

Systems analysis reveals that Chlamydomonas reinhardtii responds flexibly to an increase in light intensity. Rising metabolite levels and posttranslation regulation facilitate a rapid increase in the rate of carbon fixation and a slightly delayed increase in the rate of growth, while slower changes in protein abundance adjust allocation and relieve potential bottlenecks under the new conditions.
Abstract
We investigated the systems response of metabolism and growth after an increase in irradiance in the nonsaturating range in the algal model Chlamydomonas reinhardtii. In a three-step process, photosynthesis and the levels of metabolites increased immediately, growth increased after 10 to 15 min, and transcript and protein abundance responded by 40 and 120 to 240 min, respectively. In the first phase, starch and metabolites provided a transient buffer for carbon until growth increased. This uncouples photosynthesis from growth in a fluctuating light environment. In the first and second phases, rising metabolite levels and increased polysome loading drove an increase in fluxes. Most Calvin-Benson cycle (CBC) enzymes were substrate-limited in vivo, and strikingly, many were present at higher concentrations than their substrates, explaining how rising metabolite levels stimulate CBC flux. Rubisco, fructose-1,6-biosphosphatase, and seduheptulose-1,7-bisphosphatase were close to substrate saturation in vivo, and flux was increased by posttranslational activation. In the third phase, changes in abundance of particular proteins, including increases in plastidial ATP synthase and some CBC enzymes, relieved potential bottlenecks and readjusted protein allocation between different processes. Despite reasonable overall agreement between changes in transcript and protein abundance (R2 = 0.24), many proteins, including those in photosynthesis, changed independently of transcript abundance.
INTRODUCTION
Growth is a complex emergent output of cellular physiology and is generated through interactions between resource availability, energy metabolism, anabolism, cellular growth processes, the cell cycle, and, in multicellular organisms, developmental programs as well as the interplay between tissues and organs. Systems approaches in which transcriptomics, proteomics, and metabolomics are combined with measurements of physiological processes can provide new insights into the regulation of metabolism and growth (Castrillo et al., 2007; Brauer et al., 2008; Fendt et al., 2010; Gutteridge et al., 2010; Oliveira et al., 2012; Pir et al., 2012). In such approaches, one or more inputs that are required for growth are modified and the dynamic response of the system is investigated at multiple levels. The resulting data sets can be used to identify molecular parameters that are linked to growth and to search for connectivity across different functional levels, both in individual processes and between processes that contribute to growth. This approach has, for example, been applied by subjecting yeast (Saccharomyces cerevisiae) to a change in nutrient supply (Castrillo et al., 2007; Brauer et al., 2008; Gutteridge et al., 2010; Oliveira et al., 2012; Pir et al., 2012) or, in an orthogonal approach, by changing the expression level of genes encoding pathway enzymes or regulatory proteins (Fendt et al., 2010; Pir et al., 2012). Integrated analysis of the transcriptome and the proteome has revealed the importance of posttranscriptional regulation (Castrillo et al., 2007) and identified novel examples of posttranscriptional regulation (Gutteridge et al., 2010). The integration of quantitative proteomics and metabolomics data has shown that the regulation of metabolic flux is distributed between different functional levels and is fine-tuned at the metabolic level (Fendt et al., 2010; Oliveira et al., 2012). This systems approach can also be applied to investigate growth of a photosynthetic organism, as understanding their growth has important implications in agriculture and ecology (Field et al., 1998; Farquhar et al., 2001; Zhu et al., 2010).
In photosynthetic cells, growth ultimately depends on photosynthetic carbon (C) fixation. The C supply for growth can be readily altered by changing the light intensity. Under low irradiance and saturating CO2, algae and plants need to absorb a minimum of 9 to 10 quanta to fix a molecule of CO2. This matches the stoichiometry predicted from the pathways of photosynthesis (Edwards and Walker, 1983; Heldt and Piechulla, 2010; Kramer and Evans, 2011). However, only 3 to 5% of the energy in these quanta is captured in biomass, raising many questions about why this is so and whether the energy conversion efficiency of photosynthesis and plant growth can be increased (Zhu et al., 2008; Amthor, 2010; Blankenship et al., 2011; Stitt, 2013). There are many reasons for this low conversion efficiency. Some are a consequence of pathway structure, including how light energy is absorbed and transformed, thermodynamic considerations, and wasteful side reactions. Others relate to the context in which photosynthesis operates; these include the need to deal with a fluctuating light environment and the complex coupling between photosynthetic CO2 fixation and growth (Long et al., 2006; Zhu et al., 2010; Blankenship et al., 2011; Stitt, 2013).
In a natural environment, photosynthetic cells have to cope with changes in light intensity across a wide range of time scales, including temporary shading due to canopy movement, clouds, the day‐night cycle, seasonal change, slow changes in canopy structure, and sun spots. A change in light intensity will result in changes in the rate and regulation of light harvesting, the photochemical reactions, electron transport, the formation of ATP and NADPH, the use of ATP and NADPH in the Calvin-Benson cycle (CBC), the synthesis of end products including sucrose, starch, and amino acids, and their use to drive growth or the accumulation of reserves. These processes have vastly different time scales, ranging from picoseconds for light harvesting, nano- to milliseconds for events in electron transport, to seconds for ATP and NADPH turnover and operation of the CBC, and hours to days for long-term acclimation and growth processes (Geiger and Servaites, 1994; Eberhard et al., 2008; Arrivault et al., 2009; Szecowka et al., 2013).
The short-term response to an increased irradiance (seconds to minutes) includes production of reactive oxygen species (Asada, 2006), nonphotochemical quenching of excess excitation energy as heat (Iwai et al., 2007), state transitions (reviewed in Rochaix, 2011), changes in metabolite levels (Stitt et al., 1980; Badger et al., 1984; Stitt et al., 1984a; Seemann et al., 1987; Leegood and Von Caemmerer, 1989; Servaites et al., 1991), and posttranslational regulation of proteins (Scheibe, 1991; McKim and Durnford, 2006; Liu et al., 2012). Mid- to long-term photoacclimation includes changes in transcriptional regulation, enzyme activation (minutes to hours), and protein synthesis, changes in protein stoichiometry (hours to days) (reviewed in Li et al., 2009; Foyer et al., 2012), as well as slow changes in leaf morphology that unfold over days in growing tissues (Lichtenthaler et al., 1981).
Increased rates of photosynthesis lead to an increased supply of carbohydrates and, indirectly, increased rates of assimilation of inorganic nutrients like nitrate and sulfate. In Arabidopsis thaliana, cytosolic polysome loading and, by implication, the rate of protein synthesis track the sucrose content within a time frame of 10 to 15 min (Stitt and Zeeman, 2012; Pal et al., 2013; Sulpice et al., 2014). However, little is known at the molecular level about the links between photosynthesis and growth. Although plants contain canonical signaling pathways like Target of Rapamycin (TOR) and sucrose nonfermenting-1-related protein kinases (SnRKs), it is unclear how these interact with plant-specific signaling pathways to regulate growth (Smeekens et al., 2010; Robaglia et al., 2012). Furthermore, while an increased rate of C fixation has the potential to support an increase in growth, this is not always realized (Stitt and Schulze, 1994). The minor stimulation of plant growth to elevated CO2 (Ainsworth et al., 2008) implies that the response of growth to an increased supply of carbon is often constrained by developmental processes or the availability of other resources (Long et al., 2006; Kirschbaum, 2011; Stitt, 2013). The regulation of the transport and metabolic processes that transform the end products of photosynthesis into biomass is poorly understood (Stitt, 2013). The relation between photosynthetic C fixation and growth is further complicated by the fact that a substantial proportion of the newly fixed C is not immediately used for growth, but is instead accumulated as reserves, like starch, to support future growth (Stitt and Zeeman, 2012; Sulpice et al., 2014). There is also strong evidence that feedback mechanisms are activated to decrease the momentary rate (Kramer et al., 2003; Baker et al., 2007) and capacity for photosynthesis when the rate of photosynthesis exceeds the capacity for use of C for growth (Stitt, 1991; Paul and Foyer, 2001; Rolland et al., 2006; Smeekens et al., 2010).
One important decision for a systems analysis of the relation between photosynthesis and growth is the choice of the model organism. In vascular plants, many factors complicate this analysis, including complex irradiance gradients within a leaf (Vogelmann et al., 1989), the presence of different cell types of which many are not involved in photosynthesis (Jellings and Leech, 1982; Pollock et al., 2003; Mustroph et al., 2009), implying that measurements and estimations of many parameters will not reflect the values in the photosynthetic cells (Szecowka et al., 2013), and the spatial separation of photosynthesis in mature leaves from the use of C for growth in a large number of discrete growing organs (reviewed in Poorter and Van der Werf, 1998; Stitt, 2013). Indeed, a major difficulty in studying growth in a vascular plant on a systems level is to define a unique parameter for growth (Poorter and Van der Werf, 1998). We therefore chose to conduct our study with the unicellular green alga Chlamydomonas reinhardtii. This model organism has already been used for many studies of photosynthesis and growth in different nutrient conditions (Rochaix, 1995, 2001; Wykoff et al., 1998; Grossman et al., 2007; Renberg et al., 2010; Bonente et al., 2012; Blaby et al., 2013; Schmollinger et al., 2014). The availability of a genomic sequence now opens the door for systems biology approaches (Merchant et al., 2007; Castruita et al., 2011; Chang et al., 2011). Another important advantage of using algal cells is that, as in yeast, experiments can be performed in a bioreactor from which sequential samples can be taken for molecular analysis, while the change in optical density of the algal suspension over time provides a simple nonintrusive measure for growth.
A second important decision is the choice of the intervention. We chose to use an increase in light intensity within the range where irradiance remains limiting for photosynthesis. The dynamics of transcript levels and photosynthetic parameters after transfer from low to saturating light have been studied in many species including Arabidopsis (Kimura et al., 2003; Vanderauwera et al., 2005), rice (Oryza sativa) (Murchie et al., 2005), C. reinhardtii (Im et al., 2003), and other algae (Ritz et al., 2000; Nymark et al., 2009; Deblois et al., 2013). The main aim of these studies was to understand how plants adapt the photosynthetic apparatus and its use to light intensities in excess of those required to saturate photosynthesis, i.e., minimization of stress or its avoidance. Such studies have shown that saturating light induces general stress responses and a wide range of energy dissipation mechanisms, resulting in a decrease in photosynthetic efficiency (Murchie et al., 2005; Eberhard et al., 2008; Nymark et al., 2009; Bonente et al., 2012; Deblois et al., 2013). This will overlay the response of metabolism and growth to an increase in the rate of photosynthesis. On the other hand, the use of relatively small changes in light intensity in the nonsaturating range, and hence the rate of photosynthesis, will require very precise control of the growth conditions in order to obtain reproducible biological material during and between experiments.
In the large-scale experiment reported here, we developed a bioreactor to allow reproducible growth of C. reinhardtii in continuous culture. After growth at low irradiance, the cells were shifted to a higher, but still limiting, irradiance. Photosynthetic rate, photosynthetic subreactions, metabolite levels, transcript levels, protein abundance, polysome loading, and growth were monitored in low irradiance and for 8 h after the shift to higher irradiance. The resulting multilayer data set reveals how changes at different functional levels interact to facilitate the increase in photosynthesis and the resulting increase in biomass accumulation. In particular, we investigate the quantitative relationship between metabolite levels and the kinetic properties and abundance of CBC enzymes, assess the contribution of transcriptional, translational, and posttranslational processes to the short- and mid-term adjustment of photosynthesis, and ask how closely photosynthesis is coupled to biosynthetic pathways and cellular growth processes.
RESULTS
Experimental Design and Setup
To facilitate systems analyses in unicellular photosynthetic organisms, we designed a bioreactor that allows the growth of algal cultures under fully controlled conditions. For salient features of the bioreactor, see Methods and Supplemental Figure 1. C. reinhardtii CC-1690 wild-type cells were grown in the bioreactor for 3 d at high CO2 (5%) in continuous light at a strongly limiting intensity (41 μmol photons m−2 s−1; light intensity was measured in the bioreactor containing cells; see Methods) at an optical density of 3 to 4⋅106 cells⋅mL−1. The optical density was kept constant by automatic dilution to ensure that irradiance and nutrient availability did not change during the experiment (see Methods). At time point 0, the light intensity was increased by just over 3-fold (145 μmol photons m−2 s−1). The higher light intensity was not saturating, as shown by the fact that relative electron transport rate (ETR) only saturated at a light intensity of more than 500 μmol photons m−2 s−1 and that the light-saturated ETR was more than twice as high as the photosynthetic rate at 145 μmol photons m−2 s−1 (Supplemental Figure 2).
Harvest times were chosen to combine a linear and logarithmic scale and cover both fast responses and slower acclimation processes (Supplemental Figure 3). Experimental replicates were obtained by collecting sets of samples from two (for proteome analysis and analysis of metabolites by liquid chromatography–tandem mass spectrometry [LC-MS/MS]) or three (for transcript analysis, physiological analyses, and analysis of metabolites by gas chromatography–mass spectrometry [GC-MS] and lipids) independent bioreactor runs. Per bioreactor run, one (for transcriptome analysis and analysis of metabolites by GC-MS and lipids) or two (for proteome analysis and analysis of metabolites by LC-MS/MS) samples at each time point were collected. As a control, samples were collected before the light shift and from independent bioreactor runs with a constant low light intensity (41 μmol photons m−2 s−1) during the entire experiment (for details, see Supplemental Figure 3).
Rate of Cell Growth Increased after a Short Delay
In a chemostat bioreactor, the dilution rate of the culture is equivalent to the specific growth rate (Figure 1A; see Methods). It captures both changes in cell division rate and cell size. The measurements of optical density showed some variability immediately after the increase in light intensity (Figure 1A), possibly due to changes in light scattering after a change in the light intensity. The dilution rate remained low (∼0.02 h−1) for the first 5 to 10 min, increased by 20 min (0.04 h−1), reached a maximum by 40 min (0.08 h−1) after the increase in light intensity, and remained high for the remainder of the higher light treatment (see later for more data).
Figure 1.

Changes in Doubling Time and Photosynthesis Rate of C. reinhardtii Cells Shifted to an Increased Light Intensity.
C. reinhardtii CC-1690 cells were grown in a bioreactor at 24°C, 5% CO2, and 41 μmol photons m−2 s−1 and shifted to 145 μmol photons m−2 s−1 at time point zero (dashed line).
(A) During the whole experiment, the optical density was kept constant. The dilution of the culture over time allowed calculating the dilution rate [h−1] (n = 2 ± sd) (inset), which is equivalent to the specific growth rate μ for a steady state chemostat (see Methods).
(B) The oxygen evolution was measured in a closed cuvette with an optical sensor at the low-light intensity (41 μmol photons m−2 s−1) and after transfer at time point zero to a higher light intensity (145 μmol photons m−2 s−1). The rate of O2 evolution was estimated from the slope. The figure shows an overlay of five independent measurements.
(C) ETR was measured in an open cuvette via fluorometry at the low light intensity (41 μmol photons m−2 s−1) and after transfer to a higher light intensity (145 μmol photons m−2 s−1, filled circles) at time point zero or kept at the initial light intensity for 480 min (open circles) (n = 3 ± sd). A pairwise t test between control and treatment samples and P value correction for multiple sampling by Bonferroni correction was performed (three asterisks, P < 0.001).
[See online article for color version of this figure.]
Rapid Increase of Photosynthesis
The net rate of photosynthesis was determined in two ways (Figures 1B and 1C). To obtain maximal temporal resolution during the light shift, samples were transferred from the bioreactor to a closed cuvette where O2 evolution was measured using an optical sensor (Figure 1B). The net rate of photosynthesis was 47 ± 2.4 μmol O2 h−1 mg chlorophyll−1 at the lower light intensity and increased 2.3-fold within 20 s to 106 ± 3.6 μmol O2 h−1 mg chlorophyll−1 after increasing the light intensity. This is similar to published data for the C. reinhardtii CC-125 wild-type strain, where photosynthesis rates of ∼36 and 94 μmol O2 h−1 mg chlorophyll−1 were measured at 40 and 147 μmol photons m−2 s−1, respectively (Wykoff et al., 1998). To measure photosynthesis at later time points, the ETR at the corresponding light intensities was monitored in the bioreactor before the light shift and 60, 120, 240, and 480 min after the light shift (Figure 1C). Relative ETR increased 2.6-fold between the 0 time point and 60 min after the light shift and showed no further significant change until 480 min. The O2 concentration measured in the outlet air of the bioreactor also remained stable from 60 min onwards (Supplemental Figure 4). Relative ETR was unchanged (P value > 0.05) between 0, 60, 120, 240, and 480 min in the control bioreactors (Figure 1C).
In the high CO2 concentrations used in our experiment, photorespiration is largely suppressed and the net photosynthetic rate will be approximated by the rate of electron transport and oxygen release; it may represent a slight overestimate of the gross rate of photosynthetic O2 evolution and CO2 fixation if respiratory O2 uptake and CO2 release continues in the light. For the remainder of the analysis, the measured net rates of O2 evolution were therefore used as an estimate of flux in the light reactions and the CBC.
Transcript, Protein, and Metabolite Abundance Show Very Different Temporal Kinetics
RNA was analyzed for 108 plastid- and mitochondria-encoded transcripts using a custom-made organellar microarray and for 11,455 nucleus-encoded transcripts using a genomic microarray (for details, see Methods, Supplemental Figure 3, and Supplemental Data Set 1). Proteins were analyzed by shotgun proteomics (Methods) with 644 proteins (belonging to 520 distinct protein groups) quantified across all time points. A total of 120 intermediary metabolites were analyzed by LC-MS/MS, GC-MS, and coupled enzyme assays, and 98 lipids by LC-MS (for details, see Methods, Supplemental Figure 3, and Supplemental Data Set 1).
These data sets were first subjected to a global analysis to provide an overview of the magnitude and temporal kinetics of the response at each functional level. Principle component (PC) analyses of the nuclear and plastid-encoded transcript, protein, metabolite, and lipid data sets are shown in Figures 2A, 2C, 2E, 2G, and 2I and frequency plots of the changes in transcript, protein, metabolite and lipid levels at different times after the shift in Figures 2B, 2D, 2F, 2H, and 2J.
Figure 2.

Overall Changes in Nuclear Transcripts, Organellar Transcripts, Protein Groups, Metabolites, and Lipids after Shift to a Higher Light Intensity.
C. reinhardtii CC-1690 cells were grown in a bioreactor at 24°C, 5% CO2, and 41 μmol photons m−2 s−1. At time point zero, the light was either kept at the initial light intensity (C; control) or shifted to 145 μmol photons m−2 s−1 (T; treatment). CX, TX, and X indicate the minutes relative to time point zero. For further details of analyses, see Methods.
(A), (C), (E), (G), and (I) PC analysis of log2-normalized nucleus-encoded transcript (A), plastid- and mitochondria-encoded transcript (C), and protein (E), metabolite (G), and lipid (I) levels measured at low light (orange; C5 to C240 are shown in bright orange) and higher light (red) intensity.
(B), (D), (F), (H), and (J) Histogram of the log2-fold changes of different time points after the light shift compared with the control time points of nucleus-encoded transcripts (B), plastid- and mitochondria-encoded transcripts (D), and protein (F), metabolite (H), and lipid (J) levels.
In the PC analysis of the nuclear-encoded transcript data set, the control data and low light treatment points grouped together. The most strongly separated time point from the controls was at 40 min after the light shift (Figure 2A). PC analysis of plastid- and mitochondria-encoded transcripts did not clearly separate the higher light treatment from the control treatments (Figure 2C). For the proteomics data set, early time points were not separated, while later time points (from 120 to 480 min after the light shift) were more clearly separated from the control time points (Figure 2E). For metabolites, the 5-min point was already clearly separated (Figure 2G). For lipids, the 5- and 10-min data sets were not separated, but subsequent time points were increasingly separated from the control data sets (Figure 2I). For all data sets except for the plastid- and mitochondria-encoded transcripts, PC1 accounted for 50 to 67% of the variance and captured the responses between low light and the time point in higher light when changes of the trait set were largest. PC2 accounted for <24% of the variance.
The frequency plots reveal a similar picture. Nuclear-encoded transcripts showed large changes by 40 min (Figure 2B), only a few plastid-encoded transcripts showed changes outside the range seen in the control and low light treatments (Figure 2D), and protein abundance changed slowly (Figure 2F). The amplitude of global changes in transcripts levels exceeded those for protein abundances (see later for details). There were large changes in metabolite levels within 5 min (Figure 2H) and somewhat slower changes in lipid levels (Figure 2J). There was a consistent trend to an increase in metabolite levels, but not of transcript, protein abundance, and lipid levels. The total protein concentration per culture volume also remained unaltered after the shift to higher light (Supplemental Figure 5).
Rapid Increase in the Abundance of Nucleus-Encoded Transcripts for Photosynthesis and Growth
We then analyzed the responses of transcript, protein, and metabolite abundance in more detail. Transcripts were considered to show significant changes between the two conditions if they were significantly differentially expressed (P value < 0.05 from ANOVA, corrected for multiple hypotheses testing by the Benjamini-Hochberg correction procedure) and had absolute values of fold changes of at least 2 (Supplemental Figures 6A and 6C). In total, 2205 of 11,455 detected nucleus-encoded transcripts showed significant changes after the increase in light intensity (Supplemental Figure 6A). The combined values at 0 and 480 min in the control bioreactor, in which the light intensity remained at 41 μmol photons m−2 s−1 and at 0 min in the treatment bioreactor were used as reference time points in the comparisons (for details, see Supplemental Figure 6C). Only a small number (38) of transcripts changed between the three reference samples (Supplemental Figures 6A and 6C). After the increase in light intensity, 1782 transcripts were significantly changed, with >2-fold changes at 40 min and 866 transcripts at 480 min, equivalent to 15.3 and 7.3% of all transcripts, respectively. The difference between 40 and 480 min is mainly due to a set of transcripts that were more strongly repressed at 40 min than at 480 min (Figure 2B).
Overrepresentation analysis was performed using Fisher test in the PageMan application (Usadel et al., 2006) (Supplemental Figure 7). Upregulated genes were overrepresented in MapMan bins for “amino acid activation,” “protein targeting,” “protein folding,” and “protein degradation” at 40, 120, and 480 min after the light shift (P value < 0.05), for upregulated genes in the bin “photosynthesis” at 120 min after the light shift (P value < 0.05), and for downregulated genes in the bin “cell motility” at 40, 120, and 480 min after the light shift. This analysis points to a transient transcriptional upregulation of photosynthesis and a sustained upregulation of growth processes including protein synthesis, but also of protein degradation. The nucleus-encoded transcripts that underwent the largest change between the low light and higher light intensity (Supplemental Figure 8A) included transcripts encoding members of the light harvesting complex (LHC) superfamily (LHCSR1), two genes implicated in CO2 concentration mechanisms (CCP2 and LCI1), and a nucleoredoxin (NRX3; NRX2 also increased, although less strongly; Supplemental Data Set 1), pointing to changes related to light capture and dissipation, CO2 concentration, and redox regulation.
Few Changes in the Abundance of Plastid- and Mitochondria-Encoded Transcript Levels
There were few significant changes in the abundance of organelle-encoded transcripts. Nine of the 137 chloroplast or mitochondrial transcripts (6.6%) changed significantly (P < 0.05, Wilcoxon rank sum test corrected for multiple hypotheses testing by the Benjamini-Hochberg correction procedure; Supplemental Data Set 1). Two transcripts involved in chlorophyll biosynthesis (chlB and chlN, subunits of the dark operative protochlorophyllide oxidoreductase) (Bröcker et al., 2010), rbcL, and a mitochondrial rRNA species (rrnL-2a) were all expressed at a lower level in higher light, whereas three transcripts, the chloroplastidial orf271 and orf140 (both derived from the WENDY transposon; Fan et al., 1995) and the mitochondrial reverse transcriptase-like (Boer and Gray, 1988) transcript, increased. The three transcripts that increased in abundance are of transposon origin and have unknown functions.
Slow Changes in Proteome Composition
Relative changes in protein abundance over time were determined by normalizing each time point to a 15N-proteome that was spiked into each sample before protein extraction (Mühlhaus et al., 2011; see Methods). A total of 148 proteins of the 520 distinct protein groups showed significant changes after transfer to higher light, with 72 and 133 changing by 60 and 480 min, respectively. However, a relatively large number of proteins (24) showed changes between the control treatments (Supplemental Figure 6B); of these, the majority were due to changes during the incubation in the control bioreactor rather than to differences between bioreactors (Supplemental Figures 6B and 6D). Compared with the number of total detected proteins, 14 and 26% showed significant changes at 40 and 480 min, respectively. The proportion of the detected proteins that show significant changes at 480 min is higher than the value for transcripts at 480 min but is similar to the value for transcripts at 40 min, when the maximum response is seen in the transcriptome. This points to a scenario in which many transcripts show large early changes that lead to time-delayed changes in protein abundance.
The MapMan annotation (Usadel et al., 2005) was used to visualize significantly up- and downregulated proteins in central metabolism at 60 and 480 min (Figure 3). MapMan bin enrichment was determined by the hypergeometric test (P value < 0.05) with respect to all quantified proteins. The enriched bins were mainly related to central metabolism (photosynthesis, CBC, and tricarboxylic acid [TCA] cycle) and posttranslational processes (chromatin structure and protein degradation). In the next section, changes in photosynthesis-related proteins are analyzed in greater detail.
Figure 3.

