

Abstract
Chronic hepatitis B virus (HBV) infection is an important cause of morbidity and mortality in subjects coinfected with HIV. Tenofovir disoproxil fumarate (TDF) and adefovir dipivoxil (ADV) are licensed for the treatment of HIV-1 and HBV infection, respectively, but both have in vivo and in vitro activity against HBV. This study evaluated the anti-HBV activity of TDF compared to ADV in HIV/HBV-coinfected subjects. ACTG A5127 was a prospective randomized, double-blind, placebo-controlled trial of daily 10 mg of ADV versus 300 mg of TDF in subjects with HBV and HIV coinfection on stable ART, with serum HBV DNA ≥ 100,000 copies/mL, and plasma HIV-1 RNA ≤ 10,000 copies/mL. This study closed early based on results of a prespecified interim review, as the primary noninferiority end point had been met without safety issues. Fifty-two subjects were randomized. At baseline, 73% of subjects had a plasma HIV-1 RNA < 50 copies/mL, 86% were HBeAg positive, 94% were 3TC resistant, median serum ALT was 52IU/L, and 98% had compensated liver disease. The mean time-weighted average change in serum HBV DNA from baseline to week 48 (DAVG48) was −4.44 log10 copies/mL for TDF and −3.21 log10 copies/mL for ADV. There was no difference in toxicity between the 2 treatment arms, with 11 subjects (5 ADV and 6 TDF) experiencing elevations of serum ALT on treatment. In conclusion, over 48 weeks, treatment with either ADV or TDF resulted in clinically important suppression of serum HBV DNA. Both drugs are safe and efficacious for patients coinfected with HBV and HIV.
Chronic hepatitis B virus (HBV) infection affects 400 million individuals worldwide and accounts for 1 million deaths annually. Persistent infection occurs in 20% of adults with HIV who become infected1 and is an important cause of morbidity and mortality in these coinfected patients. The incidence of HBV in HIV-infected individuals varies from 5%-10% in the United States2,3 up to 20% to 30% in Asia and parts of sub-Saharan Africa.4,5 In addition, up to 90% of patients treated with 3TC as antiret-roviral therapy (ART) develop resistance to HBV after 4 years of therapy.6
Adefovir dipivoxil (ADV; 10 mg) is active against both wild-type and 3TC-resistant HBV.7-9 Tenofovir disoproxil fumarate (TDF; 300 mg) is licensed for the treatment of HIV-1 but has been shown to have in vitro activity against both wild-type and 3TC-resistant HBV.10,11 In addition, there are a number of case series of the activity of TDF in monoinfected and coinfected HBV patients.12,13 Our study evaluated whether TDF was not inferior to ADV for treatment of coinfected individuals and is a prospective randomized, controlled trial of TDF and ADV in chronic HBV infection.
Patients and Methods
Study Design
The AIDS Clinical Trials Group (ACTG) protocol A5127 was a prospective randomized, double blind, placebo-controlled trial evaluating whether TDF was not inferior to ADV for treatment of HBV in subjects coinfected with HIV and HBV. Subjects were randomized to 10 mg of ADV plus TDF placebo daily or 300 mg of TDF plus ADV placebo daily with stratification by Child-Pugh-Turcotte (CPT) score (screening CPT score < 7 or decompensated liver disease with screening CPT score ≥ 7) and by CD4 count (<200 or ≥200 cells/mm3). Treatment assignments were generated by computer using permuted blocks within a stratum, and site personnel received a unique blinded treatment identifier by computer link. Site pharmacists identified assigned treatments from site-specific master lists. Clinical and research personnel and subjects were kept blinded to the treatment assignment until all subjects were off study. Subjects were seen every 4 weeks with laboratory evaluations. Subjects with an on-study decline in CPT by 2 points or more were eligible to cross over to the alternative regimen in a blinded fashion.
Initially, change in serum HBV DNA from baseline to week 48 was the primary end point, and subjects were required to be 3TC resistant clinically at entry, with a serum HBV DNA ≥ 1 × 106 copies/mL on 3TC. After the development of this study, subsequent information from 2 large placebo-controlled studies in HBV-monoinfected subjects7,8 and a pilot study in HBV/HIV-coinfected subjects14 indicated the time-weighted average (DAVG) may be a better surrogate for long-term histologic improvement. DAVG48 is the difference between the average postbaseline log serum HBV DNA and baseline log serum HBV DNA, where the postbaseline average is time weighted and calculated using the normalized area under the curve of log10 HBV DNA from the first postbaseline measurement to week 48. A protocol amendment (December 8, 2003) redesigned the end point to utilize DAVG48 without any examination of the old (change from baseline) or new (DAVG) end-point data. In addition, the redesign allowed entry of HBV treatment-naive subjects with serum HBV DNA ≥ 1 × 105 copies/mL. Noninferiority was defined as a tolerance of − 1 log10 copies/mL in the difference between the DAVG48 of ADV and that of TDF. Secondary objectives were to assess the clinical response to ADV versus that to TDF, defined as changes in clinical disease by CPT score; to evaluate the safety and tolerability of ADV and TDF; to evaluate seroconversion from HBeAg positive to HBeAg negative and HBeAb positive; and to evaluate the relationship between alanine aminotransferase (ALT) and serum HBV DNA. An ALT flare was defined as elevation of serum ALT level of 3 times the baseline ALT level. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki: the research protocol and amendments were approved by the relevant institutional review boards or ethics committees, and all subjects gave written informed consent.
