

Abstract
When the brain or spinal cord is injured, glial cells in the damaged area undergo complex morphological and physiological changes resulting in the formation of the glial scar. This scar contains reactive astrocytes, activated microglia, macrophages and other myeloid cells, meningeal cells, proliferating oligodendrocyte precursor cells (OPCs), and a dense extracellular matrix. Whether the scar is beneficial or detrimental to recovery remains controversial. In the acute phase of recovery, scar-forming astrocytes limit the invasion of leukocytes and macrophages, but in the subacute and chronic phases of injury the glial scar is a physical and biochemical barrier to axonal regrowth. The signals that initiate the formation of the glial scar are unknown. Both canonical and noncanonical signaling Wnts are increased after spinal cord injury (SCI). Because Wnts are important regulators of OPC and oligodendrocyte development, we examined the role of canonical Wnt signaling in the glial reactions to CNS injury. In adult female mice carrying an OPC-specific conditionally deleted β-catenin gene, there is reduced proliferation of OPCs after SCI, reduced accumulation of activated microglia/macrophages, and reduced astrocyte hypertrophy. Using an infraorbital optic nerve crush injury, we show that reducing β-catenin-dependent signaling in OPCs creates an environment that is permissive to axonal regeneration. Viral-induced expression of Wnt3a in the normal adult mouse spinal cord induces an injury-like response in glia. Thus canonical Wnt signaling is both necessary and sufficient to induce injury responses among glial cells. These data suggest that targeting Wnt expression after SCI may have therapeutic potential in promoting axon regeneration.
Keywords: glia, inflammation, repair, Wnt
Introduction
When the brain or spinal cord is injured, glial cells at the injury site undergo biochemical and morphological changes that result in the formation of a glial scar (Silver and Miller, 2004). One common characteristic of the glial reactions to different types of injury is the activation of a population of unusual glial cells known alternatively as oligodendrocyte precursor cells (OPCs), polydendrocytes (Nishiyama et al., 2009), or NG2 cells (Levine et al., 2001). This activation includes a shortening of the cell cycle (Simon et al., 2011), the withdrawal of long multibranched processes into shorter thick processes, enlargement of the cell body, and an accumulation of the cells into a dense cellular plaque surrounding the injury site. OPCs function as precursors cells to oligodendrocytes and participate in the formation of new myelin throughout the life of the animal (Young et al., 2013). Since they are postsynaptic targets for glutaminergic and GABAergic input in the hippocampus and cerebellum (Paukert and Bergles, 2006), they likely carry out other functions that have yet to be fully characterized. Their functions during injury and wound healing are also not well understood, although they secrete or shed growth-inhibitory chondroitin sulfate proteoglycans that are a factor in the inhibition of axonal regeneration (Tan et al., 2006; Donnelly et al., 2012; Petrosyan et al., 2013).
The injury-induced changes in OPCs occur precisely at the injury site suggesting that some feature of the local environment may trigger this activation. What these features are remains unknown. Multiple members of the Wnt family of developmental morphogens and their receptors are rapidly increased at sites of brain and spinal cord injury (SCI; Liu et al., 2008a; Miyashita et al., 2009; Fernández-Martos et al., 2011; Gonzalez et al., 2012). While mostly known for their role as developmental regulators, Wnts have multiple functions in the adult brain (Inestrosa and Arenas, 2010).
Here we asked whether canonical Wnt signaling plays a role in the activation of OPCs after contusive SCI. Canonical Wnt signaling is dependent on cytoplasmic β-catenin. In the absence of Wnt ligands, β-catenin is destroyed by a complex of axin, adenomatous polyposis coli, and active glycogen synthase kinase 3β (GSK3β; Kimelman and Xu, 2006). After binding of Wnts to a frizzled-LRP receptor complex, GSK3β activity is inhibited allowing β-catenin to accumulate in the cytoplasm and subsequently translocate to the nucleus where it binds to TCF/LEF transcription factors and activates gene transcription (Cadigan and Liu, 2006). Here, we conditionally knocked out β-catenin specifically in OPCs before injuring the thoracic spinal cord. The results indicate that canonical Wnt signaling is required for the injury-induced activation of OPCs. Surprisingly, attenuation of the OPC response also attenuated microglial activation and astrocyte hypertrophy. Furthermore, knocking out β-catenin allows retinal ganglion cell axons to regenerate beyond the injury zone in the crushed optic nerve. Thus, OPC activation is a critical component of glial scar formation and requires canonical Wnt signaling.
