

Abstract
Accumulating evidence points to a role for Janus kinase/signal transducers and activators of transcription (STAT) immune signaling in neuronal function; however, its role in experience-dependent plasticity is unknown. Here we show that one of its components, STAT1, negatively regulates the homeostatic component of ocular dominance plasticity in visual cortex. After brief monocular deprivation (MD), STAT1 knock-out (KO) mice show an accelerated increase of open-eye responses, to a level comparable with open-eye responses after a longer duration of MD in wild-type (WT) mice. Therefore, this component of plasticity is abnormally enhanced in KO mice. Conversely, increasing STAT1 signaling by IFNγ treatment in WT mice reduces the homeostatic component of plasticity by impairing open-eye responses. Enhanced plasticity in KO mice is accompanied by sustained surface levels of GluA1 AMPA receptors and increased amplitude and frequency of AMPA receptor-mediated mEPSCs, which resemble changes in WT mice after a longer duration of MD. These results demonstrate a unique role for STAT1 during visual cortical plasticity in vivo through a mechanism that includes AMPA receptors.
Keywords: AMPA receptors, homeostatic, IFNγ, ocular dominance plasticity, STAT1, visual cortex
Introduction
There is growing evidence that classical immune molecules contribute to neuronal function, although the function of these molecules in the developing brain, particularly in modulating experience-dependent synaptic plasticity, is not well understood. Ocular dominance (OD) plasticity in primary visual cortex (V1) induced by visual monocular deprivation (MD) is a well established model of experience-dependent plasticity (Hubel and Wiesel, 1970; Gordon and Stryker, 1996; Antonini et al., 1999). OD plasticity in mouse V1 is composed of two components: (1) an initial decrease of responses from the deprived eye that occurs after a short period (3–4 d) of MD, followed by (2) an increase of responses from the open eye after a longer period (5–7 d) of MD (Frenkel and Bear, 2004; Kaneko et al., 2008; Sato and Stryker, 2008; McCurry et al., 2010). The latter component of plasticity is noteworthy; it involves a change in eye-specific cortical drive despite no change in visual drive and is thus an internally driven “homeostatic” response of cortical synapses and circuits to visual deprivation (Turrigiano and Nelson, 2004; Keck et al., 2013).
A previous mRNA microarray screen showed that a component of immune signaling cascades, Janus kinase (JAK) and signal transducers and activators of transcription (STAT) signaling, was regulated in V1 after long-term MD during the critical period (Tropea et al., 2006). JAK/STAT signaling is initiated by a cytokine binding to its corresponding receptor, which then activates the JAK family of kinases. The JAKs (JAK1–JAK3 and tyrosine kinase TYK2) in turn mediate phosphorylation of STATs (STAT1–STAT4, STAT5a, STAT5b, and STAT6), which then translocate to the nucleus to induce transcription of immune-related genes, including STAT genes themselves (Darnell et al., 1994; Kisseleva et al., 2002). Here, we investigated the role of STAT1 in experience-dependent cortical plasticity in mouse V1 and found a novel role and mechanism for STAT1 in regulating the homeostatic component of OD plasticity.
Materials and Methods
Animals.
STAT1 knock-out (KO) mice on a 129/SvEv background were purchased (Taconic) when they were 3 weeks old and were maintained in their home cage until experiments. Age- and strain-matched 129/SvEv mice (Taconic) were used as wild-type (WT) controls. Male mice were used throughout the study, except for protein measurements during development (Fig. 1A) in which both males and females were used. We found no difference in two WT strains, C57BL/6 (The Jackson Laboratory or Taconic) and 129/SvEv, in STAT1 protein measurements; we thus used data from both in preparing Fig. 1A.
Figure 1.