Human Subjects
All subjects were coinfected with HBV and HIV. Subject inclusion criteria were: age 18-65 years; HIV-1 infection on a stable antiretroviral regimen with HIV-1 RNA ≤ 10,000 copies/mL (Roche Amplicor 1.5 (LLQ50 copies/mL) for at least 12 continuous weeks; documented positive serum HBsAg and serum HBV DNA ≥ 1 × 105 copies/mL by Roche Amplicor CobasPCR (lower limit of quantitation [LLQ] 200 copies/mL) within 12 weeks of entry into study; ALT ≤ 10 × upper limit of normal (ULN); serum creatinine < 1.5 mg/dL; serum phosphorus ≥ 2.2 mg/dL; estimated creatinine clearance ≥ 50 mL/min; use of contraception; and serum alpha-fetoprotein (AFP) ≤ 50 ng/dL. Exclusion criteria were history of clinically significant renal dysfunction within the last 12 months; other liver disease including HCV or HDV; any active medical or psychiatric illness or alcohol or drug use; pregnancy or breast-feeding; malignancy; and receipt of systemic corticosteroids, nephrotoxic drugs, or anti-HBV drugs except for 3TC within 90 days of study entry. Sequencing from the overlapping surface antigen and domains B-E of the reverse transcriptase region of HBV was performed on baseline plasma specimens using a TruGene HBV Sequencing Kit and OpenGene DNA Sequencing Analysis System (RUO Version 1.0, Bayer Nucleic Acid Diagnostics, Norwood, MA).
Statistics
The Version 2.0 design (December 8, 2003) specified 58 subjects randomized equally between the 2 arms in order to provide 80% power to establish TDF as not inferior if the standard deviation (SD) of the change in DAVG48 HBV DNA was 1.5 log10. An interim Study Monitoring Committee (SMC) review, planned to occur when more than 50% of subjects had reached week 12, took place in August 2004. This review employed a Haybittle-Peto boundary (1-sided 0.001 level) to protect the experiment-wide type I error rate.15. At that time, the primary end point had been met without safety concerns, and the SMC recommended that accrual and follow-up be terminated. The study was closed to follow-up in December 2004, and data entry screens were closed March 4, 2005. The primary analyses of DAVG48 were performed with t tests and analysis of variance. Dichotomous variables were evaluated with Fisher's exact test. Ordered categorical data (toxicity) were evaluated with exact Wilcoxon tests adjusted for ties. Analyses were performed in SAS 8.2 (SAS Institute, Cary, NC). Because this study stopped early based on interim review, primary analyses focused on data from visits up to the SMC date and used the Haybittle-Peto boundary. All-visits data are also presented.
Results
Baseline Characteristics
Table 1 summarizes the baseline characteristics of the A5127 study subjects overall and by arm. At the time accrual was closed, 52 subjects from 17 ACTG sites had been randomized to TDF (27 patients) or ADV (25 patients), with 1 site enrolling 26% of subjects. At study baseline, 47 of the 52 patients had a YMDD mutation in rt Domain C (either M204V or M204I), and the following HBV genotypes were found: A genotype, 38 patients (73%); G genotype, 9 patients (17%); D genotype, 3 patients (6%); and F genotype, 2 patients (4%) F. There were no statistical differences between arms in baseline characteristics, including serum HBV DNA level, although those on TDF were younger (P = .011). Forty-seven subjects (94%) had received prior 3TC, with a mean duration prior to the study of 1,554 days. All 47 had YMDD mutations as determined by sequencing, and the interval between development of YMDD mutations and entry into the study varied. All subjects in the study had been on concomitant ART for at least 12 weeks prior to the study and continued on ART throughout the study as noted above. Fifty patients received 3TC during the study; the 3 subjects commencing de novo 3TC received TDF. Only 1 subject on TDF had a CPT score > 6, and only 4 subjects, 2 each on ADV and TDF, had a CD4 count < 200. As a result, the end-point analyses were not stratified.
Table 1. Baseline Characteristics of A5127 Study Participants.
| All | ADV arm | TDF arm | |
|---|---|---|---|
| Number of subjects | 52 | 25 | 27 |
| Age: median in years | 41 | 47 | 40 |
| Male, n (%) | 48 (92%) | 24 (96%) | 24 (89%) |
| Caucasian, n (%) | 29 (56%) | 14 (56%) | 15 (56%) |
| African American, n (%) | 17 (33%) | 8 (32%) | 9 (33%) |
| Prior IV drug use, n (%) | 7 (13%) | 1 (4%) | 6 (22%) |
| Serum HBV DNA log10 mean copies/mL* | 9.16 ± 1.54 | 8.85 ± 1.88 | 9.45 ± 1.1 |
| HBeAg positive, n (%) | 41 (79%) | 20 (80%) | 23 (85%) |
| 3TC experienced, n (%) | 47 (94) | 24 (96%) | 25 (93%) |
| CPT score < 7, n (%) | 51 (98%) | 25 (100%) | 26 (96%) |
| ALT (median IU/mL) | 52 | 63 | 45 |
| ALT ≤ ULN n (%) | 29 (56%) | 13 (52%) | 16 (59%) |
| AST (median IU/mL) | 42 | 46 | 38 |
| Hematocrit % (median) | 42.4 | 44.2 | 42.3 |
| White cell count (median cells × 109/L) | 5.2 | 5.4 | 5.1 |
| CD4 (median cells × 106/L) | 467 | 486 | 422 |
| CD4 count > 200 cells × 106/L n(%) | 48 (92%) | 23 (92%) | 25 (93%) |
| Bilirubin (median mg/dL) | 0.6 | 0.6 | 0.5 |
| Albumin (median g/dL) | 4.1 | 4.2 | 4.0 |
| Creatinine (mean ± SD mg/dL) | 0.94 ± 0.2 | 0.96 ± 0.2 | 0.93 ± 0.2 |
| Phosphorus (mean ±± SD mg/dL) | 3.18 ± 0.6 | 3.15 ± 0.5 | 3.2 ± 0.7 |
Baseline HBV DNA was defined as the mean of the log measurements prior to entry and at entry.
