

Abstract
Artificial antigen presenting cells (aAPC), which deliver stimulatory signals to cytotoxic lymphocytes, are a powerful tool for both adoptive and active immunotherapy. Thus far, aAPC have been synthesized by coupling T cell activating proteins such as CD3 or MHC-peptide to micron-sized beads. Nanoscale platforms have different trafficking and biophysical interaction properties and may allow development of new immunotherapeutic strategies. We therefore manufactured aAPC based on two types of nanoscale particle platforms: biocompatible iron-dextran paramagnetic particles (50–100 nm in diameter) and avidin-coated quantum dot nanocrystals, (~30 nm). Nanoscale aAPC induced antigen-specific T cell proliferation from mouse splenocytes and human peripheral blood T cells. When injected in vivo, both iron-dextran particles and quantum dot nanocrystals enhanced tumor rejection in a subcutaneous mouse melanoma model. This is the first description of nanoscale aAPC that induce antigen-specific T cell proliferation in vitro and lead to effective T cell stimulation and inhibition of tumor growth in vivo.
Keywords: Nanoparticle, Artificial Antigen Presenting Cell, Immunotherapy, T cell
Background
The induction of specific cytotoxic T lymphocyte (CTL) responses is a powerful therapy for pathogens and tumors. Specific CTL populations expand several logs to produce robust responses and generate long-term memory that can prevent recurrence of disease[1]. CTL can be directly activated in vivo, as in some vaccines [2], or generated in vitro and adoptively transferred into a patient [3–5].
We have previously developed a cell-sized T cell expansion platform by coupling proteins that deliver two necessary and sufficient T cell activation signals to 4.5 µm diameter (“microscale”) beads [6,7]. Signals present on APC that are required for T cell activation include signal 1, a cognate antigenic peptide presented in the context of major histocompatibility complex (MHC) that binds the TCR [8], and signal 2, a group of co-stimulatory receptors that modulate T cell response. In our system, signal 1 is delivered by a chimeric MHC-immunoglobulin dimer (MHC-Ig) loaded with a specific peptide, and signal 2 is either B7.1 (the natural ligand for the T cell receptor CD28) or an activating antibody against CD28. Both proteins can be directly chemically coupled to the surface of microscale beads to create artificial antigen presenting cells (aAPC).
The in vivo delivery and biodistribution of bead-based therapeutics is determined primarily by particle size [9–11]. Microscale particles have limited lymphatic drainage from their injection site and are preferentially cleared by and targeted to certain phagocytic subsets[12–14]. Nanoparticle platforms have different trafficking properties which would open new immunotherapeutic delivery strategies, but the appropriateness of nanoparticles for T cell activation has been questioned.
Studies have suggested that only beads larger than 2 microns in diameter are able to induce T cell proliferation [15,16]. As a result, nanoparticles have traditionally been developed for antigen or drug delivery [17,18], or to study biophysical aspects of TCR-MHC binding [19,20]. When T cell activation was examined directly, Steenblock et al.[21] demonstrated that polymer-based nanoparticles were much less efficient than microbeads in inducing short-term functional responses, with no reported proliferation.
Here, we present nanoscale, particle-based T cell activation platforms based on either paramagnetic iron-oxide particles 50–100 nm in diameter or quantum dot nanocrystals approximately 30 nm in diameter. We show these platforms induce antigen specific T cell proliferation and functional responses from murine and human T cells in vitro. Finally we show that nano-aAPC can prime CTL to attenuate tumor growth in vivo in a mouse melanoma model.
Methods
Mice and reagents
2C TCR transgenic mice were maintained as heterozygotes by breeding on a C57/BL6 background. pMEL TCR/Thy1a Rag−/− transgenic mice were a gift from Nicholas Restifo (National Institutes of Health, Bethesda, MD) and maintained as homozygotes. C57BL/6j and Nu/J mice were purchased from Jackson Laboratories (Bar Harbor, ME). All mice were maintained according to Johns Hopkins University’s Institutional Review Board. Fluorescently labeled monoclonal antibodies were purchased from BioLegend (San Diego, CA).
Preparation of MHC-Ig Dimers
Soluble MHC-Ig dimers, Kb-Ig and Db-Ig, were prepared and loaded with peptide as described[50]. Briefly, Kb-Ig molecules were loaded with peptide by stripping at alkaline condition (pH 11.5), and then refolded in the presence of 50 fold excess peptide. Db-Ig molecules were stripped under mildly acidic conditions (pH 6.5) and refolded in the presence of 50 fold molar excess peptide and 2-fold molar excess of human β2-microglobulin. Human A2-Ig was passively loaded in the presence of excess M1 peptide [51]. Peptides SIY (SIYRYYGL, synthetic), SIIN (SIINFEKL, derived from ovalbumin protein), GP100 (KVPRNQDWL, from melanocyte GP100 protein) ASN (ASNENMETH, from influenza A nucleoprotein), and M1 (GILGFVFTL, from influenza A M1 protein) were purchased from Genscript (Piscataway, NJ). Protein concentration was determined after labeling by size exclusion high performance liquid chromatography (HPLC).
Nano-aAPC Synthesis
Nanoscale iron-dextran aAPC were manufactured in one of two ways. 2 µM biotinylated MHC-Ig dimer and an equimolar concentration of biotinylated anti-CD28 antibody were incubated with 100 µL of anti-biotin Miltenyi Microparticles (Miltenyi Biotec) for at least 1 hour with gentle agitation at 4°C. Unbound protein was washed using a MS magnetic enrichment column (Miltenyi Biotec). Particle concentration was measured by absorbance at 405 nm using a Beckman Coulter AD340 plate reader. Alternatively, MHC-Ig dimer and B7.1-Ig were directly chemically coupled to biodegradable particles (Miltenyi Biotec). Total protein content was assessed by Bradford assay. Unless otherwise stated, “iron-dextran aAPC” refers to particles directly chemically coupled to MHC and B7.1, rather than anti-biotin coupling.
Nanoscale quantum dot aAPC were manufactured by incubating 5 µM biotinylated MHC-Ig dimer and an equimolar concentration of biotinylated anti-CD28 antibody with 100 µL of 1 µM streptavidin coated quantum dots (Life Technologies) for 2 hours at at 4°C. Quantum dots were washed and concentrated using a Sartorius Vivaspin membrane with a 300,000 molecular weight cutoff. Quantum dot concentration was measured by absorbance at 405 nm using a Beckman Coulter AD340 plate reader.