Schematic Overview of the Differentially Regulated Proteins According to Their Biochemical Pathways and Cellular Processes.
Significantly decreased (blue squares) and increased (red squares) protein levels between the control time points and 60 and 480 min after light shift according to Supplemental Figure 6B are visualized within their functional categories based on MapMan ontology using MapMan software (http://mapman.gabipd.org/web/guest/mapman; version 3.6.0RC1). Nonsignificantly changing protein groups are represented by white squares. Green borders indicate enriched functional categories determined by hypergeometric testing (P value < 0.05): bin 28 (DNA; P value = 0.0025), bin 28.1 (DNA.synthesis/chromatin structure; P value = 0.0025), bin 28.1.3 (DNA.synthesis/chromatin structure.histone; P value = 0.0025), bin 1 (photosynthesis; P value < 0.0001), bin 1.3 (PS.calvin cycle; P value = 0.0434), bin 1.1 (PS.lightreaction; P value < 0.0001), bin 1.1.2 (PS.lightreaction.photosystem I; P value = 0.0001), bin 1.1.2.2 (PS.lightreaction.photosystem I.PSI polypeptide subunits; P value = 0.0015), bin 1.1.1 (PS.lightreaction.photosystem II; P value = 0.0015), bin 1.1.1.2 (PS.lightreaction.photosystem II.PSII polypeptide subunits; P value = 0.0058), bin 8.1 (TCA/org transformation.TCA; P value = 0.0154), and bin 29.5.11.1 (protein.degradation.ubiquitin. ubiquitin; P value = 0.0335).
Decreased Abundance of Proteins for Light Harvesting and Photosystems and Increased Abundance of the Thylakoid ATP Synthase
The abundance of several subunits of photosystem II (PSII), light-harvesting complex II (LHCII), photosystem I (PSI), and light-harvesting complex I (LHCI) showed a significant (P value < 0.05 from ANOVA, corrected for multiple hypotheses testing by the Benjamini-Hochberg correction procedure) decrease after transfer to higher light (Figure 4; Supplemental Figure 9). The subunits of the cytochrome b6f complex did not show any significant changes. Abundance of plastidial ATP synthase (ATPase) subunits increased, with the increase continuing until 480 min (Figure 4).
Figure 4.

Central Tendencies of Protein Levels during the Light Shift Experiment within the Functional Categories Light Reaction, CBC, and Ribosomes.
Central tendencies are calculated using average and sd of all protein groups belonging to the same functional category based on the MapMan ontology (MapMan bin numbers are given in parentheses). For single values of protein groups, see Supplemental Figure 9.
Changes in the Rate of Electron Transport and ATPase Activity in Vivo
We used spectroscopic methods to investigate if these changes in protein abundance were accompanied by functional changes in the light reactions. After transfer to a higher light intensity, the chlorophyll content per milliliter culture decreased gradually while the chlorophyll a/b ratio remained unaltered (Figure 5A, inset). This is in agreement with the concomitant decrease in PSII, PSI, and LHC protein abundance (see above; Figure 4). The decrease in PSII subunit abundance was accompanied by a decrease in the maximum quantum efficiency of PSII (FV/FM), as measured by chlorophyll a fluorescence (Figure 5B). FV/FM decreased within one hour after the high-light shift (see also Bonente et al., 2012), and the decrease was almost complete by 120 min, showing a good match with the temporal kinetics of PSII abundance. Additionally, measurements of 77K chlorophyll a fluorescence emission spectra indicated that the antenna cross section of PSII decreases relative to that of PSI from 120 min after the light shift onwards (Supplemental Figure 10). Because the subunits of both photosystems and harvesting complexes decreased proportionally, a shift of LHCII subunits from PSII to PSI (i.e., transition from state 1 to state 2) could be responsible for the changes in antenna cross sections. State transitions have been previously shown to play an important role for high light protection in C. reinhardtii (Allorent et al., 2013). Maximum light-saturated photosynthetic electron transport remained unaltered (Figure 5C), in line with unaltered contents of the subunits of the cytochrome b6f complex (Figure 4), which catalyzes the rate-limiting step of photosynthetic electron transport (Anderson, 1992).
Figure 5.

Changes in Chlorophyll Content per Milliliter of Culture, Chlorophyll a/b Ratio, Maximum Quantum Efficiency of PSII (FV/FM), Maximal Relative ETR under Light Saturated Conditions, and Maximum ATPase Activity.
C. reinhardtii CC-1690 cells were grown in a bioreactor at 24°C, 5% CO2, and 41 µmol photons m−2 s−1. At time point zero, the light was either kept at the initial light intensity (open circles) or shifted to 145 µmol photons m−2 s−1 (filled circles). A pairwise t test between control and treatment samples and P value correction for multiple sampling by Bonferroni correction was performed (n = 4 ± sd; one asterisk, P < 0.05; three asterisks, P < 0.001).
In vivo ATPase activity was estimated in saturating light by dark-interval relaxation kinetics of the proton motive force across the thylakoid membrane (Baker et al., 2007; Rott et al., 2011). There was a 40 to 70% increase in in vivo ATPase activity, with the increase being significant from 240 min onwards (Figure 5D). This resembles the timing of the increase in ATPase protein subunit abundance (Figure 4). The larger increase of in vivo activity (70%) compared with ATPase subunit abundance (15%) implies that further mechanisms also act to increase in vivo ATPase activity (Rott et al., 2011). Overall, these spectroscopic measurements confirm the changes in protein abundance of the various complexes involved in the light reactions and indicate that they contribute to, but cannot fully explain, the changes in flux through the photosynthetic electron chain and the plastidial ATPase.
Gradual Increase in the Abundance of Several CBC Enzymes
There were significant increases of several enzymes in the CBC, including fructose-1,6-bisphosphate (FBP) aldolase 1 (FBA1; +48%), sedoheptulose-1,7-bisphosphatase (SBPase; +14%), fructose-1,6-bisphoshatase (FBPase; +13%), Rubisco large subunit (RbcL; +8%), and glyceraldehyde-3-phosphate (GAP) dehydrogenase (GAP3; +5%). There was also a large increase in Rubisco activase (RCA1; +48%) abundance. There were small decreases for phosphoribulokinase (PRK1; −4%) and phosphoglycerate kinase (PGK1; −5%) abundance (Supplemental Figure 9; the number in parentheses gives the percentage of change at 480 min after the light shift compared with the 0 min control time point), and the remainder of the proteins involved in the CBC, namely, ribose-5-phosphate (R5P) isomerase (RPI1), ribulose-5-phosphate (Ru5P) epimerase (RPE1), Rubisco small subunit (RBCS), FBP aldolase 3 (FBA3), transketolase (TRK), and triose-phosphate isomerase (TPI), did not change significantly. It should be noted that FBA1 is very likely only a minor component of the aldolase enzyme activity and that the major aldolase in the C. reinhardtii stroma is FBA3 (Blaby et al., 2013; Supplemental Table 1). FBA1 and FBA3 protein levels were summed for further analyses according to their relative abundance (for details, see Supplemental Table 1).
Rapid Increase and Slower Adjustment of the Levels of CBC Intermediates
We then turned to the responses of intermediary metabolites. We measured 120 intermediary metabolites and, of these, 22 showed significantly changed levels (ANOVA P value < 0.05, P value correction for multiple sampling by the Benjamini-Hochberg correction procedure) after increasing the light intensity, corresponding to 18% of the measured metabolites (Supplemental Data Set 1). These included a significant increase in the levels of seven CBC intermediates, five amino acids, and three organic acids.
The direct products of the light reaction are NADPH and ATP. We did not measure NADPH due to technical constraints. There were no significant changes in ATP, ADP, and AMP. The adenylate energy charge was at a constant and high (>0.9) level in low light and after the shift to higher light (Supplemental Figure 11). This implies that use of ATP is rapidly upregulated after the switch to higher light.
The CBC is the major sink for NADPH and ATP. Our LC-MS/MS platform detected all CBC intermediates except erythrose-4-phosphate (E4P). Xylulose-5-phosphate (Xu5P) and ribulose-5-phosphate (Ru5P) were detected as a single peak, subsequently referred to as “Xu5P+Ru5P.” Significant changes were identified by pairwise Student’s t test between control and treatment samples, with P value < 0.05 and P value correction for multiple sampling by the more conservative Bonferroni correction procedure. Three phases could be distinguished in the response (Figure 6). In the first phase, between 0 and 5 min after the light shift, the levels of almost all CBC intermediates increased significantly, including GAP (2-fold), dihydroxyacetone-P (DHAP; 3-fold), fructose-6-P (F6P; 2.2-fold), sedoheptulose-1,7-bisP (SBP; 1.4-fold), sedoheptulose-7-P (S7P; 3.2-fold), R5P (2-fold), Xu5P+Ru5P (1.7-fold), and ribulose-1,5-bisP (RuBP; 3.3-fold). The only exceptions were 3-phosphoglycerate (3PGA), and FBP, which showed no change from the control and decreased slightly, respectively. The magnitude of the increase of the CBC intermediates (1.4- to 3.3-fold) was in the same range as the rapid increase in the rate of O2 evolution (2.3-fold) (see Figure 1B). Many metabolites increased further between 5 and 10 min. In the second phase, between 10 and 40 to 60 min after the light shift, most of the intermediate levels were at a plateau. The only significant changes during this phase were for F6P and Xu5P+Ru5P, which showed a slight but significant decrease. During the third phase, between 40 and 480 min, the intermediates showed varied responses. Some increased significantly (F6P, +39%; GAP, +38%), some remained unchanged (DHAP and S7P) and others decreased significantly (SBP, −29%; R5P, −17%; Xu5P+Ru5P, −27%; RuBP, −71%). The slow adjustments of metabolites in the third phase occur in a similar time frame to those of many photosynthesis-related proteins.
Figure 6.

Rapid Increase of CBC Intermediates.
C. reinhardtii CC-1690 cells were grown in a bioreactor at 24°C, 5% CO2, and 41 μmol photons m−2 s−1. At time point zero, the light was either kept at the initial light intensity (open circles, n = 4 ± sd) or shifted to 145 μmol photons m−2 s−1 (filled circles, n = 4 ± sd) at time point zero. Metabolite levels were given as concentrations in algal cells (μM; see Methods). A pairwise t test between control and treatment samples with P value correction for multiple sampling by Bonferroni correction was performed (one asterisk, P < 0.05; two asterisks, P < 0.01; three asterisks, P < 0.001). For both conditions, cells were harvested for metabolite analysis by LC-MS/MS during the 8 h following the time point zero. Metabolites levels are shown for the whole time course ([A] and [C]) and the first hour ([B] and [D]). Graphs are based on Supplemental Data Set 1.
To provide independent information about the impact of light intensity on CBC operation, a separate experiment was conducted in which metabolites were measured in C. reinhardtii cells grown at 46 μmol photons m−2 s−1 and then transferred to darkness or a light intensity of 20, 34, 46, 94, or 143 μmol photons m−2 s−1 for 60 min before harvest (Supplemental Figure 12). The rate of photosynthesis, measured as O2 evolution, increased linearly with light intensity. Three groups of CBC intermediates can be distinguished: (1) GAP, DHAP, FBP, and SBP showed a >10-fold increase between darkness and the lowest light intensity (20 μmol photons m−2 s−1), stabilized (DHAP and GAP) or decreased (FBP and SBP) between 20 and 46 μmol photons m−2 s−1 and rose at higher light intensities; (2) F6P, S7P, R5P, and Xu5P+Ru5P and RuBP rose progressively as the light intensity was increased, largely tracking the rate of photosynthesis; and (3) 3PGA remained at the same level between darkness and 20 μmol photons m−2 s−1, rose between 20 and 46 μmol photons m−2 s−1, and decreased slightly at higher light intensities. These results point to the following picture: between darkness and 20 μmol photons m−2 s−1, flux is induced by an increase in ATP and, possibly, NADPH, leading to a large increase in GAP and DHAP, while 3PGA remained stable or even decreases. As the light intensity was increased further, there was a gradual progressive increase in the level of many CBC intermediates, indicating that increased CBC flux is in part driven by an increase in substrate levels. The high levels of FBP and SBP at 20 μmol photons m−2 s−1 and their decline between 20 and 46 μmol photons m−2 s−1 indicate that FBPase and SBPase are not fully activated at low light intensity and that posttranslational activation of these enzymes at higher light intensities may contribute to the increase in CBC flux at intermediate light intensities.
Overall, our results show that there is a rapid increase in the levels of CBC intermediates after an increase in light intensity (Figure 6) and indicate that this plays a major role in the initial stimulation of CBC flux. There is then a period of a quasi–steady state, followed by an adjustment in the levels CBC intermediates, which (see below for more data analysis) is a consequence of the slow changes in protein abundance. This large increase in the levels of CBC intermediates between low and high irradiance is likely to lead to a large decrease of inorganic orthophosphate (Pi). It should be noted that even though the ATP/ADP ratio did not change (see above; Supplemental Figure 11), any decrease in Pi would result in an increase in the phosphorylation potential ([ATP]*[ADP]−1*[Pi]−1) after the shift to high irradiance, which would promote the 3PGA reduction. In agreement, the GAP/3PGA and DHAP/3PGA ratios increase as the light intensity is increased (see Figure 6 and Supplemental Figure 12B).
The major sinks for fixed C in vascular plants are starch, sucrose, protein, and cell wall polysaccharides and, to a lesser extent, lipids. Sucrose is present at much lower levels in C. reinhardtii than vascular plants (Klein, 1987). In our experiment, sucrose was below detection limit of highly sensitive enzymatic measurements. We investigated the response of metabolites in the remaining pathways to gain insights into how growth is increased after an increase in light intensity.
An Increase in Light Intensity Leads to a Transient Increase in ADP-Glucose and Stimulation of Starch Synthesis
ADP-glucose (ADPG) is the direct precursor for starch synthesis (reviewed in Geigenberger et al., 2004; Zeeman et al., 2007). ADPG increased >3-fold after 5 to 20 min of increased light and then declined to close to the control value (Figure 7). Glucose-6-P (G6P) and glucose-1-P (G1P) are precursors for the synthesis of ADPG and (see below) UDP-glucose (UDPG). G6P and G1P showed a small transient increase immediately after transfer to higher light and a larger sustained increase between 240 and 480 min (Figure 7).
Figure 7.

Hexose-Phosphates, ADPG, UDPG, and Starch.
C. reinhardtii CC-1690 cells were harvested after a light shift from 41 to 145 μmol photons m−2 s−1 (filled circles) or kept at 41 μmol photons m−2 s−1 (open circles). F6P, G6P, G1P, ADPG, and UDPG were measured by LC-MS/MS as described in Methods (n = 4 ± sd). Metabolite levels were given as concentrations in algal cells (μM). Starch was measured enzymatically according to Methods (n = 3, ± sd). The rate of starch synthesis (shown as inset) was calculated as described in Methods. Pairwise t test between control and treatment and P value correction for multiple sampling by Bonferroni correction was done (one asterisk, P < 0.05; two asterisks, P < 0.01; three asterisks, P < 0.001). The enzymes catalyzing steps between compounds are given in gray. PGI, phosphoglucoisomerase; PGM, phosphoglucomutase; GBSS, granule-bound starch synthase; BE, branching enzyme; DBE, debranching enzyme; DPE, disproportionating enzyme.
Starch content was low in low light, increased rapidly in the initial period after transfer to light, and then increased more slowly (Figure 7). In a bioreactor, cells are growing and the medium is exchanged to keep the cell density constant. This means that the starch content is not a measure of the rate of starch synthesis. The rates of starch synthesis were modeled from the starch content at adjacent time points, taking the medium consumption of the bioreactor into account (Figure 7; see Methods). As this calculation involves taking the difference between two quite similar values for starch content, the modeled rates are approximations. The modeled rate of starch synthesis was 0.2 μmol glucose/min in low light rose more than 10-fold within 5 min of increasing the light intensity, declined between 10 and 60 min, and from 120 min onwards was only slightly higher than in the control bioreactor. These modeled rates closely track the changes in the level of ADPG (Figure 7).
Starch synthesis is regulated by ADPG-pyrophosphorylase (AGPase) (Zeeman et al., 2007; Stitt and Zeeman, 2012). As in vascular plants, C. reinhardtii AGPase is allosterically activated by 3PGA and inhibited by Pi (Ball et al., 1991). The 3PGA/Pi probably increase after the switch to high light because although 3PGA showed a small decrease, the large and rapid increase of all other phosphorylated CBC intermediates is likely to lead to a large decrease of Pi (see above). This might explain the rapid transient increase in ADPG levels and stimulation of starch synthesis. However, this does not explain why ADPG levels and the modeled rate of starch synthesis subsequently decline; the levels of CBC intermediates remain high, and other phosphorylated intermediates like G6P and G1P even increase further. One possible explanation might be slow release of Pi from a vacuolar Pi pool and/or polyphosphates (Ruiz et al., 2001; Werner et al., 2007).
Sustained Increase in UDP-Glucose, the Precursor for O-Glycosylated Proteins
C. reinhardtii differs from vascular plants and many green algae in lacking a cellulosic cell wall. Instead, UDPG serves as a precursor for synthesis of hemicellulose and cell wall glycoproteins (Goodenough et al., 1986; Woessner and Goodenough, 1989). UDPG was significantly increased by 30 to 50% after 5 min of increased light intensity and this increase was maintained until 120 min (Figure 7).
Stimulation of Triacylglycerol Synthesis and Changes in Membrane Lipid Compositions
Lipids that showed significant changes after the increase in light intensity are shown in Supplemental Figure 8 and Supplemental Figure 13. There was a rapid increase in 34:1 monogalactosyldiacylglycerol (MGDG), which peaked at 30 to 60 min and then decreased, slower and smaller increases of 34:3 MGDG and 36:2 diacylglycerol-N,N,N-trimethylhomoserine (DGTS), which peaked at 60 to 120 min and then partially reversed, a decrease of 34:8 DGTS, 36:7 DGTS, and 36:8 DGTS to a minimum at 240 min, and a slow and progressive increase of 34:6 digalactosyldiacylglycerol (DGDG), 34:5 DGDG, and 32:0 DGTS. The time sequence in which the various MGDGs and DGDGs peaked reflects the sequence in which these compounds are synthesized. A pioneer flux study in C. reinhardtii (Giroud and Eichenberger, 1989) identified monoene and diene fatty acids to be first radioactively labeled, with a slow shift of the label over the span of several hours to trienes and tetraenes when feeding the algae with radioactive acetate. Similarly, a recent study observed a time delay between the incorporation of radioactive label into DGDG compared with MGDG (Li et al., 2012), which is explained by MGDG being the metabolic precursor of DGDG. In the time course, the increase in 36:1 DGDG preceded the increase in 36:2 DGDG, followed by the increase of 36:3 DGDG. Likewise, the increase of 34:1 MGDG preceded that of 34:3 MGDG. In addition, changes in MGDGs occur slightly before changes in DGDG. This points to an increased flux into lipid biosynthesis, which is supported by the increased C and energy that is available from photosynthesis.
The progressive decrease in highly unsaturated DGTS species might be a consequence of the accumulation of 54:9 triacylglycerol (TAG). TAG 54:9 showed a slow but sustained increase, which was detectable by 10 min and plateaued by 120 min. Li et al. (2012) proposed a mechanism in which fatty acids are initially allocated to MGDG from which they are excised and later become part of DGTS, PE, and TAG. If DGTS and TAG compete for the same pool of fatty acids, an increased shuffling of unsaturated fatty acids to TAG could have as a consequence a reduction of these fatty acids in DGTS. This idea is supported by the fact that the decrease in highly unsaturated DGTS species and the increase in TAG occur within the same time frame.
Thus, an increase in the light intensity leads to changes in membrane lipid composition as well as accumulation of the storage lipid TAG. The amount of C accumulated in TAG cannot be quantified because the lipid analyses provided only relative data.
Progressive Increase in the Levels of Amino Acids
The assimilation of inorganic nitrogen and synthesis of amino acids is closely intertwined with organic acid metabolism (Nunes-Nesi et al., 2010). We measured the levels of 2OG, aconitate, isocitrate, and malate by LC-MS/MS, and fumarate, succinate, citrate, and many individual amino acids by GC-MS (Figure 8; Supplemental Figure 14). Their response showed three main features. First, within 5 to 10 min, there was a rapid increase of succinate, malate, and several amino acids, including alanine, aspartate, serine, threonine, and glutamate. The magnitude of increase varied depending on the metabolites. It was significant for malate, glutamate, serine, and threonine when tested using pairwise Student’s t tests and additionally for aspartate when tested across multiple time points by ANOVA (P value < 0.05; Supplemental Data Set 1). Second, while malate showed an ~50% increase between 120 and 480 min, citrate, aconitate, isocitrate, fumarate, and 2OG levels did not change significantly. 2OG is synthesized from citrate via isocitrate and aconitate and is the immediate C precursor for ammonium assimilation and glutamate synthesis via the glutamine oxoglutarate aminotransferase pathway. Third, some amino acids, including aspartate, serine, threonine, and glutamate, continued to increase during the first hour to reach a temporary plateau followed by a further increase between 120 and 480 min. This late increase was significant in case of glutamate, serine, and threonine and occurred even though the levels of precursor organic acids plateaued or declined. These results point to a rapid use of the additional fixed C for organic acid and amino acid biosynthesis, followed by a selective stimulation of amino acid biosynthesis.
Figure 8.

TCA Cycle Intermediates and Amino Acids during Light Shift.
C. reinhardtii CC-1690 cells were grown in a bioreactor at 24°C, 5% CO2, and 41 μmol photons m−2 s−1 (open circles) or shifted to 145 μmol photons m−2 s−1 at time point zero (filled circles). 2OG and malate were separated and detected by LC-MS/MS and given in μM (n = 4, ± sd). Citrate, glutamate, and threonine were separated and detected by GC-MS (n = 3, ± sd) and given in fold changes normalized to time point zero. Pairwise t test between control and treatment was done and P value correction for multiple sampling by Bonferroni correction (one asterisk, P < 0.05; two asterisks, P < 0.01; three asterisks, P < 0.001). Additionally, ANOVA analysis over the whole control and treatment time course was done with P value correction for multiple sampling by Benjamini-Hochberg correction (dagger, P > 0.05; double dagger, P < 0.05). For all measured TCA intermediates and amino acids levels, see Supplemental Figure 14.
Temporal Kinetics of Polysome Loading Reveal a Slight Delay until Protein Synthesis Is Increased
The abundance of a set of ribosomal proteins did not change during the first 60 min after the light shift and even decreased slightly between 60 and 480 min (see above; Figure 4). We reasoned that the increase in the rate of protein synthesis that is required to support the faster growth must be brought about by an increase in loading of ribosomes into polysomes. Density gradient separation of polysomes from free ribosomes is a qualitative estimate of the rate of protein synthesis (Kahlau and Bock, 2008; Piques et al., 2009; Pal et al., 2013), although it probably underestimates the actual changes in protein synthesis rate (Pal et al., 2013).
Polysome loading (Figure 9) was unchanged during the first 5 min after transfer to higher light, started to increase by 10 min, was significantly increased from 20 min onwards, and reached a first plateau by 120 min, followed by a further increase between 120 and 480 min. The increase in polysome loading from 120 min onwards coincides with a slight decline in cytosolic and plastidial ribosome abundance (Figure 4). The temporal response of polysome loading also resembles that of G6P (Figure 7) and amino acid levels (Figure 8), which increased initially and then showed increase again from 120 min onwards. Overall, the temporal kinetics of the changes in polysome loading resembles the response in growth estimated from the culture dilution rate (Figure 1).
Figure 9.