Subject Disposition
At time of SMC review, 35 subjects (67%) had completed week 48. Six subjects discontinued the study early (prior to week 48), but none for drug-related toxicity. In addition, the follow-up of 10 subjects was truncated by the early closure, 4 at entry or week 4, and 6 at week 12 or 24. These subjects provided truncated or no DAVG-Xs to the final intent-to-treat (ITT) analysis. The median follow-up was 72 weeks for those on TDF and 78 weeks for those on ADV. No subjects crossed over to the alternative treatment.
Outcome
The primary end point of this study was the noninferiority of TDF relative to ADV. Figure 1 shows the change from baseline in serum HBV DNA log10 levels over time for each treatment arm with pointwise 99.9% confidence intervals. The ADV and TDF arms were very similar over the first several sampling points, but there was a greater decrease in serum HBV DNA levels in the TDF arm over time. The early termination of this study truncated follow-up for subjects entering just prior to the SMC date. This truncation resulted in DAVG values that were very similar between arms. These values could have biased the study to erroneously conclude TDF was not inferior to ADV and could have biased the estimates of mean DAVG48. Thus, we present 3 analyses of DAVG48: ITT, which uses all subjects; modified ITT (mod-ITT), which includes only subjects with at least 2 postbaseline HBV measurements; and “complete cases,” which includes only cases in the mod-ITT analyses who completed at least 36 weeks of therapy prior to the SMC date. The mean DAVG48 estimates for the 2 arms, the difference (ADV-TDF) between these estimates, and the lower one-sided 99.9 CI for this difference are presented in Table 2. In summary, the differences (lower 99.9 CI) between the arms in the DAVG were: ITT, 0.91 (−.512) log10; modified ITT, 1.11 (−.103) log10; and “complete cases,” 1.28 (0.166) log10. A multicovariate analysis was performed to confirm the importance of treatment arm and to evaluate other predictors of DAVG48. Treatment arm (P = .007), entry serum HBV DNA (continuous: P = .001), and entry percent absolute CD4+ T-cell count (P = .005) were jointly associated with DAVG48. There was no evidence of interactions between treatment arm and sex or between treatment arm and race/ethnicity. The mean change from baseline in serum HBV DNA level in the 35 subjects for whom there was week 48 data was −4.03 log10 copies/mL for ADV and −5.74 log10 copies/mL forTDF. Serum HBV DNA was undetectable (<200 copies/mL) at weeks 36 and 48 in 8.6% and 5.7%, respectively, of those subjects receiving ADF and in 11.4% and 20%, respectively, of those subjects receiving TDF. The decrease in HBV DNA by week of therapy is shown in Table 3 by intent-to-treat analysis.
Fig. 1.
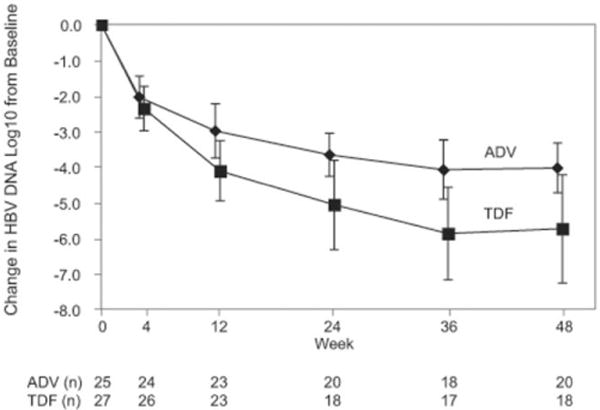
Change in HBV DNA log10 levels from baseline over 48 weeks by treatment arm (mean and 99% CI error bars). The ADV and TDF arms were very similar over the first several sampling points, but there was a greater decrease in HBV DNA in the TDF arm over time.
Table 2. Serum HBV DNA DAVG48* (log10 Copies/mL) in A5127 Study Subjects.
| At time of SMC | N | ADV arm (mean) | TDF arm (mean) | Difference | Lower CI3 |
|---|---|---|---|---|---|
| ITT† | 52 | −3.12 | −4.03 | 0.91 | −0.512 |
| Modified ITT | 47 | −3.35 | −4.46 | 1.11 | −0.103 |
| “Complete cases” | 41 | −3.48 | −4.76 | 1.28 | 0.166 |
| All data | |||||
| ITT | 52 | −3.24 | −4.49 | 1.25 | 0.069 |
| Iodified ITT | 47 | −3.33 | −4.41 | 1.08 | −0.072 |
| “Complete cases” | 41 | −3.58 | −4.78 | 1.20 | 0.124 |
DAVG, time-weighted average change from baseline.