Micro-aAPC Synthesis
Micro-aAPCs were fabricated as described previously[7] by direct chemical coupling of protein to 4.5 µm Dynal Magnetic Microbeads (Life Technologies, Carlsbad, CA). For the initial coupling step, 25 µg anti-biotin antibody (Sigma, St. Louis, MO) was added to 100 million Microbeads in 0.1 M sodium borate buffer. After washing in a magnetic column, biotin labeled MHC-Ig and CD28 were added in equimolar amounts to form aAPC.
In Vitro Cell Expansion
For murine cell culture, cells were obtained from homogenized mouse spleens followed by hypotonic lysis of RBC. Cytotoxic lymphocytes were isolated using a CD8 no-touch isolation kit and magnetic enrichment column from Miltenyi Biotec (Cologne, Germany) and if necessary labeled with carboxyfluorescein succinimidyl ester (CFSE) for 15 minutes at 37°C, then washed extensively. One million CD8+ T cells and particles at the indicated dosages were mixed and cultured in 96 well round bottom plates for 4–7 days in complete RPMI media supplemented with T cell factor (TF), a cytokine cocktail of conditioned media harvested from stimulated human PBMC [6]. CFSE fluorescence was measured on Day 4 using a BD FacsCalibur flow cytometer and analyzed in FlowJo (TreeStar). CFSE is diluted with each round of T cell division, and division thus manifests as a one half-fold decrease in CFSE fluorescence.
For human cell culture, PBMCs from healthy HLA*0201 positive donors were isolated by Ficoll-Paque PLUS gradient centrifugation following the manufacturer’s protocol (GE Healthcare). CD8+ T cells were further purified from fresh PBMC using the CD8+ T cell negative selection kit (Miltenyi Biotec). The purity of CD8+ T cells was higher than 95%, as determined by flow cytometry. Three million CD8+ T cells and particles at the indicated dosages were mixed and cultured in 96-well round bottom plates for up to 14 days in complete RPMI media supplemented with TF. On day 7 after stimulation, T cells were harvested, counted and replated at the same T cell:nano-aAPC density. Antigen specificity was determined using HLA-M1-specific, A*0201 PE or APC tetramers (Beckman Coulter) according to manufacturer’s protocol.
Near-Infrared Imaging
Anti-biotin coated micro- and nano-particles were labeled with IRDye 680 RD or 800CW Protein labeling kits from LI-COR Biosciences (Lincoln, Nebraska). Beads were washed in a magnetic column and biotin labeled MHC-Ig and anti-CD28 were added to form aAPC. pMEL T cells were labeled with CellVue NIR 815 labeling kits from LI-COR Biosciences according manufacturer’s instructions.
Nu/J mice were injected with 2 ×105B16 melanoma cells on the right flank. Four days later, 5 ×105 micro-aAPC and a fluorescence intensity equivalent amount of nano-aAPC were injected into the tail vein, ipsilateral flank, or hindlimb, as indicated. T cells were injected into tail vein. Biodistribution of injected aAPC was visualized with LI-COR Pearl Impulse imaging system (Johns Hopkins Molecular Imaging Center, Baltimore, MD). Area of distribution was calculated using thresholding of the NIR channel in the ImageJ application (NIH, Bethesda, MD).
Effect of Nano-aAPC on Subcutaneous Tumor Growth In Vivo
For QD aAPC experiment, 2 ×106 naive CD8+ pMEL T cells were adoptively transferred into 8 week old C57BL/6 male mice by tail vein injection, except for control mice which received no T cells or aAPC treatment. The same day, B16 melanoma cells (2 ×105) were injected subcutaneously into the right flank. The following day, 5 mice per group were treated with either 20 µL cognate QD aAPC, 20 µL non-cognate QD aAPC, or 20 µL PBS. Mice were treated days 3, 4, and 5 with 30,000 units intraperitoneal IL-2. Tumor growth was monitored at 2 day intervals, using digital calipers, until tumor size was ~200 mm2 at which point animals were euthanized.
For iron-dextran aAPC experiment, 2 ×106 naive CD8+ pMEL T cells were adoptively transferred as before. Four days later, mice in the treatment group received 25 µL cognate HD nano-aAPC either intravenously, iv or subcutaneously, sc, with eight mice per group. Three days later, aAPC were injected either sc or iv. B16 melanoma cells (2 ×105) were injected sc four days later, and a second injection of aAPC were given four days after tumor, either iv or sc on the ipsilateral flank. Tumor tracking and animal euthanasia proceeded as above.
Tumor growth for each mouse was summarized as Area Under Curve calculated using trapezoidal approximation. Statistical analysis was performed in GraphPad Prism.
Results
Iron-Dextran Nano-aAPC Induce Antigen Specific T Cell Expansion
Nanosized iron-oxide core, dextran coated particles produced by Miltenyi Biotec were selected as a nanoscale particle platform due to their extensive characterization and biocompatibility6. To produce nanoscale aAPC, soluble dimeric MHC-Ig loaded with an appropriate peptide (signal 1) and chimeric B7.1-Ig fusion protein (signal 2) were covalently coupled in a 1:1 ratio to the particle surface (Figure 1a). Alternatively, particles were manufactured by coupling biotinylated MHC-Ig and biotinylated anti-CD28 to an anti-biotin coated iron-dextran particle (Figure 1b).
Figure 1.

Synthesis and Characterization of Iron-Dextran Nano-aAPC . Nano-aAPC were synthesized in one of two ways: (A) Direct chemical coupling of soluble MHC-Ig dimer (signal 1) and B7.1-Ig (signal 2) in a 1:1 molar ratio to the surface of a paramagnetic iron-oxide, dextran-coated particle. (B) Binding of biotinylated MHC-Ig dimer (signal 1) and biotinylated anti-CD28 (signal 2) in a 1:1 molar ratio to anti-biotin coated particles.
(C) Nanoparticle tracking analysis confirms that nano-aAPC are a monodisperse mixture of particles with a mean diameter of 50–100 nm suspended at a concentration of 8.3 nM.
Iron-dextran aAPC were confirmed to be monodisperse with an average diameter of 50–100 nM in diameter using Nanoparticle Tracking Analysis (NTA, Figure 1c). Particles were suspended at a concentration of 8.3 nM (5×1012 particles/mL), and all subsequent volumes refer to particles at this concentration. By titrating the amount of protein present during the coupling reaction, we synthesized particles presenting a high density, HD (65 µg protein/mL of particles), or low density, LD (16 µg protein/mL of particles), of protein as measured by Bradford Assay.
