

Abstract
While numerous studies support regulation of Ras GTPases by reactive oxygen and nitrogen species, the Rho subfamily has received considerably less attention. Over the last few years, increasing evidence is emerging that supports the redox sensitivity of Rho GTPases. Moreover, as Rho GTPases regulate the cellular redox state by controlling enzymes that generate and convert reactive oxygen and nitrogen species, redox feedback loops likely exist. Here, we provide an overview of cellular oxidants, Rho GTPases, and their inter-dependence.
Keywords: cysteine oxidation, reactive oxygen species, reactive nitrogen species, Ras GTPases, free radicals, 2-electron oxidants, post-translational modifications
It has become increasingly clear that reactive oxygen and nitrogen species (ROS, RNS), once regarded simply as by-products of cellular metabolism, also function as regulators of numerous critical physiological processes, including cell survival,1 proliferation,2 differentiation,3 migration, and adhesion.4 To accomplish these activities, ROS and RNS modulate redox-sensitive proteins that control multiple kinase5 and GTPase6 signaling pathways. In particular, accumulating evidence suggests a role for ROS/RNS in the regulation of Ras and Rho family GTPases. Here, we highlight recent findings that demonstrate a role for ROS/RNS in the regulation of Rho GTPase activity.
Rho Family GTPases
Rho GTPases comprise 1 of 5 distinct classes of Ras superfamily GTPase proteins. The canonical members of the Rho subfamily are RhoA, Rac1, and Cdc42.7 Rho GTPases function in a wide variety of cellular processes, including cell growth, motility, polarity, and adhesion.8 Rho GTPases, like most Ras superfamily GTPases, cycle between the active GTP-bound and inactive GDP-bound states to modulate their binding to regulators and cellular targets (Fig. 1). The exchange of bound GDP for GTP (activation) is regulated by guanine nucleotide exchange factors (GEFs),9 which catalyze GDP dissociation and promote GTP binding due to the higher cellular ratio of GTP to GDP. Once in their active GTP-bound state, Rho GTPases interact with a number of downstream effectors to promote their biological activities. Deactivation of the GTPases is catalyzed by GTPase activating proteins (GAPs), which stimulate hydrolysis of bound GTP to GDP and inorganic phosphate.10 In addition, Rho GTPases are regulated by guanine nucleotide dissociation inhibitors (GDIs), which result in inactivation by sequestering the GDP-bound GTPase away from the membrane and stabilizing GDP binding.11,12
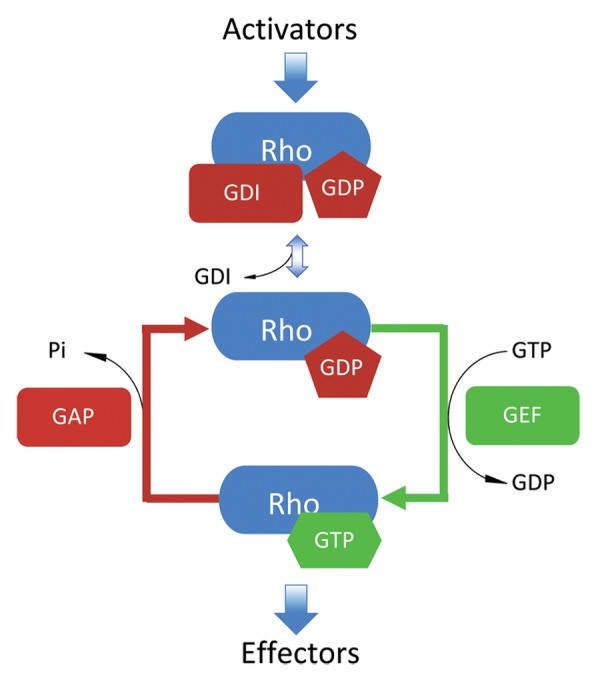
Figure 1. The GTPase cycle of Rho. Rho GTPases cycle between GTP-bound active states and GDP-bound inactive states. Guanine nucleotide dissociation inhibitors (RhoGDIs) extract lipid-modified Rho GTPases from cellular membranes to the cytosol and prevent GDP dissociation until released upon stimulus. Following signal input, the exchange of the bound GDP is facilitated by guanine nucleotide exchange factors (GEFs) that dramatically increase the dissociation rate of nucleotides. This promotes binding of GTP owing to its 10-fold higher in cellulo concentration vs. GDP. Once GTP-bound, active Rho is capable of binding downstream effectors and executing its biological functions. Rho remains active until GTP is converted to GDP due to its intrinsic hydrolytic capacity or through catalysis by GTPase activating proteins (GAPs).
Rho GTPases can be modified by a variety of post-translational modifications (PTMs) that drive differences in localization and activity.13 One such PTM is lipid modification. Rho proteins associate with cellular membranes upon isoprenoid lipid modification at their carboxyl-terminal CAAX (C, cysteine; A, aliphatic residue; X, variable residue) motifs.14,15 RhoA, Rac1, and Cdc42 are all geranylgeranylated at the CAAX motif, which is required for their association with the inner leaflet of the plasma membrane and/or with internal membranes.16 As membrane association is critical for downstream signaling,17 dynamic PTMs and protein:protein interactions that affect membrane association can significantly affect Rho biological activity. For example, Rho GDIs recognize and sequester the geranylgeranyl lipid moiety,18 preventing membrane association and inactivating the GTPase. Moreover, palmitoylation of Rac1 at Cys178 is critical for Rac1-mediated actin cytoskeleton remodeling.19 Mutation of Cys178 to Ser, which prevents palmitoylation, results in decreased partitioning to the plasma membrane and reduced Rac1 activation. Rho GTPase membrane association is further regulated by interaction with other proteins and other nearby PTMs. Some Rho GTPases, such as RhoA, RhoG, and Cdc42, are phosphorylated at their C-terminal membrane-targeting domains near the prenylated CAAX motif,20 which may be another mechanism of downregulation. While RhoA phosphorylation at Ser188 increases its association with GDIs, which causes release from the membrane and inactivation, the biological effect of phosphorylation at this residue in RhoG and Cdc42 is unclear.21,22
Ubiquitination, including mono-, di-, and polyubiquitination, is another PTM that has been recently observed in Rho GTPases.23 These modifications result in the addition of single or multiple ubiquitin moieties (76-residue protein) that affect protein localization, stability, and trafficking.24 Ras was one of the first GTPases shown to be mono- and diubiquitinated. While ubiquitination of H-Ras promotes endosomal localization,25,26 monoubiquitination at Lys147 in K-Ras upregulates K-Ras activity by impairing GAP-mediated GTP hydrolysis.27,28 Moreover, a subset of Rho GTPases is ubiquitinated. Of these, polyubiquitination of RhoA, Rac1, and Cdc42 result in their deactivation and degradation.29-31 Rac1 is the only Rho family GTPase shown to be monoubiquitinated, and this modification affects Rac1 localization.32 Ubiquitin is a highly regulated and reversible modification that can be removed by deubiquitinating enzymes (DUBs); however, Ras- and Rho-specific DUBs have yet to be discovered.
Cysteine oxidation represents yet another type of PTM in Ras superfamily GTPases. Although this form of redox regulation is often described as an “emerging” or “novel” mechanism of GTPase regulation, it was originally identified in Ras over 15 years ago.33 What makes cysteine oxidation a unique PTM is that oxidation can occur absent direct enzymatic action. Rather, reactive and/or solvent exposed cysteines can be modified directly by reactive nitrogen (RNS) and oxygen (ROS) species (oxidants capable of cysteine oxidation are depicted in Fig. 2); however, whether enzymes can assist or catalyze thiol oxidation remains undetermined. While a variety of enzymes, including nitric oxide synthase (NOS), NADPH oxidase (NOX), and superoxide dismutase (SOD), produce oxidants that promote protein oxidation, the oxidants themselves have long been viewed as nonspecific hallmarks of oxidative stress. However, it has become increasingly apparent that specific proteins containing redox-sensitive cysteine residues are targeted under physiological conditions by both RNS and ROS, including nitrogen dioxide (NO2•), nitric oxide (NO•), superoxide (O2•-), peroxynitrite (ONOO-), and peroxide (H2O2).34-36
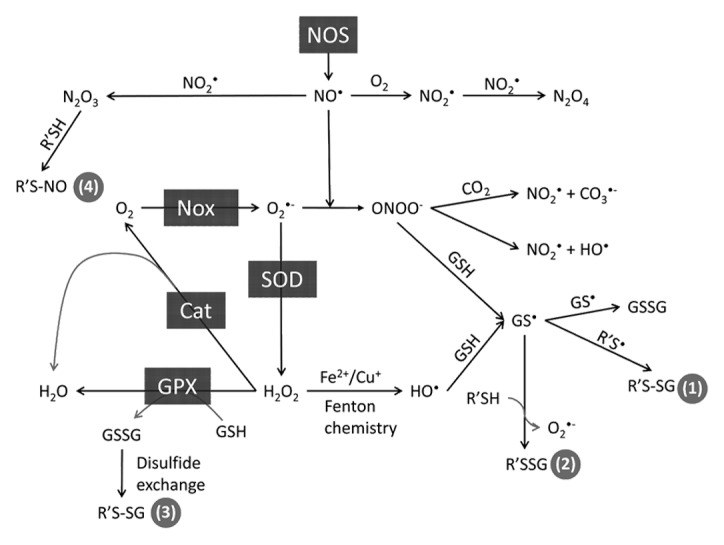
Figure 2. Generation and interconversion of ROS and RNS in cells. Select oxidation end products are presented, including radical-mediated glutathiolation (1), glutathiolation with O2•- production (2), disulfide exchange-mediated glutathiolation (3), and nitrosation by a non-radical pathway (4). Not all reactions shown are chemically balanced. Key: Cat, catalase; GPX, glutathione peroxidase; NOS, nitric oxide synthase; Nox, NADPH oxidase; SOD1, superoxide dismutase 1.
Ras GTPases were among the first GTPases shown to be sensitive to RNS and ROS in cells.33 Ras proteins contain a redox-sensitive NKCD motif. While covalent modification of this cysteine (Cys118) by oxidants does not alter Ras activity,37,38 oxidants capable of generating a thiyl radical at Cys118 (e.g., NO2•) cause oxidation of the guanine nucleotide and enhance nucleotide exchange under physiological or mild-oxidizing conditions.39,40 Given the >10-fold higher levels of cellular GTP compared with GDP, this enhanced exchange likely activates Ras.41 In support of these findings, radical-mediated activation of Ras at Cys118 contributes to tumor maintenance in a pancreatic cancer cell line and in a severe combined immunodeficiency/beige mouse model.42 However, a subset of Rho family GTPases has been observed to be sensitive to oxidants.43,44 In Rho GTPases, the redox-sensitive motif has been determined to be located adjacent to the phosphoryl-binding loop (GXXXXGK[S/T]C) motif. As with Ras, Rho GTPases are sensitive to free radical-mediated oxidation and regulation; however, because the redox-sensitive thiols in Rho GTPases make direct contact with the bound nucleotide, 2-electron oxidation of the phosphoryl-binding loop thiols can also regulate Rho GTPase activity.
