

SUMMARY
Aerial spores, crucial for propagation and dispersal of the Kingdom Fungi, are commonly the initial inoculum of pathogenic fungi. Natural products (secondary metabolites) have been correlated with fungal spore development and enhanced virulence in the human pathogen Aspergillus fumigatus but mechanisms for metabolite deposition in the spore are unknown. Metabolomic profiling of A. fumigatus deletion mutants of fumiquinazoline (Fq) cluster genes reveal that the first two products of the Fq cluster, FqF and FqA, are produced to comparable levels in all fungal tissues but the final enzymatically-derived product, FqC, predominantly accumulates in the fungal spore. Loss of the sporulation-specific transcription factor, BrlA, yields a strain unable to produce FqA or FqC. Fluorescence microscopy showed FmqD, the oxidoreductase required to generate FqC, was secreted via the Golgi apparatus to the cell wall in an actin-dependent manner. In contrast, all other members of the Fq pathway including the putative transporter, FmqE – which had no effect on Fq biosynthesis – were internal to the hyphae. The coordination of BrlA-mediated tissue specificity with FmqD secretion to the cell wall presents a previously undescribed mechanism to direct localization of specific secondary metabolites to spores of the differentiating fungus.
Keywords: fumiquinazoline, cell wall, BrlA, signal peptide, spore
INTRODUCTION
Fungi produce a wide range of bioactive natural products whose assembly typically requires multiple biosynthetic steps. With a few exceptions, localization of these steps in the fungal thallus – whether tissue specificity or cellular localization - is largely unknown. A few studies have shown that certain secondary metabolites localize to specific tissues, such as endocrocin and fumigaclavines to asexual spores (conidia) of the human opportunistic pathogen Aspergillus fumigatus (1, 2). The mechanism(s) resulting in placement of metabolites into spores is not known although the study on fumigaclavine synthesis suggests a need for BrlA, the Aspergillus transcription factor required for asexual sporulation (1). Existing studies addressing subcellular localization of individual steps have implicated the peroxisome as one synthesis site of certain steps of several secondary metabolites including aflatoxin, penicillin, paxilline and AK-toxin (3–6). Aflatoxin synthesis initiates in the peroxisome (4) but the last steps are housed in large, fused vesicle/vacuole compartments termed aflatoxisomes (7, 8). The terminal step(s) in fungal metabolite synthesis is assumed to involve some type of export, as most secondary metabolites are secreted molecules. However, despite this similar end destination, mechanisms of secretion are largely unknown.
Many fungal secondary metabolite clusters contain putative transporters, which seem like excellent candidates for secretion. Following this line of reasoning, some cluster transporters have been deleted and fungal strains examined for impact on product formation. The results are inconsistent: some transporter deletions show no phenotype (e.g. aflatoxin, (9)), some do not impact production but afford self-protection towards the endogenous metabolite (e.g. sirodesmin and gliotoxin, (10)), whereas others are required for product formation (e.g. zearalenone, (11)). Thus the presence of a cluster transporter does not necessarily present the mechanism of small molecule secretion. Exocytosis is also theorized to be involved in release of secondary metabolites, as is hypothesized for aflatoxin secretion from aflatoxisomes (8). Taken together, these studies suggest there are additional, unknown mechanisms involved in the secretion of fungal secondary metabolites.
One type of secondary metabolite, well characterized in terms of biochemistry, is the fumiquinazolines (Fqs), which comprise a related, sequentially generated family of cytotoxic peptidyl alkaloids that are signature metabolites of A. fumigatus (12, 13). Originally isolated from a marine A. fumigatus isolate, Fqs were found in over forty wild type (WT) isolates (13, 14). A recent study also reported the isolation of fumiquinazoline C (FqC) from the conidial extracts of A. fumigatus (15). The Fq framework is built by non-ribosomal peptide synthetase (NRPS) enzymatic machinery with anthranilate as a key non-proteinogenic amino acid building block (16). Bioinformatic analyses followed by heterologous expression and purification in E. coli of A. fumigatus proteins predicted to be involved in Fq biosynthesis led to the identification of a four-enzymatic reaction process that builds increasingly complex Fq scaffolds, starting with the trimodular NRPS FmqA (AFUA_6G12080) and ending with the FAD-dependent oxidoreductase FmqD (AFUA_6G12070) ((16–18) and depicted in Fig. 1A and 1B).
Figure 1.

Fumiquinazoline biosynthesis in A. fumigatus. A) Fumiquinazoline gene cluster in A. fumigatus. B) Proposed biosynthetic route of fumiquinazolines in A. fumigatus. C) Fq profiles of single deletion mutants in AF293. Fq profiling was done with total extracts of solid growth culture. Left panel: UV trace overlay (from 254nm detection). Right panel: Overlay of extracted ion chromatogram (EIC) for the Fq metabolites FqA (RT = 7.1 min), FqC (RT = 7.4 min), and FqF (RT = 7.9 min). The Fqs were identified with the Agilent MassHunter software using the “Find Compound by Formula” option.
Despite the detailed knowledge of the biosynthetic pathway, neither the encoding gene cluster nor tissue/subcellular location of the Fq metabolites/enzymes has been characterized in vivo. Here we provide in vivo validation to the Fq biosynthetic framework and elucidate the subcellular localization of the enzymes responsible for generating each step of the Fq pathway. The cluster contains four biosynthetic enzyme-encoding genes termed fmqA-D and one transporter gene termed fmqE (AFUA_6G12040). Whereas the first two intermediates in the Fq pathway, FqF and FqA, are found equally in all fungal tissues, the final enzymatic product FqC predominates in conidial tissues. FqC accumulation requires localization of FmqD, the oxidoreductase responsible for FqC synthesis, to spore cell walls. Deletion of the FmqD N-terminal signal peptide eliminates localization of the oxidoreductase to the cell wall and yields a similar chemical profile as a ΔfmqD strain. BrlA is also required for FqC and FqA syntheses as a ΔbrlA strain only produces FqF. This work presents a prototype for the networking of both spore-specific (BrlA) and secretion mechanisms (FmqD cell wall localization) in deposition and selective accumulation of specific natural products to spores of the differentiating fungus.
RESULTS
The fumiquinazoline cluster consists of four biosynthetic genes fmqA – fmqD
To validate the activities observed for FmqA, FmqB (AFUA_6G12060), FmqC (AFUA_6G12050) and FmqD in the heterologous E. coli system (16–18), single deletants in each of these genes were examined for Fq synthesis (Fig. 1C). Mutants were created in the Af293 background and some also assessed in the CEA17 ΔKU80 background as noted below. Extracts from the fmqA, fmqB, fmqC and fmqD deletion mutants corroborate the findings obtained with purified enzymes published previously (16–18).
The trimodular NRPS, FmqA is required for production of all Fq metabolites in A. fumigatus. Inactivation of FmqA (ΔfmqA) renders a strain deficient for all fumiquinazolines (Fig. 1C). Deletion of the FAD-dependent monooxygenase, FmqB, responsible for oxidization of FqF, which then is acted upon by the monomodular NRPS, FmqC, to form FqA, resulted in loss of both FqA and FqC (Fig. 1C). This mutant also showed a 19-fold accumulation in FqF production over WT further supporting FqF being the biological precursor to FqA (Fig. 1C). Deletion of the monomodular NRPS, FmqC, also abolished FqA and FqC production (Fig. 1C). However, unlike the ΔfmqB strain, the ΔfmqC strain did not accumulate FqF (or any detectable amounts of products from FmqB activity such as oxidized forms of FqF, e.g. FqF-diol, (18)) but rather produced FqF levels similar to that of WT.
Interestingly, deletion of fmqD encoding the last enzymatic step of the pathway resulted in a significant decrease but not complete loss of FqC production when the strain is grown on solid media (Fig. 1C). The ΔfmqD mutant accumulates FqA (81-fold increase over WT) and FqF (15-fold increase over WT) but decreased FqC (10-fold decrease compared to WT). In contrast to the small amount of FqC produced in this strain grown on solid agar culture, FqC production is not observed when the ΔfmqD mutant is grown under liquid shake conditions (data not shown). CEA17 ΔKU80 mutants gave similar profiles as the Af293 mutants (data not shown). We also thought it is possible that fmqE (Fig. 1A), encoding a putative MFS transporter, could be involved in Fq metabolism and transport. However, loss of this gene had no impact on Fq production under conditions tested (data not shown).