Increase in Polysome Loading upon Increased Light Intensity.
C. reinhardtii CC-1690 cells were grown in a bioreactor at 24°C, 5% CO2, and 41 μmol photons m−2 s−1 and kept at this light intensity (open symbols) or shifted to 145 μmol photons m−2 s−1 at time point zero (closed symbols). For each time point and both treatments, the percentage of free ribosomes (squares) and ribosomes in polysomes (cycles) was calculated based on the ribosome profile (see Methods for details; n = 3 ± sd).
In the next step of our analysis, we integrated data sets across different functional levels to obtain insights into the relationship between changes in transcript and protein abundance, the mechanisms by which an increase in intermediate levels leads to an increase in flux in the CBC, and the reasons for the 10- to 15-min delay before an increase in the rate of photosynthesis translates into an increase in the rate of growth.
The Relationship between Responses of Transcripts and Proteins after Transfer to Higher Light Depends on the Protein Classes
Nucleus-encoded transcript data were available for 462 of the 644 quantified proteins, and, of these 462 pairs, both transcript and the protein changed significantly in 141 pairs after the light shift. For the purpose of testing whether these transcripts and proteins show a similar response, we calculated the average log2-fold change of transcript and protein abundance for these pairs at all times after the shift to higher light and compared them in a scatterplot (Supplemental Figure 15). There was a highly significant positive correlation between the average log2-fold change of transcript and protein abundance (R2 = 0.24, P value < 0.001; Supplemental Figure 15A). A similar correlation was found when transcript abundance at 40 min was compared with protein abundance at 240 (R2 = 0.22, P value < 0.001) or 480 min (R2 = 0.20, P value < 0.001).
However, the relationship depended strongly on the protein class (Supplemental Figure 15B). For example, MapMan bin 13.1 (amino acid synthesis) showed a very high Pearson correlation (>0.8) between transcript and protein abundance (Supplemental Figure 15C). Transcript abundance showed a transient change with a maximum or minimum at 40 min after the light shift, and the proteins showed a continuous increase or decrease, respectively, from 120 min after light shift onwards (Supplemental Figures 15D and 15E). Furthermore, this increase in protein abundance coincides with the increase in the levels of several amino acids from 120 min onwards (see above). This points to transcriptional regulation making a contribution to the slow adjustments in amino acid metabolism.
Other protein classes had poor or even negative correlations between transcript-protein pairs, including proteins involved in photosynthesis (e.g., LHCII, LHCI, PSI, and ATPase subunits and CBC enzymes) (Supplemental Figure 15B). This reveals that the changes in protein abundance in the photosynthetic machinery are largely due to posttranscriptional mechanisms.
An analogous analysis was not possible for plastid-encoded transcripts and proteins because very few of the transcripts showed significant changes. Comparison of the averaged absolute transcript and protein abundance did reveal a significant correlation between organelle-encoded transcript levels with protein levels (Supplemental Figure 16). However, this analysis also provided evidence for strong translational or posttranslational regulation; for example, the abundance of plastid ribosomal subunits varies 7-fold even though there was no difference in the corresponding transcript abundances.
Comparison of Changes in the Proteome and the Metabolome
The initial 2.5-fold increase in the rate of photosynthesis after transfer to higher light occurred within the first 5 min (see Figures 1B and 1C) and was accompanied by rapid and large increases in the level of many CBC intermediates (Figure 6), but in the absence of any changes in the abundance of CBC enzymes (Figure 4). While several CBC enzymes did show an increase in abundance, this occurred much later and was much smaller than the increase in the rate of photosynthesis. These slow changes in enzyme abundance were accompanied by small changes in the levels of several pathway intermediates. These results point to a response in which rapid changes of metabolites facilitate the initial rapid increase in CBC flux and slower changes in protein abundance facilitate a readjustment of metabolism. We therefore asked the following questions: First, are the observed changes in metabolites adequate to increase CBC flux by 2.5-fold? Second, can the changes in metabolites later in the time course be explained as a consequence of the slow adjustments in protein abundance? In the following sections, we address these questions using our comprehensive data set to calculate for each enzyme and time point (1) changes in Gibbs free energy of reactions (ΔrG), (2) changes in the in vivo saturation of enzymes, (3) the relationship between metabolite concentration and enzyme binding site concentration, and, for the later time points, (4) the relationship between the slow changes in CBC enzyme abundance and intermediate levels.
Calculation of Gibbs Free Energy Allows the Identification of Reversible and Irreversible Reactions in the CBC
An in vivo Gibbs free energy of reaction (ΔrG) close to zero shows that a reaction is close to equilibrium, while highly negative ΔrG values reveal which reactions are so far removed from equilibrium that they can be effectively considered irreversible in vivo. Changes in ΔrG in time or between conditions reveal if a given enzyme exerts a varying restriction on pathway flux.
Details of the calculation of ΔrG are given in the legend of Figure 10 and Supplemental Data Set 2. The level of E4P was estimated assuming that the TRK reaction with E4P and GAP is at equilibrium, and individual levels of Xu5P and Ru5P were estimated by assuming that RPE is at equilibrium. The ΔrG for the combined reactions of GAPDH and PGK was estimated using measured values of 3PGA and GAP, assuming an NADPH/NADP ratio of 1 (Heineke et al., 1991) and a Pi concentration of 0.002 M (Pratt et al., 2009). Nonaqueous fractionation (Dietz and Heber, 1984) and subcellular fractionation by membrane filtration of chloroplasts have shown that the ATP/ADP ratios in the plastid are much lower than those in the cytosol (Stitt et al., 1982; Gardeström and Wigge, 1988). We therefore used an ATP/ADP ratio of 3 (Gardeström and Wigge, 1988) rather than the overall ratio measured in C. reinhardtii cells.
Figure 10.

Gibbs Free Energy of Reaction (ΔrG) of CBC Enzymes.
ΔrG based on metabolite data shown in Figure 6 of C. reinhardtii CC-1690 cells harvested 1 h before the light shift and at time point zero (light gray) and after the light shift (dark gray). The time points proceed from left to right: 1 h before light shift, time point zero, and 5, 10, 20, 40, 60, 120, 240, and 480 min after light shift. For comparison, the ΔrG values calculated by Bassham and Krause (1969) based on metabolites measured in C. pyrenoidosa are shown (white). This figure is a graphic representation of data presented in Supplemental Data Set 2. a[CO2] under 5% CO2 conditions was assumed to be 0.202 M. bNADPH/NADP+ = 1 (Heineke et al., 1991). cATP/ADP = 3 (Gardeström and Wigge, 1988). d[Pi] = 0.002 M (Pratt et al., 2009). eThe TRK reaction was assumed to be at equilibrium to estimate the in vivo concentration of E4P (K = 0.084, Bassham and Krause 1969). fThe RPE reaction was assumed to be at equilibrium to estimate individual levels of Xu5P and Ru5P (K = 0.667; Bassham and Krause, 1969).
[See online article for color version of this figure.]
The CBC reactions fall into two groups based on the estimated values for ΔrG (Figure 10): (1) four reactions with highly negative ΔrG, catalyzed by Rubisco (−40 kJ⋅mol−1), SBPase (−29 kJ⋅mol−1), FBPase (−28 kJ⋅mol−1), and PRK (−16 kJ⋅mol−1) (PGK+GAPDH will be discussed below) and (2) all other reactions with ΔrG close to zero, including TPI, FBA, SBA, and RPI. The TRK reaction with Ru5P and GAP (ΔrG = −6 kJ⋅mol−1) has a slightly negative ΔrG. The ΔrG of RPE and TRK reaction with GAP and F6P are zero by definition as these reactions were set to equilibrium for the estimation of E4P, Xu5P, and Ru5P levels.
The ΔrG values were not greatly affected by the shift from low to higher light. In the first 5 to 10 min after the shift, the estimated ΔrG for FBA and SBA became positive but small. It should be noted that small errors in the assumed chloroplast volume will lead to a systematic shift in the estimates of ΔrG for second-order reactions like the FBA and SBA reaction; it is therefore possible that the calculated ΔrG values for such second-order reactions may be slightly overestimated. Between 10 and 480 min, there were further small shifts in ΔrG values. The ΔrG values for FBA, FBPase, SBPase, and PRK increased slightly but remained negative, and ΔrG values for TRK and Rubisco decreased to even more negative values. These gradual changes point to a slow adjustment to the higher light regime (see below). A similar set of calculations was performed using metabolite data from cells exposed for 60 min to darkness or five progressively higher light intensities (Supplemental Figure 12D). These calculations confirmed that Rubisco, FBPase, SBPase, and PRK catalyze irreversible reactions, that the other CBC reactions are close to equilibrium, and that ΔrG for FBA, SBA, and TRK become progressively more negative as the light intensity and CBC flux increases.
Modeling the in Vivo Substrate Saturation of CBC Enzymes Reveals That Some Reactions Are Strongly Substrate Limited and Other Are Close to Saturation in Vivo
Fendt et al. (2010) distinguished three types of relationships between enzyme and substrate abundance in vivo: (1) metabolites are extremely limiting and enzyme capacities are in excess, (2) metabolites are limiting and enzyme capacities are in relatively small excess, and (3) metabolites are in excess and enzymes capacities are limiting. This is illustrated in Figure 11A for the simplest case of a reaction catalyzed by an enzyme with Michaelis-Menten kinetics.
Figure 11.

Calculated v/Vmax Values for CBC Reactions during Light Shift.
(A) Enzyme kinetics plotted according to Fendt et al. (2010) showing three different relationships (I-III) between enzyme abundances and metabolite concentrations alterations based on Michaelis-Menten enzyme kinetic: v = Vmax*[S]*(Km+[S])−1. Note that the x axis is shown in log10-scale.
(B) Enzyme kinetics were calculated over a range of substrate concentration for each CBC enzymes except RPE and the first TRK. For PGK, TPI, FBA, SBA, TRK, and RPI, the Michaelis-Menten kinetic equation for reversible reactions was applied. For GAPDH, FBPase, SBPase, and PRK, the Michaelis-Menten kinetic equation for irreversible reaction was applied taking into account known competitive inhibitors for the FBPase, SBPase, and PRK reactions. For Rubisco, the Michaelis-Menten-like kinetic equation based on Farquhar (1979) was used (Von Caemmerer, 2000). This was done for time point zero (0 min, thin dashed line), 20 min (intermediate dashed line), and 480 min (thick dashed line) after the light shift. Additionally, the actual measured substrate concentration at three representative time points during the light shift is shown at time point zero (yellow), 20 min (red), and 480 min (orange) after the light shift. Enzyme names are given with their corresponding substrate in parentheses. For details of the calculation, see Methods, for rate equations, see Supplemental Table 2, and for Km and KI values, see Supplemental Table 3. For FBA, SBA, and TRK, the v/Vmax modeled at different time points changes due to changes in the level of the second substrates GAP (FBA and TRK) and DHAP (FBA and SBA). Note that for FBPase and SBPase, the Km for FBP and SBP, respectively, of the reduced (red) and the oxidized (ox) enzyme is used for modeling the substrate saturation. Note that the x axes are shown in log10-scale. Values of the y axis <0 are shaded in gray and indicate that the reaction works in the opposite direction for the corresponding substrate level.
To test which of these relationships hold for the various CBC reactions, we modeled the in vivo substrate saturation curve for each enzyme using our measured metabolite levels and full rate equations that were parameterized with Km and KI values from the literature (Supplemental Tables 2 and 3). Two enzymes were omitted from this analysis (one of the reactions catalyzed by TRK and RPE because we set these reactions to equilibrium to calculate E4P, Xu5P, and Ru5P). For the carboxylation reaction catalyzed by Rubisco, a widely used Michaelis-Menten–like kinetic equation was applied (Farquhar, 1979; Von Caemmerer, 2000). Equations based on Michaelis-Menten kinetics for irreversible reactions (Segel, 1975) were used for the reactions catalyzed by GAPDH, FBPase, SBPase, and PRK and based on Michaelis-Menten kinetics for reversible reactions (Cleland, 1963) for the reactions catalyzed by PGK, TPI, FBA, SBA, TRK, and RPI. For details of the calculation, see Methods and Supplemental Tables 2 and 3. In Figure 11, the y axis shows velocity for Rubisco and the v/Vmax ratio for the other enzymes.
The modeled in vivo substrate saturation curves for Rubisco, PGK, GAPDH, TPI, FBA, FBPase, SBA, SBPase, TRK, RPI, and PRK are shown in Figure 11B. Three simulations were performed using metabolite concentrations in low light and 20 and 480 min after the switch to higher light. The latter correspond to the times when metabolite levels have reached their maximum (20 min) and after the slower adjustments to higher light (480 min). The modeled substrate saturation curves are similar in all simulations, except for TRK, FBA, and SBA. In these second-order reactions, changes in the level of one substrate leads to a change in the saturation kinetics or maximal catalytic rate for the other substrate. The colored circles on the curve indicate the in vivo substrate levels in low light (zero time point) and 20 and 480 min after the irradiance was increased.
This analysis of the relation between the predicted substrate saturation response and the estimated in vivo substrate concentration(s) divides the CBC enzymes into two groups (Figure 11B): (1) GAPDH, TPI, FBA, SBA, RPI, and PRK lie at the border between area I (where the substrate is limiting) and area II (where the substrate as well as enzyme could be limiting). For these enzymes, an increase in flux could easily be generated by an increase in substrate level. (2) Rubisco, FBPase, and SBPase lie close to area III where the enzyme is substrate saturated. For such enzymes, an increase in flux will require an increase in enzyme abundance and/or posttranslational activation and/or relaxation of feedback inhibition to allow either a decrease in Km and/or an increase in Vmax. PGK and TRK showed intermediate results. PGK was in area II in low light and closer to area III in higher light, while TRK was in area II in low light and closer to area III in higher light for S7P and in area I in low light and area II in higher light for GAP (Figure 11B). This implies that for PGK and TRK, a moderate increase in flux might be achieved by a change in substrate levels, but a large increase in flux will require an increase in enzyme abundance or activation of the enzyme, for example, by posttranslational activation which might lead to a decrease of Km or an increase of Vmax. It should also be noted that TRK will be strongly stimulated when there is a simultaneous increase in the levels of both substrates. This division matches the classification of enzymes based on their calculated ΔrG values (Figure 10) except for PRK. PRK is subject to feedback regulation by multiple metabolites and operates at fraction of its full capacity (Laing et al., 1981).
Several CBC enzymes are subject to light-dependent redox modification by thioredoxin. The above calculation used kinetic parameters for the reduced, active form of the enzyme that predominates in the light. For FBPase and SBPase, literature values are available for the Km of the oxidized and reduced state (130 and 6 μM for FBPase and 180 and 50 μM for SBPase, respectively; Cadet and Meunier, 1988). We used these values to model the substrate saturation curves for both enzyme forms (Figure 11B). While the substrate level were near-saturating for the reduced forms (area III), the oxidized forms gave an intermediate result with the measured substrate levels lying in area II of the substrate saturation curve and a corresponding 2-fold decrease in the predicted fluxes that are catalyzed by the enzymes. As already noted, the FBP level actually drops slightly in the first 5 to 10 min after the shift to higher light. The required increase in flux could be obtained if redox activation of FBPase increased between low and higher light. An analogous but smaller response is seen for SBPase. For both, there was a large increase in product/substrate ratio (Supplemental Figure 17), which is consistent with activation of FBPase and SBPase after the switch to high light. Furthermore, as already mentioned, FBP and SBP levels rise to a peak at 20 μmol photons m−2 s−1 and then decline, which is consistent with incomplete activation of FBPase and SBPase at low light intensities (Supplemental Figure 12A).
Modeled in Vivo Km Values Reveals That in Vitro Km Values Strongly Underestimate in Vivo Substrate Affinities
An earlier study in Escherichia coli (Bennett et al., 2009) presented a large-scale study of the relation between measured substrate concentrations and in vitro Km values from BRENDA (Schomburg et al., 2013). They found that the majority of metabolite concentrations lie close or above the in vitro Km and some lie >10-fold above the in vitro Km and interpreted this as evidence for a trend to saturation of enzyme binding sites. However, this approach has the drawback that in vitro Km values do not take into account possible in vivo modifications of the Km due to competition between substrate and product, competitive product binding, allosteric regulation, substrate channeling, and posttranslational modification. We took advantage of the exhaustive literature information about the kinetic properties of CBC enzymes and our near-complete analysis of CBC intermediate abundance to reexamine this claim.
The estimated in vivo Km was always higher, and in some cases much higher, than the in vitro Km (Figure 12; Supplemental Figure 18). This picture was modified when the impact of the in vivo effector metabolites on the Km values were taken into account. For the enzymes that catalyze irreversible reactions, inhibitory interactions will lead to an increase in the Km in vivo. All such CBC reactions showed a marked increase in the modeled in vivo Km compared with the in vitro value including Rubisco with RuBP (30-fold), FBA with DHAP (10- to 20-fold), FBPase with FBP (3-fold), and PRK with Ru5P (10-fold) as substrate. For the enzymes that catalyze reversible reactions, the substrates and products typically bind in a competitive manner. While the in vitro Km for a substrate of a reversible reaction is typically determined using assays in which product concentration is very low, the products are usually present at appreciable concentrations in vivo. Accordingly, the modeled in vivo Km values were often higher than the in vitro Km values, including FBA with DHAP as substrate (10- to 20-fold), SBA with E4P as substrate (5-fold), and TRK with S7P as substrate (3-fold).
Figure 12.

In Vitro and in Vivo Km Values versus Substrate Concentrations [S].
The substrate levels 20 min after the light shift of CBC reactions (Figure 11) are plotted against in vitro Km (dark gray) (literature values; for details, see Supplemental Table 3) and in vivo Km (light gray) calculated based on substrate saturation curves (Figure 11). Arrows indicate >2-fold increases between in vitro and in vivo Km values. Enzymes with their substrate in parentheses: aPGK (3PGA), bTRK (S7P), cRubisco (RuBP), dFBA (DHAP), eSBA (DHAP), foxidized SBPase (SBP), greduced SBPase (SBP), hoxidized FBPase (FBP), ireduced FBPase (FBP), jRPI (R5P), kPRK (Ru5P), lSBA (E4P), mTPI (GAP), nFBA (GAP), and oGAPDH (BPGA). In Supplemental Figure 18, the corresponding plot with substrate levels at the low light intensity is shown.
[See online article for color version of this figure.]
Comparison of in vivo substrate concentrations with in vitro Km values reveals that several substrates are present at >2-fold higher concentrations than the in vitro Km values, especially when fluxes are increased in higher light (Figure 12; Supplemental Figure 18). This resembles the conclusion of Bennett et al. (2009). However, this trend is less pronounced when in vivo substrate concentrations are compared with estimated in vivo Km; only RuBP and FBP lie >2-fold above the in vivo Km. For these, it might be noted that much of the RuBP may be bound in Rubisco active sites, while recent 13C labeling experiments in Arabidopsis indicate that part of the FBP pool may not be directly involved in CBC flux (Szecowka et al., 2013).
Many CBC Enzymes Are at a Similar or Higher Abundance Than Their Substrates
We next compared the absolute concentrations of enzymes and metabolites in the CBC. We performed this comparison to test the widely spread view that enzymes are present at far lower concentrations than their substrates. Absolute protein abundances were estimated by calculating the exponentially modified protein abundance index (emPAI) (see Methods and Supplemental Table 1) (Ishihama et al., 2005). The emPAI score was summed for isoforms of the same enzyme and then used to estimate the enzyme abundance (E) and, hence, the binding site concentration. To do this, the emPAI count for a given protein was first expressed as a fraction of the total emPAI count (termed relative abundance [RA]). RA was then normalized on total measured protein and corrected for the mole mass of the protein to estimate the molarity of each enzyme. This calculation requires assumptions because only 767 of 17,038 annotated C. reinhardtii proteins were detected by our proteomics analysis. The normalization was initially performed in two ways. In the first, RA was normed to total protein as suggested by Ishihama et al. (2005). In the second, RA was normed to total protein after correcting the latter for the fraction of the total annotated C. reinhardtii proteins that were detected in our analysis (i.e., after multiplying total protein by 767/17,038). It is likely that many of the undetected proteins are at low abundance but will still represent a substantial part of the total cellular protein. The value of E for all individual proteins will be overestimated by the first approach and strongly underestimated by the second approach (Supplemental Table 4). As an example, these calculations gave values of 1006 ± 107 μM and 45 ± 5 μM for RbcL. To experimentally assess which normalization factor should be used in these calculations, we determined the level of RbcL by scanning the absorbance of the 52 kD RbcL band in Coomassie-stained SDS-PAGE gels (Supplemental Figure 19). This gave a value for RbcL abundance of 304 ± 33 μM, which is 3-fold lower than the modeled value given by our first approach. We used this value to correct the value of E for all other enzymes (Supplemental Table 4) and used these corrected values to calculate the relation between E and substrate concentration, S (Figure 13).
Figure 13.