There is no known consensus sequence that can be used to predict which cysteines are redox-sensitive.45 A cysteine residue is generally considered reactive to oxidants, or “redox-sensitive,” if it has a depressed pKa and is solvent accessible.46,47 In general, a free cysteine has a pKa of approximately 8.5, which populates the thiol form at physiological pH, and is less sensitive to oxidation than the thiolate (S-) form.48 For example, while peroxynitrite can oxidize the cysteine thiol, it is more reactive with the thiolate. The radical-mediated breakdown products of peroxynitrite are even more reactive and likely to oxidize cellular thiols.49 Moreover, nitric oxide and peroxide are believed to react primarily with the thiolate form of cysteines as they react too slowly with thiols to be physiologically relevant.50 However, an altered pKa is not always a prerequisite for redox sensitivity, especially for radical-mediated reactions, as Ras Cys118 does not have an altered pKa despite its regulation by free radical-mediated oxidants.51 Furthermore, as oxidants are generally short-lived and confined to the compartment in which they are generated, localization of the enzymes that generate oxidants with their target proteins is often critical for regulation by ROS and RNS.
In this review, we present fundamental concepts associated with redox signaling, with a specific emphasis on cysteine oxidation, and describe the current state of the field regarding the redox regulation of Rho GTPases.
Generation of ROS and RNS in Cells and Antioxidant Defense
Varying levels of oxidative stress can occur in cells when the cellular redox potential becomes oxidizing. Dysregulation of the cellular redox potential is correlated with elevated levels of ROS and RNS, which contributes to and promotes a variety of diseases, including cancer,52 neurodegeneration,53 atherosclerosis,54 diabetes,55 and aging.56,57 As such, ROS and RNS are often considered damaging to cellular components, including proteins, lipids, and DNA. To provide appropriate background for the reader on the currently understood role of ROS and RNS in the regulation of Rho GTPases, we will first give a brief overview of cellular ROS/RNS production and the cellular antioxidant defense system.
Nitric oxide is perhaps the best-known cellular RNS and is produced by a class of enzymes called nitric oxide synthases (NOSs). There are 3 NOS isoforms: endothelial NOS (eNOS), neuronal NOS (nNOS), and inducible NOS (iNOS), all of which function as homodimers to generate nitric oxide (NO•) from l-arginine.58 Some functions of nitric oxide produced by NOS include the regulation of blood pressure by controlling vasodilation, inflammation, and erectile function.58 Endothelial NOS is constitutively expressed and localized to cell membranes.59 When activated, eNOS can produce low levels of nitric oxide. Neuronal NOS is a cytosolic protein that is found primarily in the central nervous system.60 Activation of eNOS and nNOS is regulated by calcium-bound calmodulin,61 interactions with various effector proteins,62 and phosphorylation.63 Lastly, iNOS expression is induced in macrophages and other cell types; however, unlike eNOS or nNOS, iNOS is constitutively active once expressed.64 The functional role of iNOS is to produce nitric oxide to regulate the immune response, including inhibition of iron-sulfur proteins, oxidation of invading pathogen DNA, and oxidative damage of tumor cells.
A primary source of cellular ROS exists within the mitochondria. All aerobic organisms require oxygen for oxidative phosphorylation, and the mitochondria are central organelles that generate a number of cellular oxidants.65 Moreover, nitric oxide regulates oxygen consumption, metabolism, and ROS production in the mitochondria66 and can be generated through NOS-dependent and -independent pathways.67 One of the most common ROS generated by the mitochondria is superoxide (O2•-), which is produced due to the incomplete reduction of molecular oxygen (O2). Superoxide is a charged molecule and does not readily diffuse across membranes,68,69 which limits where it can react with cellular biomolecules. Furthermore, in aqueous solution, superoxide undergoes a fast dismutation reaction (rate constant of 5 × 105 M−1s−1 at pH 7.0) to peroxide (H2O2).70 Superoxide dismutase, an enzyme capable of reducing superoxide to peroxide, speeds up this reaction nearly 104-fold.71 In the mitochondria, SOD2 is maintained at a high concentration.72,73 However, the product of this reaction, H2O2, can easily cross the membrane and move into the cytoplasm.74
Phagocytes are a common source of cellular ROS and can produce superoxide in ‘bursts’ due to the action of NOXs.75 There are currently 7 known NOX enzymes (Nox1/2/3/4/5, Duox1, and Duox2). NOX1, NOX2, and NOX3 all require the Rac1 GTPase for activation.76 NOX isoforms are highly regulated and tissue-specific. In phagocytes, cytochrome b559 (a heterodimer consisting of NOX2 ([formerly gp91phox] and p22phox) combines with p47phox, p67phox, and Rac1 to form the membrane-associated NOX complex, which catalyzes the 1-electron reduction of molecular oxygen to superoxide. A major role of the NOX complex is to eliminate invading pathogens by oxidation.77 In addition, superoxide generated in the arteries by NOX2 and NOX4 compete with the antihypertensive effects of nitric oxide by reacting with nitric oxide to form peroxynitrite.78
The cell has also evolved a network of antioxidant enzymes and small molecules to protect the cell from oxidative damage. SOD is an abundant antioxidant enzyme.79 Three distinct isoforms of SOD exist: SOD1, a dimeric protein that binds copper and zinc and is localized in the cytosol;80,81 SOD2, a homotetrameric protein that binds 1 manganese ion per subunit and is localized in the mitochondria;82 and SOD3, a homotetrameric protein that binds copper and zinc and is anchored to the extracellular matrix.83,84 SODs efficiently reduce superoxide to peroxide.80 Peroxide, in turn, can be reduced to water by catalases and glutathione peroxidases (GSH-Pxs) (Fig. 2).85 In addition, GSH-Pxs can reduce lipid peroxides to their respective lipid alcohols.86,87 Other antioxidant enzymes include thioredoxin reductases, peroxiredoxins, and glutaredoxins, all of which function in the removal of peroxide.88
Common cellular small molecule antioxidants include glutathione, ascorbic acid, tocopherol, and β-carotene. Glutathione is a tripeptide that contains a cysteine thiol group and is the major cellular antioxidant. The cellular concentration of glutathione in cells ranges between 0.5 and 10 mM, whereas oxidized glutathione is reported to range from 5–50 μM.89 Proper regulation of the ratio of reduced to oxidized glutathione (GSH:GSSG) is critical for managing the cellular redox potential.90 GSH functions with GSH-Pxs to reduce peroxides in the cell and can reduce other oxidized species. Ascorbic acid, more commonly known as Vitamin C, scavenges free radicals in the cytosol, whereas tocopherol (vitamin E) protects cellular membranes from oxidative damage.91 Β-carotene serves to scavenge radicals as well, including peroxyl, hydroxyl, and superoxide radicals.92
Reactive Oxygen and Nitrogen Species as Second Messengers
While ROS and RNS are generally grouped into a single category of reactive intermediates, it is important to understand that there are 2 classes of oxidants, 2-electron and 1-electron oxidants. A predominant cellular 2-electron oxidant is hydrogen peroxide.47 One-electron oxidants, such as nitrogen dioxide, generate thiyl radical intermediates. In general, free radical oxidants have a high redox potential and react faster with cellular thiols than 2-electron (non-radical) oxidants. Oxidation can result in a variety of cysteine oxidative byproducts, thereby altering the cellular redox state or protein function (Fig. 2).93,94 Importantly, ROS and RNS can interact with cell signaling proteins (phosphatases, kinases, etc.) and function as second messengers downstream of a variety of signaling stimuli.35 However, it is important to note that oxidation observed in vitro does not always correlate with oxidation in vivo, as in vitro experiments tend to have few substrates in solution, which leaves the oxidant little competition. Moreover, reaction of protein thiols with cellular oxidants is dependent on thiol accessibility, reactivity, spatial and temporal localization to oxidants, and enzymes that catalyze thiol oxidation and reduction.95
Some examples of common ROS are peroxide (H2O2), superoxide (O2•-), and hydroxyl radical (HO•). Examples of common RNS are nitric oxide (NO•), nitrogen dioxide (NO2•), peroxynitrite (ONOO-), and dinitrogen trioxide (N2O3; Fig. 2 and 7; see Glossary). For these molecules to function as second messengers in cellular signaling, they must be generated in response to a specific stimulus, be short-lived, specifically target effectors, and transiently and reversibly affect signaling. Superoxide and peroxide are considered to function as second messengers but are less reactive than some other ROS, such as the hydroxyl radical. The hydroxyl radical lacks specificity as it reacts with virtually all biomolecules at the rate of diffusion. Aside from peroxynitrite and dinitrogen trioxide (N2O3), other cellular RNS that oxidize thiols are free radicals. Nitrogen dioxide, rather than nitric oxide, is thought to be the major radical-mediated cellular RNS. The thiyl radical, once formed, can react with nitric oxide to generate nitrosothiols (SNO). Peroxynitrite undergoes homolytic cleavage to generate nitrogen dioxide and carbonate radicals (CO3•-) in the presence of CO2, which can directly oxidize thiols; however, dinitrogen trioxide and the nitrosonium ion (NO+) are capable of nitrosating thiols through slower 2-electron mechanisms96 to generate nitrosothiols (RSNO). For a detailed review of the interconversion of ROS and RNS, we direct readers to a previous review.97
Protein thiols in cysteine residues are among the most common sites of protein oxidation. Reversible oxidation states of protein thiols in vivo include oxidation to sulfenic acid (RSO-), nitrosation (RSNO), glutathiolation (RSSG; mixed disulfide bonds), and disulfide bonds (RSSR’; intramolecular disulfide bonds).98 Sulfinic acid (RSO2-), which has been regarded as an irreversible modification, has been shown to be reduced by an enzyme class termed sulfiredoxins; however, sulfiredoxins have only been shown to reduce sulfinic acids in specific peroxiredoxins.99 Sulfonic acid (RSO3-) is another consequence of thiol oxidation; however, this oxidation state is irreversible and often results in proteasome-mediated degradation.100
Redox Regulation by Rac
Small GTPases of the Rho family both regulate and are regulated by ROS and RNS. It has been well established that Rac GTPases modulate the cellular redox state through regulating the assembly and activation of the NOX complex.101 In particular, Rac1 and Rac2 are involved in the activation of NOX1, NOX2, and NOX3.101-103 This function of Rac, first demonstrated in neutrophils and other phagocytic cells and later expanded to non-phagocytic cells,104 is important in both physiological105 and pathophysiological106-108 settings.
