0198-078. [DOI] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109:75–86. doi: 10.1016/s0092-8674(02)00678-5. [DOI] [PubMed] [Google Scholar]
- Mandell JW, Gocan NC, Vandenberg SR. Mechanical trauma induces rapid astroglial activation of ERK/MAP kinase: Evidence for a paracrine signal. Glia. 2001;34:283–295. doi: 10.1002/glia.1062. [DOI] [PubMed] [Google Scholar]
- Mandell JW, VandenBerg SR. ERK/MAP kinase is chronically activated in human reactive astrocytes. Neuroreport. 1999;10:3567–3572. doi: 10.1097/00001756-199911260-00019. [DOI] [PubMed] [Google Scholar]
- Marta CB, Adamo AM, Soto EF, Pasquini JM. Sustained neonatal hyperthyroidism in the rat affects myelination in the central nervous system. J Neurosci Res. 1998;53:251–259. doi: 10.1002/(SICI)1097-4547(19980715)53:2<251::AID-JNR14>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Martinez G, Carnazza ML, Di Giacomo C, Sorrenti V, Vanella A. Expression of bone morphogenetic protein-6 and transforming growth factor-beta1 in the rat brain after a mild and reversible ischemic damage. Brain Res. 2001;894:1–11. doi: 10.1016/s0006-8993(00)03140-1. [DOI] [PubMed] [Google Scholar]
- Martinez G, Di Giacomo C, Sorrenti V, Carnazza ML, Ragusa N, Barcellona ML, Vanella A. Fibroblast growth factor-2 and transforming growth factor-beta1 immunostaining in rat brain after cerebral postischemic reperfusion. J Neurosci Res. 2001;63:136–142. doi: 10.1002/1097-4547(20010115)63:2<136::AID-JNR1005>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Mason JL, Suzuki K, Chaplin DD, Matsushima GK. Interleukin-1beta promotes repair of the CNS. J Neurosci. 2001;21:7046–7052. doi: 10.1523/JNEUROSCI.21-18-07046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JL, Toews A, Hostettler JD, Morell P, Suzuki K, Goldman JE, Matsushima GK. Oligodendrocytes and progenitors become progressively depleted within chronically demyelinated lesions. Am J Pathol. 2004;164:1673–1682. doi: 10.1016/S0002-9440(10)63726-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JL, Ye P, Suzuki K, D’Ercole AJ, Matsushima GK. Insulin-like growth factor-1 inhibits mature oligodendrocyte apoptosis during primary demyelination. J Neurosci. 2000;20:5703–5708. doi: 10.1523/JNEUROSCI.20-15-05703.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura I, Taniguchi J, Hata K, Saeki N, Yamashita T. BMP inhibition enhances axonal growth and functional recovery after spinal cord injury. J Neurochem. 2008;105:1471–1479. doi: 10.1111/j.1471-4159.2008.05251.x. [DOI] [PubMed] [Google Scholar]
- Matthieu JM, Comte V, Tosic M, Honegger P. Myelin gene expression during demyelination and remyelination in aggregating brain cell cultures. J Neuroimmunol. 1992;40:231–234. doi: 10.1016/0165-5728(92)90138-b. [DOI] [PubMed] [Google Scholar]
- Matyash V, Kettenmann H. Heterogeneity in astrocyte morphology and physiology. Brain Res Rev. 2010;63:2–10. doi: 10.1016/j.brainresrev.2009.12.001. [DOI] [PubMed] [Google Scholar]
- McKinnon RD, Matsui T, Dubois-Dalcq M, Aaronson SA. FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron. 1990;5:603–614. doi: 10.1016/0896-6273(90)90215-2. [DOI] [PubMed] [Google Scholar]
- McTigue DM, Tripathi RB. The life, death, and replacement of oligodendrocytes in the adult CNS. J Neurochem. 2008;107:1–19. doi: 10.1111/j.1471-4159.2008.05570.x. [DOI] [PubMed] [Google Scholar]
- Meijer DH, Kane MF, Mehta S, Liu H, Harrington E, Taylor CM, Stiles CD, Rowitch DH. Separated at birth? The functional and molecular divergence of OLIG1 and OLIG2. Nat Rev Neurosci. 2012;13:819–831. doi: 10.1038/nrn3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekki-Dauriac S, Agius E, Kan P, Cochard P. Bone morphogenetic proteins negatively control oligodendrocyte precursor specification in the chick spinal cord. Development. 2002;129:5117–5130. doi: 10.1242/dev.129.22.5117. [DOI] [PubMed] [Google Scholar]
- Mela A, Goldman JE. CD82 blocks cMet activation and overcomes hepatocyte growth factor effects on oligodendrocyte precursor differentiation. J Neurosci. 2013;33:7952–7960. doi: 10.1523/JNEUROSCI.5836-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menn B, Garcia-Verdugo JM, Yaschine C, Gonzalez-Perez O, Rowitch D, Alvarez-Buylla A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci. 2006;26:7907–7918. doi: 10.1523/JNEUROSCI.1299-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messersmith DJ, Murtie JC, Le TQ, Frost EE, Armstrong RC. Fibroblast growth factor 2 (FGF2) and FGF receptor expression in an experimental demyelinating disease with extensive remyelination. J Neurosci Res. 2000;62:241–256. doi: 10.1002/1097-4547(20001015)62:2<241::AID-JNR9>3.0.CO;2-D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi S, Hu B, Hahm K, Luo Y, Kam Hui ES, Yuan Q, Wong WM, Wang L, Su H, Chu TH, et al. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat Med. 2007;13:1228–1233. doi: 10.1038/nm1664. [DOI] [PubMed] [Google Scholar]