Materials and Methods
Animals
B6.129-Ctnnb1tm2Kem/KnwJ (β-cateninf/f) (Brault et al., 2001) and B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J (Rosay/y) animals were obtained from The Jackson Laboratory. PDGFrαcreERT2/+ (Rivers et al., 2008) animals were obtained from Dr. B. Popko (University of Chicago, Chicago, IL). All strains were back-crossed onto a C57BL/6J background for >6 generations. Three breeding stocks were created: PDGFrαcreERT2/+ Rosay/y, PDGFrαcreERT2/+ Rosay/y β-cateninf/f, and Rosay/y β-cateninf/f. The PDGFrαcreERT2/+ Rosay/y and Rosay/y β-cateninf/f stocks were created by crossing PDGFrαcreERT2 and β-cateninf/f animals on Rosay/y twice. PDGFrαcreERT2/+ Rosay/y β-cateninf/f animals were generated by breeding PDGFrαcreERT2/+ animals twice on β-cateninf/f animals then crossing the PDGFrαcreERT2/+ β-cateninf/f progeny with Rosay/y β-cateninf/f to introduce the Rosa reporter. Viral overexpression experiments were conducted in C57BL/6J animals.
Tamoxifen induction
Tamoxifen was dissolved in ethanol at a concentration of 1 g/ml then dissolved in vegetable oil (19:1) with 5 min of sonication to a final concentration of 50 mg/ml and frozen. Frozen stocks of tamoxifen were warmed to 37°C and sonicated for 1 min before use. Tamoxifen was administered via oral gavage at a dose of 600 mg/kg 1× daily for 3 d.
Surgical methods
SCI.
Female mice of 6–8 weeks of age were used throughout these experiments. Animals were deeply anesthetized using isoflurane. The spinal column was exposed from T7–T9 and a laminectomy was performed at T8. The lateral processes at T7 and T9 were stabilized with immobilized forceps attached to the Infinite Horizon Impactor (Precision Systems and Instrumentation) and a contusion of 45 kdyn was delivered. Hemostasis was obtained with Gelfoam, and the deep muscles, superficial muscles, and skin were sutured in layers. During recovery, animals were given 50 mg/kg buprenorphine 2× daily for 4 d.
Viral injections.
After a laminectomy and stabilization of the cord as described above, a durotomy was performed at T8. AAV virus diluted in PBS to 1 × 1011 viral genomes/ml was slowly injected into the border of the dorsal columns and dorsal horn using a sharpened glass micropipette (4–6 μm opening) and a Picospritzer (General Valve) programmed to deliver 15–25 ms pulses at 1 Hz and 25 psi over 10 min such that a total of 0.5 μl of virus was injected. The injection was aimed by centering the needle over the spinal cord then moving laterally 300 μm. The needle was inserted 500 μm into the spinal cord then brought back to a final depth of 300 μm.
Optic nerve crush.
Animals were anesthetized locally with 0.5% proparacaine hydrochloride ophthalmic solution and generally with isoflurane. An optic nerve crush was performed as described previously (Meyer and Miotke, 1990; Meyer et al., 1994). In brief, the lateral conjunctival membrane was cut and lateral muscles bluntly dissected to expose the optic nerve. The nerve was then crushed with #5 biologie forceps (Dumont) for 5 s. No compounds were administered to either promote retinal ganglion cell (RGC) survival or to upregulate their intrinsic growth state (Leon et al., 2000; Kurimoto et al., 2010). At 19 d post crush, 2 μg of Alexa Fluor 594-conjugated cholera toxin subunit B (CTB; Invitrogen) was injected into the vitreous space of the inured eye. The animals were killed 21 d post crush when the maximum number of axons was detected distal to the injury site in previous studies (Leon et al., 2000).
All procedures using animals were approved by the