

Abstract
Human differentially expressed in chondrocytes (DEC), mouse stimulated with retinoic acid and rat split and hairy related proteins constitute a structurally distinct class of the basic helix-loop-helix proteins. DEC1is abundantly expressed in tumors and protects against apoptosis induced by serum starvation. In this study, we report that DEC1 antiapoptosis is achieved by inducing survivin, an antiapoptotic protein. In paired tumor–normal tissues, survivin and DEC1 exhibited a paralleled expression pattern. Tetracycline-induced expression of DEC1 in stable lines proportionally increased the expression of survivin. In reporter assays, DEC1 transactivated the survivin promoter but repressed the DEC2 promoter. In contrast to the repression, the activation was delayed and varied depending on serum concentrations and cycle blockers. Studies with reporter mutants located, in the survivin promoter, two Sp1 sites that supported DEC1 transactivation. Electrophoretic mobility shift assay and chromatin immunoprecipitation detected the presence of DEC1 in the survivin promoter. These findings establish that the survivin gene is a transcription target of DEC1, and induction of survivin is at least in part responsible for DEC1 antiapoptosis.
Keywords: DEC1, survivin, Sp1 element, transcription activation
Introduction
Human differentially expressed in chondrocytes (DEC), mouse stimulated with retinoic acid (STRA) and rat split and hairy related protein (SHARP) constitute a structurally distinct class of basic helix-loop-helix (bHLH) proteins (Boudjelal et al., 1997; Rossner et al., 1997; Shen et al., 1997; Fujimoto et al., 2001). In each species, two members are identified with a sequence identity of >90% in the bHLH region and ~40% in total proteins, respectively. These transcription factors are involved in various cellular events such as cell proliferation and differentiation (Boudjelal et al., 1997; Shen et al., 2002), maturation of lymphocytes (Sun et al., 2001), regulation of molecular clock (Honma et al., 2002) and lipid metabolism (Yun et al., 2002). Transfection with DEC1 decreases cell proliferation, and the decrease is proportionally correlated with the levels of DEC1 (Li et al., 2002). Chondrogenic cells expressing high levels of DEC1 undergo rapid phenotypic changes toward terminal differentiation in response to mitogenic stimuli (Shen et al., 2002). Consistent with the promotion of chondrocyte differentiation, STRA13 promotes neuronal differentiation (Boudjelal et al., 1997), and SHARPs are abundantly expressed in a subset of mature neurons (Rossner et al., 1997). STRA13 null mice, although surviving to adulthood, develop autoimmune diseases accompanied by accumulation of spontaneously activated T and B cells (Sun et al., 2001). Furthermore, these proteins are found to interact directly with clock protein BmalI and regulate the expression of biological clock regulator Per (Honma et al., 2002; Li et al., 2004).
Another characteristic of DEC/STRA/SHARPs is that their expression is rapidly induced in response to detrimental stimuli (Inuzuka et al., 1999; Shen et al., 2001; Yoon et al., 2001). In rats that undergo seizure induction by kainic acid, the levels of mRNA encoding SHARP1 or 2 are sharply elevated (Rossner et al., 1997). Similarly, the expression of DEC1 is increased in response to hypoxia, a condition that mimics the microenvironment of tumors (Ivanova et al., 2001; Miyazaki et al., 2002). These findings link DEC1, particularly its elevated expression, to oncogenesis. Several lines of evidence support this possibility. First, expression of DEC1 appears to be deregulated in tumor tissues. In paired samples from the colon, lung and kidney, DEC1 is abundantly expressed in the carcinomas but not in the adjacent normal tissues (Li et al., 2002, 2003). The level of DEC1 in all tumors tested is positively correlated with tumor grade and the abundance of angiogenic protein vascular endothelial growth factor-D (Chakrabarti et al., 2004; Currie et al., 2004; Turley et al., 2004). High levels of DEC1 mRNA are also detected in an array of cancer cell lines (Ivanova et al., 2001). Cells that lack the functional tumor suppressor VHL (von Hippel-Lindau) express higher levels of DEC1 (Ivanova et al., 2001). Second, DEC1 is antiapoptotic apparently through reducing caspase activities (Li et al., 2002). Forced expression of DEC1 effectively antagonizes apoptosis induced by serum starvation and causes marked decreases on the activity of caspases-3, 7 and 9. In contrast, a DEC1 mutant, lacking the DNA binding domain, shows neither antiapoptotic activity nor inhibitory effects on caspases (Li et al., 2002), underscoring the importance of DNA binding in conferring antiapoptotic activity.
Caspases are cysteinyl aspartate-specific proteases and normally exist as inactive forms, so called procaspases (Cryns and Yuan, 1998; Budihardjo et al., 1999). Upon apoptotic stimuli, procaspases undergo proteolytic processing in a cascading activation manner, which results in the formation of active caspases. There are two major activation pathways: the surface death receptor pathway and the mitochondrial pathway. For example, binding of FasL to its receptor results in the formation of the death-inducing signaling complex, which recruits and subsequently activates the upstream procaspase such as procaspase-8 (Ashkenazi and Dixit, 1999). In contrast, the mitochondrial pathway is initiated by intracellular death signals, leading to the formation of apoptotic protein complexes (Green and Reed, 1999). The complexes initiate the activation cascade from procapase-9 to downstream targets such as procaspase-3. Caspase activation, although following a sequential cascade, is regulated by several families of proteins. The inhibitor of apoptosis (IAP) family of proteins represents one of the major classes with potent antiapoptotic activity. Several IAP proteins are found to interact directly with and inhibit the activity of caspases (Yang and Li, 2000; Reed, 2001; Li, 2003).
In this study, we report that DEC1 is a transcriptional activator of survivin, a member of the IAP family. In the paired tumor–normal tissues, survivin and DEC1 exhibited a paralleled expression pattern. DEC1 transactivated the survivin promoter but repressed the DEC2 promoter. In contrast to the repression, the activation was delayed and varied depending on serum concentrations and cycle phase. Two Sp1 sites in the survivin promoter were found to support DEC1-mediated transactivation. These findings establish that the survivin gene is a transcription target of DEC1, and induction of survivin is at least in part responsible for DEC1 antiapoptosis.