Fumiquinazoline C selectively accumulates in conidial tissues
Comparison of individual Fq metabolite ratios from cultures grown on solid defined medium shows that both WT strains predominantly yield FqC with smaller amounts of FqA and FqF and that the relative amount of FqC from total crude extract is comparable between both WT isolates (Fig. 2A). We found it interesting that FqC was found as the major metabolite in solid grown (high sporulation) but not liquid shake (low sporulation) cultures (Fig. 2B). To further examine how FqC production was associated with spore formation, Fq production was assessed in different tissues of the fungus comprised of (i) conidiophores and conidia (spores), (ii) vegetative hyphae and some dislodged conidia and (iii) invasive hyphae and secreted metabolites (Fig. 3A). The FqC:[FqF+FqA] ratio was highest in the conidial fraction (5:1) and lowest in the invasive hyphae fraction (1:2) (Fig. 3B).
Figure 2.

LC-MS comparison of Fq metabolite profiles from solid growth and liquid shake conditions. A) Fq profile from total crude extracts of AF293 and CEA17ΔKU80 on solid growth culture. B) Fq profile from fungal tissue (mycelia) and growth filtrate (media) of AF293 grown in liquid shake. Left panel: UV trace overlay (from 254nm detection). Right panel: Overlay of extracted ion chromatogram (EIC) for the Fq metabolites FqA (RT = 7.1 min), FqC (RT = 7.4 min), and FqF (RT = 7.9 min). The Fqs were identified with the Agilent MassHunter software using the “Find Compound by Formula” option.
Figure 3.

FqC production is associated with fungal sporulation. A) Schematic of fractionation. Fungal cultures are separated into three fractions: Conidial (conidiophores and conidia), top agar (primarily vegetative hyphae, some conidiophores and dislodged conidia) and bottom agar (invasive hyphae and secreted metabolites) B) Fq profiles of the three fractions sampled from AF293 solid growth culture.
BrlA, but not AbaA and WetA, is necessary for fumiquinazoline C production
The results above show selective accumulation of FqC in asexual reproduction structures. To further examine the link between Fq production and spore development, Fq profiling was conducted in characterized mutants aberrant in conidiophore development. BrlA is a conidiation-specific transcription factor involved in the early stage of asexual development and necessary for conidiophore formation in the Aspergilli (19). Previous published results showed that FqC production is abolished in the ΔbrlA mutant (15). When we analyzed the production of other Fq metabolites in this mutant, we observed that this mutant is still capable of producing FqF but not other Fq moieties (Fig. 4A). Next we asked if AbaA and WetA, two BrlA-regulated transcription factors involved in the middle and late stages of conidiophore development and maturation, were also required for Fq production. Fq production resembles that of WT in the ΔabaA mutant and is increased in the ΔwetA mutant (Fig. 4B). We also examined Fq production in a ΔflbE mutant, FlbE being an upstream regulator of BrlA. The ΔflbE mutant is delayed in conidiophore production and is decreased in brlA expression. As expected, Fq production is decreased but not eliminated in this mutant (Fig. 4B). In all cases, enhanced accumulation of FqC is still maintained in the ΔabaA, ΔwetA, and ΔflbE mutants.
Figure 4.

BrlA regulates production of FqC. A) Fq profiles comparing wildtype AF293 and ΔbrlA mutant on solid growth culture shows that FqA and FqC production is BrlA-dependent. Left panel: UV trace overlay (from 254nm detection). Right panel: Overlay of extracted ion chromatogram (EIC) for the Fq metabolites FqA (RT = 7.1 min), FqC (RT = 7.4 min), and FqF (RT = 7.9 min). The Fqs were identified with the Agilent MassHunter software using the “Find Compound by Formula” option. B) Fq production in various conidiophore developmental mutants of A. fumigatus. y-axis represents peak area of each Fq metabolites. C) Semi-quantitative PCR of fmq cluster and border genes comparing wildtype AF293 and ΔbrlA 18 and 24 hours after induction of asexual development (conidiophore formation).
These results demonstrate that brlA expression is both necessary and sufficient for Fq biosynthesis, which remains largely normal in mutants with reproductive structure abnormalities as in ΔabaA and ΔwetA strains. To further assess the regulatory impact of BrlA on Fq production, expression analysis of the Fq cluster genes was performed comparing both WT and the ΔbrlA mutant (Fig. 4C). We observed that fmqA, fmqB, fmqC, fmqD, and fmqE transcripts are undetectable in the ΔbrlA mutant.
The N-terminal signal peptide in FmqD is important for cell wall localization and fumiquinazoline C biosynthesis
The results above demonstrated that production of FqC but not FqF or FqA is predominantly conidiophore-specific. As previous in silico analysis of FmqD (the enzyme generating FqC) identified a conserved signal sequence suggesting that the enzyme is secreted (18), we speculated that FmqD localization and/or secretion could be important for FqC tissue specificity. To examine this possibility, we created two FmqD::GFP variant strains, one where full length FmqD was fused to eGFP (FmqD::GFP) and one where FmqD deleted for the N-terminal signal peptide was fused to eGFP (FmqD[Δ1-21]::GFP). The FmqD::GFP mutant produced Fq levels comparable to that of the WT strain but the FmqD[Δ1-21]::GFP mutant abolished the selective accumulation of FqC and showed a significant decrease in FqC production (3.5-fold) coupled with increased accumulation of FqA, thus resulting in a metabolite profile with some similarity to that of the ΔfmqD strain (Fig. 5A). However, we are unable to visualize localization of either FmqD::GFP or FmqD[Δ1-21]::GFP under native promoter expression. Thus, we expressed the same two alleles using the constitutive promoter, gpdA(p). These two overexpression variants are termed OEFmqD::GFP and OEFmqD[Δ1-21]::GFP respectively. Northern analysis confirmed higher expression levels of the overexpression OEFmqD::GFP variants compared to that of the native FmqD::GFP variants (Fig. S1). In addition, we overexpressed the nuclear protein, histone H2A, fused to GFP (OEH2A::GFP) to control for the possibility of subcellular mislocalization due to artifactual overexpression of a protein. As shown in Fig. 5B, nuclear localization of histone H2A is still maintained in all stages of fungal development (conidia, germlings, hyphae) examined in this study, demonstrating that subcellular localization is still maintained in the overexpression strains.
Figure 5.

Characterization of N-terminal signal peptide involvement in FqC biosynthesis. A) Fq profiles of FmqD::GFP and FmqD[Δ1-21]::GFP on solid growth cultures. Left panel: Diode array detector (DAD) trace from 254nm detection. Right panel: Overlay of extracted ion chromatogram (EIC) for the Fq metabolites FqA (RT = 7.1 min), FqC (RT = 7.4 min), and FqF (RT = 7.9 min). The Fqs were identified with the Agilent MassHunter software using the “Find Compound by Formula” option. B) Overexpression of histone H2A still maintained nuclear localization in various developmental tissues examined in this study. From left to right: conidia, germlings, and hyphae. Top Row: GFP. Bottom Row: Merged GFP and differential interference contrast (DIC) C) FmqD is cell wall-associated. Top Panel: Conidia. Middle Panel: Hyphae. Bottom Panel: Protoplasts. Protoplasts from the OEFmqD::GFP strain lose cell wall fluorescence while those from the OEFmqD[Δ1-21]::GFP strain retained cytoplasmic fluorescence. Top Row: GFP. Bottom Row: differential interference contrast (DIC)
Examination of the overexpression FmqD::GFP strains showed that FmqD is localized to the cell wall of the conidia as seen by the ring-like fluorescence (Fig. 5C). A time-course fluorescence imaging of the germination process showed that the cell wall localization decreases during isotropic growth (swelling of spores, Fig. S2) and is regained in mature hyphae (Fig. 5C). Deletion of the N-terminal signal peptide FmqD[Δ1-21] results in loss of cell wall accumulation and localization of FmqD[Δ1-21] to the cytoplasm (Fig. 5C). In the hyphae, FmqD is localized to both the cell wall and septa while FmqD[Δ1-21] again accumulates in the cytoplasm (Fig. 5C). Cell wall localization of FmqD was confirmed via a protoplasting experiment where GFP fluorescence in the cell wall was largely lost from protoplasts of the OEFmqD::GFP mutant but is retained in the cytoplasm of protoplasts from the FmqD[Δ1-21]::GFP mutant (Fig. 5C). Taken together, the above results demonstrate that the N-terminal signal peptide is important in both directing FmqD cell wall localization and FqC production in the fungus.