- Mi S, Lee X, Hu Y, Ji B, Shao Z, Yang W, Huang G, Walus L, Rhodes K, Gong BJ, et al. Death receptor 6 negatively regulates oligodendrocyte survival, maturation and myelination. Nat Med. 2011;17:816–821. doi: 10.1038/nm.2373. [DOI] [PubMed] [Google Scholar]
- Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, Levesque M, Allaire N, Perrin S, Sands B, et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci. 2004;7:221–228. doi: 10.1038/nn1188. [DOI] [PubMed] [Google Scholar]
- Mi S, Miller RH, Lee X, Scott ML, Shulag-Morskaya S, Shao Z, Chang J, Thill G, Levesque M, Zhang M, et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci. 2005;8:745–751. doi: 10.1038/nn1460. [DOI] [PubMed] [Google Scholar]
- Miller FD, Gauthier AS. Timing is everything: making neurons versus glia in the developing cortex. Neuron. 2007;54:357–369. doi: 10.1016/j.neuron.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Miller RH. Regulation of oligodendrocyte development in the vertebrate CNS. Progr Neurobiol. 2002;67:451–467. doi: 10.1016/s0301-0082(02)00058-8. [DOI] [PubMed] [Google Scholar]
- Miller RH, Mi S. Dissecting demyelination. Nat Neurosci. 2007;10:1351–1354. doi: 10.1038/nn1995. [DOI] [PubMed] [Google Scholar]
- Min J, Singh S, Fitzgerald-Bocarsly P, Wood TL. Insulin-like growth factor I regulates G2/M progression through mammalian target of rapamycin signaling in oligodendrocyte progenitors. Glia. 2012;60:1684–1695. doi: 10.1002/glia.22387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming X, Chew LJ, Gallo V. Transgenic overexpression of Sox17 promotes oligodendrocyte development and attenuates demyelination. J Neurosci. 2013;33:12528–12542. doi: 10.1523/JNEUROSCI.0536-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ, ffrench-Constant C. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitew S, Hay CM, Peckham H, Xiao J, Koenning M, Emery B. Mechanisms regulating the development of oligodendrocytes and central nervous system myelin. Neuroscience. 2013 Nov 22; doi: 10.1016/j.neuroscience.2013.11.029. S0306–4522(13)00976–7. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Moll NM, Hong E, Fauveau M, Naruse M, Kerninon C, Tepavcevic V, Klopstein A, Seilhean D, Chew LJ, Gallo V, Oumesmar BN. SOX17 is expressed in regenerating oligodendrocytes in experimental models of demyelination and in multiple sclerosis. Glia. 2013;61:1659–1672. doi: 10.1002/glia.22547. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Krencik R, Ullian EM, Tsai HH, Deneen B, Richardson WD, Barres BA, Rowitch DH. Astrocytes and disease: a neurodevelopmental perspective. Genes & Dev. 2012;26:891–907. doi: 10.1101/gad.188326.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CS, Milner R, Nishiyama A, Frausto RF, Serwanski DR, Pagarigan RR, Whitton JL, Miller RH, Crocker SJ. Astrocytic tissue inhibitor of metalloproteinase-1 (TIMP-1) promotes oligodendrocyte differentiation and enhances CNS myelination. J Neurosci. 2011;31:6247–6254. doi: 10.1523/JNEUROSCI.5474-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morga E, Mouad-Amazzal L, Felten P, Heurtaux T, Moro M, Michelucci A, Gabel S, Grandbarbe L, Heuschling P. Jagged1 regulates the activation of astrocytes via modulation of NFkappaB and JAK/STAT/SOCS pathways. Glia. 2009;57:1741–1753. doi: 10.1002/glia.20887. [DOI] [PubMed] [Google Scholar]
- Morgenstern DA, Asher RA, Fawcett JW. Chondroitin sulphate proteoglycans in the CNS injury response. Prog Brain Res. 2002;137:313–332. doi: 10.1016/s0079-6123(02)37024-9. [DOI] [PubMed] [Google Scholar]
- Mozell RL, McMorris FA. Insulin-like growth factor I stimulates oligodendrocyte development and myelination in rat brain aggregate cultures. J Neurosci Res. 1991;30:382–390. doi: 10.1002/jnr.490300214. [DOI] [PubMed] [Google Scholar]