Results
DEC1 selectively increases the expression of survivin among IAP proteins
We previously reported that DEC1 effectively protects against serum starvation, and the protection is correlated well with decreased activities of several major caspases (Li et al., 2002). Our preliminary studies excluded the possibility that the decreased caspase activities are due to decreased expression. We then examined whether DEC1 upregulates the expression of the IAP family proteins, a class of proteins that interfere with the processing and catalysis of caspases (Reed, 2001; Li, 2003). IAPs such as survivin, like DEC1, are upregulated in a variety of malignancies (Yang and Li, 2000; Reed, 2001; Li, 2003). DEC1 stable transfectants underwent serum starvation for 36 h in the presence or absence of tetracycline to modulate the expression of DEC1, and cell lysates were analysed for the abundance of cIAP-1, cIAP-2, XIAP, and survivin (Li, 2003). As shown in Figure 1a, all IAP proteins were detected in control and tetracycline-treated cells. However, the expression of survivin but not other IAP proteins was markedly increased (Figure 1a). The increase was well correlated with the level of DEC1 (Figure 1b). Consistent observations were made with three clonal lines.
Figure 1.

Expression of procaspases and inhibitor of apoptosis proteins in DEC1 stable transfected lines (a) Abundance of IAP proteins in DEC1 stable line. DEC1 stable transfectants were seeded in six-well plate and cultured in the presence or absence of tetracycline (1 μg/ml). After reaching ~80% confluence, cells underwent serum starvation for 36 h (tetracycline was kept). Cell lysates (5 μg) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE). The immunoblot was incubated with an antibody against IAP proteins, DEC1 or β-actin. The primary antibody was then located by horseradish peroxidase-conjugated goat anti-rabbit IgG and visualized with chemiluminescent substrate. (b) Expression of survivin as a function of DEC1. DEC1 stable transfectants were seeded in six-well plate and cultured in the medium containing various concentrations of tetracycline (0–1 μg/ml). After reaching ~80% confluence, cells underwent serum starvation for 36 h (tetracycline was kept). Cell lysates (5 μg) were subjected to SDS–PAGE. The immunoblots were incubated with an antibody against survivin, DEC1 or β-actin and detected as described above. Three clonal lines were used in this study.
DEC1-mediated induction of survivin varies depending on serum concentrations
In order to gain clinical implication, we examined the expression patterns of DEC1 and survivin in paired tumor–normal tissues from the lung and kidney. Representative data from the lung and kidney are shown in Figure 2. Without exceptions, both DEC1 and survivin were expressed markedly higher in the tumors than the adjacent normal tissues (Figure 2a). Such a tumor-related increase was also detected by reverse transcription–polymerase chain reaction (RT–PCR) on both DEC1 and survivin (Figure 2b).
Figure 2.

Paralleled expression of DEC1 and survivin in paired tumor and normal tissues from the kidney and lung (a) Paralleled expression of DEC1 and survivin in paired tumor and normal samples from the kidney and lung. Tissue homogenates (50 μg) from paired tumor–normal tissues were subjected to SDS–PAGE, and the immunoblot was incubated with the antibody against DEC1, survivin or β-actin. (b) DEC1 and survivin mRNA in paired tumor–normal tissues for the kidney. Total RNA (2 μg) of carcinoma–normal paired samples from the kidney was subjected to RT–PCR analyses with a ThermoScript I kit. For PCR amplification, a master tube containing all common reagents was prepared and equally distributed to individual PCR reaction tubes (DEC1, survivin and β-actin). PCR amplification was conducted with cycling parameters as follows: 95°C for 30 s, 52°C for 30 s and 68°C for 30 or 40 s for a total of 32 cycles. The PCR-amplified products were analysed by agarose gel electrophoresis and visualized by ethidium bromide staining.
Solid tumors are poorly vascularized, thus hypoxic and deficient in nutrients (Vaupel et al., 2001). We next examined the effects of serum concentrations on the induction of survivin. DEC1 stable cells were cultured in the presence or absence of tetracycline with increasing concentrations of serum (0–5%) for 24 h. Addition of tetracycline robustly increased the expression of transfected DEC1, and the increase was comparable among the cells cultured in various serum concentrations (Figure 3a). In contrast, the levels of survivin varied depending on the concentrations of serum. Generally, higher serum concentrations supported higher expression of survivin, consistent with the observation that survivin is expressed in a cycle-regulated manner with high levels in G2/M and low levels in G1 (serum starvation arrests cells at G1) (Li et al., 1998). Conversely, the induction of survivin by DEC1 was relatively greater (~2-fold) in cells cultured in 0–2% serum than cells cultured in 5% serum (Figure 3a).
Figure 3.

Time and serum dependence of DEC1-mediated induction on survivin. (a) Effects of serum concentrations on the induction of surviving. DEC1 stable transfectants were seeded in six-well plate and cultured in the presence or absence of tetracycline (1 μg/ml). After reaching ~80% confluence, cells underwent serum starvation for 24 h (tetracycline was kept). Cells were harvested and lysates (5 μg) were subjected to SDS–PAGE. The immunoblots were incubated with an antibody against survivin, DEC1 or β-actin and detected as described above. (b) Time-course study on DEC1-mediated induction of surviving. Stable transfectants expressing DEC1 or DEC1-M (lacking the DNA binding domain) were seeded in six-well plate and cultured in the presence or absence of tetracycline (1 μg/ml). After reaching ~80% confluence, cells underwent serum starvation for 12–36 h (tetracycline was kept). Cell lysates (5 μg) were subjected to SDS–PAGE. The immunoblots were incubated with an antibody against survivin, DEC1 or β-actin and detected as described above. (c) Abundance of survivin mRNA in DEC1 and DEC1-M lines. Cells at 36 h time point were also used for RNA isolation. Total RNA (2 μg) was subjected to RT–PCR analyses with a ThermoScript I kit (Invitrogen). For PCR amplification, a master tube containing all common reagents was prepared and equally distributed to individual PCR tubes (DEC1, survivin and β-actin). PCR amplification was conducted with cycling parameters as follows: 95°C for 30 s, 52°C for 30 s and 68°C for 30 or 40 s for a total of 32 cycles. The PCR-amplified products were analysed by agarose gel electrophoresis and visualized by ethidium bromide staining.
We next performed a time-course study on the serum starvation for survivin induction in the presence or absence of tetracycline. Both DEC1 and DEC1-M (lacking the DNA binding domain) lines were used in this study. As shown in Figure 3b, little changes on the survivin level were observed in the first 12 h starvation. However, a rapid decline was detected afterward (Figure 3b). Such time-dependent changes occurred in both DEC1 and DEC1-M lines. In cells that underwent for longer starvation (24- and 36 h), however, a markedly higher level of survivin was detected (two-and fivefold, respectively) in tetracycline-treated cells. The increase occurred only in DEC1 but not DEC1-M line, suggesting that DNA binding is required for the increase. Similarly, a higher level of survivin mRNA was detected in tetracycline-treated cells with DEC1 but not DEC1-M line (Figure 3c). DEC1-mediated induction of survivin was also detected in MDA-MB-468, a breast carcinoma line. Addition of actinomycin D, an inhibitor on RNA synthesis, completely abolished DEC1-mediated increases on survivin protein and mRNA (data not shown).