To evaluate aAPC-induced T cell expansion, we utilized two TCR transgenic mouse models: 2C mice, whose T cells recognize the SIY peptide presented in the context of MHC Class I H2-Kb, and pMEL mice, whose T cells recognize a peptide derived from melanoma differentiation antigen GP100 presented in the context of MHC Class I H2-Db,. Four types of anti-biotin coupled iron-dextran particles were manufactured, presenting either Kb or Db loaded respectively with either the cognate peptide described above or a non-cognate peptide (SIIN for Kb, ASN for Db). T cells were incubated with particles and proliferation was evaluated seven days later. Particle based expansion was antigen-specific, as 2C cells only proliferated in the presence of Kb-SIY particles, and pMEL cells only proliferated in the presence of Db-GP100 particles (Figure 2A). Nano-aAPC mediated expansion is therefore antigen specific. Furthermore, both signal 1 and signal 2 were required for optimal expansion, and anti-biotin particles carrying either MHC-Ig or CD28 alone were not as effective at inducing robust T cell proliferation (Figure 2B).
Figure 2.

Nano aAPC Induced Proliferation is Antigen-Specific and Dose-Dependent
(A) Antigen specific nano-aAPC induce proliferation. T cells were counted seven days after stimulation with anti-biotin coated nano-aAPC to calculate fold expansion from day 0. TCR transgenic 2C (grey) and pMEL (white) T cells proliferated only when incubated with nanoparticles bearing cognate MHC/peptide (22-fold and 16-fold, respectively), and not in the presence of naoparticles bearing either non-cognate peptide or non-cognate MHC (<3-fold).
(B) Addition of both signal 1 and signal 2 leads to optimal T cell expansion. At a dose of 10 µL particles per 1*106 T cells, only anti-biotin particles bearing both MHC-Ig and anti-CD28 induced robust T cell expansion.
Proliferation of CD8+ CTL induced by Low Density, LD (16 µg protein/mL particles), and High Density, HD (65 µg protein/mL), particles. Results are representative of three experiments.
(C) Equivalent doses of HD and LD particles were used to stimulate pMEL T cells. Proliferation was measured by dilution of CFSE three days after stimulation. Decreased fluorescence indicates increased proliferation. Equivalent volumes of HD particles induce greater proliferation than LD particles, with 0.5 uL LD particles inducing almost no expansion.
(D) Fold expansion on day 7 of dose equivalent samples shows a similar pattern. Proliferation is dose-dependent and 2–4 fold greater for HD particles compared to an equivalent dose of LD particles (21-fold compared to 7-fold at 5 µL).
(E) Day 3 CFSE dilution of CD8+ CTL induced by LD and HD particles at equivalent protein concentrations, with approximately 5.5-fold more LD than HD at a given dose. When particle doses are normalized to equivalent protein concentrations, particles induce similar amounts of CFSE dilution.
(F) Fold expansion on day 7 demonstrates equivalent expansion for HD and LD particles at an equivalent protein dose (17-fold at 3.5 µL of HD and 20 µL of LD). A threshold of about 0.5 uL LD particles or 0.08 uL HD particles is required to induce detectable expansion.
Soluble MHC-Ig and anti-CD28 are known to mediate weak T cell expansion in vitro 25,26. To demonstrate that coupling of Signal 1 and 2 to solid support enhanced activation [27], nano-aAPC mediated T cell expansion was compared to equivalent amounts soluble MHC-Ig and anti-CD28. 10 ng of protein coupled to nano-aAPC induced greater T cell expansion than several orders of magnitude more soluble protein (Figure S1). Furthermore, soluble protein induced maximal expansion at an intermediate dose of 100 ng, whereas nano-aAPC mediated expansion was dose-dependent, reaching up to 30-fold expansion at a dose of 10 µg protein.
Both the amount [28,29] and density [30,31] of antigen presented by APC influence downstream T cell behavior such as proliferation and cell death, and may thus be important parameters for aAPC stimulation. HD and LD particles were used to evaluate the effect of antigen density on T cell expansion and both sets of particles were titrated to evaluate the effect of antigen dose. Proliferation was characterized three days after stimulation by dilution of the vital dye carboxyfluorescein succinimidyl ester (CFSE). Seven days after stimulation, T cells were counted to characterize the overall balance between proliferation and death.
Both HD and LD particles were able to induce pMEL T cell proliferation in a dose-dependent fashion (Figure 2C). As measured by CFSE dilution, HD particles induced proliferation in 79%, 98%, and 99% of cells for 0.5, 5, and 20 µLs of particles, respectively, per 1 million cells, while identical amounts of LD particles induced proliferation in 4%, 40%, and 93% of cells. By day 7, HD and LD particles had induced an overall expansion of T cells on the order of 5–30 fold, with a minimum threshold of approximately 5 µL of LD particles and less than 0.5 µL of HD particles required to induce expansion (Figure 2D). Both CFSE proliferation and cell counts demonstrated that at any given quantity of particles, HD nano-aAPC induced greater expansion than LD. For example, at 5 µL of particles, HD particles induced 21-fold expansion, while LD particles induced only 7-fold expansion.
To assess whether the increased amount of protein on HD particles fully accounted for the proliferation advantage, LD and HD particles were incubated with T cells at equal protein concentrations (that is, approximately 5-fold more LD particles at a given concentration of HD). Once aAPC were normalized for protein concentration, HD and LD particles induced similar expansion as measured by CFSE dilution on Day 3 (Figure 2E) or overall expansion on Day 7 (Figure 2F). For example, 20 uL of LD particles or 3.5 uL of HD particles both induced proliferation in 94% of cells by Day 3, and approximately 17-fold expansion after 7 days of growth. Thus, at the antigen doses and densities evaluated, expansion was driven by total protein presented on aAPC, and not particle dose or protein density.
Generating sufficient numbers of antigenic-specific T cells is a critical goal of immunotherapy. However, CTL can become anergic or even suppressive under certain stimulation conditions[32], so expanded lymphocytes must also be evaluated for their ability to produce critical effector cytokines, such as IFNγ, and to secrete cytotoxic granules, as indicated by surface expression of the degranulation marker CD107a. Seven days after particle-based stimulation, CTL were harvested and re-challenged with peptide-pulsed splenocytes and assessed for functional response by intracellular cytokine assay (Figure S2).
