

Abstract
Objective
Evaluate the influence of gender and butyrylcholinesterase (BuChE) genotype on incidence of progression to AD, rate of cognitive and functional decline, and response to rivastigmine treatment in mild cognitive impairment (MCI) subjects.
Methods
This retrospective exploratory analysis from a 3–4 year, randomized, placebo-controlled study of rivastigmine in MCI subjects included participants who consented to pharmacogenetic testing.
Results
Of 1018 total patients, 490 (253 [52%] female) were successfully genotyped for BuChE. In subjects receiving placebo, the BuChE wt/wt genotype was associated with a statistically significantly higher rate of progression to AD and functional decline in women, compared with men with the BuChE wt/wt genotype. In subjects with a BuChE-K allele receiving placebo, incidence of progression to AD and rate of functional decline were not significantly different by gender, however cognitive decline was significantly faster in men. Statistically significant benefits of rivastigmine treatment on progression to AD, functional decline, ventricular volume expansion, whole brain atrophy and white matter loss were evident in female BuChE wt/wt.
Conclusion
Gender appears to differentially influence the type of decline in MCI subjects according to BuChE genotype, with more rapid progression of cognitive decline in male BuChE-K, and more rapid progression to AD and functional decline in female BuChE wt/wt. Cognitive decline in male BuChE-K and functional decline and progression to AD in female BuChE wt/wt were significantly attenuated by rivastigmine. Rivastigmine treatment also significantly reduced ventricular expansion, whole brain atrophy rate and white matter loss in female BuChE wt/wt, suggesting a possible disease-modifying effect.
Keywords: Mild cognitive impairment, butyrylcholinesterase, rivastigmine
Introduction
The InDDEx (Investigation in Delay to Diagnosis of Alzheimer's disease with Exelon®) Study was a randomized, double-blind, placebo-controlled, 3–4 year clinical trial evaluating the effect of rivastigmine 3–12 mg/day on the time to clinical diagnosis of dementia and rate of decline on cognitive and functional measures in 1018 participants with amnestic mild cognitive impairment (MCI) [1]. The study did not demonstrate any statistically significant effects of rivastigmine on the co-primary outcomes of progression to Alzheimer's disease (AD) or composite Z-score of a cognitive test battery [1]. However, ventricular volume expansion and whole brain atrophy were significantly reduced in rivastigmine-treated women [1].
A previous retrospective analysis designed to characterize the response to rivastigmine in the overall study population and by gender used a Cox proportional hazards regression model that adjusted for all baseline variables shown to influence progression to AD. In that analysis, significantly less progression to AD was seen in rivastigmine-treated subjects in the overall population (n = 1018, p = 0.045), and in women (n = 532, p = 0.046) [2]. However, the authors noted that it was not possible to discern whether the apparently greater response to rivastigmine treatment was merely due to more rapid progression to AD in women.
Rivastigmine is a potent inhibitor of butyrylcholinesterase (BuChE), in addition to acetylcholinesterase [3]. In the InDDEx study, there were statistically significantly fewer progressions to AD in rivastigmine-treated women who lacked a BuChE-K variant allele – and were therefore assumed to almost invariably be homozygous for the wild-type butyrylcholinesterase (BuChE wt/wt) genotype – compared with those who received placebo [1]. Further, the reduction in ventricular volume expansion seen in rivastigmine-treated women in this study remained apparent in women with the BuChE wt/wt genotype but not in those with one or more BuChE-K variant alleles, which are associated with fewer circulating BuChE molecules [1]. The objective of the current retrospective analysis was to further evaluate the influence of gender on rate of progression to AD, cognitive and functional decline in BuChE wt/wt and BuChE-K allele carriers with MCI receiving placebo, and the effects of rivastigmine in these subgroups.
Methods
The InDDEx study was a 3–4 year double-blind, placebo-controlled trial of 1018 participants with MCI. The study was undertaken at 65 centers in 14 countries. A detailed description of the study has been published [1]. Primary efficacy variables were the time to clinical diagnosis of AD, and the rate of cognitive decline on the composite Z-score of a detailed neuropsychological and cognitive test battery. The secondary outcomes of the InDDEx study included the 11-item Alzheimer's Disease Assessment Scale cognitive subscale (ADAS-cog) [4], the 24-item Alzheimer's Disease Cooperative Study Activities of Daily Living scale (ADCS-ADL) [5], the individual tests from the cognitive battery [1], and magnetic resonance imaging (MRI) measures to evaluate volumetric changes of the hippocampi, ventricles, and whole brain. All procedures were in accordance with ethical standards of the responsible committees on human experimentation and with the Helsinki Declaration.
Inclusion criteria
All participants from the InDDEx study who consented to pharmacogenetic testing and were successfully genotyped for BuChE were included in the current study.
Pharmacogenetic testing
Pharmacogenetic testing was performed at all centers in consenting participants. Genomic DNA was extracted from blood samples at a central laboratory using the Puregene DNA Isolation kit (D-50K; Gentra, Minneapolis, MN, USA). Genotyping for APOE ε4 and BuChE-K was performed as described previously [6].
Assessment of CSF proteins and BuChE activity
Participants from one of the study centers had plasma and cerebrospinal fluid (CSF) samples taken at baseline to assess both plasma and CSF BuChE activity. The blood and CSF samples were centrifuged at 2,000 g for 10 minutes, and the plasma was separated from the whole blood. All samples were stored at -70°C until analysis. Specific BuChE activity was measured using the Ellman's colorimetric method [7], as modified by Darreh-Shori et al [8].
Brain volumes
MRI scans were performed at selected centers only. MRI at each site included a volumetric spoiled gradient echo (T1-weighted) sequence with slice partition thickness of 1.5 mm or less with an in-plane resolution of approximately 1 mm (0.9375 in plane) in each plane direction (e.g. 124–128 slices with 256 × 256 matrix with a field of view of the order of 24 cm × 24 cm) [1]. Hippocampal volumes were determined by region with histogram constraints in the sagittal projection with manual editing accomplished in the sagittal and coronal projections [9]. Ventricular volumes were measured using manual outlining [10]. Rates of global brain atrophy were measured from registered image analysis using the boundary shift integral [11]. For assessment of white matter volume, the MRI segmentation was performed using an atlas-based approach as previously described [9].
Statistical analyses
Study population
As for the primary analysis of the InDDEx study [1], the study population used in this post-hoc analysis was the modified intent-to-treat last observation carried forward (MITT-LOCF) population. Analysis of brain volumes was performed using the classical intent-to-treat last observation carried forward (CITT-LOCF) population. Both populations included all participants successfully genotyped for BuChE, regardless of whether study drug was received or post-baseline assessments were made. The MITT endpoint was determined as follows: if AD diagnosis was reached, the last assessment prior to AD diagnosis was used. For the CITT population, the last assessment in the double-blind phase, regardless of whether assessment was performed prior, at the time of, or after diagnosis of AD, was used. In both populations, for subjects who did not progress to AD during the study, the end of study assessment (at Year 3 or 4) was used. For participants who discontinued before the end of the double-blind study for reasons other than AD diagnosis, the last retrieved drop-out assessment was used. If this was not available, the participant's last available assessment was used. Results reported are CITT for brain volumes and MITT for all other outcomes unless stated otherwise. Both populations will be referred to as ITT in this article.
Supportive observed case (OC) analyses were also performed for all measures except brain volumetric analyses and progression to AD. The OC analysis included participants who had not discontinued from the study and were available at the time of assessment. When diagnosed with AD, participants either entered open-label treatment or discontinued the study, thus they were not included in the OC analysis after diagnosis. The OC, or “completers” population, included all participants who were available for assessment at study endpoint (Year 3, or Year 4 for protocol amendment participants).
Statistical comparisons of baseline demographics
Baseline differences were compared in the following subgroups:
The subpopulation of subjects successfully genotyped for BuChE (pharmacogenetic population) was compared with the overall InDDEx study population.
All BuChE-K carriers (both men and women) were compared with all BuChE wt/wt subjects.
Women with at least one BuChE-K allele (K/*) were compared with BuChE-K/* men.
BuChE wt/wt women were compared with BuChE wt/wt men.