Substrate per Binding Sites versus Substrate Concentrations and Enzyme Saturation of the CBC.
Substrates of CBC reactions were measured via LC-MS/MS and are shown in Figure 6. Binding sites of CBC enzymes were calculated based on proteomics data via the emPAI as described in the text and in Supplemental Table 1.
(A) Substrate per binding site values of CBC reactions are plotted against the substrate level 20 min after the light intensity was increased from 41 to 145 μmol photons m−2 s−1.
(B) Substrate per binding site values of CBC reactions are plotted against the degree of saturation according to the saturation curves shown in Figure 11, 20 min after the light intensity was increased from 41 to 145 μmol photons m−2 s−1. For a similar plot at the lower light intensity, see Supplemental Figure 20.
This analysis reveals, rather unexpectedly, that many CBC intermediates are present at concentrations that are far lower than the estimated binding site concentration of the enzymes of which they act as substrates (Figure 13A). Examples after 20 min in higher light include BPGA with GAP and PGK, GAP with TPI, TRK, and TPI, E4P with SBA and TRK, Ru5P with PRK, RPI, RPE, and R5P with RPE and TRK, and FBP with FBA as substrates. Further examples in low light include DHAP with FBA and SBP with SBA (Supplemental Figure 20A). In particular, the substrate concentrations GAP, E4P, and Ru5P are 5- to 20-fold lower than the binding site concentration of their enzymes FBA and TRK. Many other metabolites are at similar or only slightly higher concentrations than the corresponding enzyme binding site concentrations. We realize that these calculations involve approximations, but even a 5-fold larger value for E would still leave many substrates at comparable or lower concentrations than the respective enzymes.
Two CBC enzymes catalyze more than one reaction. Aldolase catalyzes the reversible interconversion of GAP + DHAP and FBP, and GAP + E4P and SBP, while TRK catalyzes the reversible interconversion of F6P + GAP with E4P + Xu5P, and S7P + GAP with R5P + Ru5P. However, after taking these alternative reactions into account, there is still a large excess of enzyme binding site over some of the intermediates. There are also large imbalances in the availability of the alternative substrates for a given enzyme. For TRK reactions, F6P and S7P are well in excess of, and GAP is below, the estimated TRK binding site concentration. For the FBA reaction, leading to SBP, both substrates (GAP and E4P) are predicted to be at much lower concentrations than the FBA binding site, whereas for the alternative reaction leading to FBP, one is very low (GAP) but the other can be higher (DHAP). This raises questions about how coordinated fluxes are achieved at different sites in the CBC, but also indicates that one mechanism may be that many reactions are likely to respond very sensitively to changes in the levels of these low-abundant substrates. It is also noteworthy that FBP and SBP are in excess of FBPase and SBPase binding sites, as are 3PGA for PGK and RuBP for Rubisco. We next compared the in vivo substrate saturation (Figure 11) with the ratio between S and E (Figure 13B). The enzyme-substrate pairs fall into two groups. For one set (including GAPDH, TPI, FBA/SBA, RPI, and PRK), S was much lower than the in vivo Km and was similar to or lower than E. For the other set (Rubisco, FBPase, and SBPase), S was at or above the in vivo Km and was also higher than E. Compared with low light, in higher light, there was a shift to increased saturation (compared with Figure 13B and Supplemental Figure 20B). TRK and PGK took an intermediate position, but it should be noted that a combined increase in the levels of both substrates of TRK will lead to a substantial increase in flux.
These plots summarize the preceding sections and show that, for one set of CBC enzymes, flux can easily be increased by an increase in substrate concentration, whereas for another set (Rubisco, FBPase, SBPase, and to a certain extent TRK and PGK) an increase in flux will require an increase in enzyme abundance. This analysis also reveals that, at least in C. reinhardtii, the CBC does not operate in the mode assumed for enzymes in central metabolism, with metabolite concentrations being well in excess of enzyme concentrations. Rather, there are high concentrations of many enzyme binding sites and low concentrations of many metabolites.
The Gradual Increase in CBC Enzyme Abundances Contributes to the Slow Changes in Intermediate Levels
In their studies with wild-type and mutant yeast lines, Fendt et al. (2010) showed that there are often reciprocal changes in enzyme abundance and the abundances of their substrates and concluded that the changes in enzyme abundance often drive changes in fluxes. We tested whether any of the changes in enzyme abundance between 20 and 480 min (Figure 3) were accompanied by reciprocal changes in metabolite levels (Supplemental Figure 21). Overall, the correlation between the change in protein abundance and substrate level was weak and nonsignificant (R2 = 0.0115, P value = 0.715). For Rubisco, SBPase, and GAPDH, the enzyme and substrate levels showed opposite trends; therefore, for these reactions, the increase in enzyme abundance could make a substantial contribution of the changes in metabolite levels seen between 20 and 480 min. Thus, our analyses of enzyme kinetics, substrate levels, and enzyme abundance (see previous sections) pinpoint three enzymes as potential bottlenecks in the CBC. Two (Rubisco and SBPase) increased in abundance between 20 and 480 min accompanied by a decrease of their substrate levels, showing that the increase in Rubisco and SBPase abundance has a perceptible effect on the operation of the CBC. The third of these enzymes, FBPase, also showed an increase in abundance, but FBP levels did not fall, possibly because of the concomitant increase in DHAP and GAP, which will lead to an increase in FBP levels. The 48% increase in RCA abundance noted above will also contribute to maintaining Rubisco activity (see Discussion).
Turnover Times of CBC Intermediates and Sequestration of Carbon after Transfer to Higher Light
We next turned to the temporal relationship between photosynthesis and growth. As already noted, while photosynthesis rates rose within 15 to 20 s, there was a lag of 10 to 15 min before growth increased. We asked whether this lag could be accounted for by the increase of metabolites in the CBC and elsewhere in central metabolism.
The measured photosynthesis rates, the metabolite levels and known pathway stoichiometry were used to calculate half-times of the CBC intermediates (Supplemental Table 5). The half-times of most metabolites were in the range of one second or below. 3PGA, DHAP, F6P, S7P, and RuBP had slightly higher turnover times, reflecting their large pool sizes relative to other CBC intermediates. Overall, the turnover times of CBC intermediates in C. reinhardtii are similar to those in Arabidopsis leaves (Arrivault et al., 2009). RuBP, F6P, and S7P formed the biggest pools, while 3PGA and DHAP pools were smaller than in C. reinhardtii and, correspondingly, had even lower turnover times.
We used these half-times to estimate how many seconds of photosynthesis would be required to fix enough carbon to account for the observed increase in the levels of CBC intermediates after the increase in light intensity (Supplemental Table 5). The difference between the pool size in low light and after 5 min in high irradiance was calculated for each metabolite, multiplied by the number of C atoms in the metabolite, divided by the increase in carboxylation rate, and then summed across all metabolites. This calculation reveals that it would take ∼32 s of photosynthesis at the new higher light intensity to generate the observed increase in intermediate levels. This is in quite good agreement with the ∼20 s it took the photosynthesis rate to rise to a new steady value, based on the measurements of O2 evolution (Figure 1B). This supports the idea that the increase in CBC metabolite pool sizes is needed to allow increased fluxes around the CBC (and, hence, to consume the additional ATP and NADPH that is available from the light reactions).
Modeling of the Kinetics of Growth by Carbon Balance Analysis
While photosynthesis increased within 20 s of increasing the light intensity, analyses of optical density (Figure 1A) indicated that there is a lag of over 10 min until growth starts to increase. During this period, the additional newly fixed C presumably accumulates in intermediate pools or C reserves. We first calculated the additional fixed C per time interval from the increment in photosynthesis between low and higher light. The metabolite levels measured before and at consecutive time points after transfer to higher light (Figures 6 to 8; Supplemental Data Set 1) and the estimated rates of starch synthesis (Figure 7) were then used to calculate how much C is sequestered in these pools in the different time intervals after increasing the light intensity (Figure 14A). The pools covered by this analysis include CBC intermediates, TCA cycle intermediates, some organic acids, amino acids, and starch (Supplemental Table 6). Total C in the metabolite pools and starch during the first 5 min in higher light increased by 3.7 × 1022 C*L cell volume−1, which is equivalent to almost 80% of the additionally fixed C in this time interval (Figure 14A). Of this, over three-quarters was sequestered into starch (78%), while the remainder was sequestered in metabolites, of which S7P and malate were the major contributors (5.4 and 4.5% of additionally fixed C, respectively; Supplemental Table 6). The remaining C may be sequestered into metabolites that were omitted from our analyses and TAG, which was not quantified absolutely. Less C was sequestered between 10 and 20 min (3.4 × 1022 C*L cell volume−1, equivalent to ∼40% of the extra C fixed in this time). Sequestration of photosynthates in starch and metabolite pools was even smaller between 40 and 60 min and essentially zero at later time points. These results show that most of the additional fixed C accumulates in intermediate pools during the first 5 to 10 min after the light shift, whereas at later times an increasing proportion is used for growth.
Figure 14.

C Sequestration after Light Shift into Metabolite Pools and Starch.
C. reinhardtii CC-1690 cells were grown in a bioreactor at 24°C, 5% CO2, and 41 μmol photons m−2 s−1 and shifted to 145 μmol photons m−2 s−1 at time point zero.
(A) Based on photosynthesis rate, the additionally fixed C in the higher light condition was calculated and set to 100% (n = 4 ± sd). Based on changes between metabolites pools measured by LC-MS/MS (Supplemental Data Set 1) and starch (Figure 7), the changes in C atoms in these pools were calculated and expressed relative to the additionally fixed C between all time points (see Methods).
(B) Based on the C sequestration shown in (A), the growth rate was modeled (for details, see Methods). The dashed line indicates the modeled growth rate at the low light intensity that was set to 1. AU, arbitrary units.
(C) Pearson correlation between the modeled growth rate shown in (B) and the specific growth rate measured by medium dilution rate (Figure 1A).
(D) Pearson correlation between the modeled growth rate shown in (B) and polysome loading (Figure 9).
We modeled growth as the difference between net C fixed and the C that is sequestered into starch and metabolite pools in a given time interval (Figure 14B). Compared with the rate in low light, the modeled growth rate was almost unaltered in the first 10 min after transfer to high irradiance and then increased gradually between 60 and 120 min to reach a maximum that was 2.5-fold higher than the initial rate. This resembles the measured increase in the rate of photosynthesis (2.3- to 2.6-fold; Figures 1B and 1C). The modeled growth rates in each time interval correlated well with the growth rate measured from the dilution rate in the chemostat culture shown in Figure 1A (R2 = 0.73, P = 0.0019; Figure 14C) and the temporal kinetics of polysome loading shown in Figure 9 (R2 = 0.67, P = 0.004; Figure 14D).
DISCUSSION
Temporal Response of Photosynthesis, of Metabolite, Transcript, and Protein Abundance, and of Growth Uncovers a Three-Phase Response to an Increase in Irradiance
We applied a systems approach to investigate how photosynthetic cells translate an increase in incoming light energy into an increase in the rate of C fixation and growth. The model green alga C. reinhardtii was grown in a custom-designed bioreactor to allow precise control of the environmental conditions and sufficient algal material for all measurements. The light intensity was increased in the nonsaturating range to avoid additional complications due to high light stress. A large multilevel data set was generated by analyzing the photosynthesis rates, electron transport, ATP synthesis, the abundance of 11,455 nuclear-encoded and 108 organellar-encoded transcripts, 644 proteins, 120 intermediary metabolites, and 98 lipids as well as polysome loading and growth, as monitored by medium exchange.
After transfer to higher light, the rate of photosynthesis increased within 20 s to a 2.5-fold higher rate that was maintained for the next 8 h. In the first 5 to 10 min, almost all the additionally fixed C accumulated in metabolites, including a 2- to 3-fold increase in the levels of many CBC intermediates, a slightly slower increase in organic acids and amino acids, and, especially, a strong transient stimulation of starch accumulation. The growth rate started to increase from ∼10 min onwards and by 40 min reached a new stable rate that was 2.5-fold higher than in low light. Similar temporal kinetics were obtained from measurements of polysome loading, which provides a qualitative proxy for the rate of protein synthesis (Rudra and Warner, 2004; Pal et al., 2013) and by modeling the growth rate from the C balance. There were widespread changes in the levels of nucleus-encoded transcripts within 40 min, while plastid-encoded transcripts showed only small changes in abundance. Protein abundance changed more slowly, with relatively few detectable changes in the first hour, but many by 4 to 8 h, which were accompanied by further changes in metabolite levels.
The rates of photosynthesis and growth increase long before there are detectable changes in protein abundance. This implies that when C. reinhardtii is grown in low irradiance, it maintains protein levels that are able to support 2- to 3-fold higher fluxes than those that actually occur in low irradiance. This surplus capacity may be important to allow rapid responses of photosynthesis and growth in a fluctuating light environment. It is possible that some proteins respond more rapidly but were not detected in our study. Current limitations in the technology for detecting and quantifying proteins restrict protein quantification to the most abundant proteins in plants (Baerenfaller et al., 2012). Nevertheless, our analyses covered a large proportion of the proteins in the photosynthetic apparatus and many proteins involved in processes that are required for protein synthesis including amino acid biosynthesis and ribosome biogenesis.
The temporal response of C. reinhardtii to an increase in irradiance can be schematized as a three-phase process (Figure 15). In the first phase, photosynthesis has increased but growth has not yet been stimulated, and C accumulates in metabolic intermediates and, especially, as starch. In the second phase, growth has increased to match the rate of C fixation and metabolite levels stabilize at a high level, whereby the magnitude of the increase depends on the particular metabolite. In the first and second phase, the increase in metabolic fluxes and growth is driven by the increase in metabolite levels and posttranslational regulation, which allow more efficient use of the existing proteins. Between the second and third phase, transcriptional and posttranscriptional regulation lead to selective changes in the abundance of particular proteins, which in turn lead to changes in the levels of metabolites. This includes an increase in the abundance of proteins that are potential bottlenecks. For example, an increase in Rubisco and SBPase protein abundance is accompanied by a partial reversal of the large initial increase in RuBP and SBP (see below for more discussion). It also includes changes in protein allocation between subprocesses. In some cases, the changes in allocation are directly related to the initial perturbation; for example, less protein is allocated to light harvesting complexes and photosystem complexes and more to thylakoid ATPase and key CBC enzymes (Figure 4). Others are probably a response to changes in metabolite levels and regulatory networks that occur as a consequence of the increase in light intensity, for example, the small decrease in abundance of ribosomal proteins (Figure 4). This slight decrease is unexpected; the higher rate of growth in high light will require higher rates of protein synthesis and might therefore be expected to require more ribosomes. However, in low irradiance, polysome loading is low (Figure 9), probably because low sugar levels lead to an inhibition of translation initiation (Smeekens et al., 2010; Robaglia et al., 2012; Pal et al., 2013). In high light, sugars increase and this inhibition is relieved, allowing a more efficient use of ribosomes for protein synthesis and a small decrease in overall ribosome abundance, although the overall rate of protein synthesis increases. It should be noted that in the first phase, metabolism is not in steady state. In the second and third phases, metabolism is in a quasi–steady state; this is possible because metabolites have much faster turnover times than proteins. In the following sections, selected aspects of this multilevel response are discussed in more detail.
Figure 15.