DEC1 stimulates the survivin promoter
The ablation on survivin induction by actinomycin D suggests that DEC1 induces survivin through transactivation. This possibility was tested by cotransfection with promoter reporters including: pSurvivin-6270, pSurvivin-268 and pDEC2-luc. These reporters contain different numbers of E-box elements, and DEC1 was shown to repress transcription through E-box elements (Figure 4a) (Li et al., 2003). The pSurvivin-268 reporter was derived from the 3′ sequence of the pSurvivin-6270 reporter, and both started from nucleotide -39 (Li and Altieri, 1999). More importantly, pSurvivin-6270 contains numerous E-box elements (none of them is CACGTG), whereas no E-box is present in pSurvivin-268. The pDEC2-luc reporter contains a 1888 bp-upstream sequence from the DEC2 gene, where two CACGTG E-boxes are present (Li, 2003). We recently showed that DEC1 represses the pDEC2-luc reporter through the E-box in the proximal promoter (Li et al., 2003).
Figure 4.

DEC1 transactivates the survivin promoter but represses the DEC2 promoter. (a) Diagrammatical presentation of pDEC2-luc, pSurvivin-6270 and -268. (b) Activation of the survivin reporters and repression of pDEC2-luc. DEC1 stable transfectants were seeded in 24-well plate and cultured in the presence or absence of tetracycline (1 μg/ml). After reaching ~80% confluence, cells were transfected again with a reporter construct (100 ng) and the pRL-TK Renilla (1 ng). The retransfected cells were cultured with serum-free medium in the presence or absence of tetracycline (1 μg/ml) for 24 h. The cells were collected, washed once with PBS and resuspended in passive lysis buffer. The reporter enzyme activities were assayed with a Dual-Luciferase Reporter Assay System. The firefly luminescence signal was normalized based on the Renilla luminescence signal. (c) Activation of the pSurvivin-268 is delayed. DEC1 stable transfectants were cultured, retransfected and underwent serum starvation as above. However, cells were harvested at various time points (2–24 h). The reporter enzyme activities were determined as described above. The reporter activities were expressed as percentages: presence over absence of tetracycline from the same time point.
Cotransfection was conducted in DEC1 stable cells. Cells were transfected and cultured in the presence or absence of tetracycline. After 24 h-incubation (no serum but with tetracycline), lysates were collected and assayed for luciferase activity. As shown in Figure 4b, DEC1 (addition of tetracycline) repressed the pDEC2-luc reporter by as much as 90%. In contrast, both pSurvivin-6270 and -268 reporters were markedly activated (Figure 4b). In addition, we examined the effect of serum concentrations and cell cycle blockers on the reporter activities in response to DEC1. The pDEC2-luc reporter was consistently repressed by as much as 90% independently of cycle blockers or serum concentrations, whereas the transactivation of the survivin reporters varied depending on serum concentrations and cycle blockers. Overall, lower serum concentrations supported higher activation of the survivin reporters in response to DEC1. Under cycle-synchronized condition, DEC1 transactivation was evident in cells treated with cycle blockers mimosine (late G1), hydroxyurea (G1/S) and thymidine (S), but not nocodazole (G2/M) (data not shown).
Time-course studies were conducted to determine whether the activation and repression have a similar onset. DEC1 cells were cotransfected with pSurvivin-268 or pDEC2-luc and cultured in the presence or absence of tetracycline (no serum). The reporter activities were expressed as percentages (presence over absence of tetracycline from the same time point). As shown in Figure 4c, activation of pSurvivin-268 was evident only after 10 h incubation. In contrast, the pDEC2-luc reporter was markedly repressed as early as 4 h, and the overall repression was rather steady during the entire period of incubation (Figure 4c). The difference on the onset between repression and activation points to two possibilities: pDEC2-luc is a more sensitive target (requiring less DEC1) than pSurvivin-268; alternatively, activation of pSurvivin-268 requires synthesis of additional factors.
Activation of pSurvivin-268 and repression of pDEC2-luc require different functional domains
We next examined whether disruption of certain functional domains differentially affects repression of pDEC2-luc and activation of pSurvivin-268. These mutants are shown in (Figure 5a, Left). The results of the reporter assays are summarized in Figure 5a (Right). Deletion mutants with DNA binding ability (DEC11–347, DEC11–270, DEC11–197 and DEC11–150) all exhibited some transactivation activities depending on the length of a mutant. For example, DEC11–347 exhibited ~60% of the activity of DEC1, whereas DEC11–150, with a longer C-terminal deletion, had only ~20% of the activity. In contrast, mutants with deletion of the DNA binding domain or an N-terminal sequence (DEC1-M, DEC1105–412, and DEC1237–412) showed little activity. These mutants were previously shown to have no DNA binding activity (Li et al., 2003). Among substitution mutants, DEC1P56A was equally active as DEC1, whereas DEC1R58P and DEC1P56A/R58P showed no activation activity. DEC1P56A but not DEC1R58P nor DEC1P56A/R58P was shown to bind to E-box (Li et al., 2003). It should be noted that comparable expression was detected with all constructs (data not shown).
Figure 5.

Differences on the requirements of functional domains to transactivate pSurvivin-268 and repress pDEC2-luc. (a) Activation of the pSurvivin-268 reporter. Cells (293, the parental line of DEC1 stable transfectants) were cultured in 24-well plates and transiently transfected with DEC1 or a mutant (100 ng), pSurvivin-268 (100 ng) and the pRL-TK Renilla (1 ng). The transfected cells were cultured in serum-free medium for 24 h, and lysates were collected and analysed for luciferase activities. Similarly, firefly luminescence signal was normalized based on the Renilla luminescence signal. (b) Comparison of requirements for functional domains between pSurvivin-268 activation and pDEC2-luc repression. Cells (293) were cultured in 24-well plates and transiently transfected with DEC1 or a mutant (100 ng), pSurvivin-268 or pDEC2-luc (100 ng) and the pRL-TK Renilla (1 ng). The transfected cells were cultured in serumfree medium for 24 h, and lysates were collected and analysed for luciferase activities. Firefly luminescence signal was normalized based on the Renilla luminescence signal.