All participants (whether randomized to rivastigmine or placebo) were included in baseline comparisons. Comparisons of baseline characteristics were performed using ANOVA for continuous variables (age and years of education) and Fisher's exact test for dichotomous variables (gender and race). Baseline outcome scores were compared using ANCOVA, with a two-tailed test adjusted for age, race and years of education. For the purpose of baseline comparison across groups, the hippocampal volume and white matter volume of each participant was normalized according to the total brain volumes of the sample group, using the formula:
Volume (normalized) = hippocampal or white matter volume (individual) × total brain volume (mean) / total brain volume (individual)
Comparisons between gender groups were not undertaken for whole brain volume or ventricle volume as these assessments were not normalized for gender.
BuChE activity and biomarker analysis
Differences in plasma and CSF BuChE activity at baseline were compared between men and women only. An ANCOVA model was used, adjusting for age, years of education and BuChE and APOE genotype.
Statistical analysis of efficacy outcome measures (placebo-treated subjects)
The Cox's proportional hazards regression model used to analyze the time to diagnosis of AD has been described previously [1]. Kaplan-Meier estimates were performed as supporting non-parametric analyses, with significance values calculated using the log-rank test.
Differences between men and women in change from baseline to endpoint on the composite Z-score of the cognitive test battery, ADAS-cog, ADCS-ADL and brain volumes were also compared in each gender and BuChE genotype subgroup using an ANCOVA model. Age, race, years of education and baseline values of the outcome measure being assessed were included as covariates.
Statistical analysis of efficacy outcome measures (treatment effect of rivastigmine versus placebo)
The interaction between gender and treatment on progression to AD by BuChE genotype was assessed using Cox's proportional hazards regression model. Age, race, and years of education were included in this model as explanatory variables. Kaplan-Meier estimates with a log-rank test were also used to compare differences in incidence of progression to AD between treatments for each of the four gender and genotype subgroups.
To assess differences in change from baseline to endpoint on the composite Z-score of the cognitive test battery, ADAS-cog, ADCS-ADL and brain volumes, rivastigmine-treated subjects were compared with placebo-treated subjects in each of the four gender and genotype subgroups using an ANCOVA model. A 2-way model analysis was also used to assess the interaction of gender and treatment on these outcome measures in each BuChE genotype group.
As the current retrospective study was intended to be exploratory and hypothesis-generating, no statistical corrections for multiple comparisons were undertaken.
Results
Subjects
Of the 1018 participants of the InDDEx study, 490 were successfully genotyped for BuChE (and thus met the inclusion criteria of the current study). Of these, 153 men and 181 women lacked a BuChE-K allele, and 84 men and 72 women had one or more BuChE-K alleles. No significant deviation from Hardy-Weinberg equilibrium was observed for genotype frequencies of BuChE. BuChE activity was determined in a small number of plasma (36 women and 27 men) and CSF (18 women and 14 men) baseline samples (from a single center).
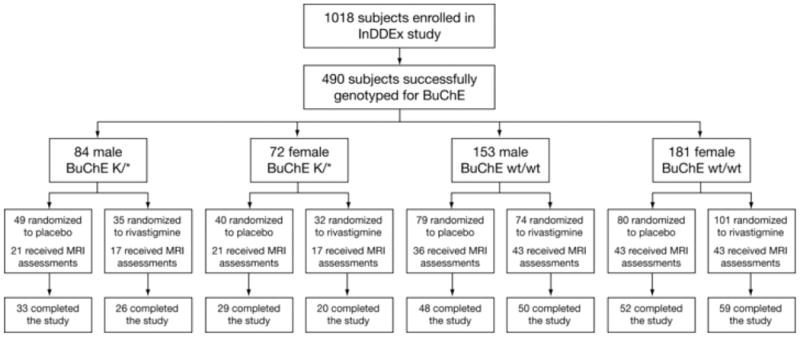
Figure 1 provides the flow of subjects through the study, and shows the numbers of subjects from each subgroup randomized to rivastigmine and placebo, the number of subjects who received MRI assessments, and the number of subjects who completed the study. The rate of discontinuation from the study was similar in each subgroup.
Figure 1. Patient flow.
Baseline comparisons
Baseline demographics are summarized in Table 1. No significant differences were observed between this pharmacogenetic subpopulation (n = 490) and the overall study population (n = 1018). Similar to findings in the full InDDEx study population [1], women relative to men in this pharmacogenetic subpopulation had more impaired cognitive function at baseline on the MMSE (26.1 ± 3.4 versus 27.2 ± 2.4, p = 0.003), cognitive test battery (-2.0 ± 7.5 versus 0.8 ± 6.2, p = 0.025) and ADAS-cog (11.4 ± 5.5 versus 10.1 ± 4.5, p = 0.081), and impaired functional ability on the ADCS-ADL (58.6 ± 7.9 versus 57.6 ± 8.6, p = 0.008). They also had fewer years of education compared with men (9.9 ± 3.8 versus 12.4 ± 4.6, p < 0.0001).
Table 1. Demographic and baseline characteristics by gender and BuChE genotype in MCI participants in the current study (pharmacogenetic population) and in the overall study population (ITT analysis).
| Variables | Men | Women | Overall | |||
|---|---|---|---|---|---|---|
| BuChE-K/* | BuChE wt/wt | BuChE-K/* | BuChE wt/wt | Pharmacogenetic population | Study population | |
| n = 84 | n = 153 | n = 72 | n = 181 | n = 490 | n = 1018 | |
| Age in years, mean (SD) | 70.74 (8.02) | 71.21 (6.67) | 70.89 (7.35) | 71.28 (7.51) | 71.11 (7.31) | 70.46 (7.48) |
| Gender, % women | 51.63 | 52.26 | ||||
| Race, % Caucasian | 95.24 | 95.42 | 100 | 93.92 | 95.51 | 97.05 |
| Years of education, mean (SD) | 12.30 (3.94) | 12.50 (4.95) | 9.60 (3.69) | 10.02 (3.86) | 11.12 (4.40) | 11.03 (4.08) |
| MMSE, mean (SD) | 27.25 (2.62) | 27.21 (2.21) | 26.00 (3.27) | 26.08 (3.41) | 26.62 (2.97) | 26.96 (2.71) |
| Composite Z-score, mean (SD) | 0.42 (6.70) | 0.94 (5.84) | -2.34 (6.99) | -1.92 (7.66) | -0.69 (6.99) | 0 (6.83) |
| ADAS-cog, mean (SD) | 9.75 (4.63) | 10.26 (4.38) | 11.67 (5.74) | 11.28 (5.34) | 10.76 (5.03) | 10.32 (4.91) |
| ADCS-ADL, mean (SD) | 56.12 (9.75) | 58.34 (7.76) | 58.73 (7.63) | 58.50 (7.98) | 58.07 (8.22) | 58.07 (8.15) |
| n = 38 | n = 79 | n = 38 | n = 86 | n = 241 | n = 513 | |
| Left hippocampal volume, cm3, mean (SD) | 2.00 (0.39) | 1.88 (0.36) | 1.95 (0.47) | 1.93 (0.44) | 1.93 (0.41) | 1.88 (0.47) |
| Right hippocampal volume, cm3, mean (SD) | 2.06 (0.42) | 1.94 (0.42) | 2.05 (0.42) | 1.98 (0.41) | 1.99 (0.42) | 1.94 (0.48) |
| Ventricular volume, cm3, mean (SD) | 35.44 (20.87) | 38.54 (19.92) | 33.95 (16.36) | 35.81 (25.14) | 36.35 (21.54) | 37.64 (22.90) |
| Whole brain volume, cm3, mean (SD) | 1135.21 (120.47) | 1120.03 (136.44) | 998.34 (103.67) | 996.10 (117.89) | 1059.01 (137.88) | 1062.04 (135.24) |
| Whole brain white matter volume, cm3, mean (SD) | 526.19 (67.23) | 557.99 (73.89) | 531.68 (80.38) | 548.87 (89.68) | 545.57 (80.38) | 545.32 (77.31) |
Includes both placebo- and rivastigmine-treated participants. K/*, participants with one or more BuChE-K allele; wt/wt, participants with two wild-type BuChE alleles; ADAS-cog, Alzheimer's Disease Assessment Scale Cognitive Subscale; MMSE, Mini-Mental State Examination; ADCS-ADL, Alzheimer's Disease Co-operative Study Activities of Daily Living. The hippocampal and white matter volumes of each participant were normalized according to the total brain volumes of the sample group, but no adjustment was made for the whole brain or ventricular volumes.