- Murtie JC, Zhou YX, Le TQ, Vana AC, Armstrong RC. PDGF and FGF2 pathways regulate distinct oligodendrocyte lineage responses in experimental demyelination with spontaneous remyelination. Neurobiol Dis. 2005;19:171–182. doi: 10.1016/j.nbd.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Nagy Z, Westerberg H, Skare S, Andersson JL, Lilja A, Flodmark O, Fernell E, Holmberg K, Bohm B, Forssberg H, et al. Preterm children have disturbances of white matter at 11 years of age as shown by diffusion tensor imaging. Ped Res. 2003;54:672–679. doi: 10.1203/01.PDR.0000084083.71422.16. [DOI] [PubMed] [Google Scholar]
- Nait-Oumesmar B, Decker L, Lachapelle F, Avellana-Adalid V, Bachelin C, Baron-Van Evercooren A. Progenitor cells of the adult mouse subventricular zone proliferate, migrate and differentiate into oligodendrocytes after demyelination. Eur J Neurosci. 1999;11:4357–4366. doi: 10.1046/j.1460-9568.1999.00873.x. [DOI] [PubMed] [Google Scholar]
- Nait-Oumesmar B, Picard-Riera N, Kerninon C, Decker L, Seilhean D, Hoglinger GU, Hirsch EC, Reynolds R, Baron-Van Evercooren A. Activation of the subventricular zone in multiple sclerosis: evidence for early glial progenitors. Proc Natl Acad Sci (USA) 2007;104:4694–4699. doi: 10.1073/pnas.0606835104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani H, Martin E, Hassani H, Clavairoly A, Maire CL, Viadieu A, Kerninon C, Delmasure A, Frah M, Weber M, et al. Ascl1/Mash1 promotes brain oligodendrogenesis during myelination and remyelination. J Neurosci. 2013;33:9752–9768. doi: 10.1523/JNEUROSCI.0805-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namihira M, Kohyama J, Semi K, Sanosaka T, Deneen B, Taga T, Nakashima K. Committed neuronal precursors confer astrocytic potential on residual neural precursor cells. Dev Cell. 2009;16:245–255. doi: 10.1016/j.devcel.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Namura S, Iihara K, Takami S, Nagata I, Kikuchi H, Matsushita K, Moskowitz MA, Bonventre JV, Alessandrini A. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci (USA) 2001;98:11569–11574. doi: 10.1073/pnas.181213498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA, Trapp BD. Axon-glial signaling and the glial support of axon function. Ann Rev Neurosci. 2008;31:535–561. doi: 10.1146/annurev.neuro.30.051606.094309. [DOI] [PubMed] [Google Scholar]
- Ness JK, Mitchell NE, Wood TL. IGF-I and NT-3 signaling pathways in developing oligodendrocytes: differential regulation and activation of receptors and the downstream effector Akt. Dev Neurosci. 2002;24:437–445. doi: 10.1159/000069050. [DOI] [PubMed] [Google Scholar]
- Nielsen JA, Berndt JA, Hudson LD, Armstrong RC. Myelin transcription factor 1 (Myt1) modulates the proliferation and differentiation of oligodendrocyte lineage cells. Mol Cell Neurosci. 2004;25:111–123. doi: 10.1016/j.mcn.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. 2009;10:9–22. doi: 10.1038/nrn2495. [DOI] [PubMed] [Google Scholar]
- Niu Y, Shen B, Cui Y, Chen Y, Wang J, Wang L, Kang Y, Zhao X, Si W, Li W, et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell. 2014;156:836–843. doi: 10.1016/j.cell.2014.01.027. [DOI] [PubMed] [Google Scholar]
- Nobuta H, Ghiani CA, Paez PM, Spreuer V, Dong H, Korsak RA, Manukyan A, Li J, Vinters HV, Huang EJ, et al. STAT3-mediated astrogliosis protects myelin development in neonatal brain injury. Ann Neurol. 2012;72:750–765. doi: 10.1002/ana.23670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygardas PT, Hinkkanen AE. Up-regulation of MMP-8 and MMP-9 activity in the BALB/c mouse spinal cord correlates with the severity of experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2002;128:245–254. doi: 10.1046/j.1365-2249.2002.01855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya W, Funakoshi H, Kurosawa T, Nakamura T. Hepatocyte growth factor (HGF) promotes oligodendrocyte progenitor cell proliferation and inhibits its differentiation during postnatal development in the rat. Brain Res. 2007;1147:51–65. doi: 10.1016/j.brainres.2007.02.045. [DOI] [PubMed] [Google Scholar]
- Okada A, Tominaga M, Horiuchi M, Tomooka Y. Plexin-A4 is expressed in oligodendrocyte precursor cells and acts as a mediator of semaphorin signals. Biochem Biophys Res Comm. 2007;352:158–163. doi: 10.1016/j.bbrc.2006.10.176. [DOI] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Mikami Y, Shimazaki T, Mihara M, Ohsugi Y, Iwamoto Y, Yoshizaki K, Kishimoto T, Toyama Y, Okano H. Blockade of interleukin-6 receptor suppresses reactive astrogliosis and ameliorates functional recovery in experimental spinal cord injury. J Neurosci Res. 2004;76:265–276. doi: 10.1002/jnr.20044. [DOI] [PubMed] [Google Scholar]