ITT, intent-to-treat, DAVG48 for all 52 enrolled subjects; modified ITT, all subjects with 2 postbaseline serum HBV DNA measurements; “complete cases,” all subjects with 2 postbaseline serum HBV DNA measurements with at least 36 weeks of follow-up.
Lower CI is 1-sided 99.9%.
Table 3. Serum HBV DNA Decrease in A5127 Study Subjects During Therapy With ADV or TDF.
| HBV DNA drop | w 12 | w 24 | w 36 | w 48 | |
|---|---|---|---|---|---|
| ADV (n = 25) | <2 | 3 | 0 | 0 | 1 |
| 2-4 | 17 | 15 | 11 | 8 | |
| >4 | 3 | 5 | 7 | 8 | |
| n/a | 2 | 5 | 7 | 8 | |
| TDF (n = 27) | <2 | 1 | 1 | 1 | 1 |
| 2-4 | 8 | 4 | 1 | 3 | |
| >4 | 14 | 13 | 15 | 14 | |
| n/a | 4 | 9 | 10 | 9 |
w, Week; n/a: number of subjects for whom data was not available at that time.
The percent normalization of ALT level for each week varied by treatment arm. Twelve subjects on ADV and 11 on TDF had abnormal ALT at baseline. At week 48, 25% of subjects on ADV and 36% of subjects on TDF normalized serum ALT, but these numbers were not statistically significant in this small subset. There was no evidence of the deterioration in CPT score of any subject during the study. Of the 18 subjects on ADV who were HBeAg positive at entry, 12 had data at week 48, 1 of whom had converted from HBeAg positive to HBeAg negative without an ALT flare. Of the 23 subjects on TDF who were HBeAg positive at entry, 15 had data at week 48, none of whom had converted from HBeAg positive to HBeAg negative. There was no evidence of loss of HIV control during the study.
Dose Modifications and Flares in ALT
Eleven subjects experienced ALT flares (elevation of serum ALT level to 3 times the baseline ALT level; Table 4), 5 on ADV and 6 on TDF. These flares occurred after varying durations of therapy. None of the flares was associated with decompensation in synthetic function nor were they followed by seroconversion to anti-HBe. Four subjects with flares had their dosage modified, and 4 discontinued treatment, as outlined in Table 4. No subject met the current FDA definition of ALT flare, which is ≥ 10 × ULN and ≥ 2 × baseline.
Table 4. Flares in sSerum ALT levels During Therapy of A5127 Study Subjects.
| Week | Baseline | Max | x baseline | X ULN‡ | ||
|---|---|---|---|---|---|---|
| ADV arm* | 1 | 12 | 68 | 383 | 5.6 | 6.4 |
| 2 | 14 | 95 | 319 | 3.4 | 5.3 | |
| 3 | 24 | 49 | 168 | 3.4 | 4.2 | |
| 4 | 36 | 55 | 228 | 4.1 | 5.7 | |
| 5 | 48 | 23 | 73 | 3.2 | 1.6 | |
| TDF arm† | 1 | 8 | 39 | 621 | 15.9 | 7.9 |
| 2 | 12 | 77 | 278 | 3.6 | 6.0 | |
| 3 | 16 | 120 | 355 | 3.0 | 7.1 | |
| 4 | 51 | 70 | 214 | 3.1 | 4.7 | |
| 5 | 57 | 43 | 134 | 3.1 | 2.1 | |
| 6 | 60 | 36 | 146 | 4.1 | 2.4 |
Of those subjects on ADV: subjects 1, 2, 5 completed 96 weeks of treatment; subject 3 discontinued at week 35 with HCC; subject 4 discontinued at week 57 per MD request.
Of those subjects on TDF: subjects 1, 3, 4, and 6 completed treatment; subject 2 discontinued at week 20 per patient request and subject 5 had multiple missed doses and discontinued at week 38 per patient request.
ULN: upper limit of normal: There was no central laboratory for ALT determinations. ULN were determined by each separate laboratory site with range of 33-79 U/L.
Adverse Events
Two subjects died while in the study: one on ADV at week 48 from hepatocellular carcinoma, and another on TDF at week 57 while hospitalized from cause unknown. Table 5 lists all toxicities (defined as ≥ grade 2 by ACTG criteria), including all signs and symptoms and laboratory toxicities at any time including baseline. Overall, 18 subjects in each arm showed laboratory toxicities. Three subjects on ADV had hypophosphatemia after baseline, 2 grade 2 and 1 transient grade 3. Three subjects on TDF had hypophosphatemia, all grade 2. There was no elevation in serum creatinine ≥ 0.5 mg/dL above baseline, and no change in serum creatinine was seen among those in either arm over the course of the study. Three subjects developed pancreatitis, 2 of whom received concomitant didanosine (ddI), 1 at a dose of 400 mg. Ten subjects received ddI during the study, 5 each on TDF and ADV. Four subjects had grade 1 creatinine elevation, one of whom received ddI 10 years prior to the study and another who was on concurrent ddI and did develop pancreatitis.