When all subjects with one or more BuChE-K alleles were compared with those without a BuChE-K allele, no significant differences between genotype groups in terms of baseline demographics or levels of cognitive or functional impairment were seen (data not shown). However, the proportion of women in the BuChE wt/wt group was greater than in the BuChE-K carrier group; a difference which showed a trend towards statistical significance (54.2% versus 46.2%, p = 0.097).
There were no significant differences in demographic characteristics or in levels of cognitive or functional impairment at baseline between the subgroups determined by BuChE genotype and gender (Table 1).
At baseline, mean CSF BuChE activity was numerically slightly higher in men compared with women (8.4 versus 7.6 nmol/min/ml, p = 0.614). Mean plasma BuChE activity was similar in both groups (3.4 nmol/min/ml in men versus 3.2 nmol/min/ml in women, p = 0.283). A 13% higher level of BuChE activity in BuChE-K non-carriers relative to carriers (regardless of gender) in both plasma and in CSF has previously been reported for this study population [12].
Mean daily doses of rivastigmine were 7.4 mg in men and 5.1 mg in women with the BuChE-K/* genotype, and 6.6 mg in men and 5.2 mg in women with the BuChE wt/wt genotype.
Outcome measures (placebo-treated participants)
Progression to AD
As in the overall study [1], women receiving placebo were more likely to progress to dementia than men receiving placebo in this pharmacogenetic subpopulation (30.0% versus 14.8%, p = 0.012). Significant differences in progression to AD were not seen between the two BuChE genotype groups receiving placebo. However, when gender was considered, women with the BuChE wt/wt genotype had the highest rates of progression to AD across all placebo-treated gender and genotype subgroups, while BuChE wt/wt men had the lowest rate (30.0% versus 13.9%, log-rank test p = 0.011; Table 2). The proportions of BuChE-K allele carriers receiving placebo who progressed to AD were 30.0% in women and 16.3% in men (log-rank test p = 0.192).
Table 2. Cox regression and Kaplan-Meier analyses of progression to AD by gender and BuChE genotype (ITT-LOCF analysis).
| % progression to AD | p-value3 (rivastigmine vs. placebo) | p-value4 (rivastigmine vs. placebo) | Hazard Ratio (95% CI) | |||
|---|---|---|---|---|---|---|
|
| ||||||
| BuChE genotype | Gender | Rivastigmine | Placebo | |||
| BuChE wt/wt | Men (n = 153) | 13.5 | 13.9 | 0.998 | 0.0745 | 0.369 (0.123–1.104) |
| Women (n = 181) | 12.9 | 30.0 | 0.014 | |||
|
| ||||||
| BuChE-K | Men (n = 84) | 14.3 | 16.3 | 0.735 | 0.8738 | 1.125 (0.262–4.836) |
| Women (n = 72) | 21.9 | 30.0 | 0.567 | |||
|
| ||||||
| p-value1 (men vs. women in BuChE-K) | 0.313 | 0.192 | ||||
|
| ||||||
| p-value2 (men vs. women in BuChE wt/wt) | 0.862 | 0.011 | ||||
Log-rank test from Kaplan-Meier estimate, men versus women in subjects with BuChE-K/* genotype
Log-rank test from Kaplan-Meier estimate, men versus women in subjects with BuChE wt/wt genotype
Log-rank test from Kaplan-Meier estimate, rivastigmine versus placebo in each gender and BuChE genotype subgroup
Chi-squared test from Cox proportional hazards model to test interaction between gender and treatment on progression to AD. Age, race and years of education were included in the model
Cognitive and functional measures
No significant differences between gender and genotype subgroups were seen on the total Z-score of the cognitive battery in either the ITT analysis (Table 3) or OC analysis (data not shown).
Table 3. Change from baseline in cognitive and functioning efficacy variables and brain volumes by gender and BuChE genotype (ITT-LOCF).
| Change from baseline to endpoint, mean (SD) | ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| BuChE-K/* | BuChE wt/wt | |||||||
|
| ||||||||
| Efficacy variable | men | women | men | women | p-value1 | p-value2 | p-value3 | p-value4 |
| Number of patients (ITT) | n = 84 | n = 72 | n = 153 | n = 181 | ||||
|
| ||||||||
| Total Z-score of cognitive battery | ||||||||
| Placebo | -1.1 (4.7) | -0.9 (4.9) | -0.9 (4.1) | -0.9 (4.7) | 0.535 | 0.351 | 0.469 | 0.982 |
| Rivastigmine | -0.4 (4.7) | -0.6 (4.3) | -1.2 (4.9) | -1.0 (4.1) | 0.902 | 0.835 | ||
| p-value5 | 0.274 | 0.922 | 0.767 | 0.663 | ||||
|
| ||||||||
| ADAS-cog | ||||||||
| Placebo | -4.7 (9.7) | -2.5 (8.2) | -1.2 (5.4) | -4.1 (10.8) | 0.040 | 0.299 | 0.632 | 0.499 |
| Rivastigmine | -2.2 (8.5) | -0.9 (4.6) | -1.7 (7.9) | -3.0 (8.4) | 0.135 | 0.280 | ||
| p-value5 | 0.037 | 0.111 | 0.559 | 0.664 | ||||
|
| ||||||||
| ADCS-ADL | ||||||||
| Placebo | -5.5 (12.8) | -6.0 (12.7) | -3.4 (9.1) | -10.9 (18.2) | 0.927 | 0.002 | 0.730 | 0.014 |
| Rivastigmine | -6.9 (16.8) | -4.9 (9.7) | -5.7 (11.3) | -6.1 (14.3) | 0.739 | 0.770 | ||
| p-value5 | 0.784 | 0.769 | 0.165 | 0.046 | ||||
|
| ||||||||
| Number of patients (ITT) | n = 37 | n = 38 | n = 79 | n = 86 | ||||
|
| ||||||||
| Total hippocampal volume, cm3 | ||||||||
| Placebo | -0.3 (0.5) | -0.5 (0.6) | -0.3 (0.6) | -0.4 (0.5) | 0.727 | 0.633 | 0.714 | 0.526 |
| Rivastigmine | -0.5 (0.6) | -0.5 (0.6) | -0.2 (0.5) | -0.4 (0.7) | 0.632 | 0.162 | ||
| p-value5 | 0.681 | 0.770 | 0.448 | 0.911 | ||||
|
| ||||||||
| Annualized rates of ventricular expansion, cm3/year | ||||||||
| Placebo | 3.9 (5.7) | 7.5 (6.5) | 5.2 (6.2) | 5.7 (6.4) | 0.015 | 0.893 | 0.137 | 0.206 |
| Rivastigmine | 4.1 (3.6) | 3.6 (5.1) | 5.5 (4.5) | 3.6 (5.1) | 0.673 | 0.120 | ||
| p-value5 | 0.843 | 0.116 | 0.634 | 0.044 | ||||
|
| ||||||||
| Annualized rates of brain atrophy, cm3/year | ||||||||
| Placebo | -1.1 (10.4) | -7.2 (8.7) | -2.9 (11.1) | -5.4 (13.1) | 0.160 | 0.526 | 0.327 | 0.007 |
| Rivastigmine | -5.5 (5.9) | -4.1 (22.5) | -6.0 (6.2) | -0.6 (11.2) | 0.949 | 0.003 | ||
| p-value5 | 0.188 | 0.640 | 0.060 | 0.039 | ||||
|
| ||||||||
| Whole brain white matter volume, % change | ||||||||
| Placebo | 5.2 (6.7) | 9.1 (12.5) | 7.0 (9.9) | 6.8 (10.5) | 0.204 | 0.659 | 0.098 | 0.124 |
| Rivastigmine | 8.6 (6.1) | 4.4 (9.7) | 7.1 (6.6) | 3.0 (7.7) | 0.143 | 0.019 | ||
| p-value6 | 0.321 | 0.155 | 0.947 | 0.027 | ||||
ANCOVA model. Age, race and years of education were adjusted.