Functional responses were robust and equivalent for all three particle doses. CTL of all groups expressed high levels of CD107a, with up to 90% of cells degranulating and expressing high levels of IFNγ when re-challenged with peptide (Figure S2A-C). Thus, while particle to T cell ratio and protein quantity on particles influence the degree of CTL expansion, the resulting T cells displayed similar, strong effector responses regardless of particle dose. CTL phenotype was also assessed by expression of effector and memory surface markers CD44 and CD62L. After activation with either HD or LD nano-aAPC, naive CD44lo CD62Lhi T cells upregulated CD44, forming both CD62Lhi “Central Memory” (Tcm) phenotype and CD62Llo “Effector Memory” (Tem) phenotypes (Figure S2D-E).
We next compared nano-aAPC mediated T cell expansion to our micro-aAPC platform [7], which is based on 4.5 µm diameter iron-dextran beads. The total dose of protein was normalized between micro- and nano-aAPC. Micro- and nano-aAPC mediated robust, dose-dependent, and comparable levels of proliferation of pMEL T cells with acquisition of effector phenotype during three weeks of re-stimulation in vitro (Supplementary Figure S3). After one week, micro-aAPC induced approximately 15-fold expansion, and nano-aAPC induced approximately 20-fold expansion at a high dose of aAPC. After three weeks, fold expansion of CTL as high as 650-fold expansion with nano-aAPC and 450-fold expansion with micro-aAPC was observed. Both sizes of aAPC induced upregulation of CD44 and downregulation of CD62L consistent with effector phenotypes. Total amounts of proliferation observed is consistent with previous studies [33] and illustrates that nano-aAPC induce robust proliferation comparable to micro-aAPC.
Quantum Dot Nano-aAPC
To evaluate nano-aAPC based stimulation at an even smaller scale, and to demonstrate that nano-aAPC are not platform-exclusive, we obtained commercially available quantum dot core, avidin coated nanocrystals approximately 30 nm in diameter from Life Technologies. Biotin labeled dimeric Db-GP100 (signal 1) and anti-CD28 antibody (signal 2) were bound in a 1:1 molar ratio to the nanocrystal surface to form a quantum dot nano-aAPC (QD-aAPC) (Figure 3A).
Figure 3.
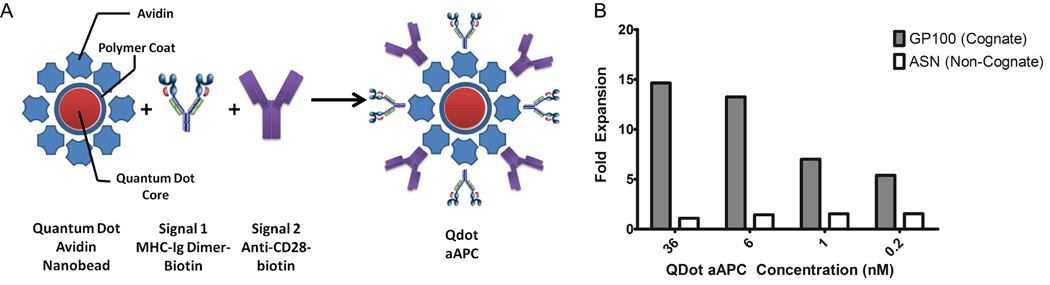
Synthesis and Characterization of Quantum Dot Nano-aAPC
(A) Quantum Dot (Qdot) Nano-aAPC were constructed by avidin-biotin mediated coupling of soluble MHC-Ig dimer (signal 1) and anti-CD28 antibody (signal 2) in a 1:1 ratio to the surface of a polymer-coated quantum dot particle.
(B) Qdot Nano-aAPC expansion in whole CD8+ T cells. Fold expansion on Day 7 is dose dependent and antigen-specific. Non cognate particles did not induce any expansion, whereas the highest dose of cognate QD aAPC (Db-GP100) induced approximately 15 fold expansion of CTL. Results are representative of 3 experiments.
QD aAPC induced dose-dependent, antigen specific T cell expansion in vitro (Figure 3B). At the highest dose evaluated, T Cells expanded 14.6 fold after 7 days, while T cells stimulated with non-cognate control QD aAPC did not expand.
Nano-aAPC Expansion of Endogenous Human T Cell Responses
Antigen-specific precursor T cells exist as low-frequency, heterogeneous populations of peripheral blood mononuclear cells (PBMC). Thus, immunotherapy ultimately depends on the expansion of antigen-reactive CTL from a polyclonal pool of endogenous precursors. Antigen-specific T cells in a polyclonal pool can be identified as cells that bind fluorescent MHC tetramer of the appropriate allele carrying the peptide of interest [34].
Anti-biotin iron-dextran aAPC were synthesized bearing the human HLA allele A2 loaded with the immunodominant T cell epitope derived from influenza protein M1 (signal 1) and anti-CD28 (signal 2). PBMC were incubated with increasing doses of nano-aAPC and antigen-specific T cell expansion was assessed by tetramer staining after two consecutive stimulations.
Before stimulation, M1 specific precursor frequency in the peripheral blood was low, with 0.4% specific CD8+ PBMC (Figure 4A, top row). Incubation with nano-aAPC for one (middle row) or two (bottom row) weeks resulted in a dose-dependent increase in the percentage of antigen specific T cells. These data are summarized in Figure 4B. The highest dose (30 µL) of nano-aAPC induced up to 44% of antigen specific T cells after one week or 80% after two weeks (left panel). This was associated with a dose-dependent increase in the total amount of antigen-specific T cells (right panel), with up to 150-fold expansion after one week and 800-fold expansion after two weeks at the highest particle dose. Nano-aAPC thus induced large populations of antigen-specific T cells from small endogenous precursor populations.
Figure 4.

Antigen-specific Human T Cell Expansion From Endogenous Precursors
(A) CD8+ T cells were isolated from PBMC by magnetic enrichment and incubated with increasing doses of iron-dextran nano-aAPC bearing A2-Ig complexes loaded with antigen derived from the immunodominant epitope of the influenza M1 protein, and assessed for antigen-specificity by tetramer staining before stimulation (PBMC, top row) and after one (middle row) or two (bottom row) weeks of stimulation. Numbers in top left represent percentage of CD8+ cells that were tetramer positive (gated).The size of the M1 specific population increases with repeated rounds of stimulation (top to bottom) and increasing dose of nano-aAPC (left to right), from 0.36% of CD8+ PBMC to 77.7% at the highest dose. Plots are representative of results from three separate experiments, summarized in panel B. (B) Percentage of CD8+ PBMC binding HLA-A2 M1 tetramers increases with repeated stimulation and increasing dose of nano-aAPC (left panel). The total number of tetramer positive cells (right panel) similarly increases with rounds of stimulation and particle dose, expanding up to 800-fold over the initial precursor population.