Schematic Summary of the Response of C. reinhardtii to an Increase in Light Intensity (for Details, See Text).
Phase 1 is not at steady state, and phase 2 and phase 3 represent quasi–steady states.
Transcriptional and Posttranscriptional Regulation Contribute to the Abundance of Organelle-Encoded Proteins
Regulation of plastid gene expression is thought to be regulated mainly at the posttranscriptional level (Eberhard et al., 2002; Marín-Navarro et al., 2007). In agreement, an increase in the light intensity in the limiting range led to only small changes in plastid transcript levels, and the decrease in rbcL transcript was not accompanied by a decrease in Rubisco protein. Nonetheless, there was a reasonable agreement between the absolute abundance of plastid- and mitochondria-encoded transcripts and the corresponding proteins when they were averaged over the entire experiment, indicating that in the long term, plastid transcript levels do influence protein levels. Furthermore, the gradual decrease in chlorophyll was accompanied by a decrease in chlB and chlN transcripts, whose gene products are involved in chlorophyll biosynthesis.
Increase in Irradiance in the Nonsaturating Range Triggers Widespread Changes in Transcript Abundance
About 15 and 7% of all detected nuclear transcripts showed significant changes in abundance after 40 and 480 min, respectively, and 6 and 4% showed >3-fold changes at 40 and 480 min. This resembles the response after a switch from limiting to saturating light in Arabidopsis, when 4% of transcripts change >3-fold (Vanderauwera et al., 2005). Transcripts with increased expression were overenriched in genes required for growth including amino acid activation, protein targeting, and protein folding, as well as photosynthesis at 120 min.
One of the most strongly increasing transcripts was LHCSR1, whose abundance increased almost 40-fold at 40 min after the light shift. The related LHCSR3 transcript was also induced. LHCSR genes are ancient member of the LHC superfamily. Peers et al. (2009) showed that the LHCSR3 protein plays a similar role as PsbS in vascular plants and is essential for de-excitation of chlorophyll molecules in PSII (nonphotochemical quenching), while LHCSR1 alone is not sufficient for nonphotochemical quenching. Previous studies reported that LHCSR transcripts are induced by a transition from limiting to saturating light in C. reinhardtii (Savard et al., 1996; Richard et al., 2000; Peers et al., 2009; Bonente et al., 2012). In our study, LHCSR1 was more strongly induced than LHCSR3, which is opposite to the response after a shift to saturating light (Peers et al., 2009). CCP2 (low-CO2-inducible chloroplast envelope protein 2) and LCI1 (low-CO2-inducible membrane protein 1) were also strongly induced by increased light. As these genes are induced by low CO2 (Burow et al., 1996; Van et al., 2001; Pollock et al., 2004), they have been proposed to be part of the inorganic carbon concentrating mechanism. High light may induce LCI1 because the resulting increase in the rate of photosynthesis leads to a temporary decrease in the internal CO2 concentration, which induces genes involved in the carbon concentrating mechanism (McGinn et al., 2003; Spijkerman, 2011). Another strongly induced transcript was NRX3, which showed a 39-fold increase 40 min after the light shift. There are three NRX (nucleoredoxin) homologs in C. reinhardtii, of which NRX2 also increased although not as strongly as NRX3. Nucleoredoxins belong to the thioredoxin family (reviewed in Funato and Miki, 2007). In maize (Zea mays) kernels, they are localized in the nucleus and may play a role in altering the reduction state of transcription factors (Laughner et al., 1998).
Some Nucleus-Encoded Proteins Are Regulated Transcriptionally, but Others, Including Many Proteins in Photosynthesis, Are Regulated Posttranscriptionally
There were widespread changes in protein abundance after 60 and 480 min, with 14 and 26%, of the detected proteins showing significant changes. The fraction of detected proteins that undergoes significant changes at 480 min is similar to the fraction of the detected transcripts that show significant changes at 40 min. However, the amplitude of the changes in protein abundance is smaller than that for transcript abundance. Our analyses may underestimate the proportion of proteins that change in abundance. A recent study in mammalian cell lines (Schwanhäusser et al., 2011) reported that rapid changes in protein abundance are more frequent for low abundant proteins like those involved in signaling pathways than for highly abundant proteins like those involved in molecular machines and central pathways. The relatively low coverage in our study means that the latter predominate. Overall, however, our data point to early changes in transcript abundances, which are followed by slower and smaller changes in protein abundance. This is the pattern expected for proteins that are not subject to rapid turnover (Stitt and Gibon, 2014).
When individual nucleus-encoded transcript-protein pairs are compared, the correlation between the changes in transcript and protein abundance is highly significant (R2 = 0.24, P value < 0.001). This resembles the values reported for comparisons of mid- to long-term changes in bacteria (Lu et al., 2007), yeast (Gygi et al., 1999; Lu et al., 2007), mammals (Gry et al., 2009; Akan et al., 2012), and plants (Baerenfaller et al., 2012; reviewed in Vogel and Marcotte, 2012). In a comparative study, R2 values of 0.41 to 0.44 were obtained for E. coli and yeast, and lower values of 0.27 to 0.29 for more complex eukaryotes including Arabidopsis and humans (de Sousa Abreu et al., 2009). Poor or even negative correlations have been reported when transcript abundance was compared with enzyme (Gibon et al., 2004) or protein (Baerenfaller et al., 2012) abundance at different times in the diurnal cycle in Arabidopsis. Interpretation of responses in vascular plants is complicated by different temporal kinetics for transcript and protein abundance and the slow turnover of many proteins in vascular plants (Stitt and Gibon, 2014).
Some individual transcript-protein pairs showed very good agreement in our data set, including proteins involved in amino acid biosynthesis, showing that transcriptional regulation contributes to the increase in protein abundance. However, other protein classes show poor agreement or even change independently of each other, including proteins involved in photosynthesis (e.g., of LHCII, LHCI, PSI, the thylakoid ATPase, and several CBC enzymes). Clearly, posttranscriptional mechanisms contribute to regulation of abundance of many proteins in the photosynthetic machinery. This includes LHCII abundance, which was previously shown to be regulated posttranscriptionally (Mussgnug et al., 2005). Another striking example is Rubisco activase (RCA1), which showed one of the largest increases of all proteins after transfer to higher light. The function of this increase will be discussed later.
Changes in Protein Abundance Contribute to the Adjustment of Photosynthesis and Growth to Higher Irradiance
The functional classes that were enriched for changes in protein abundance after transfer to higher irradiance included photosynthesis, the TCA cycle, amino acid biosynthesis, protein synthesis, protein folding, chromatin structure, and protein degradation. Overall, our results reveal that there is a transcriptional or translational decrease in proteins directly involved in the capture and use of light energy, upregulation of many other proteins involved in photosynthesis, and a sustained upregulation of proteins involved in growth processes.
Among photosynthetic proteins, there was a decrease in LHCII, PSII, LHCI, and PSI, which was accompanied by a decrease in chlorophyll and a slow increase in ATPase subunit abundance. A decrease in chlorophyll after transfer to high light was also observed by Bonente et al. (2012) in C. reinhardtii CW-15 and by Deblois et al. (2013) in various algae and cyanobacteria. Bonente et al. (2012), like us, also reported unchanged chlorophyll a/b ratios and higher ATPase protein levels at high light (400 μmol photons m−2 s−1). Thus, in high irradiance, when the higher influx of light quanta allows more efficient use of the light-harvesting and photosystem complexes, C. reinhardtii allocates protein away from these complexes and toward ATPase. The abundance of proteins in the remainder of the photosynthetic apparatus in low irradiance is nevertheless high enough to catalyze 2.5-fold higher fluxes; the abundance of the cytochrome b6f complex remained unaltered, and the rapid increase in photosynthesis occurred long before ATPase abundance increased. The immediate increase in flux at ATPase may be due to posttranslational activation through redox regulation (Nalin and McCarty, 1984; Miki et al., 1988) or dephosphorylation (Kanekatsu et al., 1998; Bunney et al., 2001; del Riego et al., 2006). The vast majority of the ATP generated by ATPase is used to reduce 3PGA to GAP (Edwards and Walker, 1983). Interestingly, the slow increase in ATPase abundance coincided with a gradual increase in GAP levels. As discussed below, this increase in GAP may be important for the operation of the CBC.
The increase in abundance of proteins for amino acid biosynthesis was accompanied by a continued increase in the levels of many amino acids, while the organic acid precursors stabilize or decline between 120 and 480 min, pointing to an important role in supplying C precursors for growth. It is also accompanied by an increase in the abundance of proteins that are required for protein synthesis, with the notable exception of ribosomal proteins, which show a slight decline. As already discussed, an increase in polysome loading allows more efficient use of the existing complement of ribosomes.
Comprehensive Estimates of Free Energy Loss Reveal That CBC Flux Increases in a Coordinated Manner
Our near-complete data set for CBC metabolite levels allowed us to estimate the in vivo change in free energy (ΔrG) for almost every reaction in the CBC. Comprehensive estimates for ΔrG values in the CBC are available from a study in Chlorella pyrenoidosa (Bassham and Krause, 1969), and a study from Dietz and Heber (1984) showed that conversion of 3PGA to GAP by GAPDH and PGK is reversible in spinach (Spinacia oleracea) leaves. Our estimates confirm that FBPase, SBPase, PRK, and Rubisco catalyze reactions that are strongly displaced from equilibrium, while the remaining enzymes are close to equilibrium. Furthermore, they show that the distribution of ΔrG between reactions in the CBC changes only slightly when the pathway flux is altered by up to 5-fold.
It has been proposed that the irreversible reactions of the CBC are regulated in a highly coordinated manner (reviewed in Raines and Lloyd, 2007; Stitt et al., 2010). This proposal was based on measurements of a small subset of metabolites (Badger et al., 1984; Stitt et al., 1984b; Servaites et al., 1991) and information about the regulatory characteristics of Rubisco (Lorimer et al., 1976), PRK (Gardemann et al., 1983), FBPase (Gardemann et al., 1986), and SBPase (Cadet and Meunier, 1988). Our more comprehensive analysis of metabolite levels in a range of irradiance regimes in C. reinhardtii provides strong experimental evidence for highly coordinated regulation of the four irreversible reactions in the CBC, except at very low light intensities.
Many CBC Enzymes Are Highly Substrate Limited and Are Present at Higher Concentrations Than Their Substrates
We combined our near-complete data sets for CBC metabolite levels and for CBC enzyme abundance with a large body of published information about the kinetic properties of CBC enzymes to investigate the in vivo relationship between substrate concentration, substrate affinity, and enzyme binding site for almost every CBC enzyme. In particular, we asked two questions. First, are substrate concentrations limiting or saturating in vivo during steady state photosynthesis? Second, is the substrate concentration higher than the enzyme binding site concentration in the CBC, as it is widely assumed to be the case in central metabolism?
Our analysis revealed that the CBC enzymes can be divided into two groups. One group includes GAPDH, TPI, aldolase (catalyzing both the FBA and SBA reaction), RPI, PRK, and, to a certain extent, TRK. For these enzymes, the substrate concentrations were (1) much lower than the modeled in vivo substrate affinity and (2) were often low relative to the estimated enzyme binding site concentration. Flux through these reactions will therefore respond very sensitively to the increase in substrate concentration in higher light. This is especially so for enzymes that utilize GAP, the immediate product of the reactions that consume most of the NADPH and ATP generated by the light reactions. An increased level of GAP and linked metabolites provides a simple mechanism to increase flux at many CBC enzymes after an increase in light intensity stimulates electron transport, and NADPH and ATP formation.
The second group of enzymes includes Rubisco, FBPase, and SBPase. For these enzymes, the substrate concentrations were similar to or slightly higher than the modeled in vivo Km values and high relative to the estimated enzyme binding site concentration. This indicates that a large increase in flux will require posttranslational activation and/or an increase in abundance of these enzymes.
FBPase and SBPase Activity Are Stimulated by Posttranslational Regulation and a Slow Increase in Abundance
FBPase and SBPase are subject to thioredoxin-mediated posttranslational activation (reviewed in Buchanan and Balmer, 2005). Light and increased substrate levels promote activation, and in vitro studies show that activation increases the substrate affinity (Scheibe, 1991; Stitt, 1996). Our modeled values for in vivo substrate saturation predict that activation leads to a considerable increase in the affinity in vivo, which will allow a large increase in flux at a given substrate concentration (Figure 11). FBP and SBP levels are high in low light and fall at higher light intensities, showing that FBPase and SBPase are progressively activated as the light intensity is increased (Supplemental Figure 12A). This explains why, compared with other CBC intermediates, FBP and SBP showed a relatively small increase after transfer from 41 to 145 μmol photons m−2 s−1. However, further experiments are needed to measure changes in reductive activation of SBPase and FBPase at the protein level.
Furthermore, FBPase and SBPase showed the largest (17 to 19%) increase in protein abundance of all CBC enzymes after 4 to 8 h in higher light. At least in the case of SBPase, this was accompanied by a decrease in its substrate (SBP) concentration, showing that the increase in SBPase abundance plays a substantial role in the adjustment of the CBC to increased irradiance (Figure 6). In the case of FBPase, the level of its substrate (FBP) did not decrease. This may be due to the concomitant increase in GAP levels (see above), which will lead via the reversible TPI and FBA reactions to an increase in FBP levels.
Rubisco Activity Is Initially Stimulated by a Large Increase in Its Substrate RuBP and Later by a Large Increase in the Abundance of Rubisco Activase
Rubisco is posttranslationally activated by Rubisco activase, which releases inhibitory sugar phosphates from the active site of Rubisco (Portis, 2003; Stotz et al., 2011). Many of these inhibitory sugar phosphates are formed as side products with a fixed probability during the catalytic cycle, like xylulose-bisphosphate (Parry et al., 2008; Whitney et al., 2011). While these findings were obtained using vascular plant Rubisco, they are likely to hold for the C. reinhardtii enzyme.
Of all the metabolites that we measured, RuBP shows the largest initial increase (almost 6-fold) after transfer to higher light and the largest reversal from 120 min onwards. At 480 min, RuBP levels were only 2-fold higher than those in the low light control, which is similar to the increase that is required to support the observed 2.5-fold increase in photosynthetic rate. These large transient changes in RuBP levels shows that Rubisco activity is restricted immediately after the increase in light intensity and that this restriction is subsequently alleviated. Our data indicate that two factors that may contribute to this transient response. One is that, despite the high CO2 concentrations in the medium, CO2 entry might transiently restrict Rubisco activity. This possibility is hinted at by the strong induction of CCP2 and LCI1, which are known to be induced by low CO2 (see above). The second is that the increased rate of catalysis by Rubisco leads to a higher rate of formation of xylulose-bisphosphate and other inhibitors and partial inactivation of Rubisco. It is striking that the decrease in RuBP between 40 and 480 min was accompanied by a large (48%) increase in Rubisco activase (RCA1) abundance. These results indicate that the abundance of RCA1 in low-light-grown C. reinhardtii is not high enough to cope with the increased rate of formation of inhibitory intermediates after transfer to higher light. They also raise questions regarding the signaling pathways that lead to the increase in RCA1 protein; intriguingly, the increase of RCA1 protein is not accompanied by an increase of the corresponding transcript (see below for more discussion).
In Vitro Km Values Strongly Underestimate in Vivo Km Values and Overestimate the Extent to Which Enzymes Are Saturated by Their Substrates in Vivo
As already mentioned, while three CBC enzymes operate close to substrate saturation, most of the others are strongly substrate-limited in vivo. Based on a large-scale comparison of literature Km values and metabolite abundance in E. coli, Bennett et al. (2009) proposed that the majority of enzymes in intermediary metabolism are near-saturated or saturated by their substrates in vivo. By combining our information about metabolite levels with detailed information about the functional characteristics of CBC enzymes, we have shown that the in vivo affinities of enzymes for their substrates are often much lower than those expected from in vitro analyses. For CBC enzymes, this difference was up to 30-fold. The reason for this discrepancy is that in vitro assays are conducted in the absence of competing products, effectors, and inhibitory posttranslational modifications.
Many CBC Enzymes Are at Higher Concentrations Than Their Substrates, with Important Implications for Operation of the CBC
It is usually assumed that enzymes in central metabolism are present at far lower concentrations than their substrates. Indeed, this is a premise of the canonical Michaelis-Menten equation, whose derivation assumes that E<<S. Our pathway-wide comparison of protein abundance and metabolite levels reveals that for many CBC enzymes, the binding site concentrations are actually similar to or higher than the concentrations of their substrates. While there may be errors in our estimates of enzyme abundance, they are unlikely to be so large that they invalidate this conclusion, in particular not for enzymes that use GAP as a substrate. Our conclusion is supported by an earlier theoretical study (Harris and Koniger, 1997). These authors used measured enzyme activities in chloroplast or leaf extracts and published data about in vitro kcat values of purified proteins to calculate enzyme concentrations and compared these with published values for metabolite concentrations. They predicted that binding site concentrations are higher than the substrate concentrations for many CBC enzymes, including Rubisco, GAPDH, aldolase, TRK, and PRK.
With the exception of Rubisco, the enzymes in the CBC have been recruited or modified from enzymes in glycolysis, gluconeogenesis, and the oxidative pentose phosphate pathway. However, the maximum flux through the CBC in high light is an order of almost a hundred above those in nonphotosynthetic metabolism (Ivanova et al., 2008; Heldt and Piechulla, 2010). This reflects the far larger input of energy that is available from sunlight, compared with uptake of organic compounds. It is difficult to conceive how a large increase in kcat could be achieved for this large set of enzymes after recruiting them to the CBC. The kcat of enzymes in central metabolism is already high, as they have been optimized over billions of years of evolution (Bar-Even et al., 2011). The increase in flux in the CBC has instead been largely achieved by increasing enzyme concentration. Thus, evolutionary pressure to maximize CBC flux to utilize the intercepted light energy may have driven protein concentrations upwards in the chloroplast stroma. The overall protein concentration in the chloroplast (600 mg mL−1; Lilley et al., 1975) is high compared with that in cytosol (2 to 300 mg mL−1; Ellis, 2001).
The finding that many CBC metabolites are at lower concentrations than enzyme binding sites raises the question whether the movement of metabolites between enzymes may become limiting. Similar issues may also arise for NADP+, NADPH, ATP, and ADP. Some in vitro experimental evidence exists for complexes between CBC enzymes (Harris and Koniger, 1997; Winkel, 2004; Sweetlove and Fernie, 2013). However, it has not been demonstrated that these putative complexes occur in vivo and some CBC enzymes are not implicated in the complexes proposed to date, including TRK, which is highlighted in our analysis because its reactions use GAP as a substrate. Furthermore, recent studies of dynamic isotope labeling did not provide evidence for microchanneling in the CBC, with the possible exception of FBP (Szecowka et al., 2013). Sweetlove and Fernie (2013) argue that movement of metabolites between enzymes is unlikely to limit the operation of glycolytic enzymes because the space occupied by each individual enzyme molecule is so small that diffusion of low molecular weight metabolites within this space will be much faster than the rate of catalysis. This argument would appear to be even more compelling for CBC enzymes, which are present at 10- to 100-fold higher concentrations (Sulpice et al., 2010).
It has been widely assumed that light-saturated photosynthesis will be limited by Rubisco due to its unfavorable substrate kinetics: low kcat and very high abundance. However, systematic application of reverse genetics has revealed that several enzymes that are involved in the regeneration of RuBP also exert control over the rate of photosynthesis in some conditions (Anderson, 1992; Kossmann et al., 1994; Haake et al., 1998, 1999; Harrison et al., 1998, 2001; Henkes et al., 2001; Miyagawa et al., 2001; Olçer et al., 2001; Lefebvre et al., 2005; Lawson et al., 2006; Rosenthal et al., 2011). Recently, evolutionary models have predicted that an increase in SBPase abundance would increase the rate of photosynthesis (Zhu et al., 2007). Such results were difficult to rationalize, given that a very minor redistribution of total protein should suffice to alleviate a limitation by any single enzyme in the regenerative part of the CBC. Our analyses indicate that operation of the CBC may be constrained by its topological structure and the high concentrations of the CBC enzymes. In particular, the supply and concentration of GAP may constrain CBC operation. GAP is the immediate product of the PGA reduction, in which the majority of the ATP and NADPH from the light reactions is used to drive the CBC. Of the five 3-carbon residues that enter the regenerative phase of the CBC, two are DHAP and three are GAP. However, due to the equilibrium constant of the TPI reaction and high activity of TPI, the GAP concentration is only 5 to 10% of that of DHAP and is very low compared with the in vivo substrate affinities and the concentrations of the enzymes that utilize GAP.
Differences in the Regulation of Phosphorylated Metabolite Levels between C. reinhardtii and Vascular Plants
The large increase in the levels of CBC intermediates in between low and higher irradiance in C. reinhardtii contrasts with previous studies in vascular plants where CBC intermediates were mostly unchanged upon an increase in light intensity (Perchorowicz et al., 1981; Perchorowicz and Jensen, 1983; Badger et al., 1984; Dietz and Heber, 1984, 1986; Paul et al., 2000). The reason for the quite stable levels of phosphorylated CBC intermediates in vascular plants is thought to be due to a tight regulation of P homeostasis, which is required to prevent Pi limitation of photosynthesis (Sharkey et al., 1986; Paul and Foyer, 2001). The large changes in the levels of phosphorylated metabolites in C. reinhardtii indicate that P homeostasis is not as critical as in vascular plants. This might be due to direct access to Pi in the surrounding medium and/or faster release of Pi from the vacuolar pool or polyphosphates, a linear polymer of Pi that is less abundant in vascular plants than in algae (Ruiz et al., 2001; Werner et al., 2007). It will be interesting to learn if this increased flexibility in P homeostasis is a general feature of algal photosynthesis.
C. reinhardtii Growing in Low Irradiance Has Excess Capacity for Growth
Interestingly, our studies did not detect any increase in transcripts for ribosomal proteins or ribosomal protein abundance; indeed, the latter declined slightly after the shift to higher light. This contrasted with genes that are required in other processes that are related to protein synthesis, like amino acid biosynthesis, amino acid activation, protein folding, and protein targeting, which showed increases of their transcripts and/or protein abundance. This apparent anomaly can be explained because ribosome use is subject to very strong posttranslational regulation. This is exerted at the step of translation initiation and can be monitored by analysis of the loading of ribosomes into polysomes, which provides a proxy measurement for protein synthesis (Rudra and Warner, 2004). Polysome loading was low in low light and after an increase in light intensity, increased after a short delay of 10 to 20 min, and then again from 120 min onwards (Figure 9). It should be noted that the polysome loading in low light may overestimate the actual rate of protein synthesis (Pal et al., 2013).
The high ribosome abundance and low polysome loading in C. reinhardtii in low irradiance contrasts with microbes in resource-limiting conditions. In microbes, ribosome loading is generally high and surplus ribosomes are rapidly degraded when resources become limiting (Deutscher, 2003; Rudra and Warner, 2004; Zundel et al., 2009). Degradation of ribosomes is under the control of the TOR complex (Deutscher, 2003; Rudra and Warner, 2004; Zundel et al., 2009; Robaglia et al., 2012). Our results indicate that these canonical signaling pathways are modified to allow C. reinhardtii to maintain an excess of ribosomes in low irradiance (Pal et al., 2013). These different strategies may be adaptations to differences in the temporal dynamics of light intensity and substrate availability in natural environments. Light intensities change more quickly and frequently fluctuate, rather than showing monotonic changes. While the synthesis and maintenance of surplus ribosomes in C. reinhardtii will require energy and investment of N and P, it will allow growth to respond almost immediately to rapid fluctuations in the light intensity.
Starch and Low Molecular Weight Metabolites Act as a Buffer between Photosynthesis and Growth