Localization of FmqD to the cell wall is dependent on ER-Golgi transport and actin polymerization
3D modeling of FmqD predicts three surface exposed N-glycosylation sites (Asn95, 257, and 284) (18). These types of modifications are often associated with protein folding and trafficking through the ER-Golgi apparatus in A. fumigatus and other organisms (17, 20–22). To address possible trafficking through the Golgi apparatus, the OEFmqD::GFP mutant was treated with Brefeldin A, a known inhibitor of ER-Golgi transport, and localization of FmqD observed at two and four hours post treatment (23). At two hours post treatment, cell wall localization of FmqD is highly reduced in the hyphae of the treated samples but rather localized to a reticulate network (ER) dispersed throughout the hyphae (Fig. 6A). At four hours post treatment, cell wall localization of FmqD is completely abolished and FmqD is mainly localized to punctate spots between the reticulate networks with low amounts still localized to the periphery of the reticulate networks (Fig. 6A). At both time points, the solvent carrier controls still maintained cell wall and septum localization (Fig. 6A). These observations demonstrate that the ER-Golgi transport is necessary to localize FmqD to the fungal cell wall. To further assess if FmqD localization to the cell wall is actin-dependent, we treated OEFmqD::GFP with cytochalasin A, a fungal compound known to inhibit actin polymerization (23). As shown in Fig. 6B, cell wall localization is abolished with cytochalasin A treatment. Rather fluorescence appeared to be slightly diffused throughout the cytoplasm and also as punctate spots throughout the fungal hyphae (Fig. 6B).
Figure 6.

Cell wall localization of FmqD is actin-dependent and requires ER-Golgi transport. A) OEFmqD::GFP mutant incubated in the presence of Brefeldin A (BFA) shows lost of cell wall accumulation and presence of reticulate network (ER proliferation) at two hours post treatment. Fluorescence is localized to punctated vesicles throughout the hyphae at four hours post treatment. B) Treatment with cytochalasin A (actin-depolymerizing agent) abolished cell wall localization of FmqD::GFP. The methanol and DMSO carrier control is depicted to the left of each time point presented. Top Row: GFP. Bottom Row: Merged GFP and differential interference contrast (DIC).
FmqD as part of the A. fumigatus secretome
As many cell wall-associated proteins from A. fumigatus are secreted proteins that are transiently localized to the cell wall prior to release into the extracellular environment (24, 25), we asked if FmqD is also part of the A. fumigatus secretome. To assess this, we performed an extraction from the cell wall of A. fumigatus WT, OEH2A::GFP, and OEFmqD::GFP strains grown under liquid shake conditions at both 48 and 72 hours post inoculation to isolate non-covalently bound proteins that are in transit through the cell wall. During the preparation of the cell wall, visualization of the cell wall fragments (hyphal ghosts) using fluorescence microscopy showed abundance of GFP fluorescence in the hyphal ghosts and residual intact hyphae of the OEFmqD::GFP strain while the OEH2A::GFP strain showed lack of GFP fluorescence in the hyphal ghosts along with minute amounts of nuclear GFP fluorescence in residual intact hyphae (data not shown). As depicted in Figure 7A, minute amounts of the nuclear-associated H2A::GFP fusion protein can be observed in the non-covalent cell wall extracts of the OEH2A::GFP at both 48 and 72 hours (lanes 2 and 6 respectively). An 82 kDa band corresponding to the size of the cell wall-associated FmqD::GFP fusion protein is observed in the OEFmqD::GFP strain at both 48 and 72 hours (lanes 3 and 7 respectively) and appears at a much higher intensity compared to the H2A::GFP bands, thus supporting the above microscopy visualization.
Figure 7.

FmqD is a non-covalvent cell wall-associated protein. A) Western blot analysis of non-covalent cell wall extracts from WT (lanes 1 and 5), OEH2A::GFP (lanes 2 and 6), and OEFmqD::GFP (lanes 3 and 7) isolated from 48 and 72 hours grown cultures using primary monoclonal GFP antibodies. Lanes 4 and 8 depict 10 ng of purified C-terminal epitope-tagged GFP. B) Western blot analysis of culture filtrate extracts from WT (lanes 1 and 4), OEH2A::GFP (lanes 2 and 5), and OEFmqD::GFP (lanes 3 and 6) isolated from 48 and 72 hours grown cultures using primary monoclonal GFP antibodies. Fusion protein sizes on blot: H2A::GFP (45 kDa), FmqD::GFP (82 kDa).
To further strengthen our observations, proteins were also isolated from the growth medium of the above cultures. As shown in Figure 7B, Western blot analysis of culture filtrates at 48 hours showed presence of the FmqD::GFP fusion protein in the OEFmqD::GFP strain (lanes 3 and 6) and absence of the nuclear-associated H2A::GFP fusion protein in the OEH2A::GFP strain (lanes 2 and 5). At 72 hours post inoculation, the accumulation of a 27 kDa band corresponding to the size of free GFP is also observed, most probably due to the cleavage/degradation of the fusion protein (lane 6).
Subcellular localization of the remaining Fmq proteins
To complete our understanding of the spatial distribution of other Fmq enzymes and Fq biosynthetic steps in the fungal body, we also examined subcellular localization of other members of the fmq gene cluster. In contrast to FmqD, none of the other enzymes are localized to the cell wall. Figure 8 shows that FmqA is associated with vesicles that varied in size but almost even spatial distribution between one another within the hyphae, FmqB and FmqC appeared evenly distributed in the cytoplasm and the putative transporter, FmqE is also localized to network-like vesicles within the hyphae but in a pattern distinct from FmqA (Fig. 8). Attempting to resolve the nature of the FmqA-associated vesicles, the OEFmqA::GFP mutant was stained with both FM4-64 and Hoechst 33342. FM4-64 is used to visualize endocytic processes, vacuoles and mitochondria in Aspergillus (26) and Hoechst 33342 labels the fungal nuclei. FmqA did not colocalize with either stain (Fig. S3) and thus, leaving the nature of the vesicles unresolved.
Figure 8.
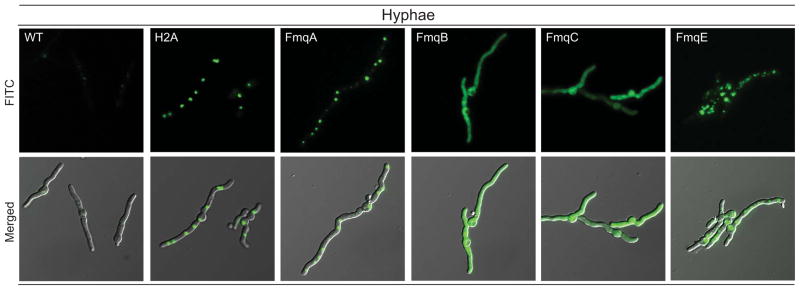
Subcellular localization of the remaining Fmq proteins in the fungal hyphae. From left to right: wildtype, OEH2A::GFP, OEFmqA::GFP, OEFmqB::GFP, OEFmqC::GFP, and OEFmqE::GFP. Both FmqA::GFP and FmqE::GFP localize to vesicles within the hyphae while FmqB::GFP and FmqC::GFP localizes to the cytoplasm. H2A::GFP control strain still maintain nuclear localization. Top Row: GFP. Bottom Row: Merged GFP and differential interference contrast (DIC).