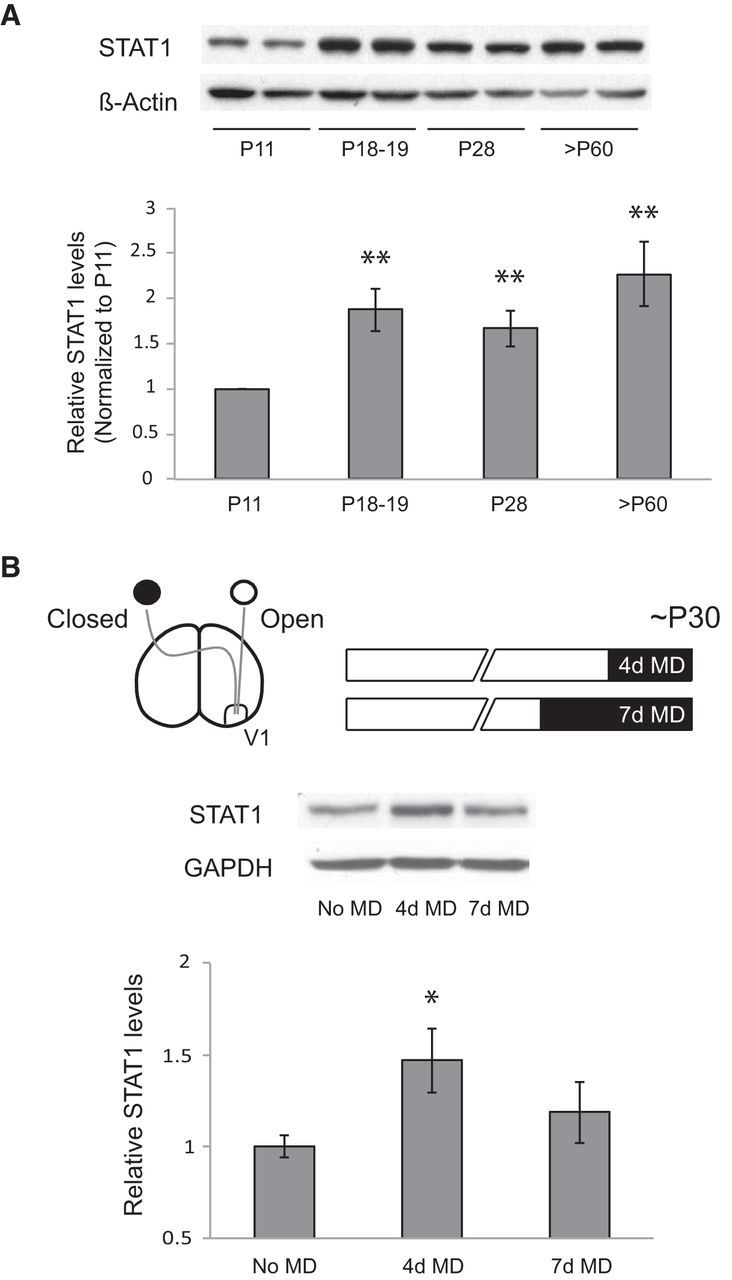
Developmental and deprivation-induced changes in STAT1 expression in visual cortex. A, STAT1 protein levels at various postnatal ages (n = 8 animals for each age; **p < 0.01 vs P11). β-Actin was used as a loading control, and the values were normalized to P11. B, Schematic drawing of an experimental paradigm for 4 and 7 d MD during the critical period (top). STAT1 protein levels after 4 or 7 d MD (n = 7–9 animals for each condition; *p < 0.05 vs no MD). GAPDH was used as a loading control, and the values were normalized to no MD. Averaged data are presented as mean ± SEM.
MD.
For MD by eyelid suture, animals [aged approximately postnatal day 23 (P23) to P26] were anesthetized with either avertin (0.016 ml/g, i.p.) or isoflurane (2–4%), and the eyelid margins were trimmed. Upper and lower lids were sutured closed, and the eyelids were regularly examined to ensure that they remained closed for the duration of the experiment. MD lasted either 4 or 7–8 d. Before the optical imaging experiment, the suture was removed, and the deprived eye was reopened while the animal was under anesthesia. For experiments in Figure 3, IFNγ (830 U/g, or 2 μg, i.p.; Sigma-Aldrich) was injected once a day for the duration (7 d) of MD, based on the concentration that has been shown to penetrate the brain (Htain et al., 1997).
Figure 3.

IFNγ reduces OD plasticity in WT mice by impairing open-eye responses. A, ODI in WT mice during the critical period with no MD, 7 d MD, or 7 d MD with additional concurrent IFNγ treatment (no MD, 7 d MD, and 7 d MD plus IFNγ, n = 10, 4, and 4 animals, respectively). B, Response amplitude for the contralateral (Contra) and ipsilateral (Ipsi) eyes in WT mice. C, ODI in STAT1 KO mice with the same treatment as in A (no MD, 7 d MD, and 7 d MD plus IFNγ, n = 10, 4, and 3 animals, respectively). D, Response amplitude for the contralateral and ipsilateral eyes in STAT1 KO mice. In A–D, individual data points are plotted as circles, and asterisks above bars show statistical significance compared with no MD in each group. Averaged data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Anterograde labeling of retinal ganglion axons.
Mice during the critical period were anesthetized with isoflurane, and 2 μl of cholera toxin subunit B (CTB) conjugated to Alexa Fluor 488 was injected into the ipsilateral eye and Alexa Fluor 594 into the contralateral eye (1 mg/ml; Invitrogen). After 6 d, animals were perfused, the brains were removed and postfixed overnight at 4°C, and 40 μm coronal sections were cut with a vibratome (VT1200S; Leica). Sections were mounted on glass slides and coverslipped for imaging on a Zeiss LSM 510 confocal microscope.
Intrinsic signal optical imaging.
Mice were anesthetized with urethane (1.5 mg/g, i.p.) and chlorprothixene (10 mg/kg, i.p.). The skin was excised and the skull was exposed over V1. A head plate was used to fix the head and minimize movements. The cortex was covered with agarose solution (1.5%) and a glass coverslip. Red light (630 nm) was used to illuminate the cortical surface, and the change of luminance was captured by a CCD camera (Cascade 512B; Roper Scientific) during the presentation of visual stimuli (custom MATLAB scripts). An elongated horizontal white bar (9° × 72°) over a uniformly gray background was drifted upward continuously through the peripheral–central dimension of the visual field. After moving to the last position, the bar would jump back to the initial position and start another cycle of movement; thus, the chosen region of visual space (72° × 72°) was stimulated in a periodic manner (12 s/cycle). Images of the visual cortex were continuously captured at the rate of 18 frames/s during each stimulus session of 22 min. A temporal high-pass filter (135 frames) was used to remove slow noise components, after which the temporal fast Fourier transform (FFT) component at the stimulus frequency (9 s−1) was calculated pixel by pixel from the entire set of images. The amplitude of the FFT component was used to measure the strength of visually driven response for each eye, and the OD index (ODI) was derived from the response of each eye (R) at each pixel as ODI = (Rcontra − Ripsi)/(Rcontra + Ripsi). The binocular zone was defined as the cortical region that is driven by both eyes. The response amplitude for each eye was defined as fractional changes in reflectance over baseline reflectance (ΔR/R × 10−3), and the top 50% pixels were analyzed to avoid background contamination.