- O’Leary MT, Hinks GL, Charlton HM, Franklin RJ. Increasing local levels of IGF-I mRNA expression using adenoviral vectors does not alter oligodendrocyte remyelination in the CNS of aged rats. Mol Cell Neurosci. 2002;19:32–42. doi: 10.1006/mcne.2001.1062. [DOI] [PubMed] [Google Scholar]
- Ong J, Plane JM, Parent JM, Silverstein FS. Hypoxic-ischemic injury stimulates subventricular zone proliferation and neurogenesis in the neonatal rat. Ped Res. 2005;58:600–606. doi: 10.1203/01.PDR.0000179381.86809.02. [DOI] [PubMed] [Google Scholar]
- Orentas DM, Hayes JE, Dyer KL, Miller RH. Sonic hedgehog signaling is required during the appearance of spinal cord oligodendrocyte precursors. Development. 1999;126:2419–2429. doi: 10.1242/dev.126.11.2419. [DOI] [PubMed] [Google Scholar]
- Pacey LK, Doering LC. Developmental expression of FMRP in the astrocyte lineage: implications for fragile X syndrome. Glia. 2007;55:1601–1609. doi: 10.1002/glia.20573. [DOI] [PubMed] [Google Scholar]
- Parras CM, Galli R, Britz O, Soares S, Galichet C, Battiste J, Johnson JE, Nakafuku M, Vescovi A, Guillemot F. Mash1 specifies neurons and oligodendrocytes in the postnatal brain. EMBO J. 2004;23:4495–4505. doi: 10.1038/sj.emboj.7600447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JR, McCandless EE, Dorsey D, Klein RS. CXCR4 promotes differentiation of oligodendrocyte progenitors and remyelination. Proc Natl Acad Sci (USA) 2010;107:11062–11067. doi: 10.1073/pnas.1006301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JR, Williams JL, Muccigrosso MM, Liu L, Sun T, Rubin JB, Klein RS. Astrocyte TNFR2 is required for CXCL12-mediated regulation of oligodendrocyte progenitor proliferation and differentiation within the adult CNS. Acta Neuropathol. 2012;124:847–860. doi: 10.1007/s00401-012-1034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard-Riera N, Decker L, Delarasse C, Goude K, Nait-Oumesmar B, Liblau R, Pham-Dinh D, Baron-Van Evercooren A. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc Natl Acad Sci (USA) 2002;99:13211–13216. doi: 10.1073/pnas.192314199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas AM, Justicia C, Soriano MA, Ferrer I. Epidermal growth factor receptor in proliferating reactive glia following transient focal ischemia in the rat brain. Glia. 1998;23:120–129. doi: 10.1002/(sici)1098-1136(199806)23:2<120::aid-glia3>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Plane JM, Liu R, Wang TW, Silverstein FS, Parent JM. Neonatal hypoxic-ischemic injury increases forebrain subventricular zone neurogenesis in the mouse. Neurobiol Dis. 2004;16:585–595. doi: 10.1016/j.nbd.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Raine CS, Bonetti B, Cannella B. Multiple sclerosis: expression of molecules of the tumor necrosis factor ligand and receptor families in relationship to the demyelinated plaque. Revue Neurologique. 1998;154:577–585. [PubMed] [Google Scholar]
- Ransohoff RM. Animal models of multiple sclerosis: the good, the bad and the bottom line. Nature neuroscience. 2012;15:1074–1077. doi: 10.1038/nn.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempe DA, Nedergaard M. Targeting glia for treatment of neurological disease. Neurotherapeutics. 2010;7:335–337. doi: 10.1016/j.nurt.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice JE, 3rd, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. 1981;9:131–141. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- Richardson WD, Kessaris N, Pringle N. Oligodendrocyte wars. Nat Rev Neurosci. 2006;7:11–18. doi: 10.1038/nrn1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson WD, Young KM, Tripathi RB, McKenzie I. NG2-glia as multipotent neural stem cells: fact or fantasy? Neuron. 2011;70:661–673. doi: 10.1016/j.neuron.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robel S, Berninger B, Gotz M. The stem cell potential of glia: lessons from reactive gliosis. Nat Rev Neurosci. 2011;12:88–104. doi: 10.1038/nrn2978. [DOI] [PubMed] [Google Scholar]
- Rolls A, Shechter R, Schwartz M. The bright side of the glial scar in CNS repair. Nat Rev Neurosci. 2009;10:235–241. doi: 10.1038/nrn2591. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Rowitch DH. Glial specification in the vertebrate neural tube. Nat Rev Neurosci. 2004;5:409–419. doi: 10.1038/nrn1389. [DOI] [PubMed] [Google Scholar]