Table 5. Adverse Events (≥ Grade 2) During ADV or TDF Treatment of A5127 Study Subjects.
| ADV arm (N = 25) | ADF arm (N = 27) | |||
|---|---|---|---|---|
|
|
|
|||
| Variable | Grade 2 | Grades 3-4 | Grade 2 | Grades 3-4 |
| Any laboratory toxicity | 6 | 12 | 2 | 16 |
| Chemistry | 6 | 2 | 5 | 3 |
| Liver* | 5 | 9 | 4 | 9 |
| Metabolic | 3 | 0 | 5 | 2 |
| Endocrine | 2 | 0 | 0 | 1 |
| Pancreatic† | 1 | 3 | 0 | 8 |
| Hematology | 0 | 0 | 1 | 0 |
| Any signs/symptoms | 2 | 5 | 4 | 3 |
| General body | 2 | 5 | 2 | 0 |
| Respiratory | 1 | 0 | 0 | 0 |
| Gastrointestinal | 1 | 3 | 2 | 0 |
| Skin | 1 | 0 | 0 | 0 |
| Neurological | 0 | 2 | 1 | 3 |
note. ACTG website for grading criteria: http://rcc.tech-res-intl.com/DAIDS%20RCC%20Forms/TB_ToxicityTables_Adult_TRP_v01a.pdf
Total bilirubin elevation after baseline was reported in one subject on TDF (grade 2) and in 5 subjects on ADV (1 grade 2, 3 grade 3, and 1 grade 4).
Postentry elevations in serum amylase or lipase were reported in 6 subjects on TDF and 3 on ADV. However, only four episodes of chemical pancreatitis were reported in three subjects. One subject was receiving 3TC, zidovudine and nevirapine; one ddI, d4T, and 3TC; and another received no ddI.
All Visits
The median follow-up time of all subject visits was 91 weeks for ADV and 81 weeks for TDF. No additional ALT flares, grade 4 adverse events, or deaths were reported for the additional visits. There were additional serum HBV DNA evaluations, primarily after week 48. However, the analysis of DAVG48 was essentially unchanged from the SMC analysis, as shown in Table 2. At the end of the study, letters were sent to sites, patients, and treating physicians recommending continued therapy for all subjects. Following the study, prescriptions were written to maintain patients on therapy, and treatment assignment was provided. All patients were continued on therapy for both HIV and HBV. No follow-up was available through the study.
Discussion
ACTG protocol A5127 was a prospective randomized, controlled study of TDF versus ADV for HBV infection and demonstrated the value of each therapeutic agent in the treatment of chronic HBV in the setting of HIV/HBV coinfection. Treatment with either ADV or TDF resulted in clinically significant reductions in serum HBV DNA levels in coinfected patients. TDF was not inferior to ADV according to the primary study end point, DAVG48. Suppression of HBV DNA level was accompanied by improvement in serum ALT, and both drugs were well tolerated. On-treatment hepatitis flares occurred but were well tolerated and not associated with clinical decompensation; however, all but 1 subject had well-compensated liver disease at entry. Most subjects were HBeAg+, genotype A, and resistant to lamivudine. Of note, ALT flares were not accompanied by seroconversion to anti-HBe, which occurs in 12% of patients without HIV on ADV after 48 weeks of therapy.8
Adefovir dipivoxil, a nucleotide analogue of adenosine monophosphate, and tenofovir disoproxil suppress viral replication through inhibition of HBV DNA polymerase and chain termination. Both ADV and TDF have been shown to be active against HBV in vivo and in vitro.16 Pivotal studies of monoinfected individuals with HBeAg-positive and -negative disease and a pilot study of HBV/HIV-coinfected individuals showed that ADV suppressed HBV DNA,7,8,14 resulting in improved liver histology, including reduced fibrosis, enhanced HBeAg seroconversion, ALT normalization, and clinical improvement. Case studies have shown that TDF has anti-HBV activity in both HBV-monoinfected and -coinfected subjects11-13 and can rescue HBV-infected patients for whom treatment with lamivudine and ADV has failed.17 In a recent retrospective study of 65 HIV/HBV-coinfected patients treated with 300 mg of TDF per day, 80% were also HBeAg positive at entry and serum HBV DNA decreased by 4.56 log10 copies/mL on TDF.18
The goals of HBV therapy are to stop or reverse progression of liver inflammation and fibrosis through sustained suppression of HBV replication. The use of 3TC as a component of ART is frequent and 94% of our enrolled subjects were 3TC experienced. However, suppression of serum HBV DNA levels is not sustained in the majority of HIV patients treated with 3TC. HBV strains containing mutations in subdomain B and/or subdomain C (YMDD region) of the gene encoding HBV DNA polymerase become detectable with increasing time on 3TC, with up to 90% of HIV/HBV-coinfected subjects exhibiting mutations after 4 years of 3TC monotherapy.6 The HBV YMDD mutant that is resistant to 3TC is sensitive to ADV and TDF both in vivo and in vitro.9,16,19
There was no evidence of differential toxicity between the 2 arms. Renal toxicity did not occur in those in either arm, and grade 3 hypophosphatemia was transient. Early treatment discontinuation because of intolerance or other causes was no more frequent than the 5%-10% the design anticipated and was equal across the study arms in both frequency and cause. The occurrence of pancreatitis in patients receiving concomitant therapy with didanosine (ddI) underscores the recommendation that this drug not be used in combination with TDF or ADV.20
The early termination of this study as a result of an interim monitoring review resulted in a number of subjects having 24 or fewer weeks of follow-up for DAVG48 based on the SMC date. The truncated DAVG48 values are very similar between the arms because the divergence of the HBV DNA levels seen in Figure 1 did not become established until week 24. Three analyses of the primary end point, noninferiority, are presented, all of which confirmed that TDF is not inferior to ADV. In fact, the DAVG48 for TDF was more negative (i.e., was more favorable in terms of HBV control) than the DAVG48 for ADV on the log10 scale. Using a strict 1-sided Haybittle-Peto lower confidence interval and considering several analyses that did and did not incorporate subjects with very short follow-up, the lower bound of the difference between the DAVG48 on ADV and TDF was clearly and conclusively more positive than the −1.0 log10 in any of the 3 analyses. Whereas TDF is clearly not inferior to ADV, any more favorable interpretation of the results—that TDF is superior—must be made with caution, as the study was designed and powered to demonstrate noninferiority, it was terminated early, and follow-up was truncated. Superiority can only be demonstrated using the confidence interval of the original design, which employed a Haybittle-Peto boundary (1-sided 0.001 level) to protect the experiment-wide type I error rate, requiring 99.9% confidence intervals to be used for all evaluations of serum HBV DNA results.