We recently reported the activities of DEC1 and its mutants toward pDEC2-luc (Li et al., 2003). In comparison, several important differences were identified between the repression of pDEC2-luc and the activation of pSurvivin-268 (Figure 5b): (a) DEC11–347 was equally active as DEC1 in repressing pDEC2-luc, whereas it had only ~60% of the activity of DEC1 in activating pSurvivin-268; (b) DEC1R58P and DEC1P56A/R58P showed no activity toward pSurvivin-268, whereas both had ~50% of the activity of DEC1 on repressing pDEC2-luc; and (c) DEC11–150 showed no repression on pDEC2-luc, whereas it caused somewhat activation (~20% of DEC1) on pSurvivin-268. Taken those together, it appears that DNA binding is essential for the activation of pSurvivin-268, but not always true for the repression of pDEC2-luc.
Both DEC1 and Sp1 bind DNA elements in the proximal promoter
The essentiality of DNA binding for effective transactivation of pSurvivin-268 points to an important possibility: DEC1 binds directly to and stimulates the survivin promoter. We and other investigators have shown that DEC1 binds to a particular form of E-box CACGTG (Zawel et al., 2002; Li et al., 2003). Motif analysis, however, identified neither this nor other E-box sequences in this region. Instead, a clustered Sp1/CDE region (cycle-dependent element) is located (Li and Altieri, 1999). Some of the Sp1 sites are slightly altered and arranged in a modified configuration (Figure 6a). In order to identify DNA sequences that act as potential binding sites for DEC1, ten oligonucleotides were synthesized to span the entire region with partial sequences overlapped on one or both ends of an oligonucleotide.
Figure 6.

DEC1 selectively increases DNA binding among Sp1 sites in the proximal promoter of surviving. (a) Diagrammatical presentation of the location of Sp1 sites. Potential Sp1 sites are boxed. (b) Expression of DEC1 selectively alters DNA binding toward Sp1 sites. DEC1 stable transfectants were seeded in six-well plate and cultured in the presence or absence of tetracycline (1 μg/ml). After reaching ~80% confluence, cells underwent serum starvation for 36 h (tetracycline was kept the same). Nuclear extracts were prepared with a nuclear extraction kit (Active Motif). Nuclear proteins (10 μg) were incubated with radiolabeled double-stranded oligonucleotides in a final volume of 10 μl containing ×1 DNA binding buffer. The protein–DNA complexes were resolved in 6% polyacrylamide gel electrophoresis and visualized by autoradiography. (c) Disruption of shifted bands by anti-DEC1 or Sp1 antibody. Nuclear extracts were incubated with radiolabeled oligonucleotides 123–158 or preincubated with an antibody against DEC1 (d), Sp1 (S) or Flag (F), and then with the probe. The protein–DNA complexes were resolved in 6% polyacrylamide gel electrophoresis and visualized by autoradiography. (d) ChIP analysis DEC1 stable transfectants (2×107) were cultured in the presence or absence of tetracycline (1 μg/ml) and then starved for 36 h. Soluble chromatins were precipitated with affinity purified antibody against DEC1 (5 μg) or the same amount of normal IgG. The antibody-bound chromatins and DNA input (10% of initial chromatins for antibody-enrichment) were analysed by PCR for the presence of the Sp1-containing fragment (lane 1) or a fragment within the transcription region (lane 2). The PCR cycling parameters are: 95°C for 30 s, 55°C for 30 s and 72°C for 30 or 40 s for a total of 36 (input template) or 40 (anti-DEC1 enriched template) cycles. The PCR-amplified products were analysed by agarose gel electrophoresis and visualized by ethidium bromide staining.
The initial focus of the DNA binding study was on whether any of the oligonucleotides are bound by nuclear extracts and whether the degree of binding is altered as a result of the presence of DEC1. DEC1 stable cells were cultured in the presence or absence of tetracycline (no serum) to modulate the expression of DEC1 and the nuclear extracts were prepared. Equal amounts of extracts from control or tetracycline-treated cells were subjected to bind to double-stranded oligonucleotides, and shifted bands were detected by non-denaturing PAGE. As shown in Figure 6b, one or more shifted bands were detected with seven out of ten oligonucleotides including oligonucleotides 41–68, 55–84, 85–114, 105–126, 115–144, 123–158, 205–234. More importantly, DEC1 differentially affected the overall binding intensity among some of these oligonucleotides. Markedly higher binding was detected with oligonucleotides 115–144, 123–158 and 205–234 in cells cultured with tetracycline. In addition, two shifted bands were detected with oligonucleotides 115–144 and 123–158, but only the upper shifted band was intensified as a result of induced expression of DEC1, suggesting that DEC1 is responsible for the increased binding.
We next examined whether DEC1 is present in the binding complex. Nuclear extracts were initially incubated with anti-DEC1 antibody and then with radio-labeled oligonucleotide 123–158. This probe contains an Sp1 complex site, therefore, anti-Sp1 antibody was also examined. As shown in Figure 6c, addition of anti-DEC1 or anti-Sp1 antibody caused disappearance of the upper shifted band. Interestingly, anti-Sp1 but not DEC1 antibody increased the intensity of the lower shifted band (Figure 6c). Anti-Flag antibody, however, caused no changes on the shifted bands, suggesting that both DEC1 and Sp1 are indeed present in the DNA binding complex with this probe. The presence of DEC1 in the survivin promoter was further established by chromatin immunoprecipitation (ChIP). DEC1 stable cells were cultured in the presence and absence of tetracycline and underwent serum starvation for 36 h. Soluble chromatins were prepared and incubated with anti-DEC1 antibody or normal IgG. The antibody bound DNAs were analysed for the presence of the Sp1-containing fragment (−268 to −78) or a fragment in the transcription region (+339 to +650). As shown in Figure 6d, with DNA input as the template, PCR produced comparable amplification for both fragments. In contrast, only the Sp1-containing fragment was robustly amplified with DNA template enriched by anti-DEC1 antibody, providing direct evidence that DEC1 occupies the survivin promoter. It should be noted that DNAs from cells cultured without tetracycline or precipitated with normal IgG produced only background amplification (data not shown).