We used our combined data set of time-resolved changes in the rate of photosynthesis and absolute metabolite levels to model the temporal relationship between photosynthetic C fixation and cellular growth and validated this model by comparing the modeled rates with the measured response of growth. An increase in irradiance is followed by a rapid increase in metabolites in biosynthetic and growth pathways, including hexose-phosphates, ADPG, UDPG, malate, and several amino acids. The large, and partly transient, increase of ADPG was accompanied by a transient increase in the rate of starch accumulation. In the first 5 to 10 min after the shift to higher light intensity, ∼60% of the additionally fixed C was sequestered into starch and another 20% in low molecular weight metabolites. This is likely to still be an underestimate, as carbon may have accumulated in other metabolites that were not analyzed in our study. This includes TAG, which also showed a transient increase in the first 40 to 60 min after the light shift. The close temporal link between photosynthesis and cellular growth has implications for bioengineering. While such close temporal links have not been reported in vascular plants, this may be partly due to their greater structural complexity, the time needed to transport photosynthate from source leaves to growing tissues, and because most measurements of growth in high plants monitor changes in physical size, which is largely due to water uptake into the vacuole in expanding cells, rather than the synthesis of cellular constituents. Rapid changes in ribosome loading after illumination or darkening were recently reported in the Arabidopsis rosette (Pal et al., 2013). The relationship between photosynthesis and growth is nevertheless clearer in algae. This will simplify modeling of growth with respect to central carbon metabolism and protein synthesis. Additionally, the ability of starch and other metabolites to act as a buffer between photosynthesis and growth could simplify the design of bioreactors, as it will decrease the impact of transient changes in light intensity inside the bioreactor on the efficiency of conversion of light energy into biomass.
Trade-Off between Protein Abundance and Flexible Response to Changes in the Environment
C. reinhardtii maintains surplus protein levels in low light, allowing photosynthesis and central metabolism to respond rapidly to an increase in light intensity. In the short interim period before growth is stimulated, much of the increment in photosynthesis accumulates in intermediary metabolites and, especially, starch. By modeling the response of enzymes in the CBC, we show that some enzymes are extremely substrate-limited in vivo and will respond sensitively to an increase in substrate levels, while for others, additional mechanisms, including posttranslational regulation, are required. The rapid changes are followed by widespread changes in transcript and, later, protein abundances. At a global level, these lead to adaptation in the photosynthetic machinery, increase the ability to cope with light and redox stress, and increase growth capacity. In specific cases, they are accompanied by changes in metabolites that point to these slower changes in protein allowing a readjustment of metabolism to a sustained increase in flux through central metabolism. Overall, C. reinhardtii is able to take almost immediate advantage of higher light to drive higher rates of photosynthesis and the accumulation of C reserves, and after a short lag, to increase the rate of growth. The surplus protein levels that allow this will carry a cost but will be beneficial for survival in a fluctuating light environment.
METHODS
Chemicals
Biochemicals, enzymes, and other special reagents were obtained from Sigma-Aldrich, Roche, and Merck. All solutions were prepared with purified deionized water (0.055 μS cm−1, PureLab plus and PureLab ultra; ELGA).
Algal Growth
Chlamydomonas reinhardtii CC-1690 wild-type strain (Sager, 1955) was obtained from the Chlamydomonas Resource Center and kept at 22°C and 55 μmol photons m−2 s−1 on TAP (Gorman and Levine, 1965) agar (1.4%) plates containing 0.4% Bacto-Yeast extract (Becton). A 50-mL preculture was grown mixotrophically on a rotary shaker at 124 rpm, 22°C, and an illumination of 55 μmol photons m−2 s−1 for 3 d in a medium containing 5 mM HEPES, 17.5 mM acetate, 1 mM K-phosphates, Beijerinck salts (7 mM NH4Cl, 0.34 mM CaCl2, and 0.41 mM MgSO4), and trace salt solution [184 μM H3BO3, 77 μM ZnSO4, 26 μM MnCl2, 18 μM FeSO4, 7 μM CoCl2, 6 μM CuSO4, and 1 μM (NH4)6Mo7O24] (Stern et al., 2009). It was used to inoculate 500 mL of the same medium. This second preculture was used to inoculate the 5-liter bioreactor BIOSTAT B-DCU (Sartorius Stedim). The medium in the bioreactor was the same as for the precultures but lacked acetate in order to grow the cells photoautotrophically. After approximately 5 d, a cell density of 3 to 5 × 106 cells mL−1 was reached and turbidity was kept constant by medium exchange for at least 2 d before the cells were harvested. The culture in the bioreactor was constantly stirred with 50 rpm at 24°C and bubbled at a rate of 200 cm3 min−1 with air enriched with 5% CO2. The internal light intensity was measured with an integrating sphere (Walz) at four different places within the bioreactor filled with algal culture and the average of the applied light intensities were 41 and 145 μmol photons m−2 s−1, respectively.
To check for the absence of microbial contaminations, 500 μL of each cell suspension was streaked on four 1.4% agar plates containing different media that promote yeast and bacterial growth. The Luria-Bertani medium contained 1% (w/v) Bacto Trypton, 1% (w/v) NaCl, and 0.5% (w/v) yeast extract (Bertani, 1951). The MacConkey medium contained 1.7% (w/v) peptone from casein, 1% (w/v) lactose, 0.5% NaCl, 0.15% (w/v) bile salts, 0.3% (w/v) peptone from meat, 0.003% (w/v) neutral red, and 0.0001% (w/v) crystal violet. YEB medium contained 0.5% (w/v) bacto-peptone, sucrose and beef extract, 0.1% (w/v) yeast extract, and 2 mM MgSO4. Standard bacterial agar contained 1.6% (w/v) bacto-peptone, 0.2% (w/v) yeast extract, 0.1% (w/v) glucose, and 0.56% (w/v) NaCl. The plates were incubated overnight at 37°C and subsequently kept at room temperature for 1 week. Additionally, cultures were regularly analyzed microscopically for the absence of contaminating microorganisms. Cultures were only used for further analysis when no bacterial or yeast growth was found on the control agar plates and no microorganisms were observed by microscopy.
Cell number and cell volumes were obtained by a Z2 Coulter Counter (Beckman Coulter) in triplicates of 100-fold diluted samples.
Operation of the Photobioreactor
A photobioreactor designed for systems biology approaches was developed (Supplemental Figure 1). The LED light system was custom-made and the self-made units were attached to a 5-liter BIOSTAT B-DCU (Sartorius Stedim). The bioreactor allowed growth of a chemostat culture under controlled conditions and was operated by the MFCS shell software (Sartorius Stedim). The cylindrical LED light system provided a homogenous light distribution within the bioreactor vessel and allowed fast changes of light intensity. A range of online probes allowed us to control and acquire data for pH (pHeasy; Hamilton), pO2 (Visiferm DO; Hamilton), temperature (resistance thermometer; Satorius Stedim), optical density (Fundalux II with a measurement range between 840 and 910 nm; Satorius Stedim), and medium consumption reported as changes in weight of fresh medium (table balance; Satorius Stedim). An online multi-valve gas chromatograph (3000A MicroGC run by EZChromElute software; Agilent Technologies) was used to measure O2 and CO2 in the outlet air of the bioreactor. Culture density was kept constant by coupling the addition of fresh medium by a digitally controlled peristaltic pump to the online measurements of the optical density. The dilution rate [h−1] was calculated based on the formula:
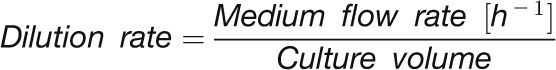 |
For steady state chemostat cultures as this one, the dilution rate is equivalent to the specific growth rate μ.
Fluorometry
Room temperature (25°C) chlorophyll a fluorescence of liquid culture was measured using a Dual-PAM-100 (Walz) under constant stirring. Samples were dark adapted for 30 min before the determination of the maximum quantum efficiency of PSII (FV/FM) by a saturating light pulse (5000 μmol photons m−2 s−1). Then, a light response curve of chlorophyll-fluorescence was measured between 0 and 2000 μmol photons m−2 s−1 actinic light intensity (20 steps, 2-min measuring time per light intensity). ETR was calculated from independent samples based on the quantum yield of PSII [ΦPSII = (FM’ − FS)/FM’] multiplied with the actinic light intensity and assuming equal distribution of excitation energy between both photosystems (as supported by parallel changes in the contents of both photosystems and their antenna system; Figure 4), without correcting data for absorbance of the sample.
77K chlorophyll a fluorescence emission was determined on cells frozen immediately after harvesting from the bioreactor using an F-6500 fluorometer (Jasco). The sample was excited at 430 nm (10-nm bandwidth). The emission spectra between 660 and 800 nm were recorded with a bandwidth of 1 nm and corrected for the instrumental response. For comparability of the different emission signals, the maximum fluorescence emission of PSII at 687 nm was normalized to 1.
Gas Exchange Measurements
The O2 evolution of the algal suspension was measured at 24°C in a temperature-controlled cuvette under continuous stirring using a Fibox-3 optical O2 sensor (PreSens Precision Sensing) (Ramírez-Aguilar et al., 2011). For light exposure, a custom-made cylindrical LED light system was used.
Chlorophyll, Total Protein Measurements, and SDS-PAGE
Chlorophyll content in algal suspensions was measured after extraction in 80% acetone (Porra et al., 1989).
Protein from algal cells was extracted by extraction buffer containing 2% Triton (50 mM HEPES, 20 μM leupeptin, 500 μM DTT, 1 mM PMSF, 17.4% glycerol, and 2% Triton) and measured by the Bradford protein assay (Bradford, 1976).
For SDS-PAGE, resuspended cells (50 mM DTT, 50 mM sodium-carbonate, 15% [w/v] sucrose, 2.5% [w/v] SDS) were ruptured by two freeze/thawing cycles and proteins were separated by SDS-PAGE (12%), and stained with Coomassie Brilliant Blue (Laemmli, 1970).
Determination of ATPase Activity from Proton Motif Force Relaxation Kinetics
ATPase activity was determined from the decay kinetic of the electrochromic shift signal, which can be used as an in vivo probe of the proton-motive force across the thylakoid membrane (Kramer et al., 2003; Takizawa et al., 2007). The signal was measured using a KLAS-100 LED array spectrophotometer (Heinz Walz). Prior to each measurement, cells were illuminated for 10 min with saturating light (688 μmol photons m−2 s−1; Supplemental Figure 1) to fully redox-activate ATPase and allow photosynthesis to reach its steady state. Then, actinic illumination was interrupted by short intervals of darkness, and the rapid decay of the ECS during the first 300 to 500 ms after the end of actinic illumination was fitted with a single exponential decay kinetic. The reciprocal value of the time constant was used as a measure of the conductivity of the thylakoid membrane for protons (gH+), which directly reflects ATPase activity (Baker et al., 2007).
Microarray Analysis of Chloroplast- and Mitochondria-Encoded Transcripts
Cells were cooled to below −20°C by adding one volume cell culture to two volumes precooled quenching solution (5 mM HEPES, 0.5 mM K2HPO4, 0.4 mM KH2PO4, 7 mM NH4Cl, 0.4 mM MgSO4, and 0.3 mM CaCl2 in 70% methanol). Cells were precipitated by centrifugation for 5 min, 4000g, −9°C. Sixty milligrams of frozen cell pellet was ground and RNA was extracted using the RNeasy Plant Mini Kit (Qiagen) according to instructions for plant tissue, including the optional on-column DNA digestion.
An oligonucleotide-based microarray (Kahlau and Bock, 2008) was designed for chloroplast- and mitochondria-encoded genes in C. reinhardtii. The synthetic oligonucleotides have lengths from 68 to 75 nucleotides, GC content 24 to 60%, and predicted melting temperatures of 64 to 80°C. Seven micrograms of total RNA mixed with 2 μL of spike RNA (SpotReport Alien Oligos; Stratagene) was used for cDNA synthesis with the Super-Script III Indirect cDNA labeling system (Invitrogen) according to the manufacturer's instructions. cDNA was purified with S.N.A.P. columns and precipitated with 75% ethanol. Labeling of cDNA was performed with the Cy3 Post-Labeling Reactive Dye Pack (GE Healthcare) according to the manufacturer's instructions. Labeled cDNA was concentrated with Microcon YM-10 centrifugal filter units (Millipore) and mixed with 130 μL 56°C hybridization buffer, heated to 95°C for 5 min, and loaded on microarrays that had been washed in 2× SSC, 0.1% sodium lauroyl sarcosinate, 2× SSC without detergent, 100°C water, −20°C ethanol, and 100°C water, heated to 75°C, and cooled to 45°C. The temperature was then decreased stepwise from 45 to 40°C in 1-h steps, and a final 12 h 40°C step. Microarrays were washed with 2× SSC, 0.1% sodium lauroyl sarcosinate, 0.2× SSC, 0.1% sodium lauroyl sarcosinate, 0.2× SSC, and then water and dried by centrifugation (1000g, 3 min, 4°C). Microarrays were scanned in a FLA-8000 scanner (Fujifilm) and spots quantified with the GeneSpotter software (MicroDiscovery). Values from spike-in RNAs were used to normalize each microarray block by a linear regression.
Microarray Analysis of Nuclear Encoded Transcripts
Total RNA extraction was performed using the RNeasy plant mini kit (Qiagen). The frozen cell pellet was homogenized in 450 μL of lysis buffer and frozen again in liquid nitrogen for 3 min. Cells were completely lysed by incubating the frozen cell pellet at 56°C for 3 min. Further steps of the RNA extraction were performed as described by the manufacturer. Genomic DNA was digested from the RNA extracts with DNase I (Qiagen), and an additional DNase treatment was performed, on the RNA eluate, using TURBO DNase (Ambion) as indicated by the manufacturer. The integrity of the total RNA was checked by electrophoresis on 2% denaturing agarose gels; RNA quality was assessed by determining the 260 nm/280 nm absorbance ratio using a Nanodrop (Thermo Scientific). Furthermore, the absence of genomic DNA in the RNA extracts was assessed by performing a quantitative PCR on a 1-μL aliquot of each sample of total RNA using primer pair annealing to an intergenic region of chromosome 16 (forward primer 5′-TGTCTTGTGAATCCTGCCCTC-3′ and reverse primer 5′-AAAGAGCTCACAAGTACAC ACCGA-3′). A minimum of 1.5 μg of total RNA was used for cDNA synthesis, quantification, labeling, and microarray hybridization by ATLAS Biolabs. Transcripts with an average SD of >40% were excluded from downstream analysis.
15N-Labeling Based Quantitative Shotgun Proteomics
Nonlabeled samples from each time point of the photoacclimation time course were harvested by centrifugation at 4°C and 3220g for 2 min, washed with 5 mM HEPES-KOH, pH 7.4, centrifuged again at 4°C and 21,500g for 2 min, resuspended in lysis buffer (50 mM NH4HCO3, 1 mM DTT, and 1 mM NaCl), frozen in liquid nitrogen, and stored at −80°C as previously described (Mühlhaus et al., 2011).
15N-Reference Culture
CC-1690 cells were grown at constant illumination (40 μmol photons m−2 s−1) in TAP medium containing 7.5 mM 15NH4Cl (>98%; Cambridge Isotope Laboratories; NLM-467) as nitrogen source for overall >10 generations to ensure a labeling efficiency >98%. Cells were harvested in exponential phase and stored as described above.
Sample Preparation and Analysis
15N-labeled reference cells were mixed with nonlabeled samples from each time point and biological replicate (n = 2) of the photoacclimation time course at a 15N/14N ratio of 0.8 based on cell count. Cells were ruptured by two freeze/thawing cycles. Broken cells were centrifuged to separate soluble and membrane-associated proteins. The gained supernatant, considered as soluble protein fraction, was recovered, while the gained pellet containing membrane-associated proteins was washed once in lysis buffer, centrifuged again, and resuspended in the same buffer. Proteins of both fractions were extracted and tryptically digested as previously described (Mühlhaus et al., 2011). The extracted peptides of each sample were three times chromatographically separated by reverse-phase separation (EASY-nLC; Proxeon Biosystems) on a monolithic capillary column (RP-18e; 150 to 0.1 mm; Merck) as already described (Mühlhaus et al., 2011). The separated peptides were directed to a linear trap quadrupole-Orbitrap mass spectrometer (Thermo Scientific) by electrospray ionization for detection.
Identification and Quantification
Proteins were identified and quantified using the in-house developed IOMIQS framework (Integration of Mass spectrometry Identification and Quantification Software) as described previously (Mühlhaus et al., 2011). In addition, peptides were classified according to their information content (Qeli and Ahrens, 2010). The Occam’s Razor approach (Nesvizhskii et al., 2003) was applied to each peptide class from high to low information content to report a minimal set of proteins and protein groups.
For protein quantification, both fractions (soluble and membrane associated proteins) were pooled, and mean and sd was calculated from two biological and three technical replicates each. Protein groups that showed significant pairwise anticorrelation between the two fractions were excluded from the data set. For single-protein and PC analyses, an additional cutoff (average sd was required to be <20%) was used (Figures 2 and 4; Supplemental Figures 8 and 9).
ATP Measurement by Enzymatic Assay
ATP was measured in freshly prepared methanol/water phase as described for the metabolite measurements by LC-MS/MS via an enzymatic assay described previously (Merlo et al., 1993).
Measurements of Metabolites by LC-MS/MS
Harvesting Procedure
A quenching solution made of 70% methanol was cooled down to < −60°C by −70°C cold ethanol, which was cooled by dry ice (Bölling and Fiehn, 2005; Kempa et al., 2009). The culture was sprayed into the quenching solution by a syringe or precooled methanol was added to the cell culture (1:2 = culture:quenching solution). Temperature was checked regularly to assure that temperature was kept below −20°C during the whole procedure. The material was frozen in liquid nitrogen and stored at −80°C or in liquid nitrogen before metabolite extraction.
Sample Extraction for Metabolite Measurements
For LC-MS/MS analysis, water-soluble metabolites were extracted using a slightly modified protocol as previously published (Lunn et al., 2006; Arrivault et al., 2009). Quenched algal material (525 μL) was added to 105 μL precooled chloroform and was vigorously vortexed in the presence of five glass beads (final ratio C:W:M = 1:5.3:2.3). Recovery experiments were performed by adding authentic analyte standards to the quenched material before adding chloroform according to Tohge et al. (2011). The cells were exposed to three cycle of thaw-freeze to break cells completely. The aqueous fraction was collected after centrifugation (5 min, 13,500 rpm, 4°C), and the chloroform fraction was washed two additional times with 560 μL ice-cold water. The three aqueous fractions were unified and freeze-dried overnight (Alpha 2-4; Christ). Samples were resuspended and filtered with Ultracel-10 multiscreen filter plates (Millipore) before application to the LC-MS/MS. A total of 70 to 80 μL of culture equivalent (cell density 3 to 5 × 106 cells mL−1) was analyzed per LC-MS/MS run.
Ion Pair Chromatography–Triple Quadrupole MS
LC-MS/MS was performed on a Dionex HPLC system coupled to a Finnigan TSQ Quantum Discovery MS-Q3 (Thermo Scientific) equipped with an electrospray ionization interface. It was operated as described by Arrivault et al. (2009) with slight modifications of the LC gradient. Chromatographic separation was performed by passing aliquots through a Gemini (C18) 4 × 2.00-mm precolumn (Phenomenex), before separation on a Gemini (C18) 150 × 2.00-mm inner diameter, 5-μm 110 Å particle column (Phenomenex) at 35°C using a multistep gradient with online-degassed eluent A (10 mM tributylamine aqueous solution, adjusted to pH 5.0 with 15 mM acetic acid and 5% methanol) and eluent B (100% methanol): 0 to 5 min, 100% A; 5 to 15 min, 100 to 95% A; 15 to 22 min, 95 to 90% A; 22 to 37 min, 90 to 85% A; 37 to 40 min, 85 to 70% A, and maintained for 3 min; 43 to 47 min, 70 to 45% A, and maintained for 3 min; 50 min, 10% A, and maintained for 8 min; 58 min, 100% A, and maintained for 8 min. The flow rate was 0.2 mL min−1 and was increased to 0.3 mL min−1 between 22 and 58 min. After separation, compounds were ionized by electrospray ionization and detected by a triple quadrupole that was operated in negative ion mode with selected reaction monitoring, using an ion spray voltage of 4000 V and a capillary temperature of 230°C. The Finnigan XCALIBUR 2.5 software (Thermo Scientific) was used for both instrument control and data acquisition. Prior to injection (100 μL), a mixture of 15 stable isotope reference compounds of known concentrations was added to the sample to correct for matrix effects on these analytes in the analysis. Metabolites were quantified by comparison of the integrated MS-Q3 signal peak area with a calibration curve obtained using authentic standards by the LCQuan software (Thermo Scientific). Further analysis was done using Microsoft Excel and R statistics software (R version 2.9.2 and 2.14.1 provided by the CRAN project, http://www.R-project.org). Metabolite levels were given as concentrations in algal cells (μM) by relating the total amount measured per milliliter of suspension to the measured total cell volume per milliliter of suspension, which was measured by Z2 Coulter Counter (Beckman Coulter) as described above. Metabolites with an average sd of >40% were excluded from downstream analyses.
Measurements of Lipids by Ultraperformance Liquid Chromatography–Mass Spectrometry
Lipids were extracted from a pellet of 20 mL of cell culture in three steps while incubating for 20 min each time (first, 1.5 mL of hot [80°C] isopropanol [0.01% BHT]; second, 1.5 mL of a mixture of hot [60°C] isopropanol/hexane/water [55:20:25] [0.01% BHT]; third, 1.5 mL of a mixture of chloroform/methanol [2:1] [0.01% BHT]). The combined extracts were dried down and resuspended in 100 μL isopropanol/hexane/water (55:20:25, upper phase). Ultraperformance liquid chromatography (UPLC) separation was performed using a Waters Acquity UPLC system, using a C8 reversed phase column (150 × 2.1-mm inner diameter, 1.8-μm particle size; Waters) at 60°C. The mobile phases consisted of 1% 1 M NH4Ac and 0.1% acetic acid in water (A) and methanol/isopropanol (5:2, 1% 1 M NH4Ac, 0.1% acetic acid) (B). The injection volume was 1 μL. The following gradient profile was applied: a linear 17-min gradient starting from 65% B to 100% B, followed by a 2.5-min isocratic period at 100% B, before the decrease back to 65% B in 0.5 min. Finally, the column was reequilibrated for 5 min, leading to a total run time of 25 min. The flow rate of the mobile phase was 350 μL min−1, and the column oven temperature was set to 60°C. The UPLC and the mass spectrometer were connected by a TriVersa NanoMate (Advion). The UPLC flow rate was split 1:1000, and a nanospray chip (type A) was used by applying a voltage of 1.8 kV. The mass spectra were acquired using a Orbitrap hybrid mass spectrometer (LTQ-Orbitrap; Thermo Scientific) using full scan mode, covering a mass range from m/z 200 to 2000, at a resolution of 30,000 at 400 m/z and with a maximal injection time of 500 ms. Relative quantification of 98 lipids were performed using Xcalibur Quan Browser (Thermo Scientific). Lipids with an average sd of >40% were excluded from downstream analyses.
Measurements of Metabolites by GC-MS
Cells were harvested by vacuum filtering 5 mL of cell culture through a polyvinylidene fluoride filter membrane (GVWP4700; Millipore) in the light. Filters were put in Falcon tubes, immediately frozen in liquid nitrogen, and stored at −80°C until extraction. The whole sampling process took ∼30 to 60 s per sample. For metabolite extraction, cells were carefully washed off from filters in three subsequent steps using 600 μL extraction buffer and combined in a new 2-mL tube. The extraction buffer consisted of methanol and chloroform (ratio 7:3) containing internal standards 13C6-sorbitol, d4-alanine, and iso-ascorbate, precooled to −20°C. The extract was vortexed for 60 min at 4°C. Afterwards, each sample was equally split into two fresh 1.5-mL tubes, to give two technical replicates and 0.5 parts of ice cold water was added. After vigorous vortexing, the cells were subjected to two freeze-thaw cycles and centrifuged 10 min at maximum speed at 0°C in a tabletop centrifuge. The upper, methanol-water phase was carefully transferred to a new tube and concentrated to dryness in a speed-vac overnight. Dry extracts were flushed with argon, sealed with Parafilm, and stored at −80°C before derivatization. Derivatization of metabolites was done manually by first adding 40 μL of methoxyamine hydrochloride (20 mg/mL in pyridine) and incubating at 30°C for 90 min with shaking. Next, 70 μL N-methyl-N-(trimethylsilyl)trifluoroacetamide and 10 μL of a mix of n-alkanes (decane, dodecane, pentadecane, octadecane, nonadecane, docosane, octacosane, dotriacontane, and hexatriacontane; 0.22 mg/mL in pyridine) were added followed by another incubation for 30 min at 37°C with shaking. Samples were centrifuged for 5 min at full speed, and 80 μL of the supernatant was transferred in GC vials. Gas chromatography–time of flight–mass spectrometry measurements of derivatized metabolites was done as described by Strehmel et al. (2011).
The ChromaTOF chromatography processing and mass spectral deconvolution software, v3.32 (LECO Instrumente) was used to preprocess raw data and to export it into netCDF file format. Preprocessing parameters of the ChromaTOF software were: baseline subtraction just above the noise (1.0), smoothing of data points (5), and exporting signal intensities with an signal-to-noise ratio >10. Further data processing was done with the TagFinder software (Luedemann et al., 2008). Metabolites were annotated manually using the Target Finder function in the TagFinder software using the Golm Metabolome Database (Kopka et al., 2005) (http://gmd.mpimp-golm.mpg.de). Criteria for manual metabolite identification were the presence of at least four specific mass fragments per compound, a match value >750, and a retention index deviation <1.0% (Strehmel et al., 2008).
The data of matched analytes were normalized by the intensity of the alkane nonadecane. Further between-sample normalization was done by probabilistic quotient normalization (Dieterle et al., 2006) with the average of all chromatograms set as reference chromatogram. Next, laboratory and reagent contaminations evaluated by non-sample control experiments were removed from the normalized data. The signals of several analytes corresponding to one single metabolite were summed up. This resulted in a normalized raw data matrix encompassing 67 known and 32 unknown metabolites. Metabolites with an average SD of >40% were excluded from downstream analysis.
Starch Measurements by Enzymatic Assay and Calculation of Starch Synthesis Rate
Starch was measured in the insoluble fraction after an ethanolic extraction as described previously (Hendriks et al., 2003). The starch was solubilized in 0.1 M NaOH for 30 min at 95°C. After neutralization, starch was digested to glucose by the addition of amyloglucosidase and α-amylase at 37°C overnight. Glucose content was then measured enzymatically to assess the starch level in the sample. The rate of starch synthesis was calculated based on measured starch levels (S) at two different time points (t1 and t0) and the volumes (V) of the chemostat culture. This was done as follows:
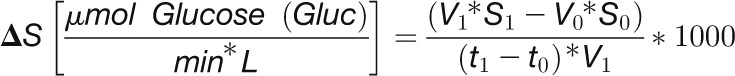 |
 |
VP is the volume of medium pumped per time interval to maintain a constant optical density.
Polysome Loading