Men versus women in subjects with BuChE-K/*
Men versus women in subjects with BuChE wt/wt
Interaction of gender and treatment in subjects with BuChE-K/* (2-way model)
Interaction of gender and treatment in subjects with BuChE wt/wt (2-way model)
Rivastigmine versus placebo in each gender and BuChE genotype subgroup
BuChE-K carrier men receiving placebo were associated with the most rapid cognitive decline on the ADAS-cog across all gender and genotype subgroups, while BuChE-K carrier women had significantly slower decline (change from baseline: -4.71 versus -2.48, p = 0.040; Table 3). Decline on the ADAS-cog in BuChE wt/wt subjects receiving placebo was -4.14 in women and -1.15 in men (p = 0.299). In the OC analysis, BuChE-K carrier men were again associated with more rapid decline compared with BuChE-K carrier women, although the difference did not reach statistical significance (-3.04 versus -0.44, p = 0.165). No significant differences were seen between men and women with the BuChE wt/wt genotype in the OC analysis (0.23 versus -0.52, p = 0.899).
BuChE wt/wt women receiving placebo were associated with the highest rates of functional decline on the ADCS-ADL across all gender and genotype subgroups, while BuChE wt/wt men showed the least decline (change from baseline: -10.91 versus -3.38, p = 0.002; Table 3). Functional decline on the ADCS-ADL in BuChE-K carriers receiving placebo was -5.95 in women and -5.45 in men (p = 0.927). In the OC population, no significant differences at study endpoint were seen between men and women with the BuChE wt/wt genotype (-2.21 in men versus -2.94 in women, p = 0.917) or the BuChE-K/* genotype (-2.20 in men versus -0.06 in women, p = 0.313).
Brain volumes
BuChE-K carrier women receiving placebo showed numerically the greatest decline in hippocampal volume, whole brain atrophy, white matter volume and ventricular expansion (Table 3). In BuChE-K carriers the increase in ventricular expansion was significantly greater in women than in men (7.50 versus 3.88 cm3/year, p = 0.015).
Effect of rivastigmine treatment
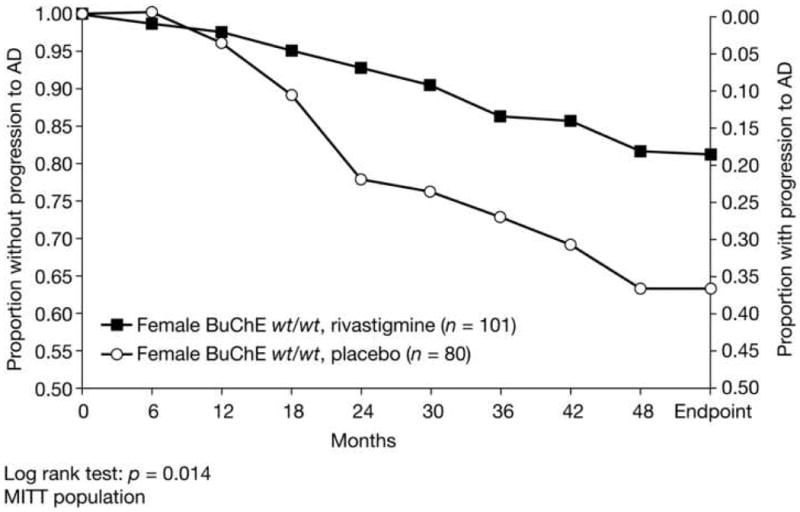
The rate of progression to AD in women with the BuChE wt/wt genotype was reduced by rivastigmine treatment compared with placebo-treated subjects (12.9% versus 30.0%; hazard ratio 0.369; chi-squared test p = 0.0745; Table 2; Figure 2). This effect was significant in the Kaplan-Meier test (log-rank test p = 0.014). No significant effects of rivastigmine on progression to AD were seen in men in either BuChE genotype group or in women with the BuChE-K allele (Table 2).
Figure 2. Kaplan-Meier estimates of progression to AD over 3–4 years by treatment group in BuChE wt/wt women with MCI (ITT).
No significant benefits of rivastigmine were seen on the Z-score of the cognitive test battery in any of the gender and genotype groups in the ITT analysis (Table 3), or the OC analysis (data not shown). Cognitive decline on the ADAS-cog was significantly reduced by rivastigmine treatment in male BuChE-K carriers compared with those receiving placebo in the ITT analysis (-2.23 versus -4.71, p = 0.037, Table 3) and the OC analysis (1.12 versus -3.04, p = 0.027). No significant benefits of treatment were seen in BuChE wt/wt men or in women with either BuChE genotype in the ITT analysis (Table 3) or the OC analysis (data not shown). No significant interaction of gender and treatment was seen in subjects of either BuChE genotype.
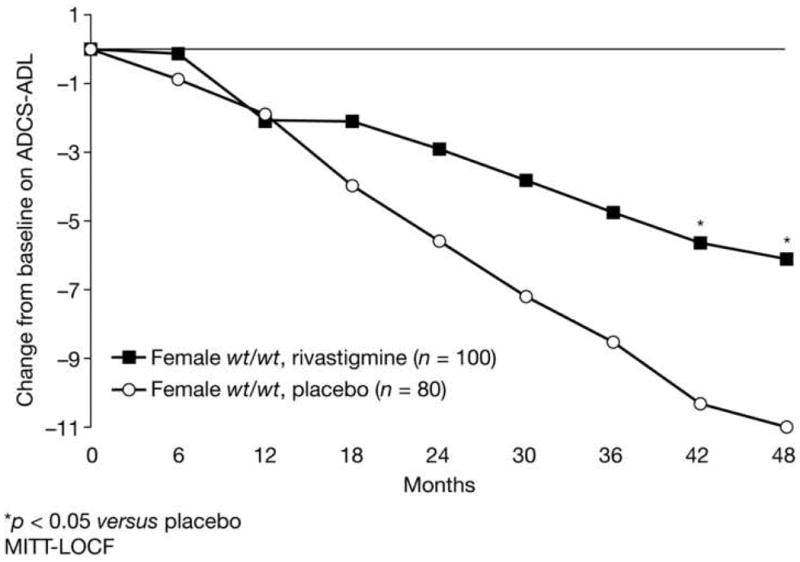
Functional decline on the ADCS-ADL was significantly reduced by rivastigmine treatment in BuChE wt/wt women compared with those receiving placebo in the ITT analysis (-6.08 versus -10.9, p = 0.046, Table 3, Figure 3), but not in the OC analysis (-2.07 versus -2.94, p = 0.550). No significant effects of rivastigmine on functional decline were seen for men in either BuChE genotype group or in women with the BuChE-K allele in the ITT analysis (Table 3) or the OC analysis (data not shown). A significant interaction between gender and treatment on the ADCS-ADL was seen in subjects with the BuChE wt/wt genotype (p = 0.014).
Figure 3. Functional decline over 3–4 years by treatment group in BuChE wt/wt women with MCI (ITT).
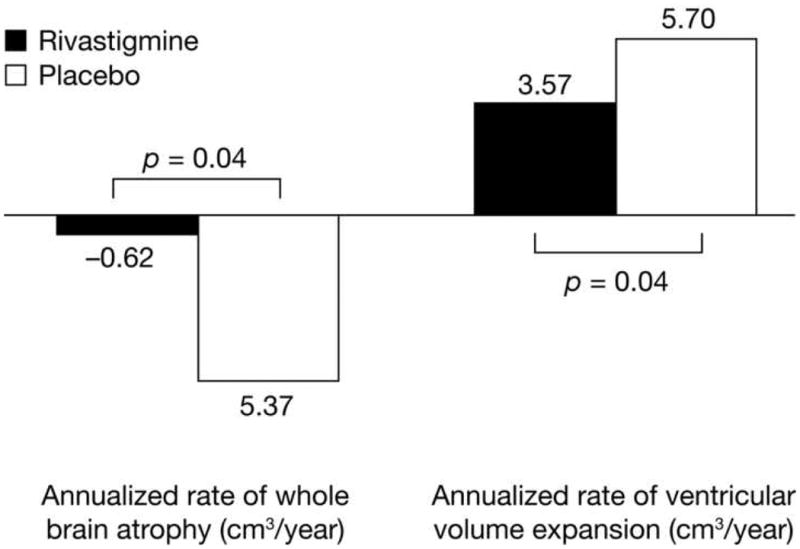
There were no significant effects of rivastigmine on hippocampal volume decline in any of the subgroups (Table 3). However, both ventricular expansion and whole brain atrophy in BuChE wt/wt women were significantly attenuated by rivastigmine treatment compared with placebo (3.57 versus 5.70 cm3/year, p = 0.044, and -0.62 versus -5.37 cm3/year, p = 0.039, respectively, Figure 4). The interaction of gender and treatment on change from baseline in whole brain atrophy was significant in subjects with the BuChE wt/wt genotype (p = 0.007), but no significant interaction of gender and treatment on ventricular volume expansion was detected. Decline in white matter volume was reduced in rivastigmine-treated women compared with placebo-treated women. The reduction was significant in female BuChE-K non-carriers (3.0 versus 6.8%, p = 0.027), but did not reach statistical significance in female BuChE-K carriers (4.4 versus 9.1%, p = 0.155). A trend for an interaction of gender and treatment on white matter volume in BuChE-K carriers was seen but this did not reach statistical significance (p = 0.098).