- Ruckh JM, Zhao JW, Shadrach JL, van Wijngaarden P, Rao TN, Wagers AJ, Franklin RJ. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell. 2012;10:96–103. doi: 10.1016/j.stem.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo JK, Aumann TD, Merlo D, Kilpatrick TJ, Cate HS. Remyelination is altered by bone morphogenic protein signaling in demyelinated lesions. J Neurosci. 2011;31:4504–4510. doi: 10.1523/JNEUROSCI.5859-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni V, Mukhopadhyay A, Tysseling V, Hebert A, Birch D, McGuire TL, Stupp SI, Kessler JA. BMPR1a and BMPR1b signaling exert opposing effects on gliosis after spinal cord injury. J Neurosci. 2010;30:1839–1855. doi: 10.1523/JNEUROSCI.4459-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmaso N, Jablonska B, Scafidi J, Vaccarino FM, Gallo V. Neurobiology of premature brain injury. Nat Neurosci. 2014;17:341–346. doi: 10.1038/nn.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta J, Burke GM, McGuire T, Pisarek AJ, Mukhopadhyay A, Mishina Y, Kessler JA. BMPR1a signaling determines numbers of oligodendrocytes and calbindin-expressing interneurons in the cortex. J Neurosci. 2007;27:7397–7407. doi: 10.1523/JNEUROSCI.1434-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta J, Kessler JA. Interactions between ID and OLIG proteins mediate the inhibitory effects of BMP4 on oligodendroglial differentiation. Development. 2004;131:4131–4142. doi: 10.1242/dev.01273. [DOI] [PubMed] [Google Scholar]
- Sanai N, Nguyen T, Ihrie RA, Mirzadeh Z, Tsai HH, Wong M, Gupta N, Berger MS, Huang E, Garcia-Verdugo JM, et al. Corridors of migrating neurons in the human brain and their decline during infancy. Nature. 2011;478:382–386. doi: 10.1038/nature10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafian TA, Montes C, Imura T, Qi J, Coppola G, Geschwind DH, Sofroniew MV. Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PloS One. 2010;5:e9532. doi: 10.1371/journal.pone.0009532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scafidi J, Fagel DM, Ment LR, Vaccarino FM. Modeling premature brain injury and recovery. Int J Dev Neurosci. 2009;27:863–871. doi: 10.1016/j.ijdevneu.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scafidi J, Hammond TR, Scafidi S, Ritter J, Jablonska B, Roncal M, Szigeti-Buck K, Coman D, Huang Y, McCarter RJ, Jr, et al. Intranasal epidermal growth factor treatment rescues neonatal brain injury. Nature. 2014;506:230–234. doi: 10.1038/nature12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer S, Meisel A, Marschenz S. Epigenetic mechanisms in cerebral ischemia. J Cereb Blood Flow Metab. 2013;33:1335–1346. doi: 10.1038/jcbfm.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CE, Wynn SL, Sesay A, Cruz C, Cheung M, Gomez Gaviro MV, Booth S, Gao B, Cheah KS, Lovell-Badge R, Briscoe J. SOX9 induces and maintains neural stem cells. Nat Neurosci. 2010;13:1181–1189. doi: 10.1038/nn.2646. [DOI] [PubMed] [Google Scholar]
- See J, Mamontov P, Ahn K, Wine-Lee L, Crenshaw EB, 3rd, Grinspan JB. BMP signaling mutant mice exhibit glial cell maturation defects. Mol Cell Neurosci. 2007;35:171–182. doi: 10.1016/j.mcn.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See J, Zhang X, Eraydin N, Mun SB, Mamontov P, Golden JA, Grinspan JB. Oligodendrocyte maturation is inhibited by bone morphogenetic protein. Mol Cell Neurosci. 2004;26:481–492. doi: 10.1016/j.mcn.2004.04.004. [DOI] [PubMed] [Google Scholar]
- See JM, Grinspan JB. Sending mixed signals: bone morphogenetic protein in myelination and demyelination. J Neuropathol Exp Neurol. 2009;68:595–604. doi: 10.1097/NEN.0b013e3181a66ad9. [DOI] [PubMed] [Google Scholar]
- Setoguchi T, Yone K, Matsuoka E, Takenouchi H, Nakashima K, Sakou T, Komiya S, Izumo S. Traumatic injury-induced BMP7 expression in the adult rat spinal cord. Brain Res. 2001;921:219–225. doi: 10.1016/s0006-8993(01)03123-7. [DOI] [PubMed] [Google Scholar]