Protein measurements.
V1 contralateral to the deprived eye was removed and snap-frozen with dry ice. The brains were homogenized in RIPA buffer (Invitrogen) containing proteinase and phosphatase inhibitors (Roche). For Western blot, equal amounts of protein were loaded on a precast gel and transferred to a PVDF membrane. The membrane was blotted with antibodies against STAT1α (ab2071; Abcam), β-actin (A1978; Sigma-Aldrich), GAPDH (ab9484; Abcam), GluA1 (04-855, clone C3T; Millipore), and pan-Cadherin (ab6529; Abcam).
Cell surface biotinylation assay.
The levels of cell surface GluA1 were measured using a biotinylation assay as described previously (Thomas-Crusells et al., 2003; McCurry et al., 2010). Briefly, mice were anesthetized using isoflurane, and the brain was rapidly dissected out. A vibratome was used to make 300 μm coronal sections containing V1. Slices were incubated in 100 μm S-NHS-SS-biotin (Sigma-Aldrich) for 45 min on ice. The superficial layers of V1 were dissected out and homogenized in RIPA buffer. The homogenate was centrifuged, and the supernatant was incubated with streptavidin beads (Thermo Fisher Scientific) overnight at 4°C. The beads were then processed for Western blotting.
Slice electrophysiology.
Mice were anesthetized with isoflurane. The brain was rapidly removed and sliced coronally at a thickness of 300 μm with a vibratome in slicing buffer (in mm: 130 choline chloride, 25 glucose, 1.25 NaH2PO4, 26 NaCHO3, 2.5 KCl, 7 MgCl2, and 0.5 CaCl2) bubbled with 95% O2 and 5% CO2. Slices were given a minimum of 60 min of incubation in room-temperature ACSF (in mm: 130 NaCl, 10 glucose, 1.25 NaH2PO4, 24 NaCHO3, 3.5 KCl, 2.5 CaCl2, and 1.5 MgCl2) before patching. For recording of AMPA receptor (AMPAR)-mediated mEPSCs, whole-cell patch clamp of layer II/III pyramidal neurons in the binocular region of V1 was performed using pipettes (4–7 MΩ resistance) filled with an internal solution [in mm: 100 K-gluconate, 20 KCl, 0.5 EGTA, 10 NaCl, 10 Na-phosphocreatine, 4 Mg-ATP, 0.3 Na-GTP, and 10 HEPES, pH 7.2–7.3 (with 1 m KOH)]. Neurons were recorded at room temperature (25°C) in ACSF containing 1 μm TTX, 50 μm AP-5, and 50 μm picrotoxin to isolate AMPAR-mediated currents and voltage clamped at a membrane potential of −70 mV. mEPSCs were recorded using a Multiclamp 700B amplifier (Molecular Devices) at 10 kHz, filtered at 2 kHz, and analyzed with Clampfit 10.2 software (Molecular Devices). Whole-cell membrane currents were recorded for 10 min. For detection of mEPSCs, a detection template for each cell was constructed from four to six events intrinsic to each recording. Traces were analyzed in template search mode in Clampfit 10.2, with a template match threshold of 4–4.5 to reduce false positives. All events were detected automatically and edited after detection by eye to remove events that were erroneous matches or duplicate events. All mEPSC events were included in the analysis of event parameters.
Statistical analyses.
A two-tailed unpaired Student's t test was used for comparisons between two means. For comparing more than two means (Figs. 2, 3), a one-way or two-way ANOVA was used, followed by post hoc pairwise comparisons with either Tukey's test (comparing three means within a group) or Bonferroni's correction (comparing two means between groups). Statistics for cumulative probability distributions were performed using the Kolmogorov–Smirnov (K–S) test. All averaged data are presented as mean ± SEM.
Figure 2.