Both therapies were associated with improvement in ALT in those with abnormal ALT at baseline, although at week 48 normalization was seen in more subjects on TDF than in those on ADV. Serum aminotransferase was normal in 57% of subjects prior to therapy, although they had elevated HBV DNA, suggesting that in this population ALT may not be a good surrogate for inflammation.
There was no evidence of loss of antiretroviral efficacy over the study period. However, subjects had relatively well-preserved immune status, with low or complete suppression of HIV replication. The proportion of subjects with undetectable serum HBV-DNA at 48 weeks was relatively low in both treatment arms. And although most were HBeAg positive, only one seroconverted to anti-HBe. These findings contrast to seroconversion rates of HIV-negative chronic hepatitis B patients.8,21 The choice of anti-HBV therapy for patients coinfected with HIV is often based on the need for concomitant therapy for the HIV infection.22 TDF is recommended as a first-line option as a component of ART.10,23 Thus, our data suggest that a TDF-containing antiretroviral regimen is preferable for HIV/HBV-coinfected patients, when treatment for HIV infection is indicated. Conversely agents with activity against both HIV and HBV should likely be avoided when only therapy for HBV is indicated because of concerns over the development of HIV drug resistance.22 In this setting, entecavir, which has no activity against HIV, should be considered for treating chronic hepatitis B when antiretroviral therapy is not needed. In HBeAg+ patients, pegylated interferon may be an equally acceptable option.24 ADV may also be used for initial HBV treatment in coinfected subjects. There is a theoretical risk for developing mutation K65R in HIV following exposure to ADV monotherapy, but this has not been demonstrated.25 In summary, further studies of initial treatment strategies are warranted in coinfected subjects without prior 3TC experience to address these unresolved questions.
Acknowledgments
Supported in part by the Adult AIDS Clinical Trials Group (ACTG) funded by the National Institute of Allergy and Infectious Diseases (AI38855); virology support funding by the NIH/NIAID and the Adult ACTG Central Group (grants U01AI38858 and P30 AI27767); the Birmingham VA Medical Center, UAB CFAR core clinic and laboratory facilities (to V.A.J.); and NIDDK UCSF Liver Center (P30 DK 26743; to M.G.P.).
Abbreviations
- HBV
hepatitis B virus
- ACTG
Adult AIDS Clinical Trials Group
- ADV
adefovir dipivoxil
- TDF
tenofovir disoproxil fumarate
- ALT
alanine aminotransferase
- CPT
Child-Pugh-Turcotte
- ITT
intent-to-treat
- DAVG
time-weighted average
Appendix
Other members of the ACTG 5127 team were Marilyn Foutes, B.S., M.T., and Lara A. Hosey, M.A. (Social & Scientific Systems); Heather L. Sprenger, M.S., and Lynne Jones, B.S. (Frontier Science and Technology Research Foundation); Ana I. Martinez, Ph.D. (Pharmaceutical Affairs Branch, NIH, NIAID, DAIDS); Patricia Lopez, R.N. (L.A. County—USC Medical Center); Diane Daria, B.S.N., A.C.R.N. (University of Cincinnati Medical Center); Philip W. Anthony and Melvin Littles (CAB Member); Graeme Currie, M.D., and William Chang (Gilead Sciences).
Supported in part by the General Clinical Research Center Units funded by the National Center for Research Resources as noted. We acknowledge the following virology laboratory technical support—for VSL Lab 54 (VAJ): J. Darren Hazelwood and Allison B Miller for HBV genotyping (UAB); Elena Bleumers and Ciera Khuu for HBV DNA testing (MGP).