Figure 4. Annualized loss of ventricular and whole brain volume over 3–4 years by treatment group in BuChE wt/wt women with MCI (ITT).
Discussion
The InDDEx study is the largest and longest term randomized controlled trial that has been undertaken to date in subjects with amnestic MCI. It has created a unique set of longitudinal placebo data that can be used in more detailed exploratory post-hoc analyses to investigate the relationships between treatment, genetic risk factors, clinical outcomes and brain volume changes.
In the current analysis, we investigated the effects of gender on rates of progression to AD, cognitive and functional decline, and MRI volumetric changes in amnestic MCI subjects with and without a BuChE-K variant allele. The BuChE-K variant allele is the only common polymorphism of the BuChE allele, and is found in up to one-third of Caucasians and Orientals [12]. Our results suggest that, in participants not receiving active treatment, the combination of female gender and the BuChE wt/wt genotype is associated with increased rates of functional decline and a greater likelihood of progression to AD. Conversely, men with one or more BuChE-K variant alleles had the highest rate of cognitive decline. The incidence of progression to AD, functional decline, whole brain atrophy, lateral ventricular volume expansion and loss of white matter were attenuated by rivastigmine treatment in women with the BuChE wt/wt genotype. BuChE-K men derived significant benefits from rivastigmine on cognition, and these benefits had no brain volume correlates.
Previous analyses of the InDDEx study population have indicated that women with MCI may have a faster rate of progression to AD than men [1, 2]. Indeed, female gender is considered to be one of the most significant of several factors that may influence progression to AD in MCI subjects [13]. Gender differences in the clinical manifestation of AD pathology have been described, with a greater likelihood that AD pathology will manifest as the clinical symptoms of AD in women than in men [14]. Research into the higher susceptibility to AD in women has focused on the roles of estrogen [15, 16], testosterone [17] and other sex hormones [18]. However, in the current analysis, the highest incidence of AD diagnosis and the fastest rate of functional decline was seen in women with the BuChE wild-type genotype, suggesting that this effect was not driven only by gender. Gender-related differences in the expression of genetic risk factors have been reported [13, 19]. Similarly, the results of the current analysis indicate that the influence of BuChE on disease progression in the MCI population is different in women and men.
The role of BuChE in progression to AD in MCI subjects is complex. A previous investigation into the synergistic effect of APOE and BuChE genotype in the placebo-treated InDDEx study population showed that the fastest progression, in terms of incidence of AD diagnosis, cognitive decline and reduction in hippocampal volume, occurred in the subgroup of subjects with both APOE ε4 and BuChE-K alleles [12]. It was suggested that the increased soluble aggregates of beta-amyloid and neurofibrillary tangle formation associated with APOE ε4 may be attenuated by the C-terminal anti-amyloid aggregating ability of BuChE, a non-enzymatic property of the protein that reduces fibril formation [20]. The BuChE-K variant allele is associated with fewer circulating BuChE molecules in serum in healthy individuals and in early AD, and possibly with an altered C-terminal conformation, and anti-Aβ aggregation may be less pronounced in subjects with the BuChE-K genotype [21]. The classic cortico-limbic Alzheimer pathology and cognitive decline associated with APOE ε4 – presumably due to aggregated Aβ-mediated toxicity to cholinergic synapses and neurons – is most prominent in subjects with both APOE ε4 and BuChE-K alleles [12]. The ability of rivastigmine to increase synaptic acetylcholine levels and enhance neurotransmission in damaged ascending cholinergic pathways suggests that those MCI subjects with both BuChE-K and APOE ε4 alleles may have been driving the cognitive improvement seen in BuChE-K carriers treated with rivastigmine in the current analysis. The expression of BuChE is influenced by many factors including sex hormones [22], age [23], glial activation [24], and genotype [25]. In patients with AD, preliminary studies have suggested that in BuChE-K heterozygotes, instability of BuChE-K can lead to compensatory over-expression of the wild-type allele, resulting in increased or similar levels of BuChE activity compared with wild-type homozygotes (Darreh-Shori et al, unpublished observations). A similar relationship has been observed in APOE ε4 heterozygotes [25]. In an autopsy study of patients with advanced dementia, BuChE-K alleles, with an apparent gene-dose effect, were associated with higher BuChE activity in the temporal cortex. While it is interesting to speculate on underlying mechanisms, further study of these complex interrelationships that affect the expression of BuChE in brain, CSF and serum along the spectrum of disease severity and by genotype is required.
In the presence of a source of Aβ, putative decreases in the non-enzymatic effects of BuChE in BuChE-K carriers result in greater pathology in cortico-limbic areas, synaptic acetylcholine deficits and more prominent cognitive decline. In contrast, decreased extracellular acetylcholine due to increases in the enzymatic effects of BuChE are more likely in those with the BuChE wt/wt genotype. Decreased extracellular acetylcholine may increase glial activation and neuroinflammation, mediating white matter damage, loss of integrity of the neural network, reduction of fronto-subcortical connectivity and more prominent functional decline, as seen in BuChE wt/wt women in the current analysis. BuChE activity is relatively high in the hippocampus, amygdala, and in thalamic nuclei that project to frontal cortical structures involved in attention, executive function, and behavior [26], but the largest pool of BuChE in the brain is found in the glia, particularly those in deeper cortical and subcortical structures that closely map the distribution of white matter [27], While BuChE may have a supportive role in the hydrolysis of synaptic acetylcholine [28], its more important role is likely to be modulation of brain parenchymal acetylcholine [29, 30], which acts as a local anti-inflammatory signaling molecule [31]. Interestingly, in the experimental autoimmune encephalomyelitis animal model, rivastigmine markedly ameliorated clinical symptoms and spatial memory deficits. It also reduced demyelination, microglial activation, the production of pro-inflammatory cytokines and axonal damage. These effects were abolished by α7 nicotinic acetylcholine receptor antagonists [32]. Excess BuChE may induce a potentially neuroinflammatory decrease in extracellular acetylcholine levels [31, 33]. Increased BuChE is not associated with benign plaques, but released by activated glia in malignant plaques associated with neuritic tissue damage and clinical dementia [27, 34-36]. Furthermore, activated glia may ‘neglect’ their crucial roles in the maintenance of myelin, and the ensuing damage to white matter induces progressive disconnection of widely-distributed neural networks that results in cognitive and functional decline [37, 38]. Disruption of white matter may be an early event in the course of AD [39], and greater age-related deterioration of white matter has been shown in females [40, 41].
Due to the neuroanatomical distribution of BuChE in the brain, the enzymatic effects of excess BuChE may be more concentrated in the white matter of the subcortical and deep cortical structures, producing a pattern of deficits reflecting loss of cortico-cortical and cortico-subcortical, and particularly fronto-subcortical, connectivity. Myelin loss may be most pronounced in more vulnerable later myelinating association regions such as frontal lobes that contain higher proportions of smaller thinly myelinated axons [42]. The BuChE wt/wt genotype has been associated with a faster rate of disease progression on assessments of information processing speed and executive function in dementias and dementia sub-types with more prominent subcortical pathology, including dementia with Lewy bodies (DLB) [43] and Parkinson's disease dementia (PDD) [44]. Corticolimbic pathology due to Aβ pathology may be ‘unmasked’ to produce symptoms at an earlier stage of disease due to loss of neural network connectivity and cognitive processing speed [44]. This may explain the greater likelihood that AD pathology will manifest as the clinical symptoms of AD in women than in men [14]. In subjects with amnestic MCI other investigators have shown that worsening on executive functions and on functional status but not the worsening on memory functions are independently associated with progression to AD [45].