- Shen S, Li J, Casaccia-Bonnefil P. Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. J Cell Biol. 2005;169:577–589. doi: 10.1083/jcb.200412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Sandoval J, Swiss VA, Li J, Dupree J, Franklin RJ, Casaccia-Bonnefil P. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat Neurosci. 2008;11:1024–1034. doi: 10.1038/nn.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada IS, Borders A, Aronshtam A, Spees JL. Proliferating reactive astrocytes are regulated by Notch-1 in the peri-infarct area after stroke. Stroke. 2011;42:3231–3237. doi: 10.1161/STROKEAHA.111.623280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada IS, LeComte MD, Granger JC, Quinlan NJ, Spees JL. Self-renewal and differentiation of reactive astrocyte-derived neural stem/progenitor cells isolated from the cortical peri-infarct area after stroke. J Neurosci. 2012;32:7926–7940. doi: 10.1523/JNEUROSCI.4303-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Kagawa T, Wada T, Muroyama Y, Takada S, Ikenaka K. Wnt signaling controls the timing of oligodendrocyte development in the spinal cord. Dev Biol. 2005;282:397–410. doi: 10.1016/j.ydbio.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Sim FJ, Zhao C, Penderis J, Franklin RJ. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci. 2002;22:2451–2459. doi: 10.1523/JNEUROSCI.22-07-02451.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Sharma G, Mishra V. Hypoxia inducible factor-1: its potential role in cerebral ischemia. Cell Mol Neurobiol. 2012;32:491–507. doi: 10.1007/s10571-012-9803-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirko S, Behrendt G, Johansson PA, Tripathi P, Costa M, Bek S, Heinrich C, Tiedt S, Colak D, Dichgans M, et al. Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog. Cell Stem Cell. 2013;12:426–439. doi: 10.1016/j.stem.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn J, Natale J, Chew LJ, Belachew S, Cheng Y, Aguirre A, Lytle J, Nait-Oumesmar B, Kerninon C, Kanai-Azuma M, et al. Identification of Sox17 as a transcription factor that regulates oligodendrocyte development. J Neurosci. 2006;26:9722–9735. doi: 10.1523/JNEUROSCI.1716-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soula C, Danesin C, Kan P, Grob M, Poncet C, Cochard P. Distinct sites of origin of oligodendrocytes and somatic motoneurons in the chick spinal cord: oligodendrocytes arise from Nkx2.2-expressing progenitors by a Shh-dependent mechanism. Development. 2001;128:1369–1379. doi: 10.1242/dev.128.8.1369. [DOI] [PubMed] [Google Scholar]
- Spassky N, de Castro F, Le Bras B, Heydon K, Queraud-LeSaux F, Bloch-Gallego E, Chedotal A, Zalc B, Thomas JL. Directional guidance of oligodendroglial migration by class 3 semaphorins and netrin-1. J Neurosci. 2002;22:5992–6004. doi: 10.1523/JNEUROSCI.22-14-05992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassky N, Heydon K, Mangatal A, Jankovski A, Olivier C, Queraud-Lesaux F, Goujet-Zalc C, Thomas JL, Zalc B. Sonic hedgehog-dependent emergence of oligodendrocytes in the telencephalon: evidence for a source of oligodendrocytes in the olfactory bulb that is independent of PDGFRalpha signaling. Development. 2001;128:4993–5004. doi: 10.1242/dev.128.24.4993. [DOI] [PubMed] [Google Scholar]
- Sriram K, Benkovic SA, Hebert MA, Miller DB, O’Callaghan JP. Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration: key signaling pathway for astrogliosis in vivo? J Biol Chem. 2004;279:19936–19947. doi: 10.1074/jbc.M309304200. [DOI] [PubMed] [Google Scholar]
- Stankoff B, Aigrot MS, Noel F, Wattilliaux A, Zalc B, Lubetzki C. Ciliary neurotrophic factor (CNTF) enhances myelin formation: a novel role for CNTF and CNTF-related molecules. J Neurosci. 2002;22:9221–9227. doi: 10.1523/JNEUROSCI.22-21-09221.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt CC, Lommes P, Sock E, Chaboissier MC, Schedl A, Wegner M. The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes & Dev. 2003;17:1677–1689. doi: 10.1101/gad.259003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimori M, Nagao M, Bertrand N, Parras CM, Guillemot F, Nakafuku M. Combinatorial actions of patterning and HLH transcription factors in the spatiotemporal control of neurogenesis and gliogenesis in the developing spinal cord. Development. 2007;134:1617–1629. doi: 10.1242/dev.001255. [DOI] [PubMed] [Google Scholar]
- Sun Y, Nadal-Vicens M, Misono S, Lin MZ, Zubiaga A, Hua X, Fan G, Greenberg ME. Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell. 2001;104:365–376. doi: 10.1016/s0092-8674(01)00224-0. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Ying W, Kauppinen TM. Astrocyte influences on ischemic neuronal death. Curr Mol Med. 2004;4:193–205. doi: 10.2174/1566524043479185. [DOI] [PubMed] [Google Scholar]
- Syed YA, Hand E, Mobius W, Zhao C, Hofer M, Nave KA, Kotter MR. Inhibition of CNS remyelination by the presence of semaphorin 3A. J Neurosci. 2011;31:3719–3728. doi: 10.1523/JNEUROSCI.4930-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchelingerian JL, Monge M, Le Saux F, Zalc B, Jacque C. Differential oligodendroglial expression of the tumor necrosis factor receptors in vivo and in vitro. J Neurochem. 1995;65:2377–2380. doi: 10.1046/j.1471-4159.1995.65052377.x. [DOI] [PubMed] [Google Scholar]