DISCUSSION
The fumiquinazolines comprise a family of cytotoxic peptidyl alkaloids that are signature metabolites of the opportunistic human pathogen A. fumigatus. This family of metabolites has received considerable interest due to their complex biochemistry and potential antitumor properties (13, 27) but until now, nothing is known of their biosynthesis or localization in vivo. We show here that deletion of the Fq cluster genes fmqA-D yield Fq intermediates as predicted by biochemical studies (16–18). Deletion of fmqA abolished all Fq production while deletions of fmqB and fmqC yielded only the first Fq precursor, FqF. Finally, fmqD mutants produced primarily FqF and FqA with trace amounts of FqC present only in crude extracts of solid grown cultures. This trace amount of FqC observed in solid grown cultures of ΔfmqD may be attributable to another promiscuous oxidoreductase with sufficient similarity to FmqD to weakly engage FqA. We find that FqC is concentrated in the conidial tissues compared to other Fq moieties and that this localization is dependent on the coordinate action of BrlA and secretion of FmqD to the cell wall. On the other hand, deletion of the putative transporter FmqE had no impact on Fq production.
The data supporting differential tissue distribution of the Fq moieties included enrichment of FqC in conidial fractions of solid grown culture, the lack of FqC enrichment in liquid grown culture (where few spores are generated) and the absence of this metabolite in the ΔbrlA mutant. Unlike many secondary metabolite clusters, the Fq cluster lacks a specific transcription factor. The regulation of such clusters is largely unknown but when identified, has been found to be due to the global regulators such as LaeA and/or trans-acting transcription factors as characterized for several gene clusters in Aspergillus spp. (28–31). We found that a previously published microarray analysis showed that loss of BrlA resulted in down regulation of fmqC and fmqD, here found to be required for FqA and FqC synthesis, respectively (32), suggestive of BrlA transcriptional control of at least part of this gene cluster.
Our work supports BrlA as a major regulator of Fq synthesis and fmq gene expression. The role of BrlA in Fq production appears twofold as it is not only required for expression of the fmq cluster genes but also provides the structure (conidiophore and associated conidia) necessary for FmqD secretion and FqC deposition. Delay of brlA expression and/or conidiophore development in the ΔflbE (33) can reduce but not eliminate Fq biosynthesis. On the other hand, once BrlA is activated, downstream mutations leading to deformed conidiophore development have no negative impact on Fq production. The wildtype levels of Fq products in the ΔabaA mutant suggests it is not the spore per se that is required for FqC accumulation as this mutant forms long chains of terminal cells on the conidiophore head unable to terminate in spores (34). We note that, to date, all BrlA-regulated A. fumigatus spore metabolites including FqC, endocrocin (2) and trypacidin (15) are also regulated by LaeA which itself regulates BrlA (35).
Aside from BrlA control, another factor important in FqC production and localization is the accumulation of FmqD to the cell wall. Figure 5 shows that FmqD localizes to both conidial and hyphal cell walls; however in this study, the FmqD::GFP allele was driven by the gpdA promoter and thus resulting in high expression in the hyphal cell wall and septum. Figures 2 and 3 support a view that FmqD primarily accumulates in the conidium cell wall. Whereas many studies have addressed the importance of subcellular localization and compartmentalization of biosynthetic enzymes and pathway intermediates in secondary metabolism in fungi (3, 5–7, 36), no study to date has demonstrated the secretion of a secondary metabolite biosynthetic enzyme. However, secretion of biotechnologically valuable fungal proteins (glucoamylases, cellulases and lipases) has been well studied in several genera including Aspergillus (37, 38). Many of these known secreted proteins are shown to first localize to the cell wall via the secretory pathway and eventually diffuse to the environment. In silico analysis of FmqD predicts that it bears attributes indicative of secretion through the classical ER-Golgi-mediated secretory pathway including an N-terminal signal peptide and putative surface exposed N-glycosylation sites that have been shown to be important in the proper folding and localization of other A. fumigatus cell wall proteins (22, 39–42). Our study supports trafficking of FmqD through this pathway as deletion of the signal peptide abolished FmqD cell wall localization and greatly reduced FqC production (despite knowing that the signal peptide is not required for enzyme activity in vitro, (17)). Treatment of OEFmqD::GFP with a known inhibitor of ER-Golgi transport (Brefeldin A) and an actin-depolymerizing agent (cytochalasin A) disrupted the localization of FmqD, further demonstrating secretion of FmqD via the classical secretory pathway in an actin-dependent manner. Taken together, this suggests that proper transport and localization of FmqD and not just enzyme activity is important for FqC synthesis and accumulation.
The presence of FqF in the ΔbrlA mutant suggests either a basal expression of fmqA independent of BrlA and undetectable by semi-quantitative PCR or participation of another NRPS in FqF synthesis. Towards this end, recent studies have shown considerable crosstalk between NRPS pathways in A. fumigatus (43, 44). Regardless of the route of production, the presence of a basal level of FqF could allow for a constant supply of the starting materials (FqF and oxidized form of FqF) in the fungal hyphae, which presumably would be readily available for conversion to FqA and FqC on the onset of conidiation. We also note that FqF is a branchpoint intermediate between the later stage fumiquinazolines and the tryptoquivaline/tryptoquialanine pathways (45, 46) and speculate that storage of this precursor metabolite may be advantageous in quick conversion to final products dependent on environmental signals to the fungus.
A major conclusion from our work is that secondary metabolite localization in the fungal thallus is dependent on several mechanisms: in this case BrlA regulation of gene expression and conidiophore formation followed by cell wall localization of FmqD, the terminal enzyme of the Fq biosynthetic pathway. It is known that spore metabolites can contribute to the virulence of pathogenic fungi such as A. fumigatus; for instance, the spore metabolites endocrocin and trypacidin/fumiquinazolines exhibit neutrophil inhibiting and cytotoxic properties respectively (15). Furthermore, the localization of FmqD protein itself to the cell wall of the conidium may have a significant implication on host-pathogen interactions by either masking or aiding in immune detection of the fungus. As FmqD is non-covalently linked to the cell wall, it is highly possible that the protein along with its secondary metabolite substrate, FqC, is being released as the spores swell both interstitially within the lung or intracellularly within resident phagocytes upon engulfment of spores. Despite being beyond the scope of this manuscript, the implications of both FmqD localization to the conidia cell wall and all Fqs on the pathogenicity of A. fumigatus will be of interest for future studies in our lab. Our findings from this work also provide a platform to explain results from previous studies where deletions of putative cluster transporters did not appear to have major impact on secondary metabolite production (9, 10, 47). Similarly, we found that deletion of the putative transporter FmqE did not affect FqC production under the conditions examined. Rather it was the localization of FmqD that impacted FqC production and presumably release to the environment.
EXPERIMENTAL PROCEDURES
Fungal strains and growth conditions
All A. fumigatus strains used in this study are listed in Table 1. Two sequenced WT isolates were used to generate the fmq mutants. Strains were maintained as glycerol stocks and activated on solid glucose minimal medium (GMM) at 37°C (48). Growth media was supplemented with five mM of uridine and uracil for pyrG auxotrophs. For Fq profiling on solid medium, A. fumigatus strains were point-inoculated onto solid GMM at 1 × 104 conidia total (in 5 μL) and cultured at 29°C or 37°C for four or six days respectively unless otherwise noted. For Fq profiling in liquid shake conditions, A. fumigatus strains were inoculated into 50 mL of liquid GMM at 1 × 106 conidia/mL and cultured at 37°C and 250 r.p.m. for three days unless otherwise noted.
Table 1.