The following persons and institutions participated in the conduct of this trial: Diane Daria, R.N., B.S.N., A.C.R.N., and Linda Hinds, R.N. (University of Cincinnati A2401); Charles Bradley Hare, M.D., and Joann Volinski, R.N. (San Francisco General Hospital A0801); Elizabeth Race, M.D., and Mamta Jain, M.D. (University of Texas, Southwestern Medical Center A3751); Susan Hulse, P.A. and Nancy Fitch, N.P. (University of California, Davis Medical Center A3851; grant 3U01AI38858-09S1); Robert Murphy, M.D. (Northwestern University A2701; grant AI25915); Sondra Middleton, P.A.-C., and Donna Mildvan, M.D. (Beth Israel Medical Center A2851; grant AI46370); Marshall Glesby, M.D., Ph.D., and Valery Hughes, F.N.P, (Cornell Clinical Trials Unit A7803; grants AI46386 and RR00047); Jeffrey Schouten, M.D., Shelia Dunaway, M.D. (University of Washington, Seattle WA); Kim Whitely, R.N., and Ann Conrad, R.N. (MetroHealth Medical Center A2503); William A. O'Brien, M.D., M.S., and Michael Reardon, R.N. (University of Texas, Galveston A6301; grant AI32782); Ilene Wiggins, R.N. (Johns Hopkins University A0201; grants AI-27668 and RR-00052); Karen Cavanagh, R.N., and Helene Lupatkin, M.D. (NYU/Bellevue A0401; grants M01-RR00096 and 5 UO1 A1027665-18); Alex Nesbit, P.A., and Jackie Kaufman, P.A. (University of North Carolina A3201; grants AI50410, AI25868, and RR00046); Beverly Putnam, R.N., N.P., and Nancy Madinger, M.D. (University of Colorado Health Sciences Center, Denver A6101; grants AI32770 and RR00051).
Footnotes
Potential conflict of interest: Dr. Peters recieved grants from Gilead. Dr. Andersen is a consultant for Tibotec. Dr. Pollard advises for Idenix. He is a consultant for Pfizer. He is on the Speakers' Bureau for Bristol Myers Squibb and Gilead. He is also a consultant for and is on the Speakers' Bureau for Boehringer Ingelheim. Dr. Rooney is an employee of and owns stocks in Gilead. He also owns stock in GlaxoSmithKline. Dr. Sherman is on the Speakers' Bureau and received grants from Roche and Schering. He is a consultant for and received grants from SciClone. He also advises for BMS. Dr. Swindells recieved grants from Bristol Myers, Novartis and Pfizer.
References
- 1.Hadler SC, Judson FN, O'Malley PM, Altman NL, Penley K, Buchbinder S, et al. Outcome of hepatitis B virus infection in homosexual men and its relation to prior human immunodeficiency virus infection. J Infect Dis. 1991;163:454–459. doi: 10.1093/infdis/163.3.454. [DOI] [PubMed] [Google Scholar]
- 2.Solomon L, Flynn C, Muck K, Vertefeuille J. Prevalence of HIV, syphilis, hepatitis B, and hepatitis C among entrants to Maryland correctional facilities. J Urban Health. 2004;81:25–37. doi: 10.1093/jurban/jth085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goedert JJ, Brown DL, Hoots K, Sherman KE. Human immunodeficiency and hepatitis virus infections and their associated conditions and treatments among people with haemophilia. Haemophilia. 2004;10(Suppl 4):205–210. doi: 10.1111/j.1365-2516.2004.00997.x. [DOI] [PubMed] [Google Scholar]
- 4.Thio CL. Hepatitis B in the human immunodeficiency virus-infected patient: epidemiology, natural history, and treatment. Semin Liver Dis. 2003;23:125–136. doi: 10.1055/s-2003-39951. [DOI] [PubMed] [Google Scholar]
- 5.Uneke CJ, Ogbu O, Inyama PU, Anyanwu GI, Njoku MO, Idoko JH. Prevalence of hepatitis-B surface antigen among blood donors and human immunodeficiency virus-infected patients in Jos, Nigeria. Mem Inst Oswaldo Cruz. 2005;100:13–16. doi: 10.1590/s0074-02762005000100002. [DOI] [PubMed] [Google Scholar]
- 6.Benhamou Y, Bochet M, Thibault V, Di MV, Caumes E, Bricaire F, et al. Long-term incidence of hepatitis B virus resistance to lamivudine in human immunodeficiency virus-infected patients. Hepatology. 1999;30:1302–1306. doi: 10.1002/hep.510300525. [DOI] [PubMed] [Google Scholar]
- 7.Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, Chang TT, Kitis G, Rizzetto M, et al. Adefovir dipivoxil for the treatment of hepatitis B e antigen-negative chronic hepatitis B. N Engl J Med. 2003;348:800–807. doi: 10.1056/NEJMoa021812. [DOI] [PubMed] [Google Scholar]
- 8.Marcellin P, Chang TT, Lim SG, Tong MJ, Sievert W, Shiffman ML, et al. Adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. N Engl J Med. 2003;348:808–816. doi: 10.1056/NEJMoa020681. [DOI] [PubMed] [Google Scholar]
- 9.Peters MG, Hann Hw H, Martin P, Heathcote EJ, Buggisch P, Rubin R, et al. Adefovir dipivoxil alone or in combination with lamivudine in patients with lamivudine-resistant chronic hepatitis B. Gastroenterology. 2004;126:91–101. doi: 10.1053/j.gastro.2003.10.051. [DOI] [PubMed] [Google Scholar]