Rac1 has been shown to associate with many enzymes necessary for maintaining redox homeostasis in cells. SOD1, but not SOD2, directly associates with Rac1 and has been proposed to regulate Rac1/NOX2 activation in a redox-dependent fashion.109 Rac1 and SOD1 can be immunoprecipitated from whole organ lysates of mice, including the brain, liver, kidney, and heart. In addition, in vitro-purified Rac1 can directly associate with SOD1.109 Intriguingly, the interaction between Rac1 and SOD1 appears dependent on both the bound guanine nucleotide and oxidation state of Rac1. In this study, Rac1 was purified from bacteria and used to pull native SOD1 from brain tissue lysates. In the absence of reducing agents, Rac1-GDP co-immunoprecipitated with SOD1, whereas in the presence of reducing agent (300 μM DTT), only Rac1-GTP co-immunoprecipitated with SOD1. While this result has interesting implications regarding redox-dependent interactions between Rac1 and SOD1, the oxidation state and sites of Rac1 oxidation were not determined. Thus, this study raised new questions but did not answer questions regarding the regulation or consequences of oxidation on Rac1/SOD1 binding.
Further interplay of Rac1 and ROS/RNS production involves the regulation of NOS activity. Selvakumar et al. observed a concomitant increase or decrease in nitric oxide and superoxide production in primary human aortic endothelial cells (HAECs) when Rac1 was activated or inhibited, respectively.110 Consistent with these observations, Rac1 can interact with and regulate the activity of the eNOS and nNOS. Rac2, however, interacts with iNOS, which is required for nitric oxide generation following phagocytosis.111
A biologically relevant downstream effect of ROS is activation of nuclear factor-κB (NFκB), a transcription factor that controls the expression of a wide spectrum of genes, especially genes involved in inflammation and the immune response.112,113 Many Rac1-induced tumorigenic properties have been ascribed to Rac1-mediated ROS production and NFκB activation.113-115 Although the majority of the experimental evidence argues for an activating role of ROS, mostly H2O2, on NFκB, inhibition of basal NFκB activity by cadmium, a heavy metal that leads to rapid and transient ROS generation, has also been reported.113 Interestingly, depending on the cellular context, NFκB reciprocally regulates ROS in a bidirectional manner (Fig. 3). During inflammation, activated NFκB facilitates the production of ROS through the induction of NOX2 to strengthen the immune defense.116 However, when cellular ROS accumulation becomes toxic, NFκB can prevent oxidative damage through the transcriptional activation of SOD2.117,118
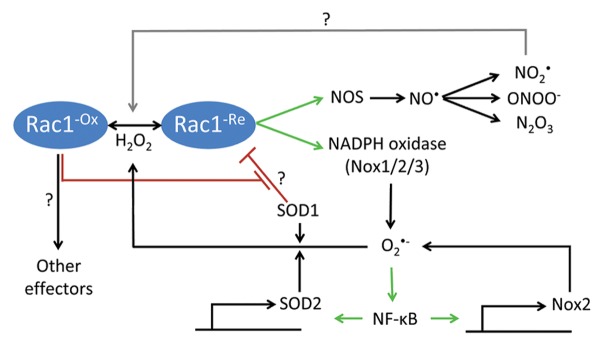
Figure 3. Rac1-mediated oxidant production. Rac1 assembles and activates NADPH oxidases 1/2/3 (Nox1/2/3), which produce superoxide (O2•-). O2•- activates nuclear factor-κB (NFκB), a transcription factor that facilitates the transcription of, among other genes, redox-related enzymes, such as Nox2 and superoxide dismutase 2 (SOD2). Nox2 can further enhance the production of oxidants, while SOD2 converts highly reactive O2•- to less reactive hydrogen peroxide (H2O2), the latter activating Rac1 via yet uncharacterized mechanisms. Under reducing conditions, SOD1 binds Rac1 and has been proposed to inhibit its GTPase activity. Oxidation of Rac1 by H2O2 uncouples SOD1-Rac1 interaction. Whether and how oxidized Rac1 further signals to other effectors is still not fully understood. In a similar vein to superoxide production, Rac1 interacts with and activates nitrogen oxide synthases (NOS), promoting the synthesis of nitric oxide (NO•). NO• can be converted into various reactive nitrogen species (RNS), including nitrogen dioxide (NO2•), peroxynitrite (ONOO-), and dinitrogen trioxide (N2O3), which possibly regulate Rac1 activity.
Redox Regulation of Rac
In contrast to the vast amount of literature documenting Rac-mediated ROS production,101-108 relatively few studies have investigated the role of ROS/RNS in regulating Rac activity. Nagase et al. have demonstrated ROS-mediated activation of Rac1 in rat cardiomyocytes. They found that L-buthionine sulfoximine (BSO), which depletes intracellular glutathione, increased the active form of Rac1, and this effect was blocked by the antioxidant N-acetylcysteine (NAC).119 Consistent with their findings with BSO, H2O2 also activated Rac1.119 Similarly, H2O2 treatment led to a robust increase in GTP-bound Rac1 in neutrophils derived from C57/BL6 control mice.120 Chronic exposure of normal mouse mammary gland epithelial (NMuMG) cells to H2O2 not only elevated Rac1 activity but also greatly enhanced cell invasiveness.121 These lines of evidence suggest that a positive feedback loop exists between Rac GTPases and ROS, which reinforces their effects on cells (Fig. 3). However, these studies have not tested whether the effects of ROS on Rac1 are direct or indirect. In addition, the effects of endogenous ROS on Rac activity have yet to be tested.
Many Rho family GTPases contain a distinct cysteine-containing motif (GXXXXGK[S/T]C) located directly adjacent to the phosphoryl-binding loop (p-loop). In Rho GTPases containing this motif, the redox-sensitive thiol (Cys18 in Rac1 and Cdc42, Cys20 in RhoA) makes direct contact with the bound nucleotide;122 therefore, any modification of this cysteine has been predicted to affect the nucleotide binding properties of these GTPases (see Fig. 4).

Figure 4. Surface rendering depicting cysteine solvent-accessible residues (yellow) in Rho family GTPases (see Table 1). (A) Left panel, Rac1 (PDB 3TH5) flipped by 180° around the y-axis. Right panel, close-up of the nucleotide-binding site with the redox-sensitive thiol (Cys18) shown. Other local thiols are labeled (note that Cys81 is not solvent-accessible). Distances are presented in Å from the reactive thiol to the nucleotide. (B) RhoA surface rendering (PDB 1A2B). Images are as in (A). The close-up of the nucleotide-binding pocket (right panel) shows the redox-sensitive thiol (Cys20) as well as other local thiols. (C) Cdc42 surface rendering (PDB 2QRZ). Images are as in (A). The close-up of the nucleotide-binding pocket (right panel) highlights Cys.18
Rho family GTPases containing this redox-sensitive motif include RhoA, RhoB, RhoC, RhoG, all Rac isoforms, and Cdc42 (for sequence comparison of the redox-sensitive motifs in Ras and Rho family GTPases, see ref. 6). The seminal papers that first identified the redox-sensitive properties of this motif in the canonical Rho family members (RhoA, Rac1, and Cdc42) showed that exposure to free radical oxidants (nitrogen dioxide and superoxide) resulted in increased nucleotide dissociation, similar to that observed for Ras.43,44 In these studies, the in vitro-purified GTPases preloaded with [3H]GDP were exposed to various ROS and RNS and monitored for their ability to retain nucleotide binding in the presence of unlabeled GDP. Whereas the non-radical oxidant peroxide stimulated guanine nucleotide dissociation in RhoA, Rac1, and Cdc42 by 10-fold under the conditions tested, the rate of guanine nucleotide dissociation was dramatically enhanced by 500- to 600-fold upon exposure to superoxide, nitrogen dioxide, and hydroxyl radicals. While these studies demonstrated that exposure of Rac1, RhoA, and Cdc42 to ROS and RNS can enhance nucleotide exchange and upregulate their function similar to the action of GEFs, oxidative modification of the GTPases was not evaluated.
We characterized the pKa of the redox-sensitive thiol in Rac1 and determined whether cysteine oxidation alters Rac1 activity in vitro. We found that Cys18 in Rac1 has a depressed pKa that populates the more reactive thiol anion (thiolate) at physiological pH, rendering Rac1 susceptible to thiol oxidation (Hobbs et al.; in submission). Consistent with these findings, Cys18 was selectively oxidized by glutathione in vitro at a mildly acidic pH of 6.5. As physiological pH ranges from 6.7–7.4,123 we used a slightly acidic pH to promote reaction with Rac1 thiol(s) that possess a depressed pKa. Rac1 glutathiolation at Cys18 caused a significant (~230-fold) enhancement in the rate of intrinsic guanine nucleotide dissociation. Further, we generated a Rac1C18D variant to mimic sulfinic acid (RSO2-) oxidation at this site and observed greatly enhanced nucleotide exchange (220-fold), similar to that observed for glutathiolated Rac1, indicating that oxidation of Cys18 can significantly accelerate nucleotide exchange. However, as exposure of Rac1 to peroxide can increase intrinsic nucleotide dissociation by 10-fold,44 we hypothesize that the effects of Rac1 oxidation are dependent on the oxidation state. The bulkier charged states, such as glutathiolation, result in greatly increased rates of nucleotide exchange and higher levels of activation relative to peroxide-mediated oxidation. Consistent with these results, RacC18D is highly activated in Swiss-3T3 cells and promotes lamellipodia formation (Hobbs et al.; in submission).
As cysteine oxidation of proteins is dependent on thiol accessibility, we used the program nAccess124 to determine the solvent accessibility of cysteine residues in RhoA, Rac1, and Cdc42. Table 1 lists the surface accessibility of all thiols in RhoA, Rac1, and Cdc42. While we find that thiol accessibility is dependent on the nucleotide bound state and interaction with regulators and effectors, we predict that Cys,18 Cys,105 and Cys178 (Rac1 numbering) are the most likely cysteine residues in Rac1 susceptible to oxidation based on solvent accessibility criterion alone. However, our data indicate that neither Cys105 nor Cys178 have a significantly altered pKa. Moreover, Cys105 does not make direct interactions with the nucleotide or regions critical for effector and/or regulator recognition; therefore, if this cysteine becomes oxidized, Rac1 activity is unlikely to be altered. As Cys178 is modified by palmitic acid in vivo, it is possible that this cysteine will be protected from ROS/RNS modification, or conversely, that oxidation may prevent Rac1 palmitoylation. This, in turn, may modulate membrane association and/or kinetics. While oxidation of the C-terminal cysteines in Ras has been shown to regulate palmitate turnover,125 the effect of oxidants on Rac1 lipidation has not been studied.