Possibly as a consequence of the additional inhibition of BuChE, rivastigmine -compared with selective AChE-inhibitor treatment - has been suggested to have a greater, longer-term treatment benefit in dementia patients likely to possess more prominent subcortical pathology, including AD patients younger than 75 years of age with the BuChE wt/wt genotype [46] and AD patients with symptoms suggestive of concomitant Lewy body disease, particularly with respect to functioning [47]. Thus the beneficial effect of rivastigmine in women with the BuChE wild-type genotype with MCI may also indicate active disease pathology in white matter in these subjects. The greater susceptibility of women to age-related decline in white matter may be of relevance [40, 41]. Lateral ventricle size is highly correlated with diminished subcortical white matter integrity [48] and with more rapid progression to AD [49]. Elsewhere, patients treated with rivastigmine, but not those treated with selective AChE inhibitors, displayed attenuated loss of white matter volume [50]. Here, interestingly, loss of white matter volume and ventricular expansion were greatest in female BuChE-K carriers receiving placebo, but were only significantly attenuated by rivastigmine treatment in female BuChE wt/wt subjects. However, there were trends that did not reach statistical significance for rivastigmine to attenuate decline in these volumes in female BuChE-K carriers. This, and the fact that white matter loss was not attenuated by rivastigmine in males with either genotype, may indicate significant additional processes inducing white matter deterioration beyond BuChE enzyme-mediated decreases in extracellular acetylcholine that are not amenable to rivastigmine treatment.
Rivastigmine has also been shown to have greater benefits in dementia patients with a more aggressive disease course [51] that could be hypothesized as due to either progressive deficits in synaptic acetylcholine reducing neuromodulatory input to cortical neurons or to lowered extracellular acetylcholine exacerbating sub-cortical neuroinflammatory processes. Although in the current study, all subgroups appeared to be at a similar stage of MCI at baseline, the more rapid progression of functional impairment during the study period, as a consequence of lowered extracellular acetylcholine, may account for the significant effects of rivastigmine seen in BuChE wt/wt women. Rivastigmine also attenuated cognitive decline in men with the BuChE-K allele, who had the most rapid rate of cognitive decline, due to increasing synaptic cholinergic neurotransmission. The different mechanisms whereby rivastigmine induced these clinical effects explains why the different populations of MCI subjects defined by gender and BuChE genotype were accompanied by dissimilar neuroimaging evidence. The neuroimaging results are likely to reflect underlying pathophysiological processes. Thus, in males with a BuChE-K allele, neuroimaging suggested only a symptomatic benefit, while a disease-modifying benefit was suggested in BuChE wt/wt women. Disease-modifying effects of rivastigmine have been suggested in previous studies [32, 46, 50, 52-57].
The potential limitations and potential for bias within this analysis should be considered. This was a retrospective investigation of a larger randomized clinical trial, with results uncorrected for multiple comparisons. The primary analysis in this study used LOCF to impute for missing data, introducing the possibility for bias due to differential attrition rates between groups. To limit bias to LOCF imputation, the InDDEx study included a retrieved drop-out phase and the retrieved drop-out assessment was used, if available. In the current study, supportive OC analyses at study endpoint were performed. In some cases these analyses showed similar trends to the LOCF analyses, and also detected a significant effect of rivastigmine on cognition in male BuChE-K carriers, strengthening confidence in the results presented. However, the censoring of clinical data from study participants after they crossed the threshold for a diagnosis of AD means that the OC analysis, which included double-blind study completers and retrieved drop-outs who did not progress to AD, only provides information on individuals who did not progress to AD during the study. In addition, within the InDDEx study the heterogeneity of the MCI sample was apparent as the rate of progression to AD of 5% per year was substantially lower than predicted [1]. Therefore, this analysis may lack sensitivity.
Further, our findings are drawn from an exclusive sample of participants who consented to pharmacogenetic testing. There were no significant differences at baseline detected between the participants in this analysis and those in the overall study population, suggesting that the two populations were similar. The lack of disequilibrium of Hardy Weinberg and the proportions of individuals within each of the genotype subgroups also offers some reassurance that the sample is representative. However, the potential for bias in the sample cannot be excluded. A three-way test of the interaction between gender, BuChE genotype and treatment in the current analysis (data not shown) did not demonstrate any statistically significant influences on progression to AD in the current study population. Were there to have been more individuals entering the study who were more proximal to a diagnosis of AD, the interactions of gender, BuChE genotype and treatment may have become more, or less, apparent. In addition, the mean daily dose of rivastigmine in both female subgroups in this study was lower than the therapeutically effective dose for AD [58], while men, particularly those with a BuChE-K allele, achieved mean daily doses of more than 6 mg/day. This was likely due to greater deficits in cholinergic neurotransmission in males with a BuChE-K allele, and to a lower average body weight in women resulting in poorer tolerability of higher doses. However, the different mean dose levels achieved may have limited the detection of treatment differences between gender and genotype subgroups in this analysis.
In summary, gender appeared to differentially influence the type of decline in MCI subjects from the InDDEx study according to their BuChE genotype, with more rapid progression of cognitive decline in men with the BuChE-K/* genotype (putatively due to progression of classic Aβ-induced cortico-limbic pathology), and more rapid progression to AD and functional decline in women with the BuChE wt/wt genotype (putatively due to neuroinflammation and loss of white matter and the integrity of the neural network). Rivastigmine-induced cognitive improvements in BuChE-K/* males appeared to be symptomatic effects due to attenuation of synaptic cholinergic deficits. The observation that rivastigmine delayed progression to AD and functional decline in women with the BuChE wt/wt genotype, supported by significant benefits on MRI measurements of ventricular volume expansion, brain atrophy and white matter loss, suggest that rivastigmine might have disease-modifying effects in these subjects that may be due to attenuation of extracellular cholinergic deficits. The role of gender and BuChE genotype in the normal, MCI and AD brain warrants further investigation.
Acknowledgments
This study was sponsored by Novartis Pharma AG, Basel, Switzerland. RL is an employee of Novartis. SF, AN and HS participated in the design of the InDDEx study. SF was a member of the InDDEx Steering Committee and has served on advisory boards for Novartis, for which he received honoraria. He also received research funds for site participation in the InDDEx trial. AN has received research grants from Novartis. AN and TD-S performed BuChE activity measurements. All authors were involved in the preparation of this manuscript; editorial assistance was provided by Alpha-Plus Medical Communications Ltd (UK). The authors thank Dr Nick Fox of the Dementia Research Centre, Institute of Neurology, University College London, London, UK, for his critical review of the manuscript.
This study was sponsored by Novartis Pharma AG, Basel, Switzerland.