Enhanced Distribution of Nano- Compared to Micro-aAPC
Nanoscale particles are expected to drain more efficiently via lymphatics than microscale particles[14,35], motivating our interest in developing a nanoscale aAPC. To assess biodistribution after subcutaneous injection, we injected near-infrared (NIR) labeled iron-dextran nano- and micro-aAPC into the right flanks of B16 melanoma bearing Nu/J (nude) mice.
Twenty-four hours after subcutaneous injection, micro-aAPC remained largely confined to the injection site (Figure 5A, left). No significant drainage was observed up to 72 hours after injection. In contrast, nano-aAPC had spread diffusely across a larger area of the right flank, with most drainage occurring within the first 24 hours (Figure 5A, right). However, neither nano- nor micro-aAPC were observed to spread to the tumor itself. Distribution was quantified by measuring the area of particle distribution in the NIR channel above a certain fluorescence threshold. Twenty-four hours post-injection, nano-aAPC were visible in a 5-fold larger area than micro-aAPC (Figure 5B).
Figure 5.

Enhanced Drainage of Nano- Compared to Micro-aAPC.
(A) Visualization of drainage of near infrared labeled micro-aAPC (left) compared to nano-aAPC (right) after subcutaneous injection on right flank. Right flank views are shown for representative mice (3 mice/group) at the indicated timepoints after injection. Legend at right relates color in image to arbitrary fluorescence units. Micro-aAPC are confined largely to injection site, whereas local drainage of Nano-aAPC is more pronounced. (B) Biodistribution is quantified as area of visible drainage at indicated timepoints. Nano-aAPC have five-fold greater area of drainage than micro-aAPC at equivalent timepoints. (C) Simultaneous NIR images of biodistribution for pMEL T cells (green) and aAPC (red). Forty-eight hours after intravenous injection, T cells are visible in axillary lymph nodes, spleen, inguinal lymph nodes (white arrows, left to right) and cervical lymph nodes (not pictured). Right flank view (top row) shows nano-aAPC which were injected in right hindlimb 48 hrs. earlier reach inguinal lymph node, whereas micro-aAPC do not. This is even more pronounced after dissection (bottom row); aAPC signal is stronger and can be seen in the area of the inguinal lymph node (white arrow) for nano- but not micro-aAPC. Images are representative of three mice.
Enhanced drainage could lead to colocalization of antigen-specific T cells and nano-aAPC in lymph nodes. To simultaneously visualize T cell and aAPC biodistribution, we labeled pMEL T cells with a NIR membrane dye and injected them in the tail veins of Nu/J mice (Figure 5c). aAPC were labeled and injected subcutaneously as before. Forty-eight hours after injection, T cells were visible in axillary, inguinal, and cervical lymph nodes, as well as spleen. Nano-APC drained broadly from the injection site and a portion of particles were localized to the inguinal lymph nodes, where they could contact cognate T cells. Micro-aAPC, in contrast, were confined to the injection site, and not present at the same location as T cells.
Nano-aAPC Inhibit Tumor Growth In Vivo
A mouse model of subcutaneous melanoma was chosen to demonstrate the functional efficacy of nanoscale aAPC for immunotherapy when injected directly in vivo. To evaluate QD-aAPC, naive TCR transgenic pMEL CTL were adoptively transferred into wild-type B6 mice, and mice were challenged the same day with B16 melanoma cells injected subcutaneously (sc) on the right flank (Figure 6A, top). The following day, mice were injected with either 20 µL of cognate QD aAPC or 20 µL of non-cognate QD aAPC or PBS as control. One injection of QD aAPC significantly inhibited tumor growth (Figure 6A, bottom). After 16 days, mice treated with T cells and cognate QD aAPC had the smallest tumor burden, with an average tumor size of 22.1 mm2 +/− (sd) 2.3, compared to 111.1 mm2 +/− 29.4 for T cell + noncognate aAPC treated mice, 141.1 mm2 +/− 9.6 for T cells alone and 133.1 mm2 +/− 7.6 for untreated mice. Total tumor growth over the course of the experiment was summarized as area under the curve (AUC). Mice treated with cognate QD-aAPC had significantly less (p=0.028) overall tumor growth by AUC (33.1 mm2 +/− 7.8) than mice treated with control, non-cognate aAPC (373.6 mm2 +/− 227.0).
Figure 6.

Nano-aAPC Inhibit Tumor Growth In Vivo
(A) QD aAPC. B16 Tumors were injected subcutaneously on day 0, with injection of naive pMEL T cells on the same day. One day later, QD aAPC were injected intravenously (iv). IL-2 was administered on days 3, 4, and 5. Tumor size was measured as surface area (mm2) on indicated days, with area under the curve (AUC) shown at right. Mice treated with pMEL T cells and cognate QD aAPC (black bars) had less tumor growth compared to no treatment (white), T cells alone (light grey), and T cells + noncognate QD aAPC (checkered) (4 mice per group). Significance was characterized over entire experiment by AUC (p<0.001 by ANOVA with Tukey’s Post-Test, * indicates significant difference from no treatment group).
(B) Iron-Dextran aAPC. Naive pMEL T cells were injected intravenously on day -7. One day later, iron-dextran aAPC were injected either iv or subcutaneously (sc) on the right flank. B16 tumors were injected sc on right flank on day 0. Mice in treatment arms were given an additional injection on day 4 post tumor injection either iv or sc, to form four treatment groups: noncognate aAPC iv (day -6) then sc (day 4) (checkered), cognate aAPC iv then iv (light grey), cognate aAPC iv then sc (dark grey), and cognate aAPC sc then sc (black). Mice treated with pMEL T cells and cognate Iron-Dextran aAPC iv/sc or sc/sc (filled squares) had less tumor growth compared to noncognate aAPC (7 mice per group, p<0.02 by ANOVA with Tukey’s Post-Test, * indicates significant difference from no treatment group).
The route of particle administration is likely to affect bead trafficking, with subcutaneously deposited beads more likely to drain to local lymph nodes [36] and intravenously injected particles more likely to be filtered by the spleen. To test the impact of route of aAPC administration as well as the in vivo efficacy of iron-dextran aAPC, particles were injected either intravenously or subcutaneously three days after pMEL adoptive transfer. B16 Tumors were injected subcutaneously on right flank four days later, and a second injection of aAPC were given four days after tumor, either iv or sc on the ipsilateral flank. Thus, there were three treatment groups: mice receiving two iv bead injections, mice receiving one iv and one sc injection, and mice receiving two sc injections (Figure 6B, top). Control mice injected with non-cognate aAPC received one iv and one sc injection.