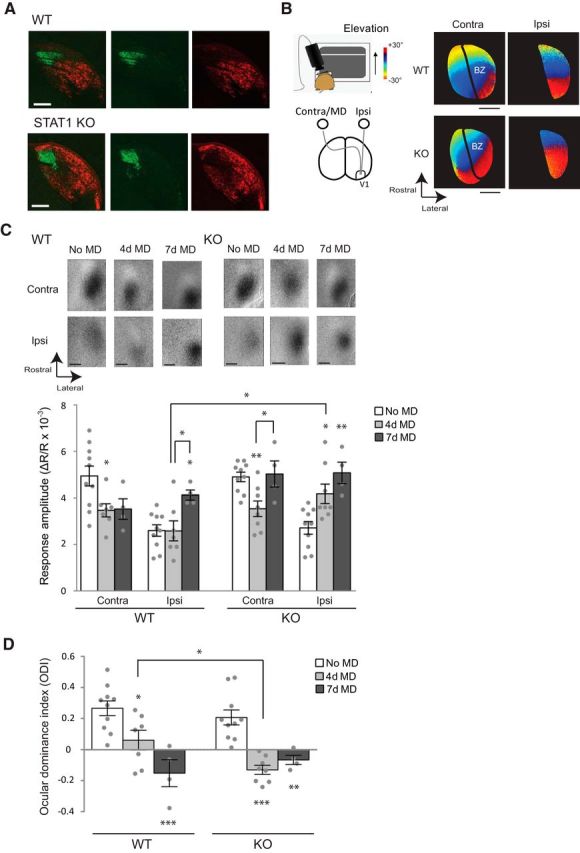
STAT1 KO mice show enhanced OD plasticity though an accelerated increase in open-eye responses. A, Projections from the retina to the dLGN after CTB injections into each eye (red, contralateral; green, ipsilateral) in WT and STAT1 KO mice. Scale bars, 200 μm. B, Schematic drawing of optical imaging (left) and images of retinotopic maps (right) in V1 in response to stimulation of the contralateral (Contra) and ipsilateral (Ipsi) eyes. BZ, Binocular zone. Scale bars, 500 μm. C, Response amplitude maps in V1 (top) and their quantification (bottom) for the contralateral and ipsilateral eyes after 4 or 7 d MD of the contralateral eye during the critical period (WT no MD, 4 d MD, and 7 d MD, n = 10, 7, and 4 animals, respectively; STAT1 no MD, 4 d MD, and 7 d MD, n = 10, 8, and 4 animals, respectively). Scale bars, 500 μm. D, ODI values in WT and STAT1 KO mice. In C and D, individual data points are plotted as circles, and asterisks above bars show statistical significance compared with no MD in each group. Averaged data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Developmental and deprivation-induced changes in STAT1 expression in visual cortex
We measured developmental changes in the expression of STAT1 protein extracted from V1. The levels of STAT1 increased after eye opening (P18–P19) and remained high during the peak of the critical period for OD plasticity (P28) through adulthood (older than P60) compared with the levels before eye opening (P11; t test, p < 0.01 vs P11; Fig. 1A), suggesting that expression of STAT1 is developmentally regulated and visual experience may drive its expression. To determine whether STAT1 is regulated by visual experience, we examined STAT1 expression after MD. During the peak of the critical period, the levels of STAT1 were significantly increased after 4 d MD (t test, p < 0.05 vs no MD) and returned to a level close to baseline after 7 d MD (Fig. 1B). These results demonstrate that STAT1 expression is rapidly altered after short-term MD. Increased expression of STAT1 after 4 d MD suggests a permissive or regulatory role for STAT1 in OD plasticity.
STAT1 KO mice show enhanced OD plasticity through an accelerated increase in open-eye responses
To determine the role of STAT1 in OD plasticity, we investigated STAT1 KO mice using optical imaging of intrinsic signals. These mice are known to have normal development, except for susceptibility to infection (Meraz et al., 1996). STAT1 KO mice showed normal retina to dLGN projections with little overlap between the contralateral and ipsilateral eye projections (Fig. 2A); the mice also had normal retinotopic maps and binocular organization in V1 (Fig. 2B). We measured eye-specific responses after suturing the contralateral eye for either 4 or 7 d during the critical period. In WT mice, 4 d MD led to a significant decrease in contralateral (closed) eye responses (one-way ANOVA, F(2,18) = 4.421, p = 0.0274, Tukey's test, p < 0.05 vs no MD) without affecting ipsilateral (open) eye responses, whereas 7 d MD led to a significantly increased response from the open eye (one-way ANOVA, F(2,18) = 4.937, p = 0.0195, Tukey's test, p < 0.05 vs no MD and vs 4 d MD; Fig. 2C); this was consistent with previous findings (Frenkel and Bear, 2004; Kaneko et al., 2008; Sato and Stryker, 2008; McCurry et al., 2010). In STAT1 KO mice, a decrease of closed-eye responses occurred as in WT mice after 4 d MD (one-way ANOVA, F(2,19) = 6.859, p = 0.0057, Tukey's test, p < 0.01 vs no MD; Fig. 2C); however, a significant increase of open-eye responses was already observed after 4 d MD (one-way ANOVA, F(2,19) = 9.608, p = 0.0013, Tukey's test, p < 0.05 vs no MD). Both WT and KO mice showed a significant shift in the ODI toward the open eye after 4 and 7 d MD: a two-way ANOVA yielded a main effect for the duration of MD (F(2,37) = 24.71, p < 0.0001; simple effects within WT, Tukey's test, p < 0.05 for no MD vs 4 d MD, p < 0.001 for no MD vs 7 d MD; simple effects within KO, Tukey's test, p < 0.001 for no MD vs 4 d MD, p < 0.01 for no MD vs 7 d MD; Fig. 2D). There was a significant difference between ODIs for WT and KO mice after 4 d MD (p < 0.05 between 4 d MD ODIs in WT and KO with Bonferroni correction; Fig. 2D) due to the increased open-eye responses in KO mice (t test, p < 0.05 between 4 d MD open-eye responses in WT and KO; Fig. 2C). Interestingly, in KO mice after 7 d MD, closed-eye responses were strengthened compared with 4 d MD to a level comparable with control (Tukey's test, p < 0.05 vs 4 d MD), suggesting that STAT1 KO mice have strong enough homeostatic plasticity to bring closed-eye responses back to pre-MD levels.