Table 1. Solvent accessibility of cysteine sulfur atoms in Rac1, RhoA, and Cdc42.
| Thiol accessibility (Rac1/Cdc42, RhoA) |
Rac1GTP PDB 3TH5 |
Rac1GDP (Zn-bound) PDB 2P2L |
RhoAGTP PDB 1A2B |
RhoAGDP PDB 1FTN |
Cdc42GTP PDB 2QRZ |
Cdc42GDP PDB 1AN0 |
|---|---|---|---|---|---|---|
| Cys6, NA | 0.0 | 0.0 | NA | NA | 0.0 | 0.0 |
| NA, Cys16 | NA | NA | 0.0 | 0.0 | NA | NA |
| Cys18, Cys20 | 8.7 | 0.0 | 4.1 | 0.0 | 0.0 | 0.0 |
| Cys81, Cys83 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Cys105, Cys107 | 9.8 | 0.56 | 3.9 | 1.9 | 0.0 | 0.05 |
| Cys157, Cys159 | 0.70 | 0.45 | 1.2 | 0.04 | 0.21 | 0.13 |
| Cys178, NA | ND | 35 | NA | NA | 6.0 | 3.7 |
Solvent accessibility (in square angstroms) of cysteine thiol atoms determined using nAccess.107 Numbering in the left column is for Rac1 and Cdc42, with numbering for RhoA listed after the comma. nAccess was executed in the presence of heteroatoms, including the bound nucleotide. Only Cys18 (Cys20) shows differential protection in the presence of nucleotide, displays nucleotide dependence, and increased accessibility in the absence of nucleotide. For comparison, the solvent accessibility of the sulfur atom in an Ala-Cys-Ala tripeptide is ~60 Å2. ND, No data; NA, Not available.
In contrast to our observations on Rac1, it has recently been observed that treatment of in vitro-purified Rac2 with a 105-fold excess of oxidized glutathione at a pH of 8.0 causes oxidation of Cys157.126 Using site-directed mutagenesis to generate individual Cys > Ala variants, the authors suggested that Rac2 was specifically glutathiolated at Cys157 in vitro. However, these experimental conditions likely result in nonselective oxidation of cysteines in Rac1. Given that Cys157 lies at the base of the nucleotide binding pocket and is inaccessible to the solvent (Table 1), it is unlikely that glutathiolation of Cys157 would be observed over Cys18 unless the protein fold was perturbed.
Redox Regulation by RhoA
Whereas Rac1 can directly stimulate ROS production via NOX activation and RNS production via NOS activation, RhoA can regulate RNS production via NOS expression levels. For example, activated RhoA negatively regulates eNOS expression in human endothelial cells,127 bovine aortic endothelial cells,128 and murine endothelial cells.129 RhoA has also been shown to suppress iNOS expression and cellular nitric oxide levels in rat vascular smooth muscle cells,130 in the normal human liver cell line AKN-1, and in the human non-small cell lung carcinoma (NSCLC) cell line A-549.131 Therefore, RhoA appears to be actively engaged in downregulating NOS expression, which will reduce nitric oxide levels. As will be discussed below, addition of exogenous peroxynitrite to bovine aortic endothelial cells can promote RhoA activation.128 If this is true in general terms, RhoA may represent a node of negative feedback on RNS production. However, whether RhoA is involved in ROS regulation is still unknown. Thus, whereas Rac1 promotes oxidant formation, Rho can downregulate a subset of oxidant-generating enzymes. Rac and Rho have long been known to have antagonistic roles and perhaps this is yet another example.
Redox Regulation of RhoA
The pioneering work of Nimnual et al. established a model whereby the cellular redox state can be coupled to actin cytoskeleton dynamics through Rho GTPases.132 In HeLa cells, the ability of constitutively active Rac1G12V to induce membrane ruffling and integrin-stimulated cell spreading was attributed to Rac1-mediated redox-dependent oxidation in addition to the inactivation of low-molecular weight protein tyrosine phosphatase (LMW-PTP), which leads to the phosphorylation and activation of p190-RhoGAP.132 The subsequent downregulation of RhoA activity thereby allowed Rac1 activity to proceed unimpeded (Fig. 5). These results required the presence of the “Rho insert” region (residues 124–135), which is necessary for Rac interaction with NOX. In contrast, addition of the antioxidant NAC, which replenishes glutathione stores, prevented Rac-mediated downregulation of RhoA-GTP, as did the NOS inhibitor diphenyleneiodonium chloride (DPI). DPI treatment prevented oxidation and inactivation of LMW-PTP as well as the phosphorylation and activation of p190-RhoGAP. In addition, DPI converted Rac1-mediated lamellipodia formation to the formation of dorsal ruffles and failed to downregulate RhoA-GTP levels. The dorsal ruffle phenotype is consistent with impairment in Rac-mediated ROS production. Finally, hydrogen peroxide was sufficient to decrease RhoA-GTP levels in a manner dependent on the insert domain of Rac, suggesting that Rac-mediated ROS generation was required. In contrast, ectopic expression of the hydrogen peroxide-scavenging enzyme catalase inhibited integrin-stimulated cell spreading. This inhibitory effect of catalase was rescued by non-oxidizable LMW-PTPC12S. Collectively, these results strongly support the hypothesis that Rac-mediated organization of the actin cytoskeleton is regulated, in part, through Rac-induced ROS. This decreases RhoA-GTP levels due to the redox-mediated activation of p190-RhoGAP, a key RhoGAP. Similar results were later obtained in K-Ras-transformed normal rat kidney cells where DPI treatment was able to restore both RhoA-GTP levels and actin stress fiber formation that had been downregulated due to K-Ras activation of Rac1/NOX1.133
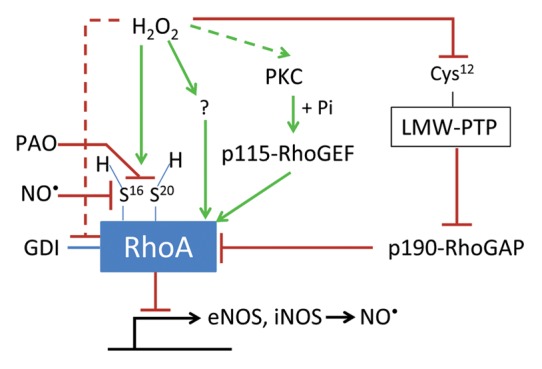
Figure 5. Redox-dependent regulation of RhoA. RhoA can be either activated or inactivated by oxidants. Reported modes of activation include: (1) PKC-dependent phosphorylation and activation of p115-RhoGEF; (2) release of GDI from RhoA; and (3) direct oxidation followed by reduction of cysteines 16 and 20. Reported modes of deactivation include: (1) phosphorylation and inactivation of p190-RhoGAP due to decreased activity of low-molecular weight protein tyrosine phosphatase (LMW-PTP) upon oxidation of an active site cysteine; (2) thiol cross-linking of cysteines 16 and 20 by phenylarsine oxide (PAO); and (3) NO•-induced RhoA S-nitrosation. However, it is still unclear whether oxidation of RhoA alters its interaction with downstream effectors.
Another RhoA deactivation mechanism was revealed by Zuckerbraun et al.,134 who showed that exogenously added nitric oxide reduced RhoA activity, as determined by decreased GTP binding in GST-Rhotekin-RBD pulldowns and by serum-stimulated stress fiber formation in rat aortic vascular smooth muscle cells. Direct modification of RhoA by S-nitrosation was verified using an anti-nitrosocysteine antibody. S-nitrosation was found to impair GTP binding, which consequently hinders RhoA activation. However, the exact site of S-nitrosation was not determined.
Finally, phenylarsine oxide (PAO) has been shown to deactivate RhoA. PAO can crosslink vicinal thiol groups and is a known phosphatase inhibitor.135 Treatment of human colonic Caco-2 cells with PAO exhibited a dose-dependent reduction in actin stress fibers, a phenotype consistent with reduced RhoA activity. Moreover, pull-down experiments revealed a PAO concentration-dependent decrease in the levels of GTP-bound RhoA. The authors performed nucleotide dissociation and association assays in vitro and observed a loss of nucleotide binding in RhoA but not Rac1 upon exposure to PAO.135 Mass spectrometry revealed that PAO crosslinked 2 vicinal cysteines in RhoA to occlude nucleotide binding. These effects were specific to RhoA but not Rac1 as only Rho GTPases (RhoA, RhoB, RhoC, and RhoE) possess vicinal cysteines (Cys16 and Cys20) within the guanine nucleotide-binding region. In contrast, Rac1 contains only a single cysteine (Cys18) and reaction with PAO does not occlude nucleotide binding. However, the binding site of PAO on Rac1 was not probed as no change in activity was observed upon exposure. Whether PAO-mediated oxidation of RhoA, which prevents nucleotide binding, is related to the function of PAO as a tyrosine phosphatase inhibitor is presently unclear.
In other contexts, a number of studies have demonstrated that ROS/RNS are capable of activating RhoA. For example, we have previously reported in vitro evidence that supports RhoA activation upon oxidation.44 Treatment of RhoA with nitrogen dioxide induced nucleotide dissociation and subsequent disulfide bond formation between Cys16 and Cys.20 The disulfide bond occluded nucleotide binding. However, nucleotide association could be observed when the radical scavenging agent ascorbate was present in the reaction. Based on these findings, we speculate that exposure of RhoA to select oxidants can promote intramolecular disulfide formation. However, under normal physiological conditions, the disulfide is likely reduced, which results in guanine nucleotide cycling and activation; under conditions of oxidative stress, disulfide reduction may not occur, thereby promoting RhoA inactivation. Thus, oxidation of RhoA could lead to activation or inactivation depending on the cellular redox state.
In a study on peroxynitrite-mediated diabetes-induced endothelial dysfunction, peroxynitrite was shown to activate RhoA. Either high glucose or exogenous peroxynitrite treatment for 18 h doubled the amount of active RhoA in bovine aortic endothelial cells, as determined by pulldown with GST-RBD (GST-Rhotekin Rho-binding domain) beads.128 The glucose-induced increase in RhoA-GTP was blocked by FeTTPS (5,10,15,20-tetrakis[4-sulfonatophenyl]porphyrinato iron [III]), which isomerizes peroxynitrite to nitrate. In human breast cancer cell lines, Lopez-Haber and Kazanietz found that the Jak2 inhibitor cucurbitacin I inhibited Rac1 activation but enhanced activation of RhoA and RhoA downstream signaling independent of Jak2. The effects of cucurbitacin I on Rac1 and RhoA were dependent on intracellular ROS generation as treatment with the ROS scavenger NAC ablated the response due to cucurbitacin I exposure.136 Yu et al. characterized the time course of RhoA activity following H2O2 stimulation in MDCK (Madin-Darby canine kidney) cells. They found a ~1.75-fold increase in RhoA-GTP levels that peaked approximately 5 min post stimulation.137 Likewise, in the human lung adenocarcinoma epithelial cell line A549, H2O2 was shown to activate RhoA by ~2.5-fold after 15 min, which was concomitant with stress fiber formation.138 As the mechanism of RhoA activation was not investigated in any of these studies, it is unclear whether RhoA activity is regulated directly by oxidants or indirectly through the redox-sensitive LMW-PTP, another GAP, GEF, or GDI.