All three treatment groups had less tumor growth than mice injected with control bead (Figure 6B, bottom). After 16 days, mice treated with one sc and one iv injection (sc/iv) showed the least tumor growth (48.0 mm2 +/− 31.16), followed by sc/sc treated (73.7 mm2 +/− 37.44), iv/iv treated (89.4 mm2 +/− 69.5), no treatment (88.4 mm2 +/− 17.8) and non-cognate treated (113 mm2 +/− 39.4). Over the entire course of the experiment, sc/iv treated mice (AUC 52.6 mm2 +/− 29.7) and sc/sc mice (AUC 73.1 mm2 +/− 36.1) showed significantly less (p<0.02) tumor growth than control mice (AUC 162.7 mm2 +/− 77.6). Mice treated with two iv injections had less total tumor burden (AUC 103.0 +/− 86.1) than control, but did not reach the significance threshold (p = 0.19). Thus, mice treated with at least one dose of nano-aAPC delivered subcutaneously had significantly less tumor than control. This was consistent with observations that sc injected nano-aAPC drained from the injection site and were retained for several days after injection, whereas iv injected nano-aAPC were more likely to be rapidly cleared by renal excretion or filtered by the liver and spleen (Figure S4).
Discussion
We have described two nanoscale T cell activation platforms, termed artificial antigen present cells (aAPCs), based on coupling signal 1, peptide-MHC, and signal 2, B7.1-Ig or anti-CD28, to iron-dextran nanoparticles and quantum dot nanocrystals. Nano-aAPC induced T cell expansion from both TCR transgenic mouse splenocytes and human, polyclonal peripheral blood T cells, generating CTL with a robust effector phenotype that inhibited tumor growth in vivo. Both 30 nm quantum nanocrystals and 50–100 nm iron oxide nanoparticles were effective aAPC platforms, indicating that bead based aAPC can be explored at a range of nano-scales.
Previous work suggested that nanoparticles were incapable of providing the robust activating signals necessary for T cell proliferation [15,16,21]. This is the first description of a nanoscale particle-based T cell stimulation platform that can effectively induce antigen-specific T cell proliferation in vitro and anti-tumor activity in vivo. Our success may be due in part to our use of MHC-Ig dimers, whose flexible hinge region and nanoscale MHC dimerization may provide more optimal TCR/MHC interactions than MHC monomer [15,37].
Microscale, cell-sized bead platforms were initially chosen as aAPC to mimic structures that form between T cells-APC conjugates during activation [38]. For example, the immune synapse is a pattern of surface receptor reorganization several microns in diameter, with centrally located TCR and peripherally located adhesion molecules. The synapse, while not absolutely required for activation, does modulate antigen search and recognition [39]. A related process, asymmetric cell division, is hypothesized to regulate memory development by providing a microscale scaffold that induces polarity during cell division [40]. It has not been shown that aAPC of any size can recapitulate these structures, which seem to at least partly depend on coordinated rearrangement of both the APC and T cell membrane. Furthermore, it is particularly unlikely that nano-aAPC will drive the formation of structures that seem to depend on microscale cell-cell interactions. The nature of T cell activation by nano-aAPC may thus have important consequences for T cell function and memory development.
Nanoscale structures, such as clusters of TCR form on the T cell membrane even prior to formation of microscale structures [41,42], are also thought to be important regulators of T cell responses. However, even on the nanoscale level, the mechanism of TCR triggering by nano-aAPC is not clear. aAPC activate T cells through specific receptor-ligand binding at the cell-bead interface. Such interactions are not well defined when one of the participants is nanoscale [43]. The geometry of the nanobead, such as high local curvature at the interface, may preclude multiple productive receptor-ligand interactions. Alternatively, nanoscale platforms may preferentially interact with nano-clustered receptors such as the TCR [44,45]. Nanoscale bead-cell interaction platforms thus represent not just a novel approach to immunotherapy, but a tool for studying the delivery of biological signals at the cell membrane [19].
Nano-aAPC are better suited than micro-aAPC for in vivo administration and thus, allow the exploration of new particle-based immunotherapy strategies. Two potential sites where aAPC might be most effective are the lymph node, where naive and memory T cells reside and the tumor itself. Nanoparticles of approximately 50–100 nm diameter can be taken up by lymphatics and transported to the lymph nodes [14,36] thus gaining access to a larger pool of T cells. Nano-aAPC were more efficiently distributed from the injection site than micro-aAPC, were found to co-localize with antigen-specific T cells in inguinal lymph nodes, and inhibited tumor growth when injected subcutaneously. This suggests drainage of nano-aAPC to lymph nodes is a potential mechanism for optimal in vivo T cell activation, and the possibility of further tuning size [46] and particle surface properties[36] to enhance lymph node trafficking.
In addition, nanoscale delivery vehicles preferentially accumulate in tumors through enhanced permeability retention due to poorly formed tumor vasculature[47,48]. In this study, nano-aAPC delivered subcutaneously were not observed to drain into the tumor environment directly, whereas future work will focus on delivery strategies that lead to intratumoral aAPC accumulation. By delivering an immunostimulatory signal in situ, aAPC in the tumor microenvironment may address one of the most prominent hurdles in cancer immunotherapy, the immunosuppressive tumor microenvironment [49]. Although our work demonstrates that nano-aAPC can induce anti-tumor effector T cells from naive populations in vivo, it does not explore the capability of nano-aAPC to mediate rejection of established tumors in highly immunosuppressive microenvironments. Thus, whether a local stimulatory signal can overcome multiple layers of tumor immunosuppresion, or whether aAPC based stimulation can synergize with immunomodulatory therapies like checkpoint blockade, remains unknown. The enhanced drainage properties of nano-aAPC compared to micro-aAPC provides a system for examining the optimal distribution of aAPC for tumor rejection, and allows the exploration of new in vivo delivery strategies.
Supplementary Material
Acknowledgements
We thank the laboratory of Dr. Jordan Green at Johns Hopkins for use of their Nanosight LM10.
Funding: This work was supported by the National Institutes of Health (P01-AI072677, R01-AI44129 and R01-CA108835) and a sponsored research agreement with Miltenyi Biotec. Karlo Perica is supported in part by a Cancer Research Institute Predoctoral Fellowship.