Ribosomes were isolated from quenched, centrifuged, and subsequently frozen and mix-milled algal material and separated in a sucrose gradient (20 to 60% w/v) as described previously (Piques et al., 2009). The sucrose gradient was fractionated into 14 fractions by a Programmable Density Gradient Fractionation System (Teledyne Isco). In each fraction, ribosome absorbance at 254 nm was recorded. The peaks corresponding to denser and lighter fractions accounted for the free ribosomes and the ribosomes in polysomes, respectively. A ribosome profile was obtained, which was used to calculate total ribosomes (total absorbance of all fractions) and the relative contribution of free ribosomes (absorbance peak of higher density fractions) and ribosomes in polysomes (absorbance peak of lighter fractions) to the total absorbance.
Data Analysis
General Statistical Analysis
After method-specific normalization to minimize technical influence, changes in transcript, protein, metabolite, and lipid level were analyzed equally for data shown in Figures 2 and 8 and Supplemental Figures 13 and 14. Data were log2-transformed and tested with a one-way ANOVA over all time points using a significance threshold (P value < 0.05) after correction for multiple hypotheses testing according to Benjamini and Hochberg (1995). To determine differences in specified time points, contrast analysis was employed in the context of the ANOVA according to Hays method for transcript and protein data for data shown in Supplemental Figure 6 (Hays, 1988).
All statistical analyses were performed using Microsoft F# functional programming language with the mathematical library Math.NET Numerics (downloaded from http://numerics.mathdotnet.com/), Microsoft Excel, and R statistics (provided by the CRAN project http://www.R-project.org).
Functional Enrichment
Proteins and transcripts were functionally annotated using MapMan (Thimm et al., 2004) for C. reinhardtii. The analysis of overrepresentation of certain functional categories within the protein data set was performed using the IOMIQS (Mühlhaus et al., 2011) functional annotation tool.
Calculation of emPAI
From proteomics data of each protein, emPAI was calculated based on Equations 1 and 2 (Ishihama et al., 2005) to estimate absolute protein concentrations (Equations 3 and 4; Ishihama et al., 2005).
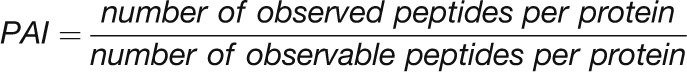 |
(1) |
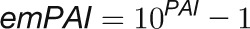 |
(2) |
 |
(3) |
 |
(4) |
The emPAI is based on the number of peptide identifications but does not cope with the fact that peptides selected for fragmentation are entering an exclusion list for a certain length of time after their fragmentation. The emPAI is therefore only an approximate measure of binding site concentration, which tends to underestimate highly abundant proteins and the derived data have to be treated with caution.
Calculation of Substrate Saturation Curves
Based on the calculation of Gibbs free energy of reaction, for FBPase, SBPase, and PRK, the Michaelis-Menten kinetics for irreversible reactions in Equations 5 and 6 were used (Segel, 1975):
 |
(5) |
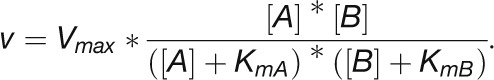 |
(6) |
Additionally, competitive inhibitions were taken into account for FBPase (F6P and Pi) (Charles and Halliwell, 1981; Gardemann et al., 1986), SBPase (Pi) (Woodrow et al., 1983), and PRK (RuBP, Pi, and ADP) (Gardemann et al., 1983) based on Equation 7:
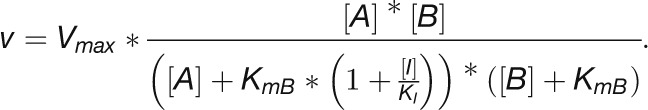 |
(7) |
Note that the known inhibitor S7P of SBPase (Schimkat et al., 1990) could not be taken into account because we lack literature KI values.
For the remaining reversible reactions (Equation 8) catalyzed by PGK, TPI, TRK, FBA, SBA, and RPI, Michaelis-Menten kinetics for reversible reaction were used (Cleland, 1963) (Equation 9):
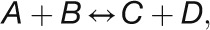 |
(8) |
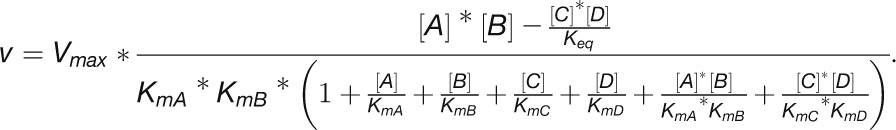 |
(9) |
For the carboxylation reaction catalyzed by Rubisco, the widely used Michaelis-Menten-like kinetic equation was applied (Equations 10 and 11) (Farquhar, 1979; Von Caemmerer, 2000). It accounts for the high RuBP level relative to binding site concentration of Rubisco as present in vascular plants and the competitive binding of 3PGA, FBP, SBP, Pi, and NADPH (Badger and Lorimer, 1981) as follows:
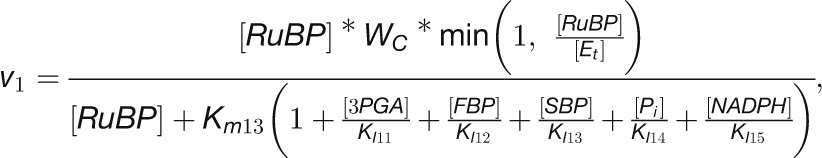 |
(10) |
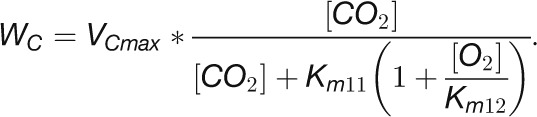 |
(11) |
Calculation of Carbon Sequestration and Modeled Growth Rate
These calculations were based on the differences between metabolites pools (measured by LC-MS/MS; Supplemental Data Set 1) and starch (Figure 7) at sequential time points. At each time point, the concentration of each metabolite and the amount of glucose in starch (μmol/g fresh weight) were multiplied by the number of C atoms per metabolite and summed to calculate the μmol C/L cell volume in soluble metabolites and starch. The difference between consecutive time points was then calculated and expressed relative to the additionally fixed C in that time interval. The absolute amount and the proportion of C sequestered into metabolites is likely to be underestimated since only a subset of metabolites of the central metabolism was measured with LC-MS/MS. However, the underestimation will be small, as all quantitatively major metabolites were measured. Based on the C sequestration (Supplemental Table 6), the growth rate was modeled assuming that the C that was not sequestered into metabolites was invested into growth based on the formula:
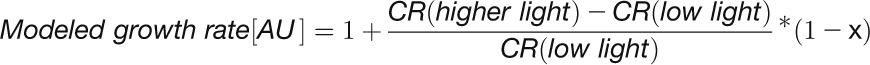 |
(12) |
where CR is the carboxylation rate and “x” denotes the fraction of the additionally fixed C sequestered into metabolites and starch.
Accession Numbers
Sequence data from this article can be found in the GenBank/EMBL libraries under the following accession numbers: CCP2, Cre04.g222750; FBA1, Cre01.g006950; FBA3, Cre05.g234550; FBP1, Cre12.g510650; GAP3, Cre01.g010900; LCI1, Cre03.g162800; LHCSR1, Cre08.g365900; LHCSR3, Cre08.g367400; NRX2, Cre02.g093750; NRX3, Cre02.g093800; PGK1, Cre22.g763250; PRK1, Cre12.g554800; rbcL, Cre-1.g2717040; RBCS1, Cre02.g120100; RBCS2, Cre02.g120150; RCA1, Cre04.g229300; RPE1, Cre12.g511900; RPI1, Cre03.g187450; SBP1, Cre03.g185550; and TRK1, Cre02.g080200. For MapMan annotations, see Supplemental Data Set 1.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Schematic Showing the Components of the Bioreactors.
Supplemental Figure 2. Light Saturation Curves of C. reinhardtii CC-1690 Cells under Low Light and during the Light Shift Experiment.
Supplemental Figure 3. Experimental Setup.
Supplemental Figure 4. Oxygen Concentration in the Outlet Air of the Bioreactor.
Supplemental Figure 5. Total Protein Content.
Supplemental Figure 6. Venn Diagrams Showing Numbers of Significantly Changed Transcripts and Proteins.
Supplemental Figure 7. Comparison of Significant Overrepresented Functional Gene Categories (and Subcategories) of Different Time Points during the Light Shift Applied to C. reinhardtii CC-1690 Cells Determined by PageMan.
Supplemental Figure 8. Top-10 Log2-Fold Changes of Transcripts, Proteins, Metabolites, and Lipids during the Light Shift Applied to C. reinhardtii CC-1690 Cells.
Supplemental Figure 9. Protein Levels in C. reinhardtii CC-1690 Cells during the Light Shift Experiment within the Functional Categories “Light Reaction,” “CBC,” and “Ribosomes.”
Supplemental Figure 10. 77K Fluorescence Emission Spectra during Light Shift.
Supplemental Figure 11. Adenylate Energy Charge (AEC) during Light Shift.
Supplemental Figure 12. CBC Intermediate Levels Change with Increasing Light Intensity.
Supplemental Figure 13. Heat Map of Lipid Levels during Light Shift in C. reinhardtii CC-1690 Cells.
Supplemental Figure 14. Amino Acids and Organic Acids.
Supplemental Figure 15. Correlation between Average Changes in Transcript and Protein Group Levels.
Supplemental Figure 16. Correlation between Protein Levels and Transcript Levels for Plastid-Encoded Genes in C. reinhardtii CC-1690 Cells.
Supplemental Figure 17. Calculated Metabolite Ratios.
Supplemental Figure 18. In Vitro and in Vivo Km Values versus Substrate Concentrations [S].
Supplemental Figure 19. Amount of RbcL Protein in C. reinhardtii Cells.
Supplemental Figure 20. Substrate per Binding Sites versus Substrate Concentrations and Enzyme Saturation of CBC Enzymes.
Supplemental Figure 21. Percentage Change for Enzyme-Substrate Pairs of the CBC between 20 min and 480 min after the Light Shift Applied to C. reinhardtii CC-1690 Cells.
Supplemental Table 1. Binding Site Concentration of CBC Enzymes Based on emPAI Analysis of Proteomics Data.
Supplemental Table 2. The Rate Equations of the CBC Reactions.
Supplemental Table 3. Km and KI Values for Different Substrates and Inhibitors of CBC Enzymes.
Supplemental Table 4. Binding Site Concentration of CBC Enzymes Based on emPAI Analysis of Proteomics Data with Two Different Methods.
Supplemental Table 5. Estimated Turnover Rates of CBC Intermediates in C. reinhardtii CC-1690 Cells.
Supplemental Table 6. Carbon Sequestration into Metabolites after Transfer to Higher Light.
Supplemental Data Set 1. Data of Organellar and Nuclear Transcript Levels, Protein, Metabolite, and Lipid Levels, and MapMan Annotations.
Supplemental Data Set 2. Calculations of Gibbs Free Energy of Reactions for CBC Enzymes.
Supplementary Material
Acknowledgments
We thank Clemens Posten and Christian Steinweg from the University of Karlsruhe for constructing the LED light system for the bioreactor. We also thank Dirk Walter and Kenny Billiau from the Max Planck Institute for Molecular Plant Physiology in Potsdam-Golm, Germany, for bioinformatical support. The research was funded by the Federal Ministry of Education and Research (BMBF), Germany, within the frame of the GoFORSYS Research Unit for Systems Biology (FKZ 0313924).
AUTHOR CONTRIBUTIONS
M. Schroda, M. Stitt, M.-A.S., B.M.-R., Z.N., L.W., and R.B. designed the research. T. Mettler, T. Mühlhaus, D.H., J.R., F.V.W., D. Veyel, D. Vosloh, S.K.P., L.Y.-R., F.S., A.I., M.-A.S., M. Schroda, U.K., M.G., A.E., and B.S. performed research. T. Mettler, T. Mühlhaus, D.H., A.I., A.A., J.R., S.A., M.L., S.S., A.B., M.-A.S., S.K.P., D. Veyel, A.E., and D. Vosloh analyzed data. M. Stitt and T. Mettler wrote the article.
Glossary
- CBC
Calvin-Benson cycle
- ETR
electron transport rate
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- GC-MS
gas chromatography–mass spectrometry
- PC
principle component
- TCA
tricarboxylic acid
- ATPase
ATP synthase
- MGDG
monogalactosyldiacylglycerol
- DGTS
diacylglycerol-N,N,N-trimethylhomoserine
- DGDG
digalactosyldiacylglycerol
- TAG
triacylglycerol
- emPAI
exponentially modified protein abundance index
- RA
relative abundance
- UPLC
ultraperformance liquid chromatography
Footnotes
Some figures in this article are displayed in color online but in black and white in the print edition.
Online version contains Web-only data.
Articles can be viewed online without a subscription.
References
- Ainsworth E.A., et al. (2008). Next generation of elevated [CO2] experiments with crops: a critical investment for feeding the future world. Plant Cell Environ. 31: 1317–1324. [DOI] [PubMed] [Google Scholar]
- Akan P., Alexeyenko A., Costea P.I., Hedberg L., Solnestam B.W., Lundin S., Hällman J., Lundberg E., Uhlén M., Lundeberg J. (2012). Comprehensive analysis of the genome transcriptome and proteome landscapes of three tumor cell lines. Genome Med. 4: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allorent G., et al. (2013). A dual strategy to cope with high light in Chlamydomonas reinhardtii. Plant Cell 25: 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amthor J.S. (2010). From sunlight to phytomass: on the potential efficiency of converting solar radiation to phyto-energy. New Phytol. 188: 939–959. [DOI] [PubMed] [Google Scholar]
- Anderson J.M. (1992). Cytochrome b6f complex: Dynamic molecular organization, function and acclimation. Photosynth. Res. 34: 341–357. [DOI] [PubMed] [Google Scholar]
- Arrivault S., Guenther M., Ivakov A., Feil R., Vosloh D., van Dongen J.T., Sulpice R., Stitt M. (2009). Use of reverse-phase liquid chromatography, linked to tandem mass spectrometry, to profile the Calvin cycle and other metabolic intermediates in Arabidopsis rosettes at different carbon dioxide concentrations. Plant J. 59: 826–839. [DOI] [PubMed] [Google Scholar]
- Asada K. (2006). Production and scavenging of reactive oxygen species in chloroplasts and their functions. Plant Physiol. 141: 391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badger M.R., Lorimer G.H. (1981). Interaction of sugar phosphates with the catalytic site of ribulose-1,5-bisphosphate carboxylase. Biochemistry 20: 2219–2225. [DOI] [PubMed] [Google Scholar]
- Badger M.R., Sharkey T.D., von Caemmerer S. (1984). The relationship between steady-state gas exchange of bean leaves and the levels of carbon-reduction-cycle intermediates. Planta 160: 305–313. [DOI] [PubMed] [Google Scholar]
- Baerenfaller K., et al. (2012). Systems-based analysis of Arabidopsis leaf growth reveals adaptation to water deficit. Mol. Syst. Biol. 8: 606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N.R., Harbinson J., Kramer D.M. (2007). Determining the limitations and regulation of photosynthetic energy transduction in leaves. Plant Cell Environ. 30: 1107–1125. [DOI] [PubMed] [Google Scholar]
- Ball S., Marianne T., Dirick L., Fresnoy M., Delrue B., Decq A. (1991). A Chlamydomonas reinhardtii low-starch mutant is defective for 3-phosphoglycerate activation and orthophosphate inhibition of ADP-glucose pyrophosphorylase. Planta 185: 17–26. [DOI] [PubMed] [Google Scholar]
- Bar-Even A., Noor E., Savir Y., Liebermeister W., Davidi D., Tawfik D.S., Milo R. (2011). The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 50: 4402–4410. [DOI] [PubMed] [Google Scholar]
- Bassham J.A., Krause G.H. (1969). Free energy changes and metabolic regulation in steady-state photosynthetic carbon reduction. Biochim. Biophys. Acta 189: 207–221. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. Ser. B 57: 289–300. [Google Scholar]
- Bennett B.D., Kimball E.H., Gao M., Osterhout R., Van Dien S.J., Rabinowitz J.D. (2009). Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 5: 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertani G. (1951). Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62: 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaby I.K., et al. (2013). Systems-level analysis of nitrogen starvation-induced modifications of carbon metabolism in a Chlamydomonas reinhardtii starchless mutant. Plant Cell 25: 4305–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship R.E., et al. (2011). Comparing photosynthetic and photovoltaic efficiencies and recognizing the potential for improvement. Science 332: 805–809. [DOI] [PubMed] [Google Scholar]
- Boer P.H., Gray M.W. (1988). Genes encoding a subunit of respiratory NADH dehydrogenase (ND1) and a reverse transcriptase-like protein (RTL) are linked to ribosomal RNA gene pieces in Chlamydomonas reinhardtii mitochondrial DNA. EMBO J. 7: 3501–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bölling C., Fiehn O. (2005). Metabolite profiling of Chlamydomonas reinhardtii under nutrient deprivation. Plant Physiol. 139: 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonente G., Pippa S., Castellano S., Bassi R., Ballottari M. (2012). Acclimation of Chlamydomonas reinhardtii to different growth irradiances. J. Biol. Chem. 287: 5833–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M.M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248–254. [DOI] [PubMed] [Google Scholar]
- Brauer M.J., Huttenhower C., Airoldi E.M., Rosenstein R., Matese J.C., Gresham D., Boer V.M., Troyanskaya O.G., Botstein D. (2008). Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol. Biol. Cell 19: 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bröcker M.J., Schomburg S., Heinz D.W., Jahn D., Schubert W.D., Moser J. (2010). Crystal structure of the nitrogenase-like dark operative protochlorophyllide oxidoreductase catalytic complex (ChlN/ChlB)2. J. Biol. Chem. 285: 27336–27345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan B.B., Balmer Y. (2005). Redox regulation: a broadening horizon. Annu. Rev. Plant Biol. 56: 187–220. [DOI] [PubMed] [Google Scholar]
- Bunney T.D., van Walraven H.S., de Boer A.H. (2001). 14-3-3 protein is a regulator of the mitochondrial and chloroplast ATP synthase. Proc. Natl. Acad. Sci. USA 98: 4249–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burow M.D., Chen Z.Y., Mouton T.M., Moroney J.V. (1996). Isolation of cDNA clones of genes induced upon transfer of Chlamydomonas reinhardtii cells to low CO2. Plant Mol. Biol. 31: 443–448. [DOI] [PubMed] [Google Scholar]
- Cadet F., Meunier J.C. (1988). pH and kinetic studies of chloroplast sedoheptulose-1,7-bisphosphatase from spinach (Spinacia oleracea). Biochem. J. 253: 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrillo J.I., et al. (2007). Growth control of the eukaryote cell: a systems biology study in yeast. J. Biol. 6: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castruita M., Casero D., Karpowicz S.J., Kropat J., Vieler A., Hsieh S.I., Yan W., Cokus S., Loo J.A., Benning C., Pellegrini M., Merchant S.S. (2011). Systems biology approach in Chlamydomonas reveals connections between copper nutrition and multiple metabolic steps. Plant Cell 23: 1273–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang R.L., Ghamsari L., Manichaikul A., Hom E.F.Y., Balaji S., Fu W., Shen Y., Hao T., Palsson B.O., Salehi-Ashtiani K., Papin J.A. (2011). Metabolic network reconstruction of Chlamydomonas offers insight into light-driven algal metabolism. Mol. Syst. Biol. 7: 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles S.A., Halliwell B. (1981). Light activation of fructose bisphosphatase in photosynthetically competent pea chloroplasts. Biochem. J. 200: 357–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland W.W. (1963). The kinetics of enzyme-catalyzed reactions with two or more substrates or products. I. Nomenclature and rate equations. Biochim. Biophys. Acta 67: 104–137. [DOI] [PubMed] [Google Scholar]
- Deblois C.P., Marchand A., Juneau P. (2013). Comparison of photoacclimation in twelve freshwater photoautotrophs (chlorophyte, bacillaryophyte, cryptophyte and cyanophyte) isolated from a natural community. PLoS ONE 8: e57139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Riego G., Casano L.M., Martín M., Sabater B. (2006). Multiple phosphorylation sites in the beta subunit of thylakoid ATP synthase. Photosynth. Res. 89: 11–18. [DOI] [PubMed] [Google Scholar]
- de Sousa Abreu R., Penalva L.O., Marcotte E.M., Vogel C. (2009). Global signatures of protein and mRNA expression levels. Mol. Biosyst. 5: 1512–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher M.P. (2003). Degradation of stable RNA in bacteria. J. Biol. Chem. 278: 45041–45044. [DOI] [PubMed] [Google Scholar]
- Dieterle F., Ross A., Schlotterbeck G., Senn H. (2006). Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem. 78: 4281–4290. [DOI] [PubMed] [Google Scholar]
- Dietz K.J., Heber U. (1984). Rate-limiting factors of leaf photosynthesis I. Carbon fluxes in the Calvin cycle. Biochim. Biophys. Acta 767: 432–443. [Google Scholar]
- Dietz K.J., Heber U. (1986). Light and CO2 limitation of photosynthesis and states of the reactions regenerating ribulose-1,5-bisphosphate or reducing 3-phosphoglycerate. Biochim. Biophys. Acta 848: 392–401. [Google Scholar]
- Eberhard S., Drapier D., Wollman F.A. (2002). Searching limiting steps in the expression of chloroplast-encoded proteins: relations between gene copy number, transcription, transcript abundance and translation rate in the chloroplast of Chlamydomonas reinhardtii. Plant J. 31: 149–160. [DOI] [PubMed] [Google Scholar]
- Eberhard S., Finazzi G., Wollman F.A. (2008). The dynamics of photosynthesis. Annu. Rev. Genet. 42: 463–515. [DOI] [PubMed] [Google Scholar]
- Edwards, G., and Walker, D. (1983). C3, C4: Mechanisms and Cellular and Environmental Regulation of Photosynthesis. (Oxford, UK: Blackwell Scientific Publications). [Google Scholar]
- Ellis R.J. (2001). Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 26: 597–604. [DOI] [PubMed] [Google Scholar]
- Fan W.H., Woelfle M.A., Mosig G. (1995). Two copies of a DNA element, ‘Wendy’, in the chloroplast chromosome of Chlamydomonas reinhardtii between rearranged gene clusters. Plant Mol. Biol. 29: 63–80. [DOI] [PubMed] [Google Scholar]
- Farquhar G.D. (1979). Models describing the kinetics of ribulose biphosphate carboxylase-oxygenase. Arch. Biochem. Biophys. 193: 456–468. [DOI] [PubMed] [Google Scholar]
- Farquhar G.D., von Caemmerer S., Berry J.A. (2001). Models of photosynthesis. Plant Physiol. 125: 42–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt S.M., Buescher J.M., Rudroff F., Picotti P., Zamboni N., Sauer U. (2010). Tradeoff between enzyme and metabolite efficiency maintains metabolic homeostasis upon perturbations in enzyme capacity. Mol. Syst. Biol. 6: 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field C.B., Behrenfeld M.J., Randerson J.T., Falkowski P. (1998). Primary production of the biosphere: integrating terrestrial and oceanic components. Science 281: 237–240. [DOI] [PubMed] [Google Scholar]
- Foyer C.H., Neukermans J., Queval G., Noctor G., Harbinson J. (2012). Photosynthetic control of electron transport and the regulation of gene expression. J. Exp. Bot. 63: 1637–1661. [DOI] [PubMed] [Google Scholar]
- Funato Y., Miki H. (2007). Nucleoredoxin, a novel thioredoxin family member involved in cell growth and differentiation. Antioxid. Redox Signal. 9: 1035–1057. [DOI] [PubMed] [Google Scholar]
- Gardemann A., Schimkat D., Heldt H.W. (1986). Control of CO2 fixation regulation of stromal fructose-1,6-bisphosphatase in spinach by pH and Mg(2+) concentration. Planta 168: 536–545. [DOI] [PubMed] [Google Scholar]
- Gardemann A., Stitt M., Heldt H.W. (1983). Control of CO2 fixation. Regulation of spinach ribulose-5-phosphate kinase by stromal metabolite levels. Biochim. Biophys. Acta 722: 51–60. [Google Scholar]
- Gardeström P., Wigge B. (1988). Influence of photorespiration on ATP/ADP ratios in the chloroplasts, mitochondria, and cytosol, studied by rapid fractionation of barley (Hordeum vulgare) protoplasts. Plant Physiol. 88: 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geigenberger P., Stitt M., Fernie A.R. (2004). Metabolic control analysis and regulation of the conversion of sucrose to starch in growing potato tubers. Plant Cell Environ. 27: 655–673. [Google Scholar]
- Geiger D.R., Servaites J.C. (1994). Diurnal regulation of photosynthetic carbon metabolism in C3 plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 45: 235–256. [Google Scholar]
- Gibon Y., Blaesing O.E., Hannemann J., Carillo P., Höhne M., Hendriks J.H.M., Palacios N., Cross J., Selbig J., Stitt M. (2004). A Robot-based platform to measure multiple enzyme activities in Arabidopsis using a set of cycling assays: comparison of changes of enzyme activities and transcript levels during diurnal cycles and in prolonged darkness. Plant Cell 16: 3304–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giroud C., Eichenberger W. (1989). Lipids of Chlamydomonas reinhardtii: incorporation of 14C-acetate, palmitate and oleate into different lipids and evidence for lipid-linked desaturation of fatty acids. Plant Cell Physiol. 30: 121–128. [Google Scholar]
- Goodenough U.W., Gebhart B., Mecham R.P., Heuser J.E. (1986). Crystals of the Chlamydomonas reinhardtii cell wall: polymerization, depolymerization, and purification of glycoprotein monomers. J. Cell Biol. 103: 405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman D.S., Levine R.P. (1965). Cytochrome f and plastocyanin: their sequence in the photosynthetic electron transport chain of Chlamydomonas reinhardi. Proc. Natl. Acad. Sci. USA 54: 1665–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman A.R., Croft M., Gladyshev V.N., Merchant S.S., Posewitz M.C., Prochnik S., Spalding M.H. (2007). Novel metabolism in Chlamydomonas through the lens of genomics. Curr. Opin. Plant Biol. 10: 190–198. [DOI] [PubMed] [Google Scholar]
- Gry M., Rimini R., Strömberg S., Asplund A., Pontén F., Uhlén M., Nilsson P. (2009). Correlations between RNA and protein expression profiles in 23 human cell lines. BMC Genomics 10: 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutteridge A., Pir P., Castrillo J.I., Charles P.D., Lilley K.S., Oliver S.G. (2010). Nutrient control of eukaryote cell growth: a systems biology study in yeast. BMC Biol. 8: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygi S.P., Rochon Y., Franza B.R., Aebersold R. (1999). Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19: 1720–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haake V., Geiger M., Walch-Liu P., Engels C., Zrenner R., Stitt M. (1999). Changes in aldolase activity in wild-type potato plants are important for acclimation to growth irradiance and carbon dioxide concentration, because plastid aldolase exerts control over the ambient rate of photosynthesis across a range of growth conditions. Plant J. 17: 479–489. [Google Scholar]
- Haake V., Zrenner R., Sonnewald U., Stitt M. (1998). A moderate decrease of plastid aldolase activity inhibits photosynthesis, alters the levels of sugars and starch, and inhibits growth of potato plants. Plant J. 14: 147–157. [DOI] [PubMed] [Google Scholar]
- Harris G.C., Koniger M. (1997). The 'high' concentrations of enzymes within the chloroplast. Photosynth. Res. 54: 5–23. [Google Scholar]
- Harrison E.P., Olcer H., Lloyd J.C., Long S.P., Raines C.A. (2001). Small decreases in SBPase cause a linear decline in the apparent RuBP regeneration rate, but do not affect Rubisco carboxylation capacity. J. Exp. Bot. 52: 1779–1784. [DOI] [PubMed] [Google Scholar]
- Harrison E.P., Willingham N.M., Lloyd J.C., Raines C.A. (1998). Reduced sedoheptulose-1,7-bisphosphatase levels in transgenic tobacco lead to decreased photosynthetic capacity and altered carbohydrate accumulation. Planta 204: 27–36. [Google Scholar]
- Hays, W.L. (1988). Statistics, 4th ed. (New York: Holt, Rinehart, & Winston). [Google Scholar]
- Heineke D., Riens B., Grosse H., Hoferichter P., Peter U., Flügge U.I., Heldt H.W. (1991). Redox transfer across the inner chloroplast envelope membrane. Plant Physiol. 95: 1131–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldt, H.-W., and Piechulla, B. (2010). Plant Biochemistry (London: Academic Press). [Google Scholar]
- Hendriks J.H.M., Kolbe A., Gibon Y., Stitt M., Geigenberger P. (2003). ADP-glucose pyrophosphorylase is activated by posttranslational redox-modification in response to light and to sugars in leaves of Arabidopsis and other plant species. Plant Physiol. 133: 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkes S., Sonnewald U., Badur R., Flachmann R., Stitt M. (2001). A small decrease of plastid transketolase activity in antisense tobacco transformants has dramatic effects on photosynthesis and phenylpropanoid metabolism. Plant Cell 13: 535–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im C.S., Zhang Z., Shrager J., Chang C.W., Grossman A.R. (2003). Analysis of light and CO(2) regulation in Chlamydomonas reinhardtii using genome-wide approaches. Photosynth. Res. 75: 111–125. [DOI] [PubMed] [Google Scholar]
- Ishihama Y., Oda Y., Tabata T., Sato T., Nagasu T., Rappsilber J., Mann M. (2005). Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics 4: 1265–1272. [DOI] [PubMed] [Google Scholar]
- Ivanova H., Keerberg H., Parnik T., Keerberg O. (2008). Components of CO2 exchange in leaves of C3 species with different ability of starch accumulation. Photosynthetica 46: 84–90. [Google Scholar]
- Iwai M., Kato N., Minagawa J. (2007). Distinct physiological responses to a high light and low CO2 environment revealed by fluorescence quenching in photoautotrophically grown Chlamydomonas reinhardtii. Photosynth. Res. 94: 307–314. [DOI] [PubMed] [Google Scholar]
- Jellings A.J., Leech R.M. (1982). The importance of quantitative anatomy in the interpretation of whole leaf biochemistry in species of Triticum, Hordeum, and Avena. New Phytol. 92: 39–48. [Google Scholar]