Sp1 sites-127 and -226 additively support DEC1 transactivation
In order to determine whether binding to Sp1 site-127 or -226 is responsible for the activation of the survivin reporter, mutants on these sites were tested for the responsiveness to DEC1. Similarly, the DEC1 stable line was transfected with a reporter mutant and cultured in the presence or absence to modulate the expression of DEC1 (no serum). As shown in Figure 7, deletion or mutagenic disruption of the Sp1 site-226 decreased the responsiveness to DEC1 by 30%. An additional 40% decrease was detected with the substitution mutant that had Sp1 site-127 disrupted (pSurvivin-154M). Therefore, binding to these Sp1 sites is responsible for as much as 70% of the overall activation by DEC1 (Figure 7).
Figure 7.
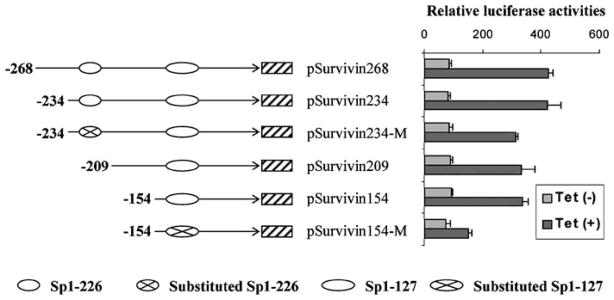
Sp1 sites-127 and -226 support DEC1-mediated activation of survivin. DEC1 stable transfectants were seeded in 24-well plate and cultured in the presence or absence of tetracycline (1 μg/ml). After reaching ~80% confluence, cells were transfected again with pSurvivin-268 or a reporter mutant (100 ng) and the pRL-TK Renilla (1 ng). The retransfected cells were cultured with serum-free medium in the presence or absence of tetracycline (1 μg/ml) for 24 h. The reporter enzyme activity was assayed with a Dual-Luciferase Reporter Assay System as described in the legend of Figure 4.
Discussion
We have demonstrated that DEC1 effectively protects against serum starvation (Li et al., 2002). This study reports that DEC1 transcriptionally upregulates the expression of antiapoptotic protein survivin, providing a molecular mechanism on its cyto-protective effect. In addition to DEC1, several other transcription factors are found to be activators of survivin transcription, notably, β-catenin/TCF (T-cell factor) (Ma et al., 2005), NF-κB (nuclear factor-κB) (Kawakami et al., 2005), E2F family members (Jiang et al., 2004) and STAT3 (signal transducer and activator of transcription-3) (Kanda et al., 2004). Consistent with abundant expression of survivin in tumors, the activity of these factors is markedly increased in a variety of malignancies (Jiang et al., 2004; Kanda et al., 2004; Kawakami et al., 2005; Ma et al., 2005). The increased transcription activity is achieved by overexpression (e.g., E2F1), post-translational modification (e.g., STAT3) or nuclear translocation (e.g., β-catenin). Studies with deletion mutants have shown that these factors stimulate the survivin promoter by binding respective elements in the proximal region (Jiang et al., 2004; Kawakami et al., 2005; Ma et al., 2005). In addition to transactivation, this region also supports transrepression by tumor suppressor p53 (Hoffman et al., 2002). Interestingly, the p53 element overlaps with a functional E2F element (Hoffman et al., 2002). Although there are conflicting reports, it is generally accepted that p53 represses the survivin promoter by direct DNA binding or induction of p21, an inhibitor of cyclin-dependent kinases. This inhibitor prevents phosphorylation of retinoblastoma (RB) family proteins. Hypophosphorylated RB binds to E2F family proteins and converts them from transcriptional activators to repressors, thus decreasing survivin expression (Hoffman et al., 2002; Mirza et al., 2002; Löhr et al., 2003).
The RB/E2F pathway arrests cells at G1 and is presumably responsible for G1-dependent repression of survivin (Jiang et al., 2004; Halaban, 2005). Cycle synchronization by G1 blocker mimosine recaptures the repression (Li et al., 1998). In contrast, nocodazole, a G2/M cycle blocker, markedly induces survivin and effectively stimulates the survivin promoter (Li et al., 1998). Interestingly, transfection of DEC1 does not enhance nocodazole-mediated stimulation on the survivin promoter, although these two mechanisms use different cis-DNA elements. In this study, Sp1 sites-127 and -226 are found to contribute as much as 70% to DEC1 transactivation (Figure 7). In contrast, nocodazole-mediated activation requires a 40 bp-proximal sequence containing two CDE and one cycle homology region (CHR) (Li et al., 1998). Mutagenic disruption of one or more of these elements abolishes the ability to respond to nocodazole. It remains to be determined whether the CDE/CHR region affects DEC1 transactivation, or the Sp1 sites-127 and -226 affect nocodazole-mediated activity. It should be emphasized that other Sp1 sites (−151 and −171) in the survivin promoter are shown to support constitutive activation of survivin in HeLa cells (Li and Altieri, 1999). However, no DNA binding to these sites is detected with 293 nuclear extracts (Figure 6b, oligonucleotides 145–174 and 175–204), suggesting that these sites represent cell type-specific regulation.
Binding to Sp1 sites-127 and -226 may not be the only event in DEC1 transactivation of the survivin promoter. We have shown that marked induction of survivin occurs only in cells cultured at relatively low serum concentrations (Figure 3a), suggesting that the presence of DEC1 alone is not sufficient to effectively upregulate the expression of survivin. Similarly, less serum supports higher DEC1 transactivation of the survivin promoter (unpublished data). Even with serum-free medium, the activation of the survivin reporter is minimal during the first 10 h incubation (Figure 4c). In contrast, the pDEC2-luc reporter is repressed by 65% as early as 4 h (Figure 4c). The delayed onset on the activation of the survivin reporter suggests that DEC1 initiates more than one event, and these events collectively determine the onset and magnitude of the activation.
Given the fact that DEC1 transactivation of the survivin promoter is largely achieved through Sp1 sites-127 and -226, it is conceivable that increased expression of Sp1 is one of the events responsible for the delayed onset. It remains to be determined whether DEC1 actually induces Sp1, however, electrophoretic mobility shift assay (EMSA) demonstrates that nuclear extracts from DEC1 transfected cells increase the binding activity toward these Sp1 sites (Figure 6b). Interestingly, increased Sp1 binding occurs only with Sp1 sites-127 and -226 but not with five others present in this region (Figure 6a), although some of the other Sp1 sites are found to bind to Sp1 and related proteins (Li and Altieri, 1999). Alternatively, Sp1 interacts with other proteins and the resultant complexes collectively determine DNA binding selectivity. In support of this possibility, mSHARP, a DEC1 related mouse protein, has been shown to interact with Sp1 (Azmi et al., 2003). In this study, we have demonstrated that anti-DEC1 antibody completely disrupts the shifted bands with Sp1 site-127 (Figure 6c), suggesting that DEC1 is part of the Sp1–DNA complex. It should be noted that Sp1–mSHARP complex represses the promoter of STRA13 (Azmi et al., 2003), whereas Sp1–DEC1 complex results in the activation of the survivin promoter (this study).