Strains used in this study
| Fungal Strain | Genotype | Source |
|---|---|---|
| AF293 | WT | (56) |
| AF293.1 | pyrG1 | (56) |
| CEA17 KU80 pyrG+ | ΔakuB pyrG+ | (57) |
| CEA17 KU80 pyrG− | ΔakuB pyrG− | (57) |
| TFYL10.1 | ΔfmqA::A. parasiticus pyrG; pyrG1 | This study |
| TFYL11.1 | ΔfmqD::A. parasiticus pyrG; pyrG1 | This study |
| TFYL12.1 | ΔfmqB::A. parasiticus pyrG; pyrG1 | This study |
| TFYL13.1 | ΔfmqC::A. parasiticus pyrG; pyrG1 | This study |
| TFYL24.1 | ΔakuB; ΔfmqE::A. parasiticus pyrG | This study |
| TFYL25.1 | ΔakuB; ΔfmqB::A. parasiticus pyrG | This study |
| TFYL26.1 | ΔakuB; ΔfmqD::A. parasiticus pyrG | This study |
| TFYL27.1 | ΔakuB; ΔfmqA::A. parasiticus pyrG | This study |
| TFYL28.1 | ΔakuB; fmqD::eGFP::A. fumigatus pyrG | This study |
| TFYL29.1 | ΔakuB; fmqD[Δ1-21]::eGFP::A. fumigatus pyrG | This study |
| TFYL34.1 | ΔakuB::A. fumigatus pyrG; gpdA(p)::fmqD::eGFP::A. fumigatus pyrG | This study |
| TFYL33.1 | ΔakuB::A. fumigatus pyrG; gpdA(p)::fmqD[Δ1-21]::eGFP::A. fumigatus pyrG | This study |
| TFYL48.1 | ΔakuB::A. fumigatus pyrG; gpdA(p)::fmqA::eGFP::A. fumigatus pyrG | This study |
| TFYL49.1 | ΔakuB::A. fumigatus pyrG; gpdA(p)::fmqB::eGFP::A. fumigatus pyrG | This study |
| TFYL50.1 | ΔakuB::A. fumigatus pyrG; gpdA(p)::fmqC::eGFP::A. fumigatus pyrG | This study |
| TFYL51.1 | ΔakuB::A. fumigatus pyrG; gpdA(p)::fmqE::eGFP::A. fumigatus pyrG | This study |
| TFYL52.1 | ΔakuB::A. fumigatus pyrG; gpdA(p)::H2A::eGFP::A. fumigatus pyrG | This study |
| TKSS6.07 | pyrG1 flbE::A. nidulans pyrG | (33) |
| TSGa17 | pyrG1 ΔabaA::A. nidulans pyrG | (34) |
| TSGw4 | pyrG1 ΔwetA::A. nidulans pyrG | (34) |
| ΔAfbrlA7 | pyrG1 ΔbrlA::A. fumigatus pyrG | (19) |
Fumiquinazoline mutant construction
All primers used in this study are listed in Table S1 and gene deletion mutants in this study were made by targeted integration of the deletion constructs through transformation (49, 50). The deletion constructs were made using a double-joint fusion PCR (DJ-PCR) approach (49, 50). To construct the deletion cassettes, two 1.0 –1.5 kb fragments flanking the deletion region of each gene were amplified from AF293 genomic DNA template. The selectable auxotrophic marker, A. parasiticus pyrG, was amplified from plasmid pJW24 (51). The deletion constructs are verified with restriction enzyme digestion. Genomic DNA isolation, protoplast generation, and transformation procedures were carried out as previously described (49, 50). The transformants were subjected to multiplex PCR using internal primers to the gene of interest and control internal primers to A. fumigatus gpdA (Table S1). To construct the FmqD::GFP fusion strains, FmqD was fused at its C-terminus to GFP::A. fumigatus pyrG with and without the N-terminal 1–21 amino acid residues. GFP::A. fumigatus pyrG was amplified from plasmid pFNO3 (52). The OEFmqD::GFP mutants were constructed as above with the exception of the native fmqD promoter replaced with the constitutive promoter, gpdA(p), amplified from plasmid pJMP9.1 (31). The above transformants were subjected to an initial PCR screen for the presence/absence of the signal peptide and FmqD::GFP fusion construct. The constructs for all OEFmqA::GFP, OEFmqB::GFP, OEFmqC::GFP, OEFmqE::GFP are generated using an in vivo yeast recombination cloning system (53). Briefly, the vector, pYH-wA-pyrG (53), was subjected to HindIII/SacII digestion. Individual segments consisting of (1) ~1 kb 5′flanking region from wildtype genomic DNA, (2) gpdA(p) amplified from pJMP9.1, (3) fmq open reading frame from wildtype genomic DNA, (4) GFP::A. fumigatus pyrG amplified from pFNO3, and (5) ~1 kb 3′flanking region from wildtype genomic DNA were amplified by high fidelity PCR. The above segments along with the digested vector were subjected to in vivo yeast recombination as previously described (53) to create plasmids pFYL7 (OEFmqA::GFP), pFYL5 (OEFmqB::GFP), pFYL6 (OEFmqC::GFP), and pFYL8 (OEFmqE::GFP). Each of the above plasmid was linearized and transformed into strain CEA17 ΔakuB pyrG− to yield strains TFYL48.1, TFYL49.1, TFYL50.1, and TFYL51.1 respectively. For all of the transformants generated above, correct integration of the transformation construct at the locus of interest was verified via Southern analysis using two restriction digest profiles according to standard procedures (54). One representative Southern blot for all mutants can be found in Figure S4.
Fq extraction and profiling
Sample preparation for Fq profiling from solid and liquid shake conditions
Agar cores (approximately 1.5 cm in diameter) were prepared in triplicate for each strain cultured as described above (fungal strains and growth conditions). Three cores from each plate were extracted with 2 mL of methanol by briefly vortexing then incubated at room temperature for 20 min submerged in solvent. Subsequently, 0.5 mL of the sample extract was filtered using a 0.45 μm PTFE Mini-UniPrep filter vial (Agilent) and 10 μL of the filtrate injected for LC-MS analysis.
For liquid shake extracts, the mycelia (cultured as described in fungal strains and growth conditions) were removed by filtration, washed with distilled water, and lyophilized. Mycelia were extracted with 5 mL of methanol by vortexing and incubating as described in the previous paragraph. Total filtered culture media was lyophilized and also extracted with 5 mL methanol. 0.5 mL samples from mycelia or media extracts were filtered using a filter vial and 10 μL of the filtrate injected for LC-MS analysis.
Extraction of different fungal tissues for Fq production
Plates were prepared by overlaying 10 mL GMM top agar (0.75% agar) onto a solidified base layer of GMM agar (1.5% agar). Plates were point inoculated with 1 × 106 conidia and grown at 37°C for 4 days followed by 25°C for 3 days. Conidia were harvested by flooding the plate with 0.1% Tween 20 (2 × 6 mL) followed by gentle agitation with a plastic spreader. The conidial fraction was inspected under the microscope to be largely free of hyphae and conidiophores. The conidial fraction was collected by centrifugation, washed once with 6 mL 0.1% Tween 20, and extracted with 5 mL 1:1 mixture of ethyl acetate:methanol (EtOAc:MeOH). The original plate was washed (3 × 10 mL volumes of 0.1% Tween 20) and the top agar layer (containing dislodged conidiophores, vegetative mycelia, and media) was removed with a spatula and extracted with 15 mL EtOAc:MeOH (1:1). The bottom agar layer (which contained some substrate mycelia and the bulk of the growth media) was washed with 10 mL water then removed, finely chopped, and extracted with 25 mL EtOAc:MeOH (1:1). 2 mL from each fraction was dried in vacuo, the residue reconstituted in 0.5 mL of 50% acetonitrile (MeCN), filtered as described in the previous section, and 10 μL samples of each fraction injected for LC-MS analysis.