Indeed, the mechanisms by which ROS and RNS regulate RhoA activity are not yet fully understood, and emerging data suggest that they are likely diverse. Thus, the seemingly opposing observations above may be explained by differential redox regulation that varies between cell type and stimuli. Depending on the specific cellular environment, redox agents can either lead to the formation of intramolecular disulfide bonds that prevent guanine nucleotide binding,44 thereby inactivating RhoA, or can facilitate guanine nucleotide dissociation,44 thereby activating RhoA due to the higher levels of cellular GTP vs. GDP. In a separate mode of activation, indirect regulation of RhoA by oxidants has been observed as well. A study on arginase activity in endothelial cells indicated that peroxynitrite and peroxide increased PKC-mediated phosphorylation and activation of p115-RhoGEF, which subsequently increased RhoA-GTP levels.139 Thus, in at least one context, activation of a RhoGEF can lead to RhoA activation by ROS. Kondrikov et al., however, proposed a different mechanism for ROS-dependent RhoA activation in lung fibroblasts. In their model, it was proposed that release of GDI from RhoA would promote RhoGEF binding and a subsequent increase in RhoA-GTP levels.140
In contrast, few studies have determined whether ROS and RNS can act directly on Rho GTPases in cells to activate them through covalent modification of specific residues. Our group has observed direct activation of RhoA by ROS that involves the 2 redox-sensitive cysteine residues (Cys16 and Cys20) within the phosphoryl-binding loop of RhoA. By treating REF-52 cells (rat embryonic fibroblasts) with low concentrations of exogenous peroxide, we were able to show increased stress fiber formation and concurrent RhoA-GTP loading, both of which were abolished by mutating the redox-sensitive cysteines to alanine residues.141 Together, these findings imply that RhoA activation by ROS can be either direct (amino acid residue modification) or indirect (RhoA regulator modulation) depending on the cellular context, or could even be a combination of both, which has not been ruled out in any study.
Peroxide reacts with thiolates at rates dependent on both the pKa and solvent accessibility of the thiol.142 As predicted by Winterbourn et al.,48,143 the rate of peroxide oxidation of thiol-containing proteins is likely too slow to reversibly regulate GTPase activity on the timescale of most physiological events. Therefore, we predict that oxidants capable of reacting with the redox-sensitive p-loop cysteine in Rho GTPases to generate a thiyl radical (i.e., nitrogen dioxide and superoxide) are more likely to alter cellular Rho GTPase activity by promoting nucleotide exchange. As the redox-sensitive cysteine forms direct interactions with the guanine substrate, non-radical oxidation likely alters guanine nucleotide binding and leads to sustained guanine nucleotide exchange and activation (Fig. 6).

Figure 6. Thiol oxidation of Rac1 and RhoA GTPases and effects on nucleotide binding and exchange. ‘M’ represents RhoA or Rac1. Rac1 and RhoA have a redox-sensitive thiol at Cys18 and Cys,20 respectively. At physiological pH, this is populated in the reactive thiol anion form. Free radical-mediated oxidants, such as NO2•, can promote thiyl radical formation, cause nucleotide oxidation, and enhance the intrinsic rate of nucleotide dissociation by ~1000-fold, leading to activation of Rac1 or RhoA. In addition, Rac1 and RhoA are susceptible to 2-electron oxidation. Oxidation of M-S- to sulfenic acid by peroxide promotes the intrinsic rate of nucleotide dissociation by ~10-fold. Oxidation to sulfinic acid, approximated by use of a Cys > Asp mutation (Rac1C18D and RhoAC20D), shows greatly increased rates of intrinsic nucleotide dissociation (unpublished results). Further oxidation to sulfonic acid is irreversible and can lead to protein degradation, which likely occurs under oxidative stress conditions. Distinct oxidative modifications can differentially affect the activity of Rac1 and RhoA. RhoA can be inactivated upon reaction with nitric oxide (NO•) in cellulo and in vitro. Rac1 glutathiolation greatly increases nucleotide exchange, similar to the Rac1C18D oxidation mimetic. The formation of an intramolecular disulfide bond is unique to RhoA due to the additional presence of Cys16; this results in loss of nucleotide binding. However, conditions that promote reduction of the disulfide bond in RhoA can facilitate nucleotide exchange and activation.
Conclusions and Future Directions
While the guanine nucleotide binding properties of RhoA and Rac1 can be modulated directly by oxidants in vitro, few cellular studies have been conducted to determine the role of direct oxidation in regulating their activity. Nevertheless, increasing evidence has tied both RhoA and Rac1 to regulation by oxidation in cells. Given the relative reactivity of free radical oxidants compared with 2-electron oxidation, it is likely that free radical oxidation of reactive cysteines in the p-loop of Rho GTPases regulates Rho GTPase activity and signaling in vivo. Under reducing conditions, we speculate that free radical oxidants promote Rac1 and RhoA activation by facilitating nucleotide dissociation and GTP-loading, similar to the function of GEFs. However, under conditions of oxidative stress, these GTPases may become inactivated. The studies highlighted in this review raise numerous questions, several of which require technically challenging approaches to address. For example, as the redox-sensitive thiol in Rho GTPases likely has an altered pKa, which causes it to be in the thiolate state at physiological pH, the sensitivity of this site to both 1-electron and 2-electron oxidation events is increased relative to other cellular thiols that lack altered pKa values. Distinct oxidative modifications can result, such as nitrosation, glutathiolation, sulfenic, sulfinic, sulfonic acid, or disulfide formation, and each of these oxidation products may uniquely alter Rho GTPase activity. Therefore, using in vitro assays to elucidate how distinct oxidative byproducts alter Rho GTPase activity using stoichiometric and specific oxidation, as has been done with Ras,37,38 will prove valuable in interpreting cellular and in vivo data on Rho oxidation. Our preliminary findings on Rac1 suggest that distinct oxidative thiol modifications have different effects on nucleotide binding properties, but verification in vivo is required. To better understand how oxidants directly regulate redox-sensitive Rho GTPases in cellulo or in vivo, the Rho GTPase oxidation state should be determined when possible or redox-insensitive variants should be used. In addition, the vast majority of observations in cells have utilized exogenous oxidants that not only alter the levels of oxidants in a non-physiological manner but also alter the redox state of the cell. Investigation of the cellular redox state with endogenously produced oxidants will be necessary to characterize how Rho GTPases are regulated by oxidation in physiological settings. The report by Aghajanian et al.141 was one of the first to show the cellular activation of RhoAWT but not of redox-insensitive RhoA variants upon exposure to exogenously and endogenously applied oxidants and currently remains the only study that reports on the direct oxidation of RhoA in cells. This type of study is still scarce-to-nonexistent for other Rho family GTPases. Given the conservation of redox-sensitive cysteines in Rho GTPases, it is highly likely that other Rho family members are also regulated directly by RNS and ROS. Determining the specificity of ROS/RNS actions on Rho proteins, and uncovering the mechanisms behind these events, will be exciting challenges for the foreseeable future.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work has been supported by grants from the National Institutes of Health through grant numbers CA089614B to S.L.C., T32 GM008570 to A.H., and CA161494 and CA042978 to A.D.C.