IFNγ reduces OD plasticity in WT mice by impairing open-eye responses
We next examined the effect of increasing STAT1 signaling by applying interferon gamma (IFNγ), a specific activator of STAT1 signaling (Meraz et al., 1996; Ramana et al., 2002), in WT mice and observing the effect on the homeostatic component of plasticity after 7 d MD. The shift in ODI observed after 7 d MD was blocked by daily IFNγ injection for 7 d (one-way ANOVA, F(2,15) = 11.83, p = 0.0008, Tukey's test, p < 0.0001 for no MD vs 7 d MD, p < 0.05 for 7 d MD vs 7 d MD plus IFNγ; Fig. 3A). The IFNγ treatment impaired open-eye responses observed after MD (one-way ANOVA, F(2,15) = 19.47, p < 0.0001, Tukey's test, p < 0.001 for 7 d MD vs 7 d MD plus IFNγ) without affecting closed-eye responses (Fig. 3B). Interestingly, IFNγ treatment decreased open-eye responses compared with the control condition (Tukey's test, p < 0.01 vs no MD), suggesting that IFNγ has an inhibitory effect on the basal response. The effect of IFNγ was likely to be mediated through STAT1 because the same treatment did not block the ODI shift observed in STAT1 KO mice (one-way ANOVA, F(2,14) = 8.082, p = 0.0046, Tukey's test, p < 0.01 for no MD vs 7 d MD, p < 0.05 for no MD vs 7 d MD plus IFNγ; Fig. 3C), nor did it affect the response amplitudes for the closed and open eyes in these mice (Fig. 3D). These results thus demonstrate that activating STAT1 through IFNγ blocks the strengthening of open-eye responses, consistent with the proposal that STAT1 negatively regulates the homeostatic component of OD plasticity.
STAT1 KO mice show enhanced surface expression and synaptic function of AMPARs after 4 d MD
We next sought to examine the mechanism underlying STAT1 regulation of the homeostatic component of OD plasticity. AMPARs are of particular interest because of their known roles in homeostatic plasticity (Turrigiano and Nelson, 2004). AMPARs regulate plasticity via surface insertion and internalization, i.e., trafficking (Anggono and Huganir, 2012). In particular, GluA1 subunits mediate deprived-eye depression through increased internalization (McCurry et al., 2010). Conversely, it is possible that GluA1 surface insertion mediates open-eye potentiation. We thus measured surface levels of GluA1 to determine whether changes in surface GluA1 expression levels could explain enhanced plasticity in STAT1 KO mice. There was no difference in surface GluA1 levels in the basal, non-deprived condition between WT and STAT1 KO mice. As expected, WT mice showed reduced levels of surface GluA1 after 4 d MD (t test, p < 0.01 vs no MD; Fig. 4A), consistent with increased GluA1 internalization (McCurry et al., 2010). After 7 d MD, levels of GluA1 returned to control levels, consistent with the proposal that GluA1 surface insertion mediates open-eye potentiation. However, in KO mice, there was no overall change in surface GluA1 after either 4 or 7 d MD, resulting in significantly more surface GluA1 after 4 d MD compared with WT mice (t test, p < 0.05 vs WT 4 d MD; Fig. 4A). Sustained surface levels of GluA1 may lead to enhanced synaptic AMPAR function in KO mice, which may underlie enhanced open-eye potentiation and plasticity in these mice.
Figure 4.
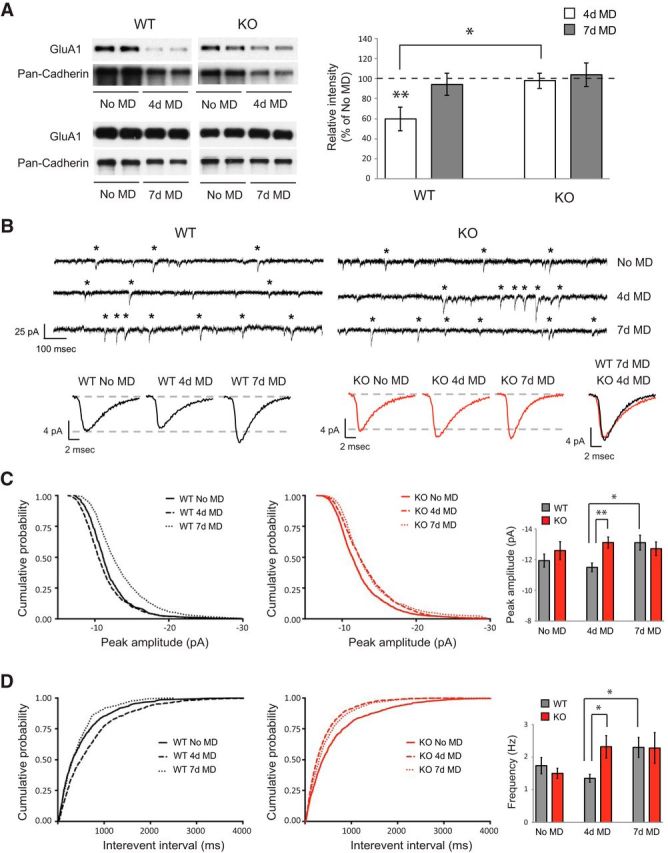
STAT1 KO mice show enhanced surface expression and synaptic function of AMPARs after 4 d MD. A, Cell surface protein levels of GluA1 (n = 4–5 animals for each condition). Pan-Cadherin was used as a loading control for surface proteins, and the values were normalized to the no MD condition of each genotype. B, Top, Representative traces of AMPAR-mediated mEPSCs. Each asterisk shows an individual mEPSC. Bottom, Average mEPSC waveforms from representative neurons in all conditions with an overlay of WT 7 d MD and KO 4 d MD at the far right. C, Left and middle, Cumulative probabilities of mEPSC peak amplitude. Right, Average peak amplitude for mEPSCs. D, Left and middle, Cumulative probabilities of mEPSC interevent interval. Right, Average frequency for mEPSCs. WT no MD, 4 d MD, and 7 d MD, n = 12 (4), 8 (4), and 15 (6) cells (animals), respectively; STAT1 no MD, 4 d MD, and 7 d MD, n = 11 (3), 9 (4), and 6 (3) cells (animals), respectively. In A, asterisks above a bar show statistical significance compared with WT no MD. Averaged data are presented as mean ± SEM. *p < 0.05, **p < 0.01.