Footnotes
Competing Financial Interests: The iron-dextran nano-aAPC is based on the Miltenyi Biotec platform technologies. MN, MA, and AR are employees of Miltenyi Biotec. Under a licensing agreement between NexImmune and the Johns Hopkins University, JPS and MO are entitled to a share of royalty received by the University on sales of products derived from this article. The terms of this arrangement are being managed by the Johns Hopkins University in accordance with its conflict of interest policies.
Author Contributions: KP wrote the manuscript, designed experiments, synthesized nano-aAPC, performed in vitro T cell expansion, in vivo tumor treatment, and particle and T cell characterization. JPS, ME, and MO conceived the nano-aAPC platform. JPS designed experiments and wrote manuscript. YLC performed human T cell expansion experiments. JGB, LS, and ALM performed murine T cell expansion experiments. MD performed quantum dot based in vivo experiments. MN, MA, AR synthesized directly conjugated iron-dextran aAPC and provided guidance on platform development.
References
- 1.Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. 2011;35(2):161–168. doi: 10.1016/j.immuni.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahlers JD, Belyakov IM. Memories that last forever: strategies for optimizing vaccine T-cell memory. Blood. 2010;115(9):1678–1689. doi: 10.1182/blood-2009-06-227546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wrzesinski C, Paulos CM, Kaiser A, et al. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. Journal of Immunotherapy. 2010;33(1):1–7. doi: 10.1097/CJI.0b013e3181b88ffc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehrotra S, Al-Khami Aa, Klarquist J, et al. A coreceptor-independent transgenic human TCR mediates anti-tumor and anti-self immunity in mice. Journal of immunology (Baltimore, Md.: 1950) 2012;189(4):1627–1638. doi: 10.4049/jimmunol.1103271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turtle CJ, Riddell SR. Genetically retargeting CD8+ lymphocyte subsets for cancer immunotherapy. Current opinion in immunology. 2011;23(2):299–305. doi: 10.1016/j.coi.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durai M, Krueger C, Ye Z, et al. In vivo functional efficacy of tumor-specific T cells expanded using HLA-Ig based artificial antigen presenting cells (aAPC) Cancer immunology, immunotherapy : CII. 2009;58(2):209–220. doi: 10.1007/s00262-008-0542-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oelke M, Schneck JP. Overview of a HLA-Ig based “Lego-like system” for T cell monitoring, modulation and expansion. Immunologic Research. 2010;47:248–256. doi: 10.1007/s12026-009-8156-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith-Garvin JE, Koretzky Ga, Jordan MS. T cell activation. Annual review of immunology. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–3666. doi: 10.1016/j.biomaterials.2010.01.065. [DOI] [PubMed] [Google Scholar]
- 10.Decuzzi P, Godin B, Tanaka T, et al. Size and shape effects in the biodistribution of intravascularly injected particles. Journal of controlled release : official journal of the Controlled Release Society. 2010;141(3):320–327. doi: 10.1016/j.jconrel.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 11.Semete B, Booysen L, Lemmer Y, et al. In vivo evaluation of the biodistribution and safety of PLGA nanoparticles as drug delivery systems. Nanomedicine : nanotechnology, biology, and medicine. 2010;6(5):662–671. doi: 10.1016/j.nano.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Sharma G, Valenta DT, Altman Y, et al. Polymer particle shape independently influences binding and internalization by macrophages. Journal of Controlled Release. 2010;147(3):408–412. doi: 10.1016/j.jconrel.2010.07.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Champion Ja, Walker A, Mitragotri S. Role of particle size in phagocytosis of polymeric microspheres. Pharmaceutical research. 2008;25(8):1815–1821. doi: 10.1007/s11095-008-9562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles target distinct dendritic cell populations according to their size. European Journal of Immunology. 2008;38(5):1404–1413. doi: 10.1002/eji.200737984. [DOI] [PubMed] [Google Scholar]
- 15.Mescher MF. Surface contact requirements for activation of cytotoxic T lymphocytes. Journal of Immunology. 1992;149(7):2402–2405. [PubMed] [Google Scholar]
- 16.Steenblock ER, Wrzesinski SH, Flavell Ra, Fahmy TM. Antigen presentation on artificial acellular substrates: modular systems for flexible, adaptable immunotherapy. Expert opinion on biological therapy. 2009;9(4):451–464. doi: 10.1517/14712590902849216. [DOI] [PubMed] [Google Scholar]
- 17.Balmert SC, Little SR. Biomimetic delivery with micro- and nanoparticles. Advanced materials (Deerfield Beach, Fla.) 2012;24(28):3757–3778. doi: 10.1002/adma.201200224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hubbell Ja, Thomas SN, Swartz Ma. Materials engineering for immunomodulation. Nature. 2009;462(7272):449–460. doi: 10.1038/nature08604. [DOI] [PubMed] [Google Scholar]
- 19.Anikeeva N, Lebedeva T, Clapp AR, et al. Quantum dot/peptide-MHC biosensors reveal strong CD8-dependent cooperation between self and viral antigens that augment the T cell response. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(45):16846–16851. doi: 10.1073/pnas.0607771103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyle S, Kolin DL, Bieler JG, Schneck JP, Wiseman PW, Edidin M. Quantum Dot Fluorescence Characterizes the Nanoscale Organization of T Cell Receptors for Antigen. Biophysical Journal. 2011;101(11):L57–L59. doi: 10.1016/j.bpj.2011.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steenblock ER, Fahmy TM. A comprehensive platform for ex vivo T-cell expansion based on biodegradable polymeric artificial antigen-presenting cells. Molecular therapy : the journal of the American Society of Gene Therapy. 2008;16(4):765–772. doi: 10.1038/mt.2008.11. [DOI] [PubMed] [Google Scholar]
- 22.Kunzmann A, Andersson B, Thurnherr T, Krug H, Scheynius A, Fadeel B. Toxicology of engineered nanomaterials: Focus on biocompatibility, biodistribution and biodegradation. Biochimica et biophysica acta. 2010;1810(3):361–373. doi: 10.1016/j.bbagen.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 23.Dobrovolskaia Ma, McNeil SE. Immunological properties of engineered nanomaterials. Nature nanotechnology. 2007;2(8):469–478. doi: 10.1038/nnano.2007.223. [DOI] [PubMed] [Google Scholar]