Induction of survivin by DEC1 likely has important pathological significance. Survivin antagonizes apoptosis and promotes cell division (Li, 2003). The expression of survivin is high in G2/M and low in G1 phase (Li et al., 1998). The dual function (antiapoptosis and cell division), along with its cycle-dependent expression, provides an effective mechanism to ensure success of cell division. In this study, forced expression of DEC1 alters the cycle-dependent activity of the survivin promoter (serum starvation). The DEC1-mediated alteration on the cycle dependence provides a mechanism that minimizes cell death during oncogenic process regardless of a cycle phase (assumed that the deregulated expression of DEC1 in tumors occurs in a cycle-independent manner). Given the fact that DEC1 has a broad tissue distribution (Shen et al., 1997), DEC1-mediated antiapoptosis may have physiological significance as well, particularly in cell proliferation and differentiation. In support of this possibility, mice deficient in STRA13 (mouse counterpart of DEC1) develop autoimmune disease featured by accumulation of spontaneously activated T and B cells (Sun et al., 2001). Although these mice are generally normal, the surface FasL is markedly reduced. Dysfunctional Fas–FasL system results in decreased apoptosis of premature lymphocytes, leading to the development of autoimmune disease. The decreased expression of FasL in STRA13-deficient mice suggests that STRA13, like its human homologue DEC1 on the survivin, acts as a transcriptional activator. It remains to be determined whether STRA13 supports the expression of FasL via STRA13–Sp1 complex. Interestingly, Sp1-dependent activation is reported to be a major pathway in FasL expression (Kavurma et al., 2002).
In summary, our work points to several important conclusions. First, DEC1 antagonizes apoptosis induced by serum starvation, and in this study, we reports that DEC1 transcriptionally upregulates the expression of the survivin, providing a molecular explanation for DEC1 antiapoptosis. Second, the activation of the survivin promoter is delayed and varies depending on serum concentration and cycle blockers, suggesting that DEC1-mediated activation requires more than one event and operates in cell type and cycle-related manner. Third, DEC1 transactivates the survivin promoter, but transrepresses the DEC2 promoter, illustrating dual transcription activities of this transcription factor. Given the fact that DEC1 has been implicated in oncogenic process (deregulated) and is generally considered to be transcriptionally repressive, transactivation of the survivin promoter provides a molecular explanation for its oncogenic involvement and represents an example that DEC1 and other members in this class can act as transcription activators depending on a target gene.
Materials and methods
Chemicals and supplies
Purvalanol A, tetracycline, and anti-FLAG antibody were from Sigma (St Louis, MO, USA). Anti-XIAP antibody was from Cell Signaling (Beverly, MA, USA). Antibody against Sp1 was from Geneka Biotechnology (Montreal, Canada). Antibodies against cIAP-1 and cIAP-2 were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibody against survivin was from Novus Biologicals (Littleton, CO, USA). Goat anti-rabbit-IgG conjugated with horseradish peroxidase and ECL substrate were from Pierce (Rockford, IL, USA). Dulbecco’s modified Eagle’s medium (DMEM) and Lipofect-AMINE were from Invitrogen (Carlsbad, CA, USA). Dual-Luciferase reagent and DNA binding buffer were from Promega (Madison, WI, USA). Unless otherwise indicated, all other reagents were purchased from Fisher Scientific (Pittsburgh, PA, USA).
Tissue collection and processing
Samples were collected from patients who underwent surgical resection for histologically confirmed adenocarcinoma. As paired controls, specimens from the adjacent, grossly normal tissues were harvested. The samples (four pairs/each organ) were collected from various organs including the kidney and lung. The age of the patients was between 23 and 68 years with four male and three female patients. The size of tumors was generally ~2 cm in diameter, and the degree of differentiation of tumors was moderate or poor as determined by pathological examination. Samples were freshly processed for RNA isolation and protein extraction. Total RNA was isolated with a Tri-reagent as described previously (Xie et al., 2003). For the preparation of protein extracts, tissues were homogenized in lysis buffer (20mM Tris-HCl, pH 7.4, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 0.2mM PMSF and 1mM dithiothreitol (DTT)). The homogenates were centrifuged at 12 000 g for 30 min to remove any insoluble precipitates. The protocol of using human pathological tissues was reviewed by the Institutional Review Board.
Reverse transcription coupled polymerase chain reaction
Total RNA (2 μg) was subjected to the synthesis of the first strand cDNA with an oligo(dT) primer and a ThermoScript reverse transcriptase. The reactions were incubated initially at 50°C for 30 min, and then at 60°C for 60 min after additional reverse transcriptase was added. The cDNAs were diluted by 100 times and then subjected to PCR amplification (10 μl of the diluted cDNAs) with cycling parameters as follows: 95°C for 30 s, 52°C for 30 s and 68°C for 30 or 40 s for a total of 32 cycles. The primers for DEC1 amplification were: 5′-GTCTGTGAGTCACTCTTCAG-3′, 5′-GAGTCTAGTTCTGTTTGAAGG-3′; the survivin primers were: 5′-TCAAGGACCACCGCATCTCTAC-3′, 5′-GCACTTTCTTCGCAGTTTCC-3′; and the β-actin primers were: 5′-GTACCCTGGCATTGCCGACAGGATG-3′, 5′-CGCAACTAAGTCATAGTCCGCCTA-3′. The PCR products were analysed by agarose gel electrophoresis.
Plasmid
Expression constructs for DEC1 and mutants as well as the DEC2 promoter reporter (pDEC2-luc) were described previously (Li et al., 2002, 2003). Survivin promoter reporters were prepared with the pGL3-basic vector (Promega). Deletion mutants of survivin reporters were prepared by PCR with pSurvivin-6270 serving the template (Li and Altieri, 1999). Substitution mutants (pSurvivin240-M and pSurvivin154-M) were also prepared by PCR with primers containing substituted nucleotides. The sequences of the primers to introduce substitutions are: 5′-CTACGCGTAATAAGGAACGAGCTGGTGATGTATCGCTGGGTGCACCGCG-3′ (pSurvivin240-M), and 5′-CTACGCGTCCCGGCACACCCCTAGTTATCAAGTTTCTACTCCCAGAAGGC-3′ (pSurvivin154-M). All mutated constructs were confirmed by sequencing analysis.