To examine this, we recorded AMPAR-mediated miniature excitatory postsynaptic currents (mEPSCs) from layer 2/3 pyramidal neurons in the binocular region of V1. WT and STAT1 KO mice showed no significant difference in the mean values of peak amplitude nor frequency at baseline (no MD). However, after 4 d MD, mEPSCs recorded from KO mice showed significant increases for both peak amplitude and frequency compared with WT mice (t test, p < 0.01 for peak amplitude vs WT 4 d MD, p < 0.05 for frequency vs WT 4 d MD; Fig. 4C,D). After 7 d MD, both mEPSC amplitude and frequency of WT neurons significantly increased compared with 4 d MD (t test < 0.05 for both peak amplitude and frequency). There was no increase in either parameter in KO neurons, indicating no additional alterations of synaptic function in KO mice after 7 d MD. The mean values and the mEPSC waveforms of KO mice after 4 d MD were similar to those of WT mice after 7 d MD (Fig. 4B–D), indicating that KO mice show enhanced AMPAR-mediated synaptic function after 4 d MD that is evident in WT mice only after 7 d MD. Rise time and decay tau kinetic parameters of mEPSCs were not significantly different between WT and KO mice comparing these (or any) conditions (Table 1). Cumulative probability distributions for mEPSC peak amplitude show that 4 d MD significantly shifted the population toward smaller amplitudes and 7 d MD toward larger amplitudes in WT mice (K–S test, p < 0.0001 for no MD vs 4 d MD and for no MD vs 7 d MD; Fig. 4C). Similarly, cumulative probability distributions for interevent interval showed a shift toward larger intervals after 4 d MD but toward smaller intervals after 7 d MD (K–S test, p < 0.0001 for no MD vs 4 d MD, p < 0.05 for no MD vs 7 d MD; Fig. 4D). In contrast, KO mice displayed a significant shift toward larger amplitudes and smaller interevent intervals after 4 d MD (K–S test, p < 0.0001 for no MD vs 4 d MD and for no MD vs 7 d MD for both peak amplitude and interevent interval; Fig. 4C,D), bypassing the reductions in amplitude and frequency observed in WT mice after 4 d MD. Collectively, the analysis of mEPSCs is consistent with STAT1 KO mice exhibiting an accelerated time course of homeostatic plasticity in response to MD.
Table 1.
Rise time and decay kinetics of mEPSCs
| Kinetics | WT no MD | WT 4 d MD | WT 7 d MD | KO no MD | KO 4 d MD | KO 7 d MD |
|---|---|---|---|---|---|---|
| Rise time (ms) | 1.52 ± 0.08 | 1.44 ± 0.10 | 1.33 ± 0.06 | 1.43 ± 0.10 | 1.40 ± 0.13 | 1.50 ± 0.05 |
| Decay tau (ms) | 8.23 ± 0.70 | 9.70 ± 1.50 | 8.0 ± 0.42 | 7.27 ± 0.96 | 8.97 ± 1.45 | 8.80 ± 0.94 |
Rise time and decay tau kinetics are not significantly different in all conditions. All data are presented as mean ± SEM.