Glossary
Abbreviations:
- BSO
L-buthionine sulfoximine
- CO3
carbonate radicals
- DPI
diphenyleneiodonium chloride
- DUB
deubiquitinating enzymes
- eNOS
endothelial NOS
- FeTTPS
5,10,15,20-tetrakis(4-sulfonatophenyl) porphyrinato iron (III)
- GAPs
GTPase activating proteins
- GDIs
guanine nucleotide dissociation inhibitors
- GEFs
guanine nucleotide exchange factors
- GSH
glutathione
- GSH-Pxs
glutathione peroxidases
- GSSG
oxidized glutathione
- H2O2
peroxide
- HAECs
human aortic endothelial cells
- iNOS
inducible NOS
- LMW-PTP
low-molecular weight protein tyrosine phosphatase
- NAC
N-acetylcysteine
- MDCK
Madin-Darby canine kidney cells
- NFκB
nuclear factor-κB
- NMuMG
normal mouse mammary gland epithelial cells
- NO
nitric oxide
- NO2
nitrogen dioxide
- nNOS
neuronal NOS
- NOS
nitric oxide synthase
- Nox
NADPH oxidase
- NSCLC
non-small cell lung carcinoma cells
- O2-
superoxide
- ONOO-
peroxynitrite
- PAO
phenylarsine oxide
- PTM
post-translational modification
- RBD
Rho-binding domain
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SNO
s-nitrosothiols
- SOD
superoxide dismutase
References
- 1.Giannoni E, Buricchi F, Grimaldi G, Parri M, Cialdai F, Taddei ML, Raugei G, Ramponi G, Chiarugi P. Redox regulation of anoikis: reactive oxygen species as essential mediators of cell survival. Cell Death Differ. 2008;15:867–78. doi: 10.1038/cdd.2008.3. [DOI] [PubMed] [Google Scholar]
- 2.Chiu J, Dawes IW. Redox control of cell proliferation. Trends Cell Biol. 2012;22:592–601. doi: 10.1016/j.tcb.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 3.Koh KP, Rao A. DNA methylation and methylcytosine oxidation in cell fate decisions. Curr Opin Cell Biol. 2013;25:152–61. doi: 10.1016/j.ceb.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hurd TR, DeGennaro M, Lehmann R. Redox regulation of cell migration and adhesion. Trends Cell Biol. 2012;22:107–15. doi: 10.1016/j.tcb.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corcoran A, Cotter TG. Redox regulation of protein kinases. FEBS J. 2013;280:1944–65. doi: 10.1111/febs.12224. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell L, Hobbs GA, Aghajanian A, Campbell SL. Redox regulation of Ras and Rho GTPases: mechanism and function. Antioxid Redox Signal. 2013;18:250–8. doi: 10.1089/ars.2012.4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci. 2005;118:843–6. doi: 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- 8.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 9.Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–80. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 10.Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13:13–22. doi: 10.1016/S0962-8924(02)00004-1. [DOI] [PubMed] [Google Scholar]
- 11.DerMardirossian C, Bokoch GM. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005;15:356–63. doi: 10.1016/j.tcb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Mata R, Boulter E, Burridge K. The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol. 2011;12:493–504. doi: 10.1038/nrm3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Visvikis O, Maddugoda MP, Lemichez E. Direct modifications of Rho proteins: deconstructing GTPase regulation. Biol Cell. 2010;102:377–89. doi: 10.1042/BC20090151. [DOI] [PubMed] [Google Scholar]
- 14.Adamson P, Marshall CJ, Hall A, Tilbrook PA. Post-translational modifications of p21rho proteins. J Biol Chem. 1992;267:20033–8. [PubMed] [Google Scholar]
- 15.Adamson P, Paterson HF, Hall A. Intracellular localization of the P21rho proteins. J Cell Biol. 1992;119:617–27. doi: 10.1083/jcb.119.3.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts PJ, Mitin N, Keller PJ, Chenette EJ, Madigan JP, Currin RO, Cox AD, Wilson O, Kirschmeier P, Der CJ. Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J Biol Chem. 2008;283:25150–63. doi: 10.1074/jbc.M800882200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bokoch GM, Bohl BP, Chuang TH. Guanine nucleotide exchange regulates membrane translocation of Rac/Rho GTP-binding proteins. J Biol Chem. 1994;269:31674–9. [PubMed] [Google Scholar]
- 18.Johnson JL, Erickson JW, Cerione RA. New insights into how the Rho guanine nucleotide dissociation inhibitor regulates the interaction of Cdc42 with membranes. J Biol Chem. 2009;284:23860–71. doi: 10.1074/jbc.M109.031815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Navarro-Lérida I, Sánchez-Perales S, Calvo M, Rentero C, Zheng Y, Enrich C, Del Pozo MA. A palmitoylation switch mechanism regulates Rac1 function and membrane organization. EMBO J. 2012;31:534–51. doi: 10.1038/emboj.2011.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hodges-Loaiza HB, Parker LE, Cox AD. Chapter 3 - Prenylation and Phosphorylation of Ras Superfamily Small GTPases. In: Christine A. Hrycyna MOB, Fuyuhiko T, eds. The Enzymes: Academic Press, 2011:43-69. [Google Scholar]
- 21.Ellerbroek SM, Wennerberg K, Burridge K. Serine phosphorylation negatively regulates RhoA in vivo. J Biol Chem. 2003;278:19023–31. doi: 10.1074/jbc.M213066200. [DOI] [PubMed] [Google Scholar]
- 22.Forget MA, Desrosiers RR, Gingras D, Béliveau R. Phosphorylation states of Cdc42 and RhoA regulate their interactions with Rho GDP dissociation inhibitor and their extraction from biological membranes. Biochem J. 2002;361:243–54. doi: 10.1042/0264-6021:3610243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de la Vega M, Burrows JF, Johnston JA. Ubiquitination: Added complexity in Ras and Rho family GTPase function. Small GTPases. 2011;2:192–201. doi: 10.4161/sgtp.2.4.16707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sadowski M, Suryadinata R, Tan AR, Roesley SN, Sarcevic B. Protein monoubiquitination and polyubiquitination generate structural diversity to control distinct biological processes. IUBMB Life. 2012;64:136–42. doi: 10.1002/iub.589. [DOI] [PubMed] [Google Scholar]
- 25.Jura N, Bar-Sagi D. Mapping cellular routes of Ras: a ubiquitin trail. Cell Cycle. 2006;5:2744–7. doi: 10.4161/cc.5.23.3532. [DOI] [PubMed] [Google Scholar]
- 26.Jura N, Scotto-Lavino E, Sobczyk A, Bar-Sagi D. Differential modification of Ras proteins by ubiquitination. Mol Cell. 2006;21:679–87. doi: 10.1016/j.molcel.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 27.Sasaki AT, Carracedo A, Locasale JW, Anastasiou D, Takeuchi K, Kahoud ER, Haviv S, Asara JM, Pandolfi PP, Cantley LC. Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci Signal. 2011;4:ra13. doi: 10.1126/scisignal.2001518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baker R, Lewis SM, Sasaki AT, Wilkerson EM, Locasale JW, Cantley LC, Kuhlman B, Dohlman HG, Campbell SL. Site-specific monoubiquitination activates Ras by impeding GTPase-activating protein function. Nat Struct Mol Biol. 2013;20:46–52. doi: 10.1038/nsmb.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei J, Mialki RK, Dong S, Khoo A, Mallampalli RK, Zhao Y, Zhao J. A new mechanism of RhoA ubiquitination and degradation: roles of SCF(FBXL19) E3 ligase and Erk2. Biochim Biophys Acta. 2013;1833:2757–64. doi: 10.1016/j.bbamcr.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nethe M, Hordijk PL. The role of ubiquitylation and degradation in RhoGTPase signalling. J Cell Sci. 2010;123:4011–8. doi: 10.1242/jcs.078360. [DOI] [PubMed] [Google Scholar]
- 31.Nethe M, Anthony EC, Fernandez-Borja M, Dee R, Geerts D, Hensbergen PJ, Deelder AM, Schmidt G, Hordijk PL. Focal-adhesion targeting links caveolin-1 to a Rac1-degradation pathway. J Cell Sci. 2010;123:1948–58. doi: 10.1242/jcs.062919. [DOI] [PubMed] [Google Scholar]
- 32.Visvikis O, Lorès P, Boyer L, Chardin P, Lemichez E, Gacon G. Activated Rac1, but not the tumorigenic variant Rac1b, is ubiquitinated on Lys 147 through a JNK-regulated process. FEBS J. 2008;275:386–96. doi: 10.1111/j.1742-4658.2007.06209.x. [DOI] [PubMed] [Google Scholar]
- 33.Lander HM, Hajjar DP, Hempstead BL, Mirza UA, Chait BT, Campbell S, Quilliam LA. A molecular redox switch on p21(ras). Structural basis for the nitric oxide-p21(ras) interaction. J Biol Chem. 1997;272:4323–6. doi: 10.1074/jbc.272.7.4323. [DOI] [PubMed] [Google Scholar]
- 34.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–28. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 35.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–90. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forman HJ, Fukuto JM, Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol. 2004;287:C246–56. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 37.Williams JG, Pappu K, Campbell SL. Structural and biochemical studies of p21Ras S-nitrosylation and nitric oxide-mediated guanine nucleotide exchange. Proc Natl Acad Sci U S A. 2003;100:6376–81. doi: 10.1073/pnas.1037299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hobbs GA, Bonini MG, Gunawardena HP, Chen X, Campbell SL. Glutathiolated Ras: characterization and implications for Ras activation. Free Radic Biol Med. 2013;57:221–9. doi: 10.1016/j.freeradbiomed.2012.10.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heo J, Prutzman KC, Mocanu V, Campbell SL. Mechanism of free radical nitric oxide-mediated Ras guanine nucleotide dissociation. J Mol Biol. 2005;346:1423–40. doi: 10.1016/j.jmb.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 40.Davis MF, Zhou L, Ehrenshaft M, Ranguelova K, Gunawardena HP, Chen X, Bonini MG, Mason RP, Campbell SL. Detection of Ras GTPase protein radicals through immuno-spin trapping. Free Radic Biol Med. 2012;53:1339–45. doi: 10.1016/j.freeradbiomed.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 42.Lim KH, Ancrile BB, Kashatus DF, Counter CM. Tumour maintenance is mediated by eNOS. Nature. 2008;452:646–9. doi: 10.1038/nature06778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heo J, Campbell SL. Mechanism of redox-mediated guanine nucleotide exchange on redox-active Rho GTPases. J Biol Chem. 2005;280:31003–10. doi: 10.1074/jbc.M504768200. [DOI] [PubMed] [Google Scholar]
- 44.Heo J, Raines KW, Mocanu V, Campbell SL. Redox regulation of RhoA. Biochemistry. 2006;45:14481–9. doi: 10.1021/bi0610101. [DOI] [PubMed] [Google Scholar]
- 45.Sanchez R, Riddle M, Woo J, Momand J. Prediction of reversibly oxidized protein cysteine thiols using protein structure properties. Protein Sci. 2008;17:473–81. doi: 10.1110/ps.073252408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spadaro D, Yun BW, Spoel SH, Chu C, Wang YQ, Loake GJ. The redox switch: dynamic regulation of protein function by cysteine modifications. Physiol Plant. 2010;138:360–71. doi: 10.1111/j.1399-3054.2009.01307.x. [DOI] [PubMed] [Google Scholar]