- Tien AC, Tsai HH, Molofsky AV, McMahon M, Foo LC, Kaul A, Dougherty JD, Heintz N, Gutmann DH, Barres BA, Rowitch DH. Regulated temporal-spatial astrocyte precursor cell proliferation involves BRAF signalling in mammalian spinal cord. Development. 2012;139:2477–2487. doi: 10.1242/dev.077214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorich B, Pasquini JM, Garcia CI, Paez PM, Connor JR. Oligodendrocytes and myelination: the role of iron. Glia. 2009;57:467–478. doi: 10.1002/glia.20784. [DOI] [PubMed] [Google Scholar]
- Tsai HH, Li H, Fuentealba LC, Molofsky AV, Taveira-Marques R, Zhuang H, Tenney A, Murnen AT, Fancy SP, Merkle F, et al. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science. 2012;337:358–362. doi: 10.1126/science.1222381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HW, Grant PA, Rissman EF. Sex differences in histone modifications in the neonatal mouse brain. Epigenetics. 2009;4:47–53. doi: 10.4161/epi.4.1.7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WA, Gangoli N, Gokina P, Kim HA, Covey M, Levison SW, Wood TL. Activation of the mammalian target of rapamycin (mTOR) is essential for oligodendrocyte differentiation. J Neurosci. 2009;29:6367–6378. doi: 10.1523/JNEUROSCI.0234-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccarino FM, Ment LR. Injury and repair in developing brain. Arch Dis Child. 2004;89:F190–192. doi: 10.1136/adc.2003.043661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallstedt A, Klos JM, Ericson J. Multiple dorsoventral origins of oligodendrocyte generation in the spinal cord and hindbrain. Neuron. 2005;45:55–67. doi: 10.1016/j.neuron.2004.12.026. [DOI] [PubMed] [Google Scholar]
- van Wijngaarden P, Franklin RJ. Ageing stem and progenitor cells: implications for rejuvenation of the central nervous system. Development. 2013;140:2562–2575. doi: 10.1242/dev.092262. [DOI] [PubMed] [Google Scholar]
- Vana AC, Lucchinetti CF, Le TQ, Armstrong RC. Myelin transcription factor 1 (Myt1) expression in demyelinated lesions of rodent and human CNS. Glia. 2007;55:687–697. doi: 10.1002/glia.20492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vondran MW, Clinton-Luke P, Honeywell JZ, Dreyfus CF. BDNF+/− mice exhibit deficits in oligodendrocyte lineage cells of the basal forebrain. Glia. 2010;58:848–856. doi: 10.1002/glia.20969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VonDran MW, Singh H, Honeywell JZ, Dreyfus CF. Levels of BDNF impact oligodendrocyte lineage cells following a cuprizone lesion. J Neurosci. 2011;31:14182–14190. doi: 10.1523/JNEUROSCI.6595-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vose LR, Vinukonda G, Jo S, Miry O, Diamond D, Korumilli R, Arshad A, Zia MT, Hu F, Kayton RJ, et al. Treatment with thyroxine restores myelination and clinical recovery after intraventricular hemorrhage. J Neurosci. 2013;33:17232–17246. doi: 10.1523/JNEUROSCI.2713-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LB, Tiwari-Woodruff S, Sofroniew MV. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci. 2009;29:11511–11522. doi: 10.1523/JNEUROSCI.1514-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sdrulla AD, diSibio G, Bush G, Nofziger D, Hicks C, Weinmaster G, Barres BA. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron. 1998;21:63–75. doi: 10.1016/s0896-6273(00)80515-2. [DOI] [PubMed] [Google Scholar]
- Wang Y, Cheng X, He Q, Zheng Y, Kim DH, Whittemore SR, Cao QL. Astrocytes from the contused spinal cord inhibit oligodendrocyte differentiation of adult oligodendrocyte precursor cells by increasing the expression of bone morphogenetic proteins. J Neurosci. 2011;31:6053–6058. doi: 10.1523/JNEUROSCI.5524-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werther GA, Russo V, Baker N, Butler G. The role of the insulin-like growth factor system in the developing brain. Horm Res. 1998;49(Suppl 1):37–40. doi: 10.1159/000053066. [DOI] [PubMed] [Google Scholar]
- Wolswijk G. Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci. 1998;18:601–609. doi: 10.1523/JNEUROSCI.18-02-00601.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XG, Hofmann HD, Deller T, Kirsch M. Induction of STAT3 signaling in activated astrocytes and sprouting septal neurons following entorhinal cortex lesion in adult rats. Mol Cell Neurosci. 2002;21:379–392. doi: 10.1006/mcne.2002.1180. [DOI] [PubMed] [Google Scholar]