Discussion
In this study, we have identified STAT1 as an important negative regulator of the homeostatic component of experience-dependent plasticity. STAT1 KO mice show enhanced OD plasticity attributable to an accelerated increase in open-eye responses after 4 d MD, whereas increasing STAT1 signaling by exogenous IFNγ in WT mice impairs open-eye responses and reduces OD plasticity after 7 d MD. Moreover, closed-eye responses return back to control levels after 7 d MD in STAT1 KO mice, suggesting that these mice have stronger homeostatic plasticity. An AMPAR-mediated mechanism contributes to enhanced plasticity in STAT1 KO mice: cell surface expression and synaptic function of AMPARs after 4 d MD in KO mice closely resemble those after 7 d MD in WT mice, when homeostatic plasticity normally becomes evident. This finding is consistent with a recent study demonstrating that GluA1 is required for the homeostatic component of OD plasticity: GluA1 KO mice lack open-eye potentiation, but do have closed-eye depression similar to WT mice (Ranson et al., 2013). STAT1 KO mice, which display an accelerated increase in open-eye responses with a normal reduction in closed-eye responses after 4 d MD, show sustained surface levels of GluA1. It is possible that both internalization and insertion of GluA1 are taking place in STAT1 KO mice, while insertion only takes place after 7 d MD in WT mice. Another interpretation is that GluA1 internalization is blocked in STAT1 KO mice. While GluA1 internalization appears to underlie closed-eye depression in WT mice, it is possible that a separate mechanism may mediate depression of closed-eye responses in GluA1 KO or STAT1 KO mice.
We found increases in both AMPAR-mediated mEPSC amplitude and frequency in KO mice after 4 d MD, which resemble those in WT mice after 7 d MD. Although an increase in mEPSC amplitude indicates more AMPARs at given postsynaptic sites, an increase in mEPSC frequency indicates either an increased number of AMPAR-containing functional synapses or an increased probability of neurotransmitter release from presynaptic sites. The former possibility is interesting: for example, PSD-95 KO mice show a reduction in mEPSC frequency, which reflects a reduced number of AMPAR-containing “functional” synapses (i.e., more “silent” synapses) rather than a reduction in the probability of neurotransmitter release (Béïque et al., 2006). It is thus possible that STAT1 KO mice after 4 d MD and WT mice after 7 d MD have an increased number of AMPAR-containing functional synapses in addition to increased AMPARs at a given synapse. However, we cannot eliminate the possibility of increased transmitter release from presynaptic sites.
How STAT1 regulates AMPAR surface expression and synaptic function needs additional investigation. A previous study identified tumor necrosis factor alpha (TNFα) as a key molecule required for the homeostatic component of OD plasticity: TNFα KO mice show impaired open-eye potentiation, resulting in reduced plasticity after 5–6 d MD (Kaneko et al., 2008), a phenotype opposite to STAT1 KO mice. Interestingly, in vitro, STAT1 has a role as a binding partner of the TNFR1 receptor complex, leading to inhibition of downstream TNFα signaling (Wang et al., 2000; Wesemann and Benveniste, 2003). In hippocampal neuronal cultures, TNFα treatment increases cell surface expression of GluA1 through TNFR1 (Stellwagen et al., 2005). Thus, one possibility is that STAT1 inhibits TNFα signaling, which then blocks GluA1 surface expression.
Previous studies have demonstrated the role of other components of immune signaling in OD plasticity. Major histocompatibility complex class I (MHC-I) is a transmembrane cell surface protein known to act in cellular recognition by the immune system; it is also expressed in neurons and regulated by neuronal activity (Corriveau et al., 1998; Huh et al., 2000). MHC-I signaling has been shown to negatively regulate OD plasticity: loss of MHC-I genes or paired Ig-like receptor B (PirB), an immune receptor for MHC-I, leads to enhancement of OD plasticity (Syken et al., 2006; Datwani et al., 2009). Whether or not STAT1 and MHC-I/PirB signaling overlap in their roles in OD plasticity is unclear. Mice deficient in MHC-I signaling have abnormal synaptic projections from retina to dLGN, in which ipsilateral projections are enlarged and show more overlap with contralateral projections, contributing to enhanced thalamocortical plasticity (Datwani et al., 2009). In contrast, retinogeniculate projections of STAT1 KO mice are normal, suggesting that the plasticity locus in STAT1 KO mice is likely to be intracortical rather than subcortical.
An involvement of STAT1 is suggested in hippocampus-dependent plasticity and learning (Feng et al., 2010). Increasing the levels of PIAS1 (protein inhibitor of activated STAT1) enhances spatial learning (Tai et al., 2011). Moreover, STAT3 is required for NMDAR-dependent LTD in the hippocampus (Nicolas et al., 2012). In our study, depression of closed-eye responses, which is known to be mediated by an LTD-like mechanism (Heynen et al., 2003), was normal in STAT1 KO mice. Although a role for STAT1 in LTD cannot be excluded, it is possible that different family members of STATs have distinct functions. Our findings provide novel evidence that STAT1 has a negative regulatory role in the homeostatic component of experience-dependent cortical plasticity in vivo, accompanied by the regulation of AMPARs.
Footnotes
This work was supported by grants from the National Institutes of Health and the Simons Foundation (M.S.), a Simons postdoctoral fellowship (I.N.), and a National Research Service Award fellowship (A.V.W.).
The authors declare no competing financial interests.