Cotransfection experiment
Cotransfection was performed with DEC1 stable transfectants or the parent line (293). Stable transfectants expressing wild-type DEC1 or a DEC1 mutant (lacking the DNA binding domain) were described previously (Li et al., 2002). Cells were plated in 24-well plates in DMEM medium supplemented with 10% fetal bovine serum at a density of 1.6×105 cells per well. Transfection was conducted with LipofectAMINE (Li et al., 2002). Transfection mixtures contained DEC1 or a mutant construct (100 ng), reporter plasmid (100 ng) and the Renilla plasmid (1 ng). If DEC1 stable line was used, DEC1 construct was omitted from the transfection mixture, but the expression of DEC1 was induced by tetracycline. Transfected cells were cultured for additional 24 h, and cell lysates were prepared (Li et al., 2003). The luciferase activities were determined with a Dual-Luciferase Reporter Assay System (firefly and Renilla luciferases) as described previously (Li et al., 2002). The firefly luminescence signal (reporter) was normalized based on the Renilla luminescence signal (internal control).
Electrophoretic mobility shift assay
DEC1 stable transfected cells were cultured in the presence or absence of tetracycline (1 μg/ml) for 36 h and nuclear extracts were prepared with a nuclear extraction kit (Active Motif). Nuclear proteins (10 μg) were incubated with radiolabeled double-stranded oligonucleotides in a final volume of 10 μl containing 1× DNA binding buffer. For competition experiments, nuclear extracts were first incubated with excess cold probe (50×) and then mixed with the corresponding probe. For super-shift assays, the nuclear extracts were preincubated with anti-DEC1 or anti-Sp1 antibody, and then with a radiolabeled probe. The protein–DNA complexes were resolved in 6% PAGE and visualized by autoradiography. The sequences of the oligonucleotides for EMSA are: 41–68: 5′-CCCCGCGGCGCGCCATTAACCGCCAGAT-3′; 55–84: 5′-GTGCGCTCCCGACATGCCCCGCGGCGCGCC-3′; 76–105: 5′-GGGGTGGACCGCCTAAGAGGGCGTGCGCTCC-3′; 85–114: 5′-GGCCGCGGGGGGTGGACCGCCTAAGAGGGC-3′; 105–126: 5′-CTACTCCCAGAAGGCCGCGGGG-3′; 115–144: 5′-CCCCGCGCCGCCCCGCCTCTACTCCCAGAA-3′; 123–158: 5′-CAACTCCCGGCACACCCCGCGCCGCCCCGCCTCTAC-3′; 145–174: 5′-CGCGGCGGGAGGACTACAACTCCCGGCACA-3′; 175–204: 5′-CTGGGTGCACCGCGACCACGGGCAGAGCCA-3′; and 205–234: 5′-TGTGGGCAGGGACGAGCTGGCGCGGCGTCG-3′. The underlined motifs are Sp1 sites.
Chromatin immunoprecipitation
ChIP experiment was performed with a ChIP-It kit (Active Motif), essentially as described previously (Song et al., 2004) and the manufacturer’s manual. Briefly, DEC1 stable cells (2×107) were cultured in the presence or absence of tetracycline (1 μg/ml) and then starved for 36 h. Soluble chromatins were prepared by enzymatic digestion (to 200–400 bp) and precipitated with affinity purified antibody against DEC1 (5 μg) or the same amount of normal IgG. The antibody-bound chromatins and DNA input were analysed by PCR for the presence of the Sp1-containing fragment with primers: 5′-CGCGTTCTTTGAAAGCAGTCGAG-3′ and 5′-AGCGCACGCCCTCTTAGGCGGTC-3′. As a control, PCR was performed with primers that recognized a segment in the transcriptional region: 5′-CCCACTGAGAACGAGCCAG AC-3′ and 5′-TAAGCATCACCAAGGCACCAG-3′.
Other analyses
Western analyses and DNA fragmentation were conducted as described previously (Li et al., 2002; Xie et al., 2002). The preparation of anti-DEC1 antibody was described elsewhere (Li et al., 2002). Protein concentration was determined with a BCA kit (Pierce) and bovine serum albumin served as the standard. Data are presented as mean±s.d. of at least four separate experiments, except where results of blots are shown in which case a representative experiment is depicted in the figures.
Acknowledgments
We thank Dr Fengzhi Li for providing the pSurvivin-6270 reporter. This work was partially supported by National Institutes of Health Grants R01ES07965, R01GM61988 and F05AT003019 (to BY).
Abbreviations
- bHLH
basic helix-loop-helix
- ChIP
chromatin immunoprecipitation
- DEC
differentially expressed in chondrocytes
- DMEM
Dulbecco’s modified Eagle’s medium
- DTT
dithiothreitol
- EMSA
electrophoretic mobility shift assay
- HLH
helix-loop-helix
- IAP
inhibitor of apoptosis
- PCR
polymerase chain reaction
- PBS
phosphate-buffered saline
- RT–PCR
reverse transcription-polymerase chain reaction
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- SHARP
split and hairy related protein
- STRA
stimulated with retinoic acid
References
- Ashkenazi A, Dixit VM. Science. 1999;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Azmi S, Sun H, Ozog A, Taneja R. J Biol Chem. 2003;278:20098–20109. doi: 10.1074/jbc.M210427200. [DOI] [PubMed] [Google Scholar]