- 47.Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–47. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 48.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–61. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 49.Trujillo M, Radi R. Peroxynitrite reaction with the reduced and the oxidized forms of lipoic acid: new insights into the reaction of peroxynitrite with thiols. Arch Biochem Biophys. 2002;397:91–8. doi: 10.1006/abbi.2001.2619. [DOI] [PubMed] [Google Scholar]
- 50.Folkes LK, Wardman P. Kinetics of the reaction between nitric oxide and glutathione: implications for thiol depletion in cells. Free Radic Biol Med. 2004;37:549–56. doi: 10.1016/j.freeradbiomed.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 51.Davis MF, Zhou L, Ehrenshaft M, Ranguelova K, Gunawardena HP, Chen X, Bonini MG, Mason RP, Campbell SL. Detection of Ras GTPase protein radicals through immuno-spin trapping. Free Radic Biol Med. 2012;53:1339–45. doi: 10.1016/j.freeradbiomed.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem. 2004;266:37–56. doi: 10.1023/B:MCBI.0000049134.69131.89. [DOI] [PubMed] [Google Scholar]
- 53.Seet RCS, Lee CYJ, Lim ECH, Tan JJH, Quek AML, Chong WL, Looi WF, Huang SH, Wang H, Chan YH, et al. Oxidative damage in Parkinson disease: Measurement using accurate biomarkers. Free Radic Biol Med. 2010;48:560–6. doi: 10.1016/j.freeradbiomed.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 54.Vogiatzi G, Tousoulis D, Stefanadis C. The role of oxidative stress in atherosclerosis. Hellenic J Cardiol. 2009;50:402–9. [PubMed] [Google Scholar]
- 55.Rains JL, Jain SK. Oxidative stress, insulin signaling, and diabetes. Free Radic Biol Med. 2011;50:567–75. doi: 10.1016/j.freeradbiomed.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gruber J, Schaffer S, Halliwell B. The mitochondrial free radical theory of ageing--where do we stand? Front Biosci. 2008;13:6554–79. doi: 10.2741/3174. [DOI] [PubMed] [Google Scholar]
- 57.Florence TM. The role of free radicals in disease. Aust N Z J Ophthalmol. 1995;23:3–7. doi: 10.1111/j.1442-9071.1995.tb01638.x. [DOI] [PubMed] [Google Scholar]
- 58.Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33:829–37, 837a-837d. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu J, Hughes TE, Sessa WC. The first 35 amino acids and fatty acylation sites determine the molecular targeting of endothelial nitric oxide synthase into the Golgi region of cells: a green fluorescent protein study. J Cell Biol. 1997;137:1525–35. doi: 10.1083/jcb.137.7.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou L, Zhu DY. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide. 2009;20:223–30. doi: 10.1016/j.niox.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 61.Cho HJ, Xie QW, Calaycay J, Mumford RA, Swiderek KM, Lee TD, Nathan C. Calmodulin is a subunit of nitric oxide synthase from macrophages. J Exp Med. 1992;176:599–604. doi: 10.1084/jem.176.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.García-Cardeña G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–4. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- 63.Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1–12. doi: 10.1152/ajpregu.00323.2002. [DOI] [PubMed] [Google Scholar]
- 64.Beck KF, Eberhardt W, Frank S, Huwiler A, Messmer UK, Mühl H, Pfeilschifter J. Inducible NO synthase: role in cellular signalling. J Exp Biol. 1999;202:645–53. doi: 10.1242/jeb.202.6.645. [DOI] [PubMed] [Google Scholar]
- 65.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48:158–67. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giulivi C, Kato K, Cooper CE. Nitric oxide regulation of mitochondrial oxygen consumption I: cellular physiology. Am J Physiol Cell Physiol. 2006;291:C1225–31. doi: 10.1152/ajpcell.00307.2006. [DOI] [PubMed] [Google Scholar]
- 67.Basu S, Azarova NA, Font MD, King SB, Hogg N, Gladwin MT, Shiva S, Kim-Shapiro DB. Nitrite reductase activity of cytochrome c. J Biol Chem. 2008;283:32590–7. doi: 10.1074/jbc.M806934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lynch RE, Fridovich I. Permeation of the erythrocyte stroma by superoxide radical. J Biol Chem. 1978;253:4697–9. [PubMed] [Google Scholar]
- 69.Salvador A, Sousa J, Pinto RE. Hydroperoxyl, superoxide and pH gradients in the mitochondrial matrix: a theoretical assessment. Free Radic Biol Med. 2001;31:1208–15. doi: 10.1016/S0891-5849(01)00707-9. [DOI] [PubMed] [Google Scholar]
- 70.Fridovich I. Superoxide radical: an endogenous toxicant. Annu Rev Pharmacol Toxicol. 1983;23:239–57. doi: 10.1146/annurev.pa.23.040183.001323. [DOI] [PubMed] [Google Scholar]
- 71.Forman HJ, Fridovich I. Superoxide dismutase: a comparison of rate constants. Arch Biochem Biophys. 1973;158:396–400. doi: 10.1016/0003-9861(73)90636-X. [DOI] [PubMed] [Google Scholar]
- 72.Weisiger RA, Fridovich I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. J Biol Chem. 1973;248:4793–6. [PubMed] [Google Scholar]
- 73.Karnati S, Lüers G, Pfreimer S, Baumgart-Vogt E. Mammalian SOD2 is exclusively located in mitochondria and not present in peroxisomes. Histochem Cell Biol. 2013;140:105–17. doi: 10.1007/s00418-013-1099-4. [DOI] [PubMed] [Google Scholar]
- 74.Antunes F, Cadenas E. Estimation of H2O2 gradients across biomembranes. FEBS Lett. 2000;475:121–6. doi: 10.1016/S0014-5793(00)01638-0. [DOI] [PubMed] [Google Scholar]
- 75.Apostol I, Heinstein PF, Low PS. Rapid Stimulation of an Oxidative Burst during Elicitation of Cultured Plant Cells : Role in Defense and Signal Transduction. Plant Physiol. 1989;90:109–16. doi: 10.1104/pp.90.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Touyz RM, Briones AM, Sedeek M, Burger D, Montezano AC. NOX isoforms and reactive oxygen species in vascular health. Mol Interv. 2011;11:27–35. doi: 10.1124/mi.11.1.5. [DOI] [PubMed] [Google Scholar]
- 77.Dang PM, Cross AR, Quinn MT, Babior BM. Assembly of the neutrophil respiratory burst oxidase: a direct interaction between p67PHOX and cytochrome b558 II. Proc Natl Acad Sci U S A. 2002;99:4262–5. doi: 10.1073/pnas.072345299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cuzzocrea S, Mazzon E, Dugo L, Di Paola R, Caputi AP, Salvemini D. Superoxide: a key player in hypertension. FASEB J. 2004;18:94–101. doi: 10.1096/fj.03-0428com. [DOI] [PubMed] [Google Scholar]
- 79.Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337–49. doi: 10.1016/S0891-5849(02)00905-X. [DOI] [PubMed] [Google Scholar]
- 80.McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–55. [PubMed] [Google Scholar]
- 81.Tainer JA, Getzoff ED, Beem KM, Richardson JS, Richardson DC. Determination and analysis of the 2 A-structure of copper, zinc superoxide dismutase. J Mol Biol. 1982;160:181–217. doi: 10.1016/0022-2836(82)90174-7. [DOI] [PubMed] [Google Scholar]
- 82.Borgstahl GE, Parge HE, Hickey MJ, Beyer WF, Jr., Hallewell RA, Tainer JA. The structure of human mitochondrial manganese superoxide dismutase reveals a novel tetrameric interface of two 4-helix bundles. Cell. 1992;71:107–18. doi: 10.1016/0092-8674(92)90270-M. [DOI] [PubMed] [Google Scholar]
- 83.Marklund SL, Holme E, Hellner L. Superoxide dismutase in extracellular fluids. Clin Chim Acta. 1982;126:41–51. doi: 10.1016/0009-8981(82)90360-6. [DOI] [PubMed] [Google Scholar]
- 84.Antonyuk SV, Strange RW, Marklund SL, Hasnain SS. The structure of human extracellular copper-zinc superoxide dismutase at 1.7 A resolution: insights into heparin and collagen binding. J Mol Biol. 2009;388:310–26. doi: 10.1016/j.jmb.2009.03.026. [DOI] [PubMed] [Google Scholar]
- 85.Gaetani GF, Galiano S, Canepa L, Ferraris AM, Kirkman HN. Catalase and glutathione peroxidase are equally active in detoxification of hydrogen peroxide in human erythrocytes. Blood. 1989;73:334–9. [PubMed] [Google Scholar]
- 86.McCray PB, Gibson DD, Fong KL, Hornbrook KR. Effect of glutathione peroxidase activity on lipid peroxidation in biological membranes. Biochim Biophys Acta. 1976;431:459–68. doi: 10.1016/0005-2760(76)90212-5. [DOI] [PubMed] [Google Scholar]
- 87.Montazerifar F, Hashemi M, Karajibani M, Sanadgol H, Dikshit M. Evaluation of lipid peroxidation and erythrocyte glutathione peroxidase and superoxide dismutase in hemodialysis patients. Saudi J Kidney Dis Transpl. 2012;23:274–9. [PubMed] [Google Scholar]
- 88.Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5:9–19. doi: 10.1097/WOX.0b013e3182439613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kosower NS, Kosower EM. The glutathione status of cells. Int Rev Cytol. 1978;54:109–60. doi: 10.1016/S0074-7696(08)60166-7. [DOI] [PubMed] [Google Scholar]
- 90.Anderson ME. Glutathione: an overview of biosynthesis and modulation. Chem Biol Interact. 1998;111-112:1–14. doi: 10.1016/S0009-2797(97)00146-4. [DOI] [PubMed] [Google Scholar]
- 91.Buettner GR. The pecking order of free radicals and antioxidants: lipid peroxidation, alpha-tocopherol, and ascorbate. Arch Biochem Biophys. 1993;300:535–43. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- 92.Kennedy TA, Liebler DC. Peroxyl radical scavenging by beta-carotene in lipid bilayers. Effect of oxygen partial pressure. J Biol Chem. 1992;267:4658–63. [PubMed] [Google Scholar]
- 93.Wardman P, von Sonntag C. Kinetic factors that control the fate of thiyl radicals in cells. Methods Enzymol. 1995;251:31–45. doi: 10.1016/0076-6879(95)51108-3. [DOI] [PubMed] [Google Scholar]
- 94.Ross D, Norbeck K, Moldéus P. The generation and subsequent fate of glutathionyl radicals in biological systems. J Biol Chem. 1985;260:15028–32. [PubMed] [Google Scholar]
- 95.Salsbury FR, Jr., Knutson ST, Poole LB, Fetrow JS. Functional site profiling and electrostatic analysis of cysteines modifiable to cysteine sulfenic acid. Protein Sci. 2008;17:299–312. doi: 10.1110/ps.073096508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gaston B. Nitric oxide and thiol groups. Biochim Biophys Acta. 1999;1411:323–33. doi: 10.1016/S0005-2728(99)00023-7. [DOI] [PubMed] [Google Scholar]
- 97.Pryor WA, Houk KN, Foote CS, Fukuto JM, Ignarro LJ, Squadrito GL, Davies KJ. Free radical biology and medicine: it’s a gas, man! Am J Physiol Regul Integr Comp Physiol. 2006;291:R491–511. doi: 10.1152/ajpregu.00614.2005. [DOI] [PubMed] [Google Scholar]