References
- 1.Feldman HH, Ferris S, Winblad B, Sfikas N, Mancione L, He Y, et al. Effect of rivastigmine on delay to diagnosis of Alzheimer's disease from mild cognitive impairment: the InDDEx study. Lancet Neurol. 2007;6:501–512. doi: 10.1016/S1474-4422(07)70109-6. [DOI] [PubMed] [Google Scholar]
- 2.Feldman H, Lane R, Sfikas N, Winblad B, Farlow M, Ferris S. Time to clinical diagnosis of dementia in MCI subjects receiving rivastigmine: effect of gender. in preparation. [Google Scholar]
- 3.Darreh-Shori T, Almkvist O, Guan ZZ, Garlind A, Strandberg B, Svensson AL, et al. Sustained cholinesterase inhibition in AD patients receiving rivastigmine for 12 months. Neurology. 2002;59:563–572. doi: 10.1212/wnl.59.4.563. [DOI] [PubMed] [Google Scholar]
- 4.Mohs RC, Rosen WG, Davis KL. The Alzheimer's disease assessment scale: an instrument for assessing treatment efficacy. Psychopharmacol Bull. 1983;19:448–450. [PubMed] [Google Scholar]
- 5.Galasko D, Bennett D, Sano M, Ernesto C, Thomas R, Grundman M, et al. An inventory to assess activities of daily living for clinical trials in Alzheimer's disease. The Alzheimer's Disease Cooperative Study Alzheimer Dis Assoc Disord. 1997;11(Suppl 2):S33–39. [PubMed] [Google Scholar]
- 6.Rust S, Funke H, Assmann G. Mutagenically separated PCR (MS-PCR): a highly specific one step procedure for easy mutation detection. Nucleic Acids Res. 1993;21:3623–3629. doi: 10.1093/nar/21.16.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellman GL, Courtney KD, Andres V, Jr, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 8.Darreh-Shori T, Brimijoin S, Kadir A, Almkvist O, Nordberg A. Differential CSF butyrylcholinesterase levels in Alzheimer's disease patients with the ApoE epsilon4 allele, in relation to cognitive function and cerebral glucose metabolism. Neurobiol Dis. 2006;24:326–333. doi: 10.1016/j.nbd.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 9.Styner M, Charles H, Park J, Gerig G. Multi-site validation of image analysis methods - Assessing intra and inter-site variability. In: Sonka Milan, Fitzpatrick J Michael., editors. Proceedings of SPIE, Volume 4684 Medical Imaging: Image Processing. May, 2002. pp. 278–286. [Google Scholar]
- 10.Schott JM, Price SL, Frost C, Whitwell JL, Rossor MN, Fox NC. Measuring atrophy in Alzheimer disease: a serial MRI study over 6 and 12 months. Neurology. 2005;65:119–124. doi: 10.1212/01.wnl.0000167542.89697.0f. [DOI] [PubMed] [Google Scholar]
- 11.Fox NC, Freeborough PA. Brain atrophy progression measured from registered serial MRI: validation and application to Alzheimer's disease. J Magn Reson Imaging. 1997;7:1069–1075. doi: 10.1002/jmri.1880070620. [DOI] [PubMed] [Google Scholar]
- 12.Lane R, Feldman HH, Meyer J, He Y, Ferris SH, Nordberg A, et al. Synergistic effect of apolipoprotein E ε4 and butyrylcholinesterase K-variant on progression from mild cognitive impairment to Alzheimer's disease. Pharmacogenet Genomics. 2008;18:289–298. doi: 10.1097/FPC.0b013e3282f63f29. [DOI] [PubMed] [Google Scholar]
- 13.Fleisher A, Grundman M, Jack CR, Jr, Petersen RC, Taylor C, Kim HT, et al. Sex, apolipoprotein E epsilon 4 status, and hippocampal volume in mild cognitive impairment. Arch Neurol. 2005;62:953–957. doi: 10.1001/archneur.62.6.953. [DOI] [PubMed] [Google Scholar]
- 14.Barnes LL, Wilson RS, Bienias JL, Schneider JA, Evans DA, Bennett DA. Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch Gen Psychiatry. 2005;62:685–691. doi: 10.1001/archpsyc.62.6.685. [DOI] [PubMed] [Google Scholar]
- 15.Yue X, Lu M, Lancaster T, Cao P, Honda S, Staufenbiel M, et al. Brain estrogen deficiency accelerates Abeta plaque formation in an Alzheimer's disease animal model. Proc Natl Acad Sci USA. 2005;102:19198–19203. doi: 10.1073/pnas.0505203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manly JJ, Merchant CA, Jacobs DM, Small SA, Bell K, Ferin M, et al. Endogenous estrogen levels and Alzheimer's disease among postmenopausal women. Neurology. 2000;54:833–837. doi: 10.1212/wnl.54.4.833. [DOI] [PubMed] [Google Scholar]
- 17.Sherwin BB. Steroid hormones and cognitive functioning in aging men: a mini-review. J Mol Neurosci. 2003;20:385–393. doi: 10.1385/JMN:20:3:385. [DOI] [PubMed] [Google Scholar]
- 18.Webber KM, Casadesus G, Marlatt MW, Perry G, Hamlin CR, Atwood CS, et al. Estrogen bows to a new master: the role of gonadotropins in Alzheimer pathogenesis. Ann N Y Acad Sci. 2005;1052:201–209. doi: 10.1196/annals.1347.020. [DOI] [PubMed] [Google Scholar]
- 19.Miyashita A, Arai H, Asada T, Imagawa M, Matsubara E, Shoji M, et al. Genetic association of CTNNA3 with late-onset Alzheimer's disease in females. Hum Mol Genet. 2007;16:2854–2869. doi: 10.1093/hmg/ddm244. [DOI] [PubMed] [Google Scholar]
- 20.Diamant S, Podoly E, Friedler A, Ligumsky H, Livnah O, Soreq H. Butyrylcholinesterase attenuates amyloid fibril formation in vitro. Proc Natl Acad Sci USA. 2006;103:8628–8633. doi: 10.1073/pnas.0602922103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altamirano CV, Bartels CF, Lockridge O. The butyrylcholinesterase K-variant shows similar cellular protein turnover and quaternary interaction to the wild-type enzyme. J Neurochem. 2000;74:869–877. doi: 10.1046/j.1471-4159.2000.740869.x. [DOI] [PubMed] [Google Scholar]
- 22.Bradamante V, Kunec-Vajic E. Effect of glucocorticoids on plasma cholinesterase in the rat. Biomed Biochim Acta. 1987;46:439–443. [PubMed] [Google Scholar]
- 23.Perry EK, Perry RH, Blessed G, Tomlinson BE. Changes in brain cholinesterases in senile dementia of Alzheimer type. Neuropathol Appl Neurobiol. 1978;4:273–277. doi: 10.1111/j.1365-2990.1978.tb00545.x. [DOI] [PubMed] [Google Scholar]
- 24.Hu J, Van Eldik LJ. Glial-derived proteins activate cultured astrocytes and enhance beta amyloid-induced glial activation. Brain Res. 1999;842:46–54. doi: 10.1016/s0006-8993(99)01804-1. [DOI] [PubMed] [Google Scholar]
- 25.Darreh-Shori T, Forsberg A, Andreasen N, Blennow K, Wall A, Kamil C, et al. Interaction betweem amyloid-beta peptides with butyrylcholinesterase and APOE proteins: mechanism of amyloid fibrils deposition in the brain of patients with Alzheimer's disease. Poster Presented at the 11th International Conference on Alzheimer's Disease; Chicago, IL, USA. 26–31 July 2008. [Google Scholar]
- 26.Darvesh S, Hopkins DA. Differential distribution of butyrylcholinesterase and acetylcholinesterase in the human thalamus. J Comp Neurol. 2003;46:25–43. doi: 10.1002/cne.10751. [DOI] [PubMed] [Google Scholar]
- 27.Wright CI, Geula C, Mesulam MM. Neurological cholinesterases in the normal brain and in Alzheimer's disease: relationship to plaques, tangles, and patterns of selective vulnerability. Ann Neurol. 1993;34:373–384. doi: 10.1002/ana.410340312. [DOI] [PubMed] [Google Scholar]
- 28.Mesulam M, Guillozet A, Shaw P, Quinn B. Widely spread butyrylcholinesterase can hydrolyze acetylcholine in the normal and Alzheimer brain. Neurobiol Dis. 2002;9:88–93. doi: 10.1006/nbdi.2001.0462. [DOI] [PubMed] [Google Scholar]
- 29.Greig NH, Utsuki T, Ingram DK, Wang Y, Pepeu G, Scali C, et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc Natl Acad Sci USA. 2005;102:17213–17218. doi: 10.1073/pnas.0508575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cerbai F, Giovannini MG, Melani C, Enz A, Pepeu G. N1 phenethyl-norcymserine, a selective butyrylcholinesterase inhibitor, increases acetylcholine release in rat cerebral cortex: a comparison with donepezil and rivastigmine. Eur J Pharmacol. 2007;572:142–150. doi: 10.1016/j.ejphar.2007.06.053. [DOI] [PubMed] [Google Scholar]