- 24.Nune SK, Gunda P, Majeti BK, Thallapally PK, Forrest ML. Advances in lymphatic imaging and drug delivery. Advanced drug delivery reviews. 2011 doi: 10.1016/j.addr.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Motta I, Lone YC, Kourilsky P. In vitro induction of naive cytotoxic T lymphocytes with complexes of peptide and recombinant MHC class I molecules coated onto beads: role of TCR/ligand density. European journal of immunology. 1998;28(11):3685–3695. doi: 10.1002/(SICI)1521-4141(199811)28:11<3685::AID-IMMU3685>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 26.Ge Q, Stone JD, Thompson MT, et al. Soluble peptide-MHC monomers cause activation of CD8+ T cells through transfer of the peptide to T cell MHC molecules. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(21):13729–13734. doi: 10.1073/pnas.212515299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curtsinger J, Deeths MJ, Pease P, Mescher MF. Artificial cell surface constructs for studying receptor-ligand contributions to lymphocyte activation. Journal of immunological methods. 1997;209(1):47–57. doi: 10.1016/s0022-1759(97)00146-4. [DOI] [PubMed] [Google Scholar]
- 28.Hosken N, Shibuya K, Heath A, KM The effect of antigen dose on CD4+ T helper cell phenotype development in a T cell receptor-alpha beta-transgenic model. The Journal of. 1995 Nov;182:20–22. doi: 10.1084/jem.182.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexander-Miller Ma, Leggatt GR, Sarin A, Berzofsky Ja. Role of antigen, CD8, and cytotoxic T lymphocyte (CTL) avidity in high dose antigen induction of apoptosis of effector CTL. The Journal of experimental medicine. 1996;184(2):485–492. doi: 10.1084/jem.184.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.González Pa, Carreño LJ, Coombs D, et al. T cell receptor binding kinetics required for T cell activation depend on the density of cognate ligand on the antigen-presenting cell. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(13):4824–4829. doi: 10.1073/pnas.0500922102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bullock TNJ, Mullins DW, Engelhard VH. Antigen density presented by dendritic cells in vivo differentially affects the number and avidity of primary, memory, and recall CD8+ T cells. Journal of Immunology. 2003;170(4):1822–1829. doi: 10.4049/jimmunol.170.4.1822. [DOI] [PubMed] [Google Scholar]
- 32.Mescher MF, Popescu FE, Gerner M, Hammerbeck CD, Curtsinger JM. Activation-induced non-responsiveness (anergy) limits CD8 T cell responses to tumors. Seminars in cancer biology. 2007;17(4):299–308. doi: 10.1016/j.semcancer.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oelke M, Maus MV, Didiano D, June CH, Mackensen A, Schneck JP. Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig-coated artificial antigen-presenting cells. Nature medicine. 2003;9(5):619–625. doi: 10.1038/nm869. [DOI] [PubMed] [Google Scholar]
- 34.Altman J, Davis M. MHC Peptide Tetramers to Visualize Antigen Specific T Cells. Current Protocols in Immunology. 2003:1–33. doi: 10.1002/0471142735.im1703s53. [DOI] [PubMed] [Google Scholar]
- 35.Fifis T, Gamvrellis A, Crimeen-Irwin B, et al. Size-dependent immunogenicity: therapeutic and protective properties of nano-vaccines against tumors. Journal of Immunology. 2004;173(5):3148–3154. doi: 10.4049/jimmunol.173.5.3148. [DOI] [PubMed] [Google Scholar]
- 36.Cai S, Yang Q, Bagby TR, Forrest ML. Lymphatic drug delivery using engineered liposomes and solid lipid nanoparticles. Advanced drug delivery reviews. 2011;63(10–11):901–908. doi: 10.1016/j.addr.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lebowitz MS, O’Herrin SM, Hamad a R, et al. Soluble, high-affinity dimers of T-cell receptors and class II major histocompatibility complexes: biochemical probes for analysis and modulation of immune responses. Cellular Immunology. 1999;192(2):175–184. doi: 10.1006/cimm.1999.1441. [DOI] [PubMed] [Google Scholar]
- 38.Moon JJ, Huang B, Irvine DJ. Engineering nano- and microparticles to tune immunity. Advanced materials (Deerfield Beach, Fla.) 2012;24(28):3724–3746. doi: 10.1002/adma.201200446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dustin ML. T-cell activation through immunological synapses and kinapses. Immunol. Rev. 2008;221:77–89. doi: 10.1111/j.1600-065X.2008.00589.x. [DOI] [PubMed] [Google Scholar]
- 40.Chang JT, Palanivel VR, Kinjyo I, et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007;315(5819):1687–1691. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 41.Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity. 2006;25:117–127. doi: 10.1016/j.immuni.2006.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lillemeier BF, Mörtelmaier Ma, Forstner MB, Huppa JB, Groves JT, Davis MM. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nature Immunology. 2010;11(1):90–96. doi: 10.1038/ni.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nel AE, Mädler L, Velegol D, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nature materials. 2009;8(7):543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 44.Fahmy TM, Bieler JG, Edidin M, Schneck JP. Increased TCR avidity after T cell activation: a mechanism for sensing low-density antigen. Immunity. 2001;14(2):135–143. [PubMed] [Google Scholar]
- 45.Kumar R, Ferez M, Swamy M, et al. Increased Sensitivity of Antigen-Experienced T Cells through the Enrichment of Oligomeric T Cell Receptor Complexes. Immunity. 2011;35(3):375–387. doi: 10.1016/j.immuni.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 46.Reddy ST, Van der Vlies AJ, Simeoni E, et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nature biotechnology. 2007;25(10):1159–1164. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 47.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Advances in enzyme regulation. 2001;41(00):189–1207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 48.Greish K. Enhanced permeability and retention of macromolecular drugs in solid tumors: a royal gate for targeted anticancer nanomedicines. Journal of drug targeting. 2007;15(7– 8):457–464. doi: 10.1080/10611860701539584. [DOI] [PubMed] [Google Scholar]
- 49.Rabinovich Ga, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annual review of immunology. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schneck JP, Slansky JE, O’Herrin SM, Greten TF. Monitoring antigen-specific T cells using MHC-Ig dimmers. Chapter 17. Current protocols in immunology / edited by John E. Coligan … [et al.] 2001 doi: 10.1002/0471142735.im1702s35. Unit 17.2. [DOI] [PubMed] [Google Scholar]
- 51.Chiu Y-L, Schneck JP, Oelke M. HLA-Ig based artificial antigen presenting cells for efficient ex vivo expansion of human CTL. Journal of visualized experiments : JoVE. 2011;(50):1–5. doi: 10.3791/2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.