- 98.Biswas S, Chida AS, Rahman I. Redox modifications of protein-thiols: emerging roles in cell signaling. Biochem Pharmacol. 2006;71:551–64. doi: 10.1016/j.bcp.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 99.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–4. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 100.Ying J, Clavreul N, Sethuraman M, Adachi T, Cohen RA. Thiol oxidation in signaling and response to stress: detection and quantification of physiological and pathophysiological thiol modifications. Free Radic Biol Med. 2007;43:1099–108. doi: 10.1016/j.freeradbiomed.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453–62. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- 102.Ueyama T, Geiszt M, Leto TL. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Mol Cell Biol. 2006;26:2160–74. doi: 10.1128/MCB.26.6.2160-2174.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Leto TL, Morand S, Hurt D, Ueyama T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid Redox Signal. 2009;11:2607–19. doi: 10.1089/ars.2009.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Werner E. GTPases and reactive oxygen species: switches for killing and signaling. J Cell Sci. 2004;117:143–53. doi: 10.1242/jcs.00937. [DOI] [PubMed] [Google Scholar]
- 105.Diebold I, Djordjevic T, Petry A, Hatzelmann A, Tenor H, Hess J, Görlach A. Phosphodiesterase 2 mediates redox-sensitive endothelial cell proliferation and angiogenesis by thrombin via Rac1 and NADPH oxidase 2. Circ Res. 2009;104:1169–77. doi: 10.1161/CIRCRESAHA.109.196592. [DOI] [PubMed] [Google Scholar]
- 106.Myant KB, Cammareri P, McGhee EJ, Ridgway RA, Huels DJ, Cordero JB, Schwitalla S, Kalna G, Ogg EL, Athineos D, et al. ROS production and NFκB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell. 2013;12:761–73. doi: 10.1016/j.stem.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Barth BM, Stewart-Smeets S, Kuhn TB. Proinflammatory cytokines provoke oxidative damage to actin in neuronal cells mediated by Rac1 and NADPH oxidase. Mol Cell Neurosci. 2009;41:274–85. doi: 10.1016/j.mcn.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ferraro D, Corso S, Fasano E, Panieri E, Santangelo R, Borrello S, Giordano S, Pani G, Galeotti T. Pro-metastatic signaling by c-Met through RAC-1 and reactive oxygen species (ROS) Oncogene. 2006;25:3689–98. doi: 10.1038/sj.onc.1209409. [DOI] [PubMed] [Google Scholar]
- 109.Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, Nelson K, Luo M, Paulson H, Schöneich C, et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–70. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Selvakumar B, Hess DT, Goldschmidt-Clermont PJ, Stamler JS. Co-regulation of constitutive nitric oxide synthases and NADPH oxidase by the small GTPase Rac. FEBS Lett. 2008;582:2195–202. doi: 10.1016/j.febslet.2008.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jyoti A, Singh A, Dubey M, Kumar S, Saluja R, Keshari RS, Verma A, Chandra T, Kumar A, Bajpai VK, et al. Interaction of inducible nitric oxide synthase with Rac2 regulates reactive oxygen and nitrogen species generation in the human neutrophil phagosomes: Implication in microbial killing. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2012.4970. [DOI] [PubMed] [Google Scholar]
- 112.Morgan MJ, Liu ZG. Reactive oxygen species in TNFalpha-induced signaling and cell death. Mol Cells. 2010;30:1–12. doi: 10.1007/s10059-010-0105-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nakajima S, Kitamura M. Bidirectional regulation of NFκB by reactive oxygen species: a role of unfolded protein response. Free Radic Biol Med. 2013;65:162–74. doi: 10.1016/j.freeradbiomed.2013.06.020. [DOI] [PubMed] [Google Scholar]
- 114.Tobar N, Cáceres M, Santibáñez JF, Smith PC, Martínez J. RAC1 activity and intracellular ROS modulate the migratory potential of MCF-7 cells through a NADPH oxidase and NFkappaB-dependent mechanism. Cancer Lett. 2008;267:125–32. doi: 10.1016/j.canlet.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 115.Binker MG, Binker-Cosen AA, Gaisano HY, de Cosen RH, Cosen-Binker LI. TGF-β1 increases invasiveness of SW1990 cells through Rac1/ROS/NFκB/IL-6/MMP-2. Biochem Biophys Res Commun. 2011;405:140–5. doi: 10.1016/j.bbrc.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 116.Anrather J, Racchumi G, Iadecola C. NFkappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem. 2006;281:5657–67. doi: 10.1074/jbc.M506172200. [DOI] [PubMed] [Google Scholar]
- 117.Djavaheri-Mergny M, Javelaud D, Wietzerbin J, Besançon F. NFkappaB activation prevents apoptotic oxidative stress via an increase of both thioredoxin and MnSOD levels in TNFalpha-treated Ewing sarcoma cells. FEBS Lett. 2004;578:111–5. doi: 10.1016/j.febslet.2004.10.082. [DOI] [PubMed] [Google Scholar]
- 118.Jones PL, Ping D, Boss JM. Tumor necrosis factor alpha and interleukin-1beta regulate the murine manganese superoxide dismutase gene through a complex intronic enhancer involving C/EBP-beta and NFkappaB. Mol Cell Biol. 1997;17:6970–81. doi: 10.1128/mcb.17.12.6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nagase M, Ayuzawa N, Kawarazaki W, Ishizawa K, Ueda K, Yoshida S, Fujita T. Oxidative stress causes mineralocorticoid receptor activation in rat cardiomyocytes: role of small GTPase Rac1. Hypertension. 2012;59:500–6. doi: 10.1161/HYPERTENSIONAHA.111.185520. [DOI] [PubMed] [Google Scholar]
- 120.Kuiper JW, Sun C, Magalhães MA, Glogauer M. Rac regulates PtdInsP₃ signaling and the chemotactic compass through a redox-mediated feedback loop. Blood. 2011;118:6164–71. doi: 10.1182/blood-2010-09-310383. [DOI] [PubMed] [Google Scholar]
- 121.Mori K, Shibanuma M, Nose K. Invasive potential induced under long-term oxidative stress in mammary epithelial cells. Cancer Res. 2004;64:7464–72. doi: 10.1158/0008-5472.CAN-04-1725. [DOI] [PubMed] [Google Scholar]
- 122.Ihara K, Muraguchi S, Kato M, Shimizu T, Shirakawa M, Kuroda S, Kaibuchi K, Hakoshima T. Crystal structure of human RhoA in a dominantly active form complexed with a GTP analogue. J Biol Chem. 1998;273:9656–66. doi: 10.1074/jbc.273.16.9656. [DOI] [PubMed] [Google Scholar]
- 123.Gillies RJ, Martinez-Zaguilan R, Martinez GM, Serrano R, Perona R. Tumorigenic 3T3 cells maintain an alkaline intracellular pH under physiological conditions. Proc Natl Acad Sci U S A. 1990;87:7414–8. doi: 10.1073/pnas.87.19.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hubbard SJ, Thornton JM. NACCESS. 1993:Computer Program, Department of Biochemistry and Molecular Biology, University College, London. [Google Scholar]
- 125.Baker TL, Booden MA, Buss JE. S-Nitrosocysteine increases palmitate turnover on Ha-Ras in NIH 3T3 cells. J Biol Chem. 2000;275:22037–47. doi: 10.1074/jbc.M001813200. [DOI] [PubMed] [Google Scholar]
- 126.Kil IS, Shin SW, Park JW. S-glutathionylation regulates GTP-binding of Rac2. Biochem Biophys Res Commun. 2012;425:892–6. doi: 10.1016/j.bbrc.2012.07.169. [DOI] [PubMed] [Google Scholar]
- 127.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–71. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 128.El-Remessy AB, Tawfik HE, Matragoon S, Pillai B, Caldwell RB, Caldwell RW. Peroxynitrite mediates diabetes-induced endothelial dysfunction: possible role of Rho kinase activation. Exp Diabetes Res. 2010;2010:247861. doi: 10.1155/2010/247861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Laufs U, Endres M, Stagliano N, Amin-Hanjani S, Chui DS, Yang SX, Simoncini T, Yamada M, Rabkin E, Allen PG, et al. Neuroprotection mediated by changes in the endothelial actin cytoskeleton. J Clin Invest. 2000;106:15–24. doi: 10.1172/JCI9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Muniyappa R, Xu R, Ram JL, Sowers JR. Inhibition of Rho protein stimulates iNOS expression in rat vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2000;278:H1762–8. doi: 10.1152/ajpheart.2000.278.6.H1762. [DOI] [PubMed] [Google Scholar]
- 131.Delarue FL, Taylor BS, Sebti SM. Ras and RhoA suppress whereas RhoB enhances cytokine-induced transcription of nitric oxide synthase-2 in human normal liver AKN-1 cells and lung cancer A-549 cells. Oncogene. 2001;20:6531–7. doi: 10.1038/sj.onc.1204801. [DOI] [PubMed] [Google Scholar]
- 132.Nimnual AS, Taylor LJ, Bar-Sagi D. Redox-dependent downregulation of Rho by Rac. Nat Cell Biol. 2003;5:236–41. doi: 10.1038/ncb938. [DOI] [PubMed] [Google Scholar]
- 133.Shinohara M, Shang WH, Kubodera M, Harada S, Mitsushita J, Kato M, Miyazaki H, Sumimoto H, Kamata T. Nox1 redox signaling mediates oncogenic Ras-induced disruption of stress fibers and focal adhesions by down-regulating Rho. J Biol Chem. 2007;282:17640–8. doi: 10.1074/jbc.M609450200. [DOI] [PubMed] [Google Scholar]
- 134.Zuckerbraun BS, Stoyanovsky DA, Sengupta R, Shapiro RA, Ozanich BA, Rao J, Barbato JE, Tzeng E. Nitric oxide-induced inhibition of smooth muscle cell proliferation involves S-nitrosation and inactivation of RhoA. Am J Physiol Cell Physiol. 2007;292:C824–31. doi: 10.1152/ajpcell.00592.2005. [DOI] [PubMed] [Google Scholar]
- 135.Gerhard R, John H, Aktories K, Just I. Thiol-modifying phenylarsine oxide inhibits guanine nucleotide binding of Rho but not of Rac GTPases. Mol Pharmacol. 2003;63:1349–55. doi: 10.1124/mol.63.6.1349. [DOI] [PubMed] [Google Scholar]
- 136.Lopez-Haber C, Kazanietz MG. Cucurbitacin I inhibits Rac1 activation in breast cancer cells by a reactive oxygen species-mediated mechanism and independently of Janus tyrosine kinase 2 and P-Rex1. Mol Pharmacol. 2013;83:1141–54. doi: 10.1124/mol.112.084293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yu W, Beaudry S, Negoro H, Boucher I, Tran M, Kong T, Denker BM. H2O2 activates G protein, α 12 to disrupt the junctional complex and enhance ischemia reperfusion injury. Proc Natl Acad Sci U S A. 2012;109:6680–5. doi: 10.1073/pnas.1116800109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dada LA, Novoa E, Lecuona E, Sun H, Sznajder JI. Role of the small GTPase RhoA in the hypoxia-induced decrease of plasma membrane Na,K-ATPase in A549 cells. J Cell Sci. 2007;120:2214–22. doi: 10.1242/jcs.003038. [DOI] [PubMed] [Google Scholar]
- 139.Chandra S, Romero MJ, Shatanawi A, Alkilany AM, Caldwell RB, Caldwell RW. Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway. Br J Pharmacol. 2012;165:506–19. doi: 10.1111/j.1476-5381.2011.01584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kondrikov D, Caldwell RB, Dong Z, Su Y. Reactive oxygen species-dependent RhoA activation mediates collagen synthesis in hyperoxic lung fibrosis. Free Radic Biol Med. 2011;50:1689–98. doi: 10.1016/j.freeradbiomed.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Aghajanian A, Wittchen ES, Campbell SL, Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One. 2009;4:e8045. doi: 10.1371/journal.pone.0008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Barton JP, Packer JE, Sims RJ. Kinetics of Reaction of Hydrogen-Peroxide with Cysteine and Cysteamine. J Chem Soc Perk T. 1973;2:1547–9. doi: 10.1039/p29730001547. [DOI] [Google Scholar]
- 143.Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27:322–8. doi: 10.1016/S0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]