- 10.Yeni PG, Hammer SM, Hirsch MS, Saag MS, Schechter M, Carpenter CC, et al. Treatment for adult HIV infection: 2004 recommendations of the International AIDS Society-USA Panel. JAMA. 2004;292:251–265. doi: 10.1001/jama.292.2.251. [DOI] [PubMed] [Google Scholar]
- 11.Lada O, Benhamou Y, Cahour A, Katlama C, Poynard T, Thibault V. In vitro susceptibility of lamivudine-resistant hepatitis B virus to adefovir and tenofovir. Antivir Ther. 2004;9:353–363. [PubMed] [Google Scholar]
- 12.van Bommel F, Wunsche T, Mauss S, Reinke P, Bergk A, Schurmann D, et al. Comparison of adefovir and tenofovir in the treatment of lamivudine-resistant hepatitis B virus infection. Hepatology. 2004;40:1421–1425. doi: 10.1002/hep.20464. [DOI] [PubMed] [Google Scholar]
- 13.Ristig MB, Crippin J, Aberg JA, Powderly WG, Lisker-Melman M, Kessels L, et al. Tenofovir disoproxil fumarate therapy for chronic hepatitis B in human immunodeficiency virus/hepatitis B virus-coinfected individuals for whom interferon-alpha and lamivudine therapy have failed. J Infect Dis. 2002;186:1844–1847. doi: 10.1086/345770. [DOI] [PubMed] [Google Scholar]
- 14.Benhamou Y, Bochet M, Thibault V, Calvez V, Fievet MH, Vig P, et al. Safety and efficacy of adefovir dipivoxil in patients co-infected with HIV-1 and lamivudine-resistant hepatitis B virus: an open-label pilot study. Lancet. 2001;358:718–723. doi: 10.1016/s0140-6736(01)05840-8. [DOI] [PubMed] [Google Scholar]
- 15.Jennison C, Turnbull BW. Group Sequential Methods with Applications to Clinical Trials. Boca Raton, FL: Chapman & Hall/CRC; 2000. [Google Scholar]
- 16.Ying C, De Clercq E, Nicholson W, Furman P, Neyts J. Inhibition of the replication of the DNA polymerase M550V mutation variant of human hepatitis B virus by adefovir, tenofovir, L-FMAU, DAPD, penciclovir and lobucavir. J Viral Hepat. 2000;7(2):161–165. doi: 10.1046/j.1365-2893.2000.00210.x. [DOI] [PubMed] [Google Scholar]
- 17.van Bommel F, Zollner B, Sarrazin C, Spengler U, Huppe D, Moller B, et al. Tenofovir for patients with lamivudine-resistant hepatitis B virus (HBV) infection and high HBV DNA level during adefovir therapy. Hepatology. 2006;44:318–325. doi: 10.1002/hep.21253. [DOI] [PubMed] [Google Scholar]
- 18.Benhamou Y, Fleury H, Trimoulet P, Pellegrin I, Urbinelli R, Katlama C, et al. Anti-hepatitis B virus efficacy of tenofovir disoproxil fumarate in HIV-infected patients. Hepatology. 2006;43:548–555. doi: 10.1002/hep.21055. [DOI] [PubMed] [Google Scholar]
- 19.Xiong X, Flores C, Yang H, Toole JJ, Gibbs CS. Mutations in hepatitis B DNA polymerase associated with resistance to lamivudine do not confer resistance to adefovir in vitro. Hepatology. 1998;28:1669–1673. doi: 10.1002/hep.510280629. see comments. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Benayas T, Rendon AL, Rodriguez-Novoa S, Barrios A, Maida I, Blanco F, et al. Higher risk of hyperglycemia in HIV-infected patients treated with didanosine plus tenofovir. AIDS Res Hum Retroviruses. 2006;22:333–337. doi: 10.1089/aid.2006.22.333. [DOI] [PubMed] [Google Scholar]
- 21.Lau GK, Piratvisuth T, Luo KX, Marcellin P, Thongsawat S, Cooksley G, et al. Peginterferon alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N Engl J Med. 2005;352:2682–2695. doi: 10.1056/NEJMoa043470. [DOI] [PubMed] [Google Scholar]
- 22.Alberti A, Clumeck N, Collins S, Gerlich W, Lundgren J, Palu G, et al. Short statement of the first European consensus conference on the treatment of chronic hepatitis B and C in HIV co-infected patients. J Hepatol. 2005;42:615–624. doi: 10.1016/j.jhep.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Department of Health and Human Services. Guidelines for the use of antiretroviral agents in HIV-1-Infected adults and adolescents. Washington, DC: Department of Health and Human Services; 2005. Available at: http://AIDSinfo.nih.gov/guidelines/adult/AA_040705.pdf.4-7-2005. [Google Scholar]
- 24.Soriano V, Puoti M, Bonacini M, Brook G, Cargnel A, Rockstroh J, et al. Care of patients with chronic hepatitis B and HIV co-infection: recommendations from an HIV-HBV International Panel. AIDS. 2005;19:221–240. [PubMed] [Google Scholar]
- 25.Sheldon JA, Corral A, Rodes B, Mauss S, Rockstroh J, Berger F, et al. Risk of selecting K65R in antiretroviral-naive HIV-infected individuals with chronic hepatitis B treated with adefovir. AIDS. 2005;19:2036–2038. doi: 10.1097/01.aids.0000189563.79976.05. [DOI] [PubMed] [Google Scholar]