- Xiang X, Zhang X, Huang QL. Plexin A3 is involved in semaphorin 3F-mediated oligodendrocyte precursor cell migration. Neurosci Lett. 2012;530:127–132. doi: 10.1016/j.neulet.2012.09.058. [DOI] [PubMed] [Google Scholar]
- Yan H, Rivkees SA. Hepatocyte growth factor stimulates the proliferation and migration of oligodendrocyte precursor cells. J Neurosci. 2002;69:597–606. doi: 10.1002/jnr.10323. [DOI] [PubMed] [Google Scholar]
- Yang Z, Levison SW. Hypoxia/ischemia expands the regenerative capacity of progenitors in the perinatal subventricular zone. Neuroscience. 2006;139:555–564. doi: 10.1016/j.neuroscience.2005.12.059. [DOI] [PubMed] [Google Scholar]
- Yao DL, Liu X, Hudson LD, Webster HD. Insulin-like growth factor I treatment reduces demyelination and up-regulates gene expression of myelin-related proteins in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci (USA) 1995;92:6190–6194. doi: 10.1073/pnas.92.13.6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Chen Y, Hoang T, Montgomery RL, Zhao XH, Bu H, Hu T, Taketo MM, van Es JH, Clevers H, et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat Neurosci. 2009;12:829–838. doi: 10.1038/nn.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, D’Ercole AJ. Insulin-like growth factor I protects oligodendrocytes from tumor necrosis factor-alpha-induced injury. Endocrinology. 1999;140:3063–3072. doi: 10.1210/endo.140.7.6754. [DOI] [PubMed] [Google Scholar]
- Ye P, Li L, Richards RG, DiAugustine RP, D’Ercole AJ. Myelination is altered in insulin-like growth factor-I null mutant mice. J Neurosci. 2002;22:6041–6051. doi: 10.1523/JNEUROSCI.22-14-06041.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes-Rapozo V, Berendonk J, Savignon T, Manhaes AC, Barradas PC. Thyroid hormone deficiency changes the distribution of oligodendrocyte/myelin markers during oligodendroglial differentiation in vitro. Int J Dev Neurosci. 2006;24:445–453. doi: 10.1016/j.ijdevneu.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Yu K, McGlynn S, Matise MP. Floor plate-derived sonic hedgehog regulates glial and ependymal cell fates in the developing spinal cord. Development. 2013;140:1594–1604. doi: 10.1242/dev.090845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung SY, Gokhan S, Jurcsak J, Molero AE, Abrajano JJ, Mehler MF. Differential modulation of BMP signaling promotes the elaboration of cerebral cortical GABAergic neurons or oligodendrocytes from a common sonic hedgehog-responsive ventral forebrain progenitor species. Proc Natl Acad Sci (USA) 2002;99:16273–16278. doi: 10.1073/pnas.232586699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi AU, Bessert DA, Ong JE, Xu H, Barks JD, Silverstein FS, Skoff RP. New oligodendrocytes are generated after neonatal hypoxic-ischemic brain injury in rodents. Glia. 2004;46:380–390. doi: 10.1002/glia.20013. [DOI] [PubMed] [Google Scholar]
- Zeger M, Popken G, Zhang J, Xuan S, Lu QR, Schwab MH, Nave KA, Rowitch D, D’Ercole AJ, Ye P. Insulin-like growth factor type 1 receptor signaling in the cells of oligodendrocyte lineage is required for normal in vivo oligodendrocyte development and myelination. Glia. 2007;55:400–411. doi: 10.1002/glia.20469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Argaw AT, Gurfein BT, Zameer A, Snyder BJ, Ge C, Lu QR, Rowitch DH, Raine CS, Brosnan CF, John GR. Notch1 signaling plays a role in regulating precursor differentiation during CNS remyelination. Proc Natl Acad Sci (USA) 2009;106:19162–19167. doi: 10.1073/pnas.0902834106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Zhao L, Du Y, Wei G, Yao WG, Lee WH. Delayed IGF-1 treatment reduced long-term hypoxia-ischemia-induced brain damage and improved behavior recovery of immature rats. Neurol Res. 2009;31:483–489. doi: 10.1179/174313208X338133. [DOI] [PubMed] [Google Scholar]
- Zhou YX, Pannu R, Le TQ, Armstrong RC. Fibroblast growth factor 1 (FGFR1) modulation regulates repair capacity of oligodendrocyte progenitor cells following chronic demyelination. Neurobiol Dis. 2012;45:196–205. doi: 10.1016/j.nbd.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo H, Nishiyama A. Polydendrocytes in development and myelin repair. Neurosci Bull. 2013;29:165–176. doi: 10.1007/s12264-013-1320-4. [DOI] [PMC free article] [PubMed] [Google Scholar]