LC-MS analysis and Fq profiling
LC-MS data were acquired on an Agilent Technologies 6520 Accurate-Mass QTOF instrument supported by the Taplin Funds for Discovery Program. Samples were injected onto a Gemini-NX C18 column (50 × 2.0 mm, 5 micron, 100 Å, Phenomenex) equilibrated in solvent system A (water + 0.1% formic acid) plus 20% B (acetonitrile + 0.1% formic acid) and the following LC method was run: 20% B for 2 min, ramp to 60% B over 10 min, ramp to 100% B over 0.5 min, and a hold at 100% B for 1 min. The column was equilibrated in 20% B for 4 minutes prior to beginning the next injection sequence. MS data were collected using electrospray ionization in positive ion detection mode (capillary voltage of 3500 V, fragmentor set to 125 V, skimmer to 70 V, 12 L/min drying gas at 295° C, 30 psig nebulizer) with real-time internal reference mass correction (API-TOF Reference Mass Solution, Agilent). UV data were collected using diode array detection. Data were analyzed using Agilent MassHunter Qualitative Analysis software. TIC, total ion current chromatogram; DAD, diode-array detector (UV254nm trace); and EIC, extracted ion chromatogram for all WT, ΔfmqA, ΔfmqB, ΔfmqC, and ΔfmqD are showed in supplemental figures S5–S9 respectively. HPLC data on the various conidiation mutants were acquired using a Perkin Elmer Flexar Instrument. Samples were injected onto a ZORBAX Eclipse XDB-C18 column (150 × 4.6 mm, 5 micron, 100 Å, Agilent) equilibrated in solvent system A (water + 0.1% formic acid) plus 20% B (acetonitrile + 0.1% formic acid) and the following HPLC method was run: 20% B for 2 min, ramp to 60% B over 10 min, ramp to 100% B over 0.5 min, and a hold at 100% B for 5 min. The column was equilibrated in 20% B for 4 minutes prior to beginning the next injection sequence.
Fluorescence Microscopy
A coverslip was placed in a small petri dish and subjected to a 15-minute exposure to a germicidal UV-C lamp in a biosafety cabinet. A. fumigatus strains were inoculated at 2 × 102 conidia/mL in liquid GMM into the above prepared petri dish and incubated at 30°C overnight. For spore germination assay, strains were cultured as above but at 37°C. Media was decanted from the petri dish and the coverslip along with adhered fungal hyphae was then mounted onto a pre-cleaned microscope slide in mounting solution (50 mM sodium phosphate buffer pH8.0, 50% glycerol, 0.1% propyl gallate), and sealed with nail polish. For staining with FM4-64 and Hoechst 33342, the coverslips were removed from the petri dishes and excess media removed. 200μL of staining solution was pipetted onto the coverslips and incubated for 10–15 minutes at room temperature. Coverslips were washed twice in PBS and mounted onto a pre-cleaned microscope slide in mounting solution. Images were taken with a Zeiss AxioImager A10 equipped with a Zeiss EC Plan-NEOFLUAR 40X/1.3 Oil DIC/∞/0.17 and Zeiss EC Plan-NEOFLUAR 100X/1.3 Oil DIC/∞/0.17 objective and a series 120 X-Cite® light source (EXFO). GFP, Hoescht, and FM4-64 were imaged using a Zeiss filter set 10, Zeiss filter set 49, and Zeiss filter set 00 respectively. Microscope settings were kept identical for all images. For imaging of spores, A. fumigatus strains were cultured at 37°C for four days, harvested in 0.1% tween 20, and subjected to the same imaging procedure as described above. For imaging of protoplasts, A. fumigatus strains were protoplasted as described previously (49, 50) and imaged as described above. Briefly, overnight grown fungal culture is subjected to treatment using VinoTaste® Pro (Novozymes) and lysing enzymes from Trichoderma harzianum (#L1412; Sigma Aldrich) to remove the cell wall. For brefeldin A and cytochalasin A treatment, liquid GMM containing wildtype, OEFmqD::GFP and OEFmqD[Δ1-21]::GFP spores were inoculated into coverslip filled petri dishes and incubated overnight at 30°C. Coverslips were removed and treated with 100 μg/ml Brefeldin A (BFA) and 80 μg/ml cytochalasin A (CA) along with a solvent carrier control (methanol and DMSO respectively). Coverslips were removed at two and four hours post BFA treatment and two hours post CA treatment, mounted onto pre-cleaned microscope slide in mounting solution, and imaged as described above.
Culture conditions and protein preparation for FmqD secretion
Growth and sample preparation from mycelium and culture filtrates
To determine if FmqD is secreted into the growth medium, A. fumigatus WT, OEH2A::GFP, and OEFmqD::GFP strains were inoculated into 50 mL of liquid GMM at 1 × 106 conidia/mL and cultured at 25°C and 250 r.p.m. Both mycelia and culture filtrate were collected at 48 and 72 hours post inoculation and separated via filtration through Miracloth. Mycelia were rinsed twice with distilled water, flash frozen in liquid nitrogen, and stored at −80°C until ready to be used. The culture filtrates were immediately centrifuged at 4°C and 4000 r.p.m. for 10 minutes to remove any remaining mycelia and filtered through 0.8 μM membrane filters (Corning), lyophilized, and stored at −80°C until ready to be used.
Preparation of non-covalently bound proteins from cell wall
To prepare non-covalent cell wall extract (NC-CWE), approximately equal amounts (wet weight) of mycelia were ground in liquid nitrogen to a fine powder using pestle and mortar. Ground mycelia were immediately reconstituted in buffer B (100 mM Tris/HCl, (pH 7.5), 300 mM NaCl, 10% glycerol, 0.1% NP-40, 1 mM EDTA, 1 mM PMSF, 1 tablet in 50 mLs of cOmplete Protease Inhibitor Cocktail (Roche)) and centrifuged at 5500 r.p.m. for 10 minutes. Supernatant containing soluble cytoplasmic proteins were carefully removed. The cell wall pellet were reconstituted in 200 mM Tris/HCl (pH 7.8) buffer containing 20 mM EDTA and 1 mM PMSF and centrifuged at 200–300 r.p.m. for 2 minutes. Upon centrifugation, the upper gradient containing hyphal ghosts was carefully separated from the lower gradient containing larger fragments of intact hyphae. This step was repeated multiple times to remove most of the intact hyphae and the sample inspected under the microscope to show mainly hyphal ghosts. The cell wall fractions were combined, centrifuged at 5500 r.p.m. for 10 minutes, and the cell wall pellets were washed 6–8 times in 200 mM Tris/HCl (pH 7.8) buffer containing 20 mM EDTA and 1 mM PMSF followed by three washes in 1 M NaCl. Non-covalently bound proteins were extracted from the cell wall by boiling the cell wall pellets twice in 50 mM Tris/HCl (pH7.4) buffer containing 50 mM EDTA, 2% SDS, and 40 mM β-mercaptoethanol for 10 minutes (25). The boiled samples were centrifuged and the supernatant containing NC-CWE were combined and stored at −80°C until ready to be used.
Protein analysis
The lyophilized culture filtrates were solubilized in Buffer B and precipitated using 20% tricholoroacetic acid (TCA). Briefly, one volume of 100% TCA was added to four volumes of the solubilized culture filtrates, incubated on ice for an hour, and centrifuged to pellet the precipitated proteins. The pellets were washed twice in pre-chilled acetone (w/v) and reconstituted in 2X LDS buffer (0.5 M Tris/HCl (pH 8.5), 20% glycerol, 4% LDS, 1 mM EDTA, 0.44 mM Coomassie, 5% β-mercaptoethanol) and stored at −20°C until ready for use. The solubilized NC-CWE materials were precipitated with 4 volumes of ethanol, reconstituted in 2X LDS buffer, and stored at −20°C until ready for use. For gel electrophoresis, the samples were heated at 95°C for 5 minutes prior to loading and resolved using a 10% Bis Tris gel in MOPS running buffer. Proteins were visualized using Coomassie blue staining.