- 31.Ben-Shaul Y, Bergman H, Soreq H. Acetylcholinesterase, cholinergic signaling and Parkinson's disease. In: Hanin I, Cacabelos R, Fisher A, editors. Recent Progress in Alzheimer's and Parkinson's diseases. London and New York: Taylor & Francis; 2005. pp. 33–44. [Google Scholar]
- 32.Nizri E, Irony-Tur-Sinai M, Faranesh N, Lavon I, Lavi E, Weinstock M, et al. Suppression of neuroinflammation and immunomodulation by the acetylcholinesterase inhibitor rivastigmine. J Neuroimmunol. 2008 Aug 8; doi: 10.1016/j.jneuroim.2008.06.018. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 33.Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–859. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 34.Geula C, Mesulam MM. Cholinesterases and the pathology of Alzheimer disease. Alzheimer Dis Assoc Disord. 1995;9(Suppl 2):23–28. doi: 10.1097/00002093-199501002-00005. [DOI] [PubMed] [Google Scholar]
- 35.Guillozet AL, Smiley JF, Mash DC, Mesulam MM. Butyrylcholinesterase in the life cycle of amyloid plaques. Ann Neurol. 1997;42:909–918. doi: 10.1002/ana.410420613. [DOI] [PubMed] [Google Scholar]
- 36.Eskander MF, Nagykery NG, Leung EY, Khelghati B, Geula C. Rivastigmine is a potent inhibitor of acetyl- and butyrylcholinesterase in Alzheimer's plaques and tangles. Brain Res. 2005;1060:144–152. doi: 10.1016/j.brainres.2005.08.039. [DOI] [PubMed] [Google Scholar]
- 37.Huang J, Friedland RP, Auchus AP. Diffusion tensor imaging of normal-appearing white matter in mild cognitive impairment and early Alzheimer disease: preliminary evidence of axonal degeneration in the temporal lobe. AJNR Am J Neuroradiol. 2007;28:1943–1948. doi: 10.3174/ajnr.A0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bartzokis G, Lu PH, Geschwind DH, Tingus K, Huang D, Mendez MF, et al. Apolipoprotein E Affects Both Myelin Breakdown and Cognition: Implications for Age-Related Trajectories of Decline Into Dementia. Biol Psychiatry. 2007;62:1380–1387. doi: 10.1016/j.biopsych.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 39.Ringman JM, O'Neill J, Geschwind D, Medina L, Apostolova LG, Rodriguez Y, et al. Diffusion tensor imaging in preclinical and presymptomatic carriers of familial Alzheimer's disease mutations. Brain. 2007;130(Pt 7):1767–1776. doi: 10.1093/brain/awm102. [DOI] [PubMed] [Google Scholar]
- 40.Kovalev V, Kruggel F. Texture anisotropy of the brain's white matter as revealed by anatomical MRI. IEEE Trans Med Imaging. 2007;26:678–685. doi: 10.1109/TMI.2007.895481. [DOI] [PubMed] [Google Scholar]
- 41.Hsu JL, Leemans A, Bai CH, Lee CH, Tsai YF, Chiu HC, et al. Gender differences and age-related white matter changes of the human brain: a diffusion tensor imaging study. Neuroimage. 2008;39:566–577. doi: 10.1016/j.neuroimage.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 42.Bartzokis G, Lu PH, Tingus K, Mendez MF, Richard A, Peters DG, et al. Lifespan trajectory of myelin integrity and maximum motor speed. Neurobiol Aging. 2008 Oct 14; doi: 10.1016/j.neurobiolaging.2008.08.015. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Brien KK, Saxby BK, Ballard CG, Grace J, Harrington F, Ford GA, et al. Regulation of attention and response to therapy in dementia by butyrylcholinesterase. Pharmacogenetics. 2003;13:231–239. doi: 10.1097/00008571-200304000-00008. [DOI] [PubMed] [Google Scholar]
- 44.Bullock R, Lane R. Executive dyscontrol in dementia, with emphasis on subcortical pathology and the role of butyrylcholinesterase. Curr Alzheimer Res. 2007;4:277–293. doi: 10.2174/156720507781077313. [DOI] [PubMed] [Google Scholar]
- 45.Rozzini L, Chilovi BV, Conti M, Bertoletti E, Delrio I, Trabucchi M, et al. Conversion of amnestic Mild Cognitive Impairment to dementia of Alzheimer type is independent to memory deterioration. Int J Geriatr Psychiatry. 2007;22:1217–1222. doi: 10.1002/gps.1816. [DOI] [PubMed] [Google Scholar]
- 46.Blesa R, Bullock R, He Y, Bergman H, Gambina G, Meyer J, et al. Effect of butyrylcholinesterase genotype on the response to rivastigmine or donepezil in younger patients with Alzheimer's disease. Pharmacogenet Genomics. 2006;16:771–774. doi: 10.1097/01.fpc.0000220573.05714.ac. [DOI] [PubMed] [Google Scholar]
- 47.Touchon J, Bergman H, Bullock R, Rapatz G, Nagel J, Lane R. Response to rivastigmine or donepezil in Alzheimer's patients with symptoms suggestive of concomitant Lewy body pathology. Curr Med Res Opin. 2006;22:49–59. doi: 10.1185/030079906x80279. [DOI] [PubMed] [Google Scholar]
- 48.Longstreth WT, Jr, Arnold AM, Manolio TA, Burke GL, Bryan N, Jungreis CA, et al. Clinical correlates of ventricular and sulcal size on cranial magnetic resonance imaging of 3,301 elderly people. The Cardiovascular Health Study. Collaborative Research Group Neuroepidemiology. 2000;19:30–42. doi: 10.1159/000026235. [DOI] [PubMed] [Google Scholar]
- 49.Carmichael OT, Kuller LH, Lopez OL, Thompson PM, Dutton RA, Lu A, et al. Ventricular volume and dementia progression in the Cardiovascular Health Study. Neurobiol Aging. 2007;28:389–397. doi: 10.1016/j.neurobiolaging.2006.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Venneri A, Lane R. Effects of cholinesterase inhibition on brain white matter volume in Alzheimer's disease. 2008 doi: 10.1097/wnr.0b013e3283207d21. submitted. [DOI] [PubMed] [Google Scholar]
- 51.Gauthier S, Vellas B, Farlow M, Burn D. Aggressive course of disease in dementia. Alzheimer Dement. 2006;2:210–217. doi: 10.1016/j.jalz.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 52.Venneri A, McGeown WJ, Shanks MF. Empirical evidence of neuroprotection by dual cholinesterase inhibition in Alzheimer's disease. Neuroreport. 2005;16:107–110. doi: 10.1097/00001756-200502080-00006. [DOI] [PubMed] [Google Scholar]
- 53.Farlow M, Potkin S, Koumaras B, Veach J, Mirski D. Analysis of outcome in retrieved dropout patients in a rivastigmine vs placebo, 26-week, Alzheimer disease trial. Arch Neurol. 2003;60:843–848. doi: 10.1001/archneur.60.6.843. [DOI] [PubMed] [Google Scholar]
- 54.Erkinjuntti T, Román G, Kalaria R, Lane R. Mechanisms of action and treatment benefits with cholinesterase inhibitors with vascular cognitive disorders. Journal of Drug Assessment. 2005;8:61–77. [Google Scholar]
- 55.Erkinjuntti T, Skoog I, Lane R, Andrews C. Potential long-term effects of rivastigmine on disease progression may be linked to drug effects on vascular changes in Alzheimer brains. Int J Clin Pract. 2003;57:756–760. [PubMed] [Google Scholar]
- 56.Doraiswamy PM, Krishnan KR, Anand R, Sohn H, Danyluk J, Hartman RD, et al. Long-term effects of rivastigmine in moderately severe Alzheimer's disease: does early initiation of therapy offer sustained benefits? Prog Neuropsychopharmacol Biol Psychiatry. 2002;26:705–712. doi: 10.1016/s0278-5846(01)00326-8. [DOI] [PubMed] [Google Scholar]
- 57.Farlow M, Anand R, Messina J, Jr, Hartman R, Veach J. A 52-week study of the efficacy of rivastigmine in patients with mild to moderately severe Alzheimer's disease. Eur Neurol. 2000;44:236–241. doi: 10.1159/000008243. [DOI] [PubMed] [Google Scholar]
- 58.Agid Y, Dubois B, International Rivastigmine Investigators. Anand R, Gharabawi G. Efficacy and tolerability of rivastigmine in patients with dementia of the Alzheimer type. Current Therapeutic Research. 1998;59:837–845. [Google Scholar]