References
- Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. 2012;22:461–469. doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonini A, Fagiolini M, Stryker MP. Anatomical correlates of functional plasticity in mouse visual cortex. J Neurosci. 1999;19:4388–4406. doi: 10.1523/JNEUROSCI.19-11-04388.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béïque JC, Lin DT, Kang MG, Aizawa H, Takamiya K, Huganir RL. Synapse-specific regulation of AMPA receptor function by PSD-95. Proc Natl Acad Sci U S A. 2006;103:19535–19540. doi: 10.1073/pnas.0608492103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corriveau RA, Huh GS, Shatz CJ. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron. 1998;21:505–520. doi: 10.1016/S0896-6273(00)80562-0. [DOI] [PubMed] [Google Scholar]
- Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- Datwani A, McConnell MJ, Kanold PO, Micheva KD, Busse B, Shamloo M, Smith SJ, Shatz CJ. Classical MHCI molecules regulate retinogeniculate refinement and limit ocular dominance plasticity. Neuron. 2009;64:463–470. doi: 10.1016/j.neuron.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13:423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel MY, Bear MF. How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron. 2004;44:917–923. doi: 10.1016/j.neuron.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Gordon JA, Stryker MP. Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J Neurosci. 1996;16:3274–3286. doi: 10.1523/JNEUROSCI.16-10-03274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heynen AJ, Yoon BJ, Liu CH, Chung HJ, Huganir RL, Bear MF. Molecular mechanism for loss of visual cortical responsiveness following brief monocular deprivation. Nat Neurosci. 2003;6:854–862. doi: 10.1038/nn1100. [DOI] [PubMed] [Google Scholar]
- Htain WW, Leong SK, Ling EA. In vivo expression of inducible nitric oxide synthase in supraventricular amoeboid microglial cells in neonatal BALB/c and athymic mice. Neurosci Lett. 1997;223:53–56. doi: 10.1016/S0304-3940(97)13387-0. [DOI] [PubMed] [Google Scholar]
- Hubel DH, Wiesel TN. The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J Physiol. 1970;206:419–436. doi: 10.1113/jphysiol.1970.sp009022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290:2155–2159. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Stellwagen D, Malenka RC, Stryker MP. Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron. 2008;58:673–680. doi: 10.1016/j.neuron.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck T, Keller GB, Jacobsen RI, Eysel UT, Bonhoeffer T, Hübener M. Synaptic scaling and homeostatic plasticity in the mouse visual cortex in vivo. Neuron. 2013;80:327–334. doi: 10.1016/j.neuron.2013.08.018. [DOI] [PubMed] [Google Scholar]
- Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285:1–24. doi: 10.1016/S0378-1119(02)00398-0. [DOI] [PubMed] [Google Scholar]
- McCurry CL, Shepherd JD, Tropea D, Wang KH, Bear MF, Sur M. Loss of Arc renders the visual cortex impervious to the effects of sensory experience or deprivation. Nat Neurosci. 2010;13:450–457. doi: 10.1038/nn.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, Carver-Moore K, DuBois RN, Clark R, Aguet M, Schreiber RD. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/S0092-8674(00)81288-X. [DOI] [PubMed] [Google Scholar]
- Nicolas CS, Peineau S, Amici M, Csaba Z, Fafouri A, Javalet C, Collett VJ, Hildebrandt L, Seaton G, Choi SL, Sim SE, Bradley C, Lee K, Zhuo M, Kaang BK, Gressens P, Dournaud P, Fitzjohn SM, Bortolotto ZA, Cho K, Collingridge GL. The Jak/STAT pathway is involved in synaptic plasticity. Neuron. 2012;73:374–390. doi: 10.1016/j.neuron.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23:96–101. doi: 10.1016/S1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- Ranson A, Sengpiel F, Fox K. The role of GluA1 in ocular dominance plasticity in the mouse visual cortex. J Neurosci. 2013;33:15220–15225. doi: 10.1523/JNEUROSCI.2078-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Stryker MP. Distinctive features of adult ocular dominance plasticity. J Neurosci. 2008;28:10278–10286. doi: 10.1523/JNEUROSCI.2451-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syken J, Grandpre T, Kanold PO, Shatz CJ. PirB restricts ocular-dominance plasticity in visual cortex. Science. 2006;313:1795–1800. doi: 10.1126/science.1128232. [DOI] [PubMed] [Google Scholar]
- Tai DJ, Hsu WL, Liu YC, Ma YL, Lee EH. Novel role and mechanism of protein inhibitor of activated STAT1 in spatial learning. EMBO J. 2011;30:205–220. doi: 10.1038/emboj.2010.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Crusells J, Vieira A, Saarma M, Rivera C. A novel method for monitoring surface membrane trafficking on hippocampal acute slice preparation. J Neurosci Methods. 2003;125:159–166. doi: 10.1016/S0165-0270(03)00050-5. [DOI] [PubMed] [Google Scholar]
- Tropea D, Kreiman G, Lyckman A, Mukherjee S, Yu H, Horng S, Sur M. Gene expression changes and molecular pathways mediating activity-dependent plasticity in visual cortex. Nat Neurosci. 2006;9:660–668. doi: 10.1038/nn1689. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wu TR, Cai S, Welte T, Chin YE. Stat1 as a component of tumor necrosis factor alpha receptor 1-TRADD signaling complex to inhibit NF-kappaB activation. Mol Cell Biol. 2000;20:4505–4512. doi: 10.1128/MCB.20.13.4505-4512.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesemann DR, Benveniste EN. STAT-1 alpha and IFN-gamma as modulators of TNF-alpha signaling in macrophages: regulation and functional implications of the TNF receptor 1:STAT-1 alpha complex. J Immunol. 2003;171:5313–5319. doi: 10.4049/jimmunol.171.10.5313. [DOI] [PubMed] [Google Scholar]