Western blot analysis
The samples along with a 10 ng of purified C-terminal epitope-tagged GFP (55) were subjected to gel electrophoresis as described above and transferred onto a PVDF membrane (Millipore) at 15V for an hour on a Trans-Blot® SD Semi-Dry transfer cell (Bio-Rad) using standard manufacturer’s protocol. The membranes were then visualized using Ponceau red staining for successful transfer and blocked for 2 hours at room temperature in TBS-T containing 5% non-fat milk. The membranes were then incubated in 1:2000 (v/v) primary monoclonal GFP antibody (NeoClone) overnight at 4°C in TBS-T containing 0.1% non-fat milk and washed four times for 15 minutes each in TBS-T. The membranes were then incubated in 1:5000 (v/v) secondary HRP-conjugated polyclonal goat anti-mouse antibody (BioLegend) in TBS-T containing 0.1% non-fat milk for 50 minutes at room temperature and washed in TBS-T as described above. Samples were then incubated in Clarity™ Western ECL substrate (Bio-Rad) for 5 minutes following manufacturer’s protocol and subjected to 2 minutes film exposure.
Supplementary Material
Acknowledgments
This research was funded by GM49338 to C.T.W. by Award F32GM090475 from the National Institute of General Medical Sciences to B.D.A and by NIH 1 R01 Al065728-01 and NIH grant GM084077 to N.P.K. The authors would like to thank Dr. Jae-Hyuk Yu at University of Wisconsin-Madison for kindly providing the conidiophore development mutants in A. fumigatus. The authors would also like to thank Drs. Richard R. Burgess and Nancy E. Thompson at the University of Wisconsin-Madison for kindly providing the purified epitope-tagged GFP for Western analysis.
References
- 1.Coyle CM, Kenaley SC, Rittenour WR, Panaccione DG. Association of ergot alkaloids with conidiation in Aspergillus fumigatus. Mycologia. 2007;99(6):804–811. doi: 10.3852/mycologia.99.6.804. [DOI] [PubMed] [Google Scholar]
- 2.Berthier E, et al. Low-Volume Toolbox for the Discovery of Immunosuppressive Fungal Secondary Metabolites. PLoS Path. 2013;9(4):e1003289. doi: 10.1371/journal.ppat.1003289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartoszewska M, Opalinski L, Veenhuis M, van der Klei IJ. The significance of peroxisomes in secondary metabolite biosynthesis in filamentous fungi. Biotechnol lett. 2011;33(10):1921–1931. doi: 10.1007/s10529-011-0664-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maggio-Hall LA, Wilson RA, Keller NP. Fundamental contribution of beta-oxidation to polyketide mycotoxin production in planta. Mol plant microbe interact. 2005;18(8):783–793. doi: 10.1094/MPMI-18-0783. [DOI] [PubMed] [Google Scholar]
- 5.Saikia S, Scott B. Functional analysis and subcellular localization of two geranylgeranyl diphosphate synthases from Penicillium paxilli. Mol genet genomics. 2009;282(3):257–271. doi: 10.1007/s00438-009-0463-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Imazaki A, et al. Contribution of peroxisomes to secondary metabolism and pathogenicity in the fungal plant pathogen Alternaria alternata. Eukaryot cell. 2010;9(5):682–694. doi: 10.1128/EC.00369-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chanda A, et al. A key role for vesicles in fungal secondary metabolism. Proc Natl Acad Sci. 2009;106(46):19533–19538. doi: 10.1073/pnas.0907416106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roze LV, Hong SY, Linz JE. Aflatoxin Biosynthesis: Current Frontiers. Annu rev food sci technol. 2012 doi: 10.1146/annurev-food-083012-123702. [DOI] [PubMed] [Google Scholar]
- 9.Chang PK, Yu J, Yu JH. aflT, a MFS transporter-encoding gene located in the aflatoxin gene cluster, does not have a significant role in aflatoxin secretion. Fungal genet and biol. 2004;41(10):911–920. doi: 10.1016/j.fgb.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 10.Gardiner DM, Jarvis RS, Howlett BJ. The ABC transporter gene in the sirodesmin biosynthetic gene cluster of Leptosphaeria maculans is not essential for sirodesmin production but facilitates self-protection. Fungal genet and biol. 2005;42(3):257–263. doi: 10.1016/j.fgb.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Lee S, Son H, Lee J, Lee YR, Lee YW. A putative ABC transporter gene, ZRA1, is required for zearalenone production in Gibberella zeae. Curr genet. 2011;57(5):343–351. doi: 10.1007/s00294-011-0352-4. [DOI] [PubMed] [Google Scholar]
- 12.Belofsky GN, Anguera M, Jensen PR, Fenical W, Kock M. Oxepinamides A-C and fumiquinazolines H--I: bioactive metabolites from a marine isolate of a fungus of the genus Acremonium. Chemistry. 2000;6(8):1355–1360. doi: 10.1002/(sici)1521-3765(20000417)6:8<1355::aid-chem1355>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi C, et al. Fumiquinazolines A-G, novel metabolites of a fungus separated from a Pseudolabrus marine fish. J Chem Soc, Perkin Trans. 1995;1(18):2345–2353. [Google Scholar]
- 14.Frisvad JC, Rank C, Nielsen KF, Larsen TO. Metabolomics of Aspergillus fumigatus. Med mycol. 2009;47(Suppl 1):53–71. doi: 10.1080/13693780802307720. [DOI] [PubMed] [Google Scholar]
- 15.Gauthier T, et al. Trypacidin, a spore-borne toxin from Aspergillus fumigatus, is cytotoxic to lung cells. PloS one. 2012;7(2):e29906. doi: 10.1371/journal.pone.0029906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ames BD, Walsh CT. Anthranilate-activating modules from fungal nonribosomal peptide assembly lines. Biochemistry. 2010;49(15):3351–3365. doi: 10.1021/bi100198y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ames BD, et al. Complexity generation in fungal peptidyl alkaloid biosynthesis: oxidation of fumiquinazoline A to the heptacyclic hemiaminal fumiquinazoline C by the flavoenzyme Af12070 from Aspergillus fumigatus. Biochemistry. 2011;50(40):8756–8769. doi: 10.1021/bi201302w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ames BD, Liu X, Walsh CT. Enzymatic processing of fumiquinazoline F: a tandem oxidative-acylation strategy for the generation of multicyclic scaffolds in fungal indole alkaloid biosynthesis. Biochemistry. 2010;49(39):8564–8576. doi: 10.1021/bi1012029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mah JH, Yu JH. Upstream and downstream regulation of asexual development in Aspergillus fumigatus. Eukaryot cell. 2006;5(10):1585–1595. doi: 10.1128/EC.00192-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen HM, Ford C, Reilly PJ. Substitution of asparagine residues in Aspergillus awamori glucoamylase by site-directed mutagenesis to eliminate N-glycosylation and inactivation by deamidation. Biochem J. 1994;301:275–281. doi: 10.1042/bj3010275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yanez E, Carmona TA, Tiemblo M, Jimenez A, Fernandez-Lobato M. Expression of the Schwanniomyces occidentalis SWA2 amylase in Saccharomyces cerevisiae: role of N-glycosylation on activity, stability and secretion. Biochem j. 1998;329:65–71. doi: 10.1042/bj3290065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin C. Protein Glycosylation in Aspergillus fumigatus Is Essential for Cell Wall Synthesis and Serves as a Promising Model of Multicellular Eukaryotic Development. Int j microbiol. 2012;2012:654251. doi: 10.1155/2012/654251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khalaj V, Brookman JL, Robson GD. A study of the protein secretory pathway of Aspergillus niger using a glucoamylase-GFP fusion protein. Fungal Genet Biol. 2001;32(1):55–65. doi: 10.1006/fgbi.2000.1245. [DOI] [PubMed] [Google Scholar]
- 24.Latge JP, et al. Specific molecular features in the organization and biosynthesis of the cell wall of Aspergillus fumigatus. Med Mycol. 2005;43(Suppl 1):S15–22. doi: 10.1080/13693780400029155. [DOI] [PubMed] [Google Scholar]
- 25.Bernard M, et al. Characterization of a cell-wall acid phosphatase (PhoAp) in Aspergillus fumigatus. Microbiology. 2002;148(Pt 9):2819–2829. doi: 10.1099/00221287-148-9-2819. [DOI] [PubMed] [Google Scholar]