- Boudjelal M, Taneja R, Matsubara S, Bouillet P, Dollè P, Chambon P. Genes Dev. 1997;11:2052–2065. doi: 10.1101/gad.11.16.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Annu Rev Cell Dev Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- Chakrabarti J, Turley H, Campo L, Han C, Harris AL, Gatter KC, et al. Br J Cancer. 2004;91:954–958. doi: 10.1038/sj.bjc.6602059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryns V, Yuan J. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- Currie MJ, Hanrahan V, Gunningham SP, Morrin HR, Frampton C, Han C, et al. J Clin Pathol. 2004;57:829–834. doi: 10.1136/jcp.2003.015644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto K, Shen M, Noshiro M, Matsubara K, Shingu S, Honda K, et al. Biochem Biophys Res Commun. 2001;280:164–671. doi: 10.1006/bbrc.2000.4133. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC. Science. 1999;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Halaban R. Cancer Metastasis Rev. 2005;24:339–356. doi: 10.1007/s10555-005-1582-z. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. J Biol Chem. 2002;277:3247–3257. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- Honma S, Kawamoto T, Takagi Y, Fujimoto K, Sato F, Noshiro M, et al. Nature. 2002;419:841–844. doi: 10.1038/nature01123. [DOI] [PubMed] [Google Scholar]
- Inuzuka H, Nanbu-Wakao R, Masuho Y, Muramatsu M, Tojo H, Wakao H. Biochem Biophys Res Commun. 1999;265:664–668. doi: 10.1006/bbrc.1999.1734. [DOI] [PubMed] [Google Scholar]
- Ivanova AV, Ivanov SV, Danilkovitch-Miagkova A, Lerman MI. J Biol Chem. 2001;276:15306–15315. doi: 10.1074/jbc.M010516200. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Saavedra HI, Holloway MP, Leone G, Altura RA. J Biol Chem. 2004;279:40511–40520. doi: 10.1074/jbc.M404496200. [DOI] [PubMed] [Google Scholar]
- Kanda N, Seno H, Konda Y, Marusawa H, Kanai M, Nakajima T, et al. Oncogene. 2004;23:4921–4929. doi: 10.1038/sj.onc.1207606. [DOI] [PubMed] [Google Scholar]
- Kavurma MM, Bobryshev Y, Khachigian LM. J Biol Chem. 2002;277:36244–36252. doi: 10.1074/jbc.M200463200. [DOI] [PubMed] [Google Scholar]
- Kawakami H, Tomita M, Matsuda T, Ohta T, Tanaka Y, Fujii M, et al. Int J Cancer. 2005;115:967–974. doi: 10.1002/ijc.20954. [DOI] [PubMed] [Google Scholar]
- Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, et al. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- Li F, Altieri DC. Biochem J. 1999;344(Part 2):305–311. doi: 10.1042/0264-6021:3440305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. J Cell Physiol. 2003;197:8–29. doi: 10.1002/jcp.10327. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhang H, Xie M, Hu M, Ge S, Yang D, et al. Biochem J. 2002;367:413–422. doi: 10.1042/BJ20020514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xie M, Song X, Gragen S, Sachdeva K, Wan Y, et al. J Biol Chem. 2003;278:16899–16907. doi: 10.1074/jbc.M300596200. [DOI] [PubMed] [Google Scholar]
- Li Y, Song X, Ma Y, Liu J, Yang D, Yan B. Biochem J. 2004;382:895–904. doi: 10.1042/BJ20040592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löhr K, Möritz C, Contente A, Dobbelstein M. J Biol Chem. 2003;278:32507–32516. doi: 10.1074/jbc.M212517200. [DOI] [PubMed] [Google Scholar]
- Ma H, Nguyen C, Lee KS, Kahn M. Oncogene. 2005;24:3619–3631. doi: 10.1038/sj.onc.1208433. [DOI] [PubMed] [Google Scholar]
- Mirza A, McGuirk M, Hockenberry TN, Wu Q, Ashar H, Black S, et al. Oncogene. 2002;21:2613–2622. doi: 10.1038/sj.onc.1205353. [DOI] [PubMed] [Google Scholar]
- Miyazaki K, Kawamoto T, Tanimoto K, Nishiyama M, Honda H, Kato Y. J Biol Chem. 2002;277:47014–47021. doi: 10.1074/jbc.M204938200. [DOI] [PubMed] [Google Scholar]
- Reed JC. J Clin Invest. 2001;108:965–969. doi: 10.1172/JCI14123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossner MJ, Dörr J, Gass P, Schwab MH, Nave KA. Mol Cell Neurosci. 1997;9:460–475. doi: 10.1006/mcne.1997.0640. [DOI] [PubMed] [Google Scholar]
- Shen M, Kawamoto T, Yan W, Nakamasu K, Tamagami M, Koyano Y, et al. Arch Biochem Biophys. 1997;236:294–298. doi: 10.1006/bbrc.1997.6960. [DOI] [PubMed] [Google Scholar]
- Shen M, Kawamoto T, Teramoto M, Makihira S, Fujimoto K, Yan W, et al. Eur J Cell Biol. 2001;80:329–334. doi: 10.1078/0171-9335-00167. [DOI] [PubMed] [Google Scholar]
- Shen M, Yoshida E, Yan W, Kawamoto T, Suardita K, Koyano Y, et al. J Biol Chem. 2002;277:50112–50120. doi: 10.1074/jbc.M206771200. [DOI] [PubMed] [Google Scholar]
- Song X, Xie M, Zhang H, Li Y, Sachdeva K, Yan B. Drug Metab Dispos. 2004;32:35–42. doi: 10.1124/dmd.32.1.35. [DOI] [PubMed] [Google Scholar]
- Sun H, Lu B, Li RQ, Flavell RA, Taneja R. Nat Immunol. 2001;2:1040–1047. doi: 10.1038/ni721. [DOI] [PubMed] [Google Scholar]
- Turley H, Wykoff CC, Troup S, Watson PH, Gatter KC, Harris AL. J Pathol. 2004;203:808–813. doi: 10.1002/path.1585. [DOI] [PubMed] [Google Scholar]
- Vaupel P, Kelleher DK, Hockel M. Semin Oncol. 2001;28:29–35. doi: 10.1016/s0093-7754(01)90210-6. [DOI] [PubMed] [Google Scholar]
- Xie M, Yang D, Liu L, Xue B, Yan B. Drug Metab Dispos. 2002;30:541–547. doi: 10.1124/dmd.30.5.541. [DOI] [PubMed] [Google Scholar]
- Xie M, Yang D, Liu L, Xue B, Yan B. Drug Metab Dispos. 2003;31:21–27. doi: 10.1124/dmd.31.1.21. [DOI] [PubMed] [Google Scholar]
- Yang YL, Li XM. Cell Res. 2000;10:169–177. doi: 10.1038/sj.cr.7290046. [DOI] [PubMed] [Google Scholar]
- Yoon DY, Buchler P, Saarikoski ST, Hines OJ, Reber HA, Hankinson O. Biochem Biophys Res Commun. 2001;288:882–886. doi: 10.1006/bbrc.2001.5867. [DOI] [PubMed] [Google Scholar]
- Yun Z, Maecker HL, Johnson RS, Giaccia AJ. Dev Cell. 2002;2:331–341. doi: 10.1016/s1534-5807(02)00131-4. [DOI] [PubMed] [Google Scholar]
- Zawel L, Yu J, Torrance CJ, Markowitz S, Kinzler KW, Vogelstein B, et al. Proc Natl Acad Sci USA. 2002;99:2848–2853. doi: 10.1073/pnas.261714999. [DOI] [PMC free article] [PubMed] [Google Scholar]