- Kahlau S., Bock R. (2008). Plastid transcriptomics and translatomics of tomato fruit development and chloroplast-to-chromoplast differentiation: chromoplast gene expression largely serves the production of a single protein. Plant Cell 20: 856–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekatsu M., Saito H., Motohashi K., Hisabori T. (1998). The beta subunit of chloroplast ATP synthase (CF0CF1-ATPase) is phosphorylated by casein kinase II. Biochem. Mol. Biol. Int. 46: 99–105. [DOI] [PubMed] [Google Scholar]
- Kempa S., Hummel J., Schwemmer T., Pietzke M., Strehmel N., Wienkoop S., Kopka J., Weckwerth W. (2009). An automated GCxGC-TOF-MS protocol for batch-wise extraction and alignment of mass isotopomer matrixes from differential 13C-labelling experiments: a case study for photoautotrophic-mixotrophic grown Chlamydomonas reinhardtii cells. J. Basic Microbiol. 49: 82–91. [DOI] [PubMed] [Google Scholar]
- Kimura M., Yamamoto Y.Y., Seki M., Sakurai T., Sato M., Abe T., Yoshida S., Manabe K., Shinozaki K., Matsui M. (2003). Identification of Arabidopsis genes regulated by high light-stress using cDNA microarray. Photochem. Photobiol. 77: 226–233. [DOI] [PubMed] [Google Scholar]
- Kirschbaum M.U.F. (2011). Does enhanced photosynthesis enhance growth? Lessons learned from CO2 enrichment studies. Plant Physiol. 155: 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein U. (1987). Intracellular carbon partitioning in Chlamydomonas reinhardtii. Plant Physiol. 85: 892–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopka J., et al. (2005). GMD@CSB.DB: the Golm Metabolome Database. Bioinformatics 21: 1635–1638. [DOI] [PubMed] [Google Scholar]
- Kossmann J., Sonnewald U., Willmitzer L. (1994). Reduction of the chloroplastic fructose-1,6-bisphosphatase in transgenic potato plants impairs photosynthesis and plant growth. Plant J. 6: 637–650. [Google Scholar]
- Kramer D.M., Evans J.R. (2011). The importance of energy balance in improving photosynthetic productivity. Plant Physiol. 155: 70–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer D.M., Cruz J.A., Kanazawa A. (2003). Balancing the central roles of the thylakoid proton gradient. Trends Plant Sci. 8: 27–32. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- Laing W.A., Stitt M., Heldt H.W. (1981). Control of CO2 fixation - changes in the activity of ribulosephosphate kinase and fructose-bisphosphatase and sedoheptulose-bisphosphatase in chloroplasts. Biochim. Biophys. Acta 637: 348–359. [Google Scholar]
- Laughner B.J., Sehnke P.C., Ferl R.J. (1998). A novel nuclear member of the thioredoxin superfamily. Plant Physiol. 118: 987–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson T., Bryant B., Lefebvre S., Lloyd J.C., Raines C.A. (2006). Decreased SBPase activity alters growth and development in transgenic tobacco plants. Plant Cell Environ. 29: 48–58. [DOI] [PubMed] [Google Scholar]
- Leegood R.C., von Caemmerer S. (1989). Some relationships between contents of photosynthetic intermediates and the rate of photosynthetic carbon assimilation in leaves of Zea mays L. Planta 178: 258–266. [DOI] [PubMed] [Google Scholar]
- Lefebvre S., Lawson T., Zakhleniuk O.V., Lloyd J.C., Raines C.A., Fryer M. (2005). Increased sedoheptulose-1,7-bisphosphatase activity in transgenic tobacco plants stimulates photosynthesis and growth from an early stage in development. Plant Physiol. 138: 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Moellering E.R., Liu B., Johnny C., Fedewa M., Sears B.B., Kuo M.H., Benning C. (2012). A galactoglycerolipid lipase is required for triacylglycerol accumulation and survival following nitrogen deprivation in Chlamydomonas reinhardtii. Plant Cell 24: 4670–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Wakao S., Fischer B.B., Niyogi K.K. (2009). Sensing and responding to excess light. Annu. Rev. Plant Biol. 60: 239–260. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler H.K., Buschmann C., Döll M., Fietz H.J., Bach T., Kozel U., Meier D., Rahmsdorf U. (1981). Photosynthetic activity, chloroplast ultrastructure, and leaf characteristics of high-light and low-light plants and of sun and shade leaves. Photosynth. Res. 2: 115–141. [DOI] [PubMed] [Google Scholar]
- Lilley R.M., Fitzgerald M.P., Rienits K.G., Walker D.A. (1975). Criteria of intactness and photosynthetic activity of spinach chloroplast preparations. New Phytol. 75: 1–10. [Google Scholar]
- Liu M.J., Wu S.H., Chen H.M., Wu S.H. (2012). Widespread translational control contributes to the regulation of Arabidopsis photomorphogenesis. Mol. Syst. Biol. 8: 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S.P., Zhu X.G., Naidu S.L., Ort D.R. (2006). Can improvement in photosynthesis increase crop yields? Plant Cell Environ. 29: 315–330. [DOI] [PubMed] [Google Scholar]
- Lorimer G.H., Badger M.R., Andrews T.J. (1976). The activation of ribulose-1,5-bisphosphate carboxylase by carbon dioxide and magnesium ions. Equilibria, kinetics, a suggested mechanism, and physiological implications. Biochemistry 15: 529–536. [DOI] [PubMed] [Google Scholar]
- Lu P., Vogel C., Wang R., Yao X., Marcotte E.M. (2007). Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 25: 117–124. [DOI] [PubMed] [Google Scholar]
- Luedemann A., Strassburg K., Erban A., Kopka J. (2008). TagFinder for the quantitative analysis of gas chromatography–mass spectrometry (GC-MS)-based metabolite profiling experiments. Bioinformatics 24: 732–737. [DOI] [PubMed] [Google Scholar]
- Lunn J.E., Feil R., Hendriks J.H., Gibon Y., Morcuende R., Osuna D., Scheible W.R., Carillo P., Hajirezaei M.R., Stitt M. (2006). Sugar-induced increases in trehalose 6-phosphate are correlated with redox activation of ADPglucose pyrophosphorylase and higher rates of starch synthesis in Arabidopsis thaliana. Biochem. J. 397: 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín-Navarro J., Manuell A.L., Wu J., P Mayfield S. (2007). Chloroplast translation regulation. Photosynth. Res. 94: 359–374. [DOI] [PubMed] [Google Scholar]
- McGinn P.J., Price G.D., Maleszka R., Badger M.R. (2003). Inorganic carbon limitation and light control the expression of transcripts related to the CO2-concentrating mechanism in the cyanobacterium Synechocystis sp. strain PCC6803. Plant Physiol. 132: 218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKim S.M., Durnford D.G. (2006). Translational regulation of light-harvesting complex expression during photoacclimation to high-light in Chlamydomonas reinhardtii. Plant Physiol. Biochem. 44: 857–865. [DOI] [PubMed] [Google Scholar]
- Merchant S.S., et al. (2007). The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318: 245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo L., Geigenberger P., Hajirezaei M., Stitt M. (1993). Changes of carbohydrates, metabolites and enzyme activities in potato tubers during development, and within a single tuber along a stolon-apex gradient. J. Plant Physiol. 142: 392–402. [Google Scholar]
- Miki J., Maeda M., Mukohata Y., Futai M. (1988). The gamma-subunit of ATP synthase from spinach chloroplasts. Primary structure deduced from the cloned cDNA sequence. FEBS Lett. 232: 221–226. [DOI] [PubMed] [Google Scholar]
- Miyagawa Y., Tamoi M., Shigeoka S. (2001). Overexpression of a cyanobacterial fructose-1,6-/sedoheptulose-1,7-bisphosphatase in tobacco enhances photosynthesis and growth. Nat. Biotechnol. 19: 965–969. [DOI] [PubMed] [Google Scholar]
- Mühlhaus T., Weiss J., Hemme D., Sommer F., Schroda M. (2011). Quantitative shotgun proteomics using a uniform ¹⁵N-labeled standard to monitor proteome dynamics in time course experiments reveals new insights into the heat stress response of Chlamydomonas reinhardtii. Mol. Cell. Proteomics 10: 004739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchie E.H., Hubbart S., Peng S., Horton P. (2005). Acclimation of photosynthesis to high irradiance in rice: gene expression and interactions with leaf development. J. Exp. Bot. 56: 449–460. [DOI] [PubMed] [Google Scholar]
- Mussgnug J.H., et al. (2005). NAB1 is an RNA binding protein involved in the light-regulated differential expression of the light-harvesting antenna of Chlamydomonas reinhardtii. Plant Cell 17: 3409–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustroph A., Zanetti M.E., Jang C.J.H., Holtan H.E., Repetti P.P., Galbraith D.W., Girke T., Bailey-Serres J. (2009). Profiling translatomes of discrete cell populations resolves altered cellular priorities during hypoxia in Arabidopsis. Proc. Natl. Acad. Sci. USA 106: 18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalin C.M., McCarty R.E. (1984). Role of a disulfide bond in the gamma subunit in activation of the ATPase of chloroplast coupling factor 1. J. Biol. Chem. 259: 7275–7280. [PubMed] [Google Scholar]
- Nesvizhskii A.I., Keller A., Kolker E., Aebersold R. (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75: 4646–4658. [DOI] [PubMed] [Google Scholar]
- Nunes-Nesi A., Fernie A.R., Stitt M. (2010). Metabolic and signaling aspects underpinning the regulation of plant carbon nitrogen interactions. Mol. Plant 3: 973–996. [DOI] [PubMed] [Google Scholar]
- Nymark M., Valle K.C., Brembu T., Hancke K., Winge P., Andresen K., Johnsen G., Bones A.M. (2009). An integrated analysis of molecular acclimation to high light in the marine diatom Phaeodactylum tricornutum. PLoS ONE 4: e7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olçer H., Lloyd J.C., Raines C.A. (2001). Photosynthetic capacity is differentially affected by reductions in sedoheptulose-1,7-bisphosphatase activity during leaf development in transgenic tobacco plants. Plant Physiol. 125: 982–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira A.P., Ludwig C., Picotti P., Kogadeeva M., Aebersold R., Sauer U. (2012). Regulation of yeast central metabolism by enzyme phosphorylation. Mol. Syst. Biol. 8: 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S.K., et al. (2013). Diurnal changes of polysome loading track sucrose content in the rosette of wild-type arabidopsis and the starchless pgm mutant. Plant Physiol. 162: 1246–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry M.A.J., Keys A.J., Madgwick P.J., Carmo-Silva A.E., Andralojc P.J. (2008). Rubisco regulation: a role for inhibitors. J. Exp. Bot. 59: 1569–1580. [DOI] [PubMed] [Google Scholar]
- Paul M.J., Foyer C.H. (2001). Sink regulation of photosynthesis. J. Exp. Bot. 52: 1383–1400. [DOI] [PubMed] [Google Scholar]
- Paul M.J., Driscoll S.P., Andralojc P.J., Knight J.S., Gray J.C., Lawlor D.W. (2000). Decrease of phosphoribulokinase activity by antisense RNA in transgenic tobacco: definition of the light environment under which phosphoribulokinase is not in large excess. Planta 211: 112–119. [DOI] [PubMed] [Google Scholar]
- Peers G., Truong T.B., Ostendorf E., Busch A., Elrad D., Grossman A.R., Hippler M., Niyogi K.K. (2009). An ancient light-harvesting protein is critical for the regulation of algal photosynthesis. Nature 462: 518–521. [DOI] [PubMed] [Google Scholar]
- Perchorowicz J.T., Jensen R.G. (1983). Photosynthesis and activation of ribulose bisphosphate carboxylase in wheat seedlings: regulation by CO2 and O2. Plant Physiol. 71: 955–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perchorowicz J.T., Raynes D.A., Jensen R.G. (1981). Light limitation of photosynthesis and activation of ribulose bisphosphate carboxylase in wheat seedlings. Proc. Natl. Acad. Sci. USA 78: 2985–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piques M., Schulze W.X., Höhne M., Usadel B., Gibon Y., Rohwer J., Stitt M. (2009). Ribosome and transcript copy numbers, polysome occupancy and enzyme dynamics in Arabidopsis. Mol. Syst. Biol. 5: 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pir P., Gutteridge A., Wu J., Rash B., Kell D.B., Zhang N., Oliver S.G. (2012). The genetic control of growth rate: a systems biology study in yeast. BMC Syst. Biol. 6: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock C., Farrar J., Tomos D., Gallagher J., Lu C., Koroleva O. (2003). Balancing supply and demand: the spatial regulation of carbon metabolism in grass and cereal leaves. J. Exp. Bot. 54: 489–494. [DOI] [PubMed] [Google Scholar]
- Pollock S.V., Prout D.L., Godfrey A.C., Lemaire S.D., Moroney J.V. (2004). The Chlamydomonas reinhardtii proteins Ccp1 and Ccp2 are required for long-term growth, but are not necessary for efficient photosynthesis, in a low-CO2 environment. Plant Mol. Biol. 56: 125–132. [DOI] [PubMed] [Google Scholar]
- Poorter, H., and Van der Werf, A. (1998). Is inherent variation in RGR determined by LAR at low irradiance and by NAR at high irradiance? A review of herbaceous species. In Inherent Variation in Plant Growth: Physiological Mechanisms and Ecological Consequences, H. Lambers, H. Poorter, and M.M.I. Van Vuuren, eds (Leiden, The Netherlands: Backhuys Publishers), pp. 309–336. [Google Scholar]
- Porra R.J., Thompson W.A., Kriedemann P.E. (1989). Determination of accurate extinction coefficients and simultaneous-equations for assaying chlorophyll a and chlorophyll b extracted with four different solvent: verification of the concentration of chlorophyll standards by atomic-absorption spectroscopy. Biochim. Biophys. Acta 975: 384–394. [Google Scholar]
- Portis A.R., Jr (2003). Rubisco activase - Rubisco’s catalytic chaperone. Photosynth. Res. 75: 11–27. [DOI] [PubMed] [Google Scholar]
- Pratt J., Boisson A.M., Gout E., Bligny R., Douce R., Aubert S. (2009). Phosphate (Pi) starvation effect on the cytosolic Pi concentration and Pi exchanges across the tonoplast in plant cells: an in vivo 31P-nuclear magnetic resonance study using methylphosphonate as a Pi analog. Plant Physiol. 151: 1646–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qeli E., Ahrens C.H. (2010). PeptideClassifier for protein inference and targeted quantitative proteomics. Nat. Biotechnol. 28: 647–650. [DOI] [PubMed] [Google Scholar]
- Raines, C.A., and Lloyd, J.C. (2007). C3 Carbon Reduction Cycle. (Chichester, UK: John Wiley & Sons). [Google Scholar]
- Ramírez-Aguilar S.J., Keuthe M., Rocha M., Fedyaev V.V., Kramp K., Gupta K.J., Rasmusson A.G., Schulze W.X., van Dongen J.T. (2011). The composition of plant mitochondrial supercomplexes changes with oxygen availability. J. Biol. Chem. 286: 43045–43053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renberg L., Johansson A.I., Shutova T., Stenlund H., Aksmann A., Raven J.A., Gardeström P., Moritz T., Samuelsson G. (2010). A metabolomic approach to study major metabolite changes during acclimation to limiting CO2 in Chlamydomonas reinhardtii. Plant Physiol. 154: 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard C., Ouellet H., Guertin M. (2000). Characterization of the LI818 polypeptide from the green unicellular alga Chlamydomonas reinhardtii. Plant Mol. Biol. 42: 303–316. [DOI] [PubMed] [Google Scholar]
- Ritz M., Thomas J.C., Spilar A., Etienne A.L. (2000). Kinetics of photoacclimation in response to a shift to high light of the red alga Rhodella violacea adapted to low irradiance. Plant Physiol. 123: 1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robaglia C., Thomas M., Meyer C. (2012). Sensing nutrient and energy status by SnRK1 and TOR kinases. Curr. Opin. Plant Biol. 15: 301–307. [DOI] [PubMed] [Google Scholar]
- Rochaix J.D. (1995). Chlamydomonas reinhardtii as the photosynthetic yeast. Annu. Rev. Genet. 29: 209–230. [DOI] [PubMed] [Google Scholar]
- Rochaix J.D. (2001). Assembly, function, and dynamics of the photosynthetic machinery in Chlamydomonas reinhardtii. Plant Physiol. 127: 1394–1398. [PMC free article] [PubMed] [Google Scholar]
- Rochaix J.D. (2011). Regulation of photosynthetic electron transport. Biochim. Biophys. Acta 1807: 375–383. [DOI] [PubMed] [Google Scholar]
- Rolland F., Baena-Gonzalez E., Sheen J. (2006). Sugar sensing and signaling in plants: conserved and novel mechanisms. Annu. Rev. Plant Biol. 57: 675–709. [DOI] [PubMed] [Google Scholar]
- Rosenthal D.M., Locke A.M., Khozaei M., Raines C.A., Long S.P., Ort D.R. (2011). Over-expressing the C3 photosynthesis cycle enzyme sedoheptulose-1,7-bisphosphatase improves photosynthetic carbon gain and yield under fully open air CO2 fumigation (FACE). BMC Plant Biol. 11: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rott M., Martins N.F., Thiele W., Lein W., Bock R., Kramer D.M., Schöttler M.A. (2011). ATP synthase repression in tobacco restricts photosynthetic electron transport, CO2 assimilation, and plant growth by overacidification of the thylakoid lumen. Plant Cell 23: 304–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudra D., Warner J.R. (2004). What better measure than ribosome synthesis? Genes Dev. 18: 2431–2436. [DOI] [PubMed] [Google Scholar]
- Ruiz F.A., Marchesini N., Seufferheld M., Govindjee, Docampo R. (2001). The polyphosphate bodies of Chlamydomonas reinhardtii possess a proton-pumping pyrophosphatase and are similar to acidocalcisomes. J. Biol. Chem. 276: 46196–46203. [DOI] [PubMed] [Google Scholar]
- Sager R. (1955). Inheritance in the green alga Chlamydomonas reinhardi. Genetics 40: 476–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savard F., Richard C., Guertin M. (1996). The Chlamydomonas reinhardtii LI818 gene represents a distant relative of the cabI/II genes that is regulated during the cell cycle and in response to illumination. Plant Mol. Biol. 32: 461–473. [DOI] [PubMed] [Google Scholar]
- Scheibe R. (1991). Redox-modulation of chloroplast enzymes: a common principle for individual control. Plant Physiol. 96: 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimkat D., Heineke D., Heldt H.W. (1990). Regulation of sedoheptulose-1,7-bisphosphatase by sedoheptulose-7-phosphate and glycerate, and of fructose-1,6-bisphosphatase by glycerate in spinach chloroplasts. Planta 181: 97–103. [DOI] [PubMed] [Google Scholar]
- Schmollinger S., et al. (2014). Systems analysis of nitrogen sparing mechanisms in Chlamydomonas. Plant Cell, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schomburg I., Chang A., Placzek S., Söhngen C., Rother M., Lang M., Munaretto C., Ulas S., Stelzer M., Grote A., Scheer M., Schomburg D. (2013). BRENDA in 2013: integrated reactions, kinetic data, enzyme function data, improved disease classification: new options and contents in BRENDA. Nucleic Acids Res. 41: D764–D772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011). Global quantification of mammalian gene expression control. Nature 473: 337–342. [DOI] [PubMed] [Google Scholar]
- Seemann J.R., Sharkey T.D., Wang J., Osmond C.B. (1987). Environmental effects on photosynthesis, nitrogen-use efficiency, and metabolite pools in leaves of sun and shade plants. Plant Physiol. 84: 796–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segel, I.H. (1975). Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems. (New York: Wiley). [Google Scholar]
- Servaites J.C., Shieh W.J., Geiger D.R. (1991). Regulation of photosynthetic carbon reduction cycle by ribulose bisphosphate and phosphoglyceric acid. Plant Physiol. 97: 1115–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharkey T.D., Stitt M., Heineke D., Gerhardt R., Raschke K., Heldt H.W. (1986). Limitation of photosynthesis by carbon metabolism II. O2-insensitive CO2 uptake results from limitation of triose phosphate utilization. Plant Physiol. 81: 1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeekens S., Ma J., Hanson J., Rolland F. (2010). Sugar signals and molecular networks controlling plant growth. Curr. Opin. Plant Biol. 13: 274–279. [DOI] [PubMed] [Google Scholar]
- Spijkerman E. (2011). The expression of a carbon concentrating mechanism in Chlamydomonas acidophila under variable phosphorus, iron, and CO2 concentrations. Photosynth. Res. 109: 179–189. [DOI] [PubMed] [Google Scholar]
- Stern, D., Harris, E.H., and Witman, G. (2009). The Chlamydomonas Sourcebook. (London: Elsevier). [Google Scholar]
- Stitt M. (1991). Rising CO2 levels and their potential significance for carbon flow in photosynthetic cells. Plant Cell Environ. 14: 741–762. [Google Scholar]
- Stitt M. (1996). Plasmodesmata play an essential role in sucrose export from leaves: a step toward an integration of metabolic biochemistry and cell biology. Plant Cell 8: 565–571. [Google Scholar]
- Stitt M. (2013). Progress in understanding and engineering primary plant metabolism. Curr. Opin. Biotechnol. 24: 229–238. [DOI] [PubMed] [Google Scholar]
- Stitt M., Gibon Y. (2014). Why measure enzyme activities in the era of systems biology? Trends Plant Sci. 19: 256–265. [DOI] [PubMed] [Google Scholar]
- Stitt M., Schulze D. (1994). Does Rubisco control the rate of photosynthesis and plant growth? An exercise in molecular ecophysiology. Plant Cell Environ. 17: 465–487. [Google Scholar]
- Stitt M., Zeeman S.C. (2012). Starch turnover: pathways, regulation and role in growth. Curr. Opin. Plant Biol. 15: 282–292. [DOI] [PubMed] [Google Scholar]
- Stitt M., Cseke C., Buchanan B.B. (1984b). Regulation of fructose 2,6-bisphosphate concentration in spinach leaves. Eur. J. Biochem. 143: 89–93. [DOI] [PubMed] [Google Scholar]
- Stitt M., Herzog B., Heldt H.W. (1984a). Control of photosynthetic sucrose synthesis by fructose 2,6-bisphosphate I. Coordination of CO2 fixation and sucrose synthesis. Plant Physiol. 75: 548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt M., Lilley R.M., Heldt H.W. (1982). Adenine nucleotide levels in the cytosol, chloroplasts, and mitochondria of wheat leaf protoplasts. Plant Physiol. 70: 971–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt M., Sulpice R., Keurentjes J. (2010). Metabolic networks: how to identify key components in the regulation of metabolism and growth. Plant Physiol. 152: 428–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt M., Wirtz W., Heldt H.W. (1980). Metabolite levels during induction in the chloroplast and extrachloroplast compartments of spinach protoplasts. Biochim. Biophys. Acta 593: 85–102. [DOI] [PubMed] [Google Scholar]
- Stotz M., Mueller-Cajar O., Ciniawsky S., Wendler P., Hartl F.U., Bracher A., Hayer-Hartl M. (2011). Structure of green-type Rubisco activase from tobacco. Nat. Struct. Mol. Biol. 18: 1366–1370. [DOI] [PubMed] [Google Scholar]
- Strehmel N., Hummel J., Erban A., Strassburg K., Kopka J. (2008). Retention index thresholds for compound matching in GC-MS metabolite profiling. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 871: 182–190. [DOI] [PubMed] [Google Scholar]
- Strehmel N., Praeger U., Konig C., Fehrle I., Erban A., Geyer M., Kopka J., van Dongen J.T. (2011). Time course effects on primary metabolism of potato (Solanum tuberosum) tuber tissue after mechanical impact. Postharvest Biol. Technol. 56: 109–116. [Google Scholar]
- Sulpice R., Flis A., Ivakov A.A., Apelt F., Krohn N., Encke B., Abel C., Feil R., Lunn J.E., Stitt M. (2014). Arabidopsis coordinates the diurnal regulation of carbon allocation and growth across a wide range of photoperiods. Mol. Plant 7: 137–155. [DOI] [PubMed] [Google Scholar]
- Sulpice R., et al. (2010). Network analysis of enzyme activities and metabolite levels and their relationship to biomass in a large panel of Arabidopsis accessions. Plant Cell 22: 2872–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweetlove L.J., Fernie A.R. (2013). The spatial organization of metabolism within the plant cell. Annu. Rev. Plant Biol. 64: 723–746. [DOI] [PubMed] [Google Scholar]
- Szecowka M., Heise R., Tohge T., Nunes-Nesi A., Vosloh D., Huege J., Feil R., Lunn J., Nikoloski Z., Stitt M., Fernie A.R., Arrivault S. (2013). Metabolic fluxes in an illuminated Arabidopsis rosette. Plant Cell 25: 694–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa K., Cruz J.A., Kanazawa A., Kramer D.M. (2007). The thylakoid proton motive force in vivo. Quantitative, non-invasive probes, energetics, and regulatory consequences of light-induced pmf. Biochim. Biophys. Acta 1767: 1233–1244. [DOI] [PubMed] [Google Scholar]
- Thimm O., Bläsing O., Gibon Y., Nagel A., Meyer S., Krüger P., Selbig J., Müller L.A., Rhee S.Y., Stitt M. (2004). MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37: 914–939. [DOI] [PubMed] [Google Scholar]
- Tohge T., Mettler T., Arrivault S., Carroll A.J., Stitt M., Fernie A.R. (2011). From models to crop species: caveats and solutions for translational metabolomics. Front. Plant Sci. 2: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usadel B., Nagel A., Steinhauser D., Gibon Y., Bläsing O.E., Redestig H., Sreenivasulu N., Krall L., Hannah M.A., Poree F., Fernie A.R., Stitt M. (2006). PageMan: an interactive ontology tool to generate, display, and annotate overview graphs for profiling experiments. BMC Bioinformatics 7: 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usadel B., et al. (2005). Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol. 138: 1195–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van K., Wang Y., Nakamura Y., Spalding M.H. (2001). Insertional mutants of Chlamydomonas reinhardtii that require elevated CO(2) for survival. Plant Physiol. 127: 607–614. [PMC free article] [PubMed] [Google Scholar]
- Vanderauwera S., Zimmermann P., Rombauts S., Vandenabeele S., Langebartels C., Gruissem W., Inzé D., Van Breusegem F. (2005). Genome-wide analysis of hydrogen peroxide-regulated gene expression in Arabidopsis reveals a high light-induced transcriptional cluster involved in anthocyanin biosynthesis. Plant Physiol. 139: 806–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C., Marcotte E.M. (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13: 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelmann T.C., Bornman J.F., Josserand S. (1989). Photosynthetic light gradients and spectral regime within leaves of Medicago sativa. Philos. Trans. R. Soc. Lond. B Biol. Sci. 323: 411–421. [Google Scholar]
- Von Caemmerer, S. (2000). Biochemical Models of Leaf Photosynthesis: Techniques in Plant Sciences, No. 2. (Collingwood, Australia: CSIRO Publishing). [Google Scholar]
- Werner T.P., Amrhein N., Freimoser F.M. (2007). Inorganic polyphosphate occurs in the cell wall of Chlamydomonas reinhardtii and accumulates during cytokinesis. BMC Plant Biol. 7: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney S.M., Houtz R.L., Alonso H. (2011). Advancing our understanding and capacity to engineer nature’s CO2-sequestering enzyme, Rubisco. Plant Physiol. 155: 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkel B.S.J. (2004). Metabolic channeling in plants. Annu. Rev. Plant Biol. 55: 85–107. [DOI] [PubMed] [Google Scholar]
- Woessner J.P., Goodenough U.W. (1989). Molecular characterization of a zygote wall protein: an extensin-like molecule in Chlamydomonas reinhardtii. Plant Cell 1: 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodrow I.E., Murphy D.J., Walker D.A. (1983). Regulation of photosynthetic carbon metabolism. The effect of inorganic phosphate on stromal sedoheptulose-1,7-bisphosphatase. Eur. J. Biochem. 132: 121–123. [DOI] [PubMed] [Google Scholar]
- Wykoff D.D., Davies J.P., Melis A., Grossman A.R. (1998). The regulation of photosynthetic electron transport during nutrient deprivation in Chlamydomonas reinhardtii. Plant Physiol. 117: 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeman S.C., Smith S.M., Smith A.M. (2007). The diurnal metabolism of leaf starch. Biochem. J. 401: 13–28. [DOI] [PubMed] [Google Scholar]
- Zhu X.G., de Sturler E., Long S.P. (2007). Optimizing the distribution of resources between enzymes of carbon metabolism can dramatically increase photosynthetic rate: a numerical simulation using an evolutionary algorithm. Plant Physiol. 145: 513–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.G., Long S.P., Ort D.R. (2008). What is the maximum efficiency with which photosynthesis can convert solar energy into biomass? Curr. Opin. Biotechnol. 19: 153–159. [DOI] [PubMed] [Google Scholar]
- Zhu X.G., Long S.P., Ort D.R. (2010). Improving photosynthetic efficiency for greater yield. Annu. Rev. Plant Biol. 61: 235–261. [DOI] [PubMed] [Google Scholar]
- Zundel M.A., Basturea G.N., Deutscher M.P. (2009). Initiation of ribosome degradation during starvation in Escherichia coli. RNA 15: 977–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.