- 26.Penalva MA. Tracing the endocytic pathway of Aspergillus nidulans with FM4-64. Fungal Genet Biol. 2005;42(12):963–975. doi: 10.1016/j.fgb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 27.Liu JF, et al. Three-component one-pot total syntheses of glyantrypine, fumiquinazoline F, and fiscalin B promoted by microwave irradiation. J org chem. 2005;70(16):6339–6345. doi: 10.1021/jo0508043. [DOI] [PubMed] [Google Scholar]
- 28.Brakhage AA, et al. Aspects on evolution of fungal beta-lactam biosynthesis gene clusters and recruitment of trans-acting factors. Phytochemistry. 2009;70(15–16):1801–1811. doi: 10.1016/j.phytochem.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 29.Yin WB, et al. An Aspergillus nidulans bZIP response pathway hardwired for defensive secondary metabolism operates through aflR. Mol microbiol. 2012;83(5):1024–1034. doi: 10.1111/j.1365-2958.2012.07986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forseth RR, et al. Homologous NRPS-like gene clusters mediate redundant small-molecule biosynthesis in Aspergillus flavus. Angewandte Chemie. 2013;52(5):1590–1594. doi: 10.1002/anie.201207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lim FY, et al. Genome-based cluster deletion reveals an endocrocin biosynthetic pathway in Aspergillus fumigatus. Appl Environ Microbiol. 2012;78(12):4117–4125. doi: 10.1128/AEM.07710-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Twumasi-Boateng K, et al. Transcriptional profiling identifies a role for BrlA in the response to nitrogen depletion and for StuA in the regulation of secondary metabolite clusters in Aspergillus fumigatus. Eukaryot Cell. 2009;8(1):104–115. doi: 10.1128/EC.00265-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwon NJ, Shin KS, Yu JH. Characterization of the developmental regulator FlbE in Aspergillus fumigatus and Aspergillus nidulans. Fungal Genet Biol. 2010;47(12):981–993. doi: 10.1016/j.fgb.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 34.Tao L, Yu JH. AbaA and WetA govern distinct stages of Aspergillus fumigatus development. Microbiology. 2011;157(Pt 2):313–326. doi: 10.1099/mic.0.044271-0. [DOI] [PubMed] [Google Scholar]
- 35.Perrin R, et al. Transcriptional regulation of chemical diversity in Aspergillus fumigatus by LaeA. PLoS Pathog. 2007;3(4):508–517. doi: 10.1371/journal.ppat.0030050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meijer WH, et al. Peroxisomes are required for efficient penicillin biosynthesis in Penicillium chrysogenum. Appl Environ Microbiol. 2010;76(17):5702–5709. doi: 10.1128/AEM.02327-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saloheimo M, Pakula TM. The cargo and the transport system: secreted proteins and protein secretion in Trichoderma reesei (Hypocrea jecorina) Microbiology. 2012;158(1):46–57. doi: 10.1099/mic.0.053132-0. [DOI] [PubMed] [Google Scholar]
- 38.Fleissner A, Dersch P. Expression and export: recombinant protein production systems for Aspergillus. Appl microbiol biotechnol. 2010;87(4):1255–1270. doi: 10.1007/s00253-010-2672-6. [DOI] [PubMed] [Google Scholar]
- 39.Kotz A, et al. Approaching the secrets of N-glycosylation in Aspergillus fumigatus: characterization of the AfOch1 protein. PloS one. 2010;5(12):e15729. doi: 10.1371/journal.pone.0015729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li K, et al. Repression of N-glycosylation triggers the unfolded protein response (UPR) and overexpression of cell wall protein and chitin in Aspergillus fumigatus. Microbiology. 2011;157(Pt 7):1968–1979. doi: 10.1099/mic.0.047712-0. [DOI] [PubMed] [Google Scholar]
- 41.Zhang L, et al. Comparative proteomic analysis of an Aspergillus fumigatus mutant deficient in glucosidase I (AfCwh41) Microbiology. 2009;155(7):2157–2167. doi: 10.1099/mic.0.027490-0. [DOI] [PubMed] [Google Scholar]
- 42.Zhang L, Zhou H, Ouyang H, Li Y, Jin C. Afcwh41 is required for cell wall synthesis, conidiation, and polarity in Aspergillus fumigatus. FEMS microbiol lett. 2008;289(2):155–165. doi: 10.1111/j.1574-6968.2008.01376.x. [DOI] [PubMed] [Google Scholar]
- 43.Yin WB, et al. A nonribosomal peptide synthetase-derived iron(III) complex from the pathogenic fungus Aspergillus fumigatus. J Am Chem Soc. 2013;135(6):2064–2067. doi: 10.1021/ja311145n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Hanlon KA, et al. Nonribosomal peptide synthetase genes pesL and pes1 are essential for Fumigaclavine C production in Aspergillus fumigatus. Appl Environ Microbiol. 2012;78(9):3166–3176. doi: 10.1128/AEM.07249-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Afiyatullov S, et al. New metabolites from the marine-derived fungus Aspergillus fumigatus. Nat prod commun. 2012;7(4):497–500. [PubMed] [Google Scholar]
- 46.Gao X, et al. Fungal indole alkaloid biosynthesis: genetic and biochemical investigation of the tryptoquialanine pathway in Penicillium aethiopicum. J am chem soc. 2011;133(8):2729–2741. doi: 10.1021/ja1101085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schrettl M, et al. Self-Protection against Gliotoxin-A Component of the Gliotoxin Biosynthetic Cluster, GliT, Completely Protects Aspergillus fumigatus Against Exogenous Gliotoxin. Plos Pathog. 2010;6(6) doi: 10.1371/journal.ppat.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shimizu K, Keller NP. Genetic involvement of a cAMP-dependent protein kinase in a G protein signaling pathway regulating morphological and chemical transitions in Aspergillus nidulans. Genetics. 2001;157(2):591–600. doi: 10.1093/genetics/157.2.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szewczyk E, et al. Fusion PCR and gene targeting in Aspergillus nidulans. Nat Protoc. 2006;1(6):3111–3120. doi: 10.1038/nprot.2006.405. [DOI] [PubMed] [Google Scholar]
- 50.Lim FY, Sanchez JF, Wang CC, Keller NP. Toward awakening cryptic secondary metabolite gene clusters in filamentous fungi. Methods Enzymol. 2012;517:303–324. doi: 10.1016/B978-0-12-404634-4.00015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Calvo AM, Bok J, Brooks W, Keller NP. veA is required for toxin and sclerotial production in Aspergillus parasiticus. Appl and Environ Microbiol. 2004;70(8):4733–4739. doi: 10.1128/AEM.70.8.4733-4739.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang L, et al. Rapid production of gene replacement constructs and generation of a green fluorescent protein-tagged centromeric marker in Aspergillus nidulans. Eukaryot cell. 2004;3(5):1359–1362. doi: 10.1128/EC.3.5.1359-1362.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yin WB, et al. Discovery of Cryptic Polyketide Metabolites from Dermatophytes Using Heterologous Expression in Aspergillus nidulans. ACS synthetic biology. 2013 doi: 10.1021/sb400048b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sambrook J, Russell DW. Molecular cloning: a laboratory manual. Cold Spring Harb Laborat Press; Cold Spring Harbor, N.Y: 2001. [Google Scholar]
- 55.Stalder ES, et al. The epitope for the polyol-responsive monoclonal antibody 8RB13 is in the flap-domain of the beta-subunit of bacterial RNA polymerase and can be used as an epitope tag for immunoaffinity chromatography. Protein Expr Purif. 2011;77(1):26–33. doi: 10.1016/j.pep.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xue T, Nguyen CK, Romans A, Kontoyiannis DP, May GS. Isogenic auxotrophic mutant strains in the Aspergillus fumigatus genome reference strain AF293. Arch microbiol. 2004;182(5):346–353. doi: 10.1007/s00203-004-0707-z. [DOI] [PubMed] [Google Scholar]
- 57.da Silva Ferreira ME, et al. The akuB(KU80) mutant deficient for nonhomologous end joining is a powerful tool for analyzing pathogenicity in Aspergillus fumigatus. Eukaryot cell. 2006;5(1):207–211. doi: 10.1128/EC.5.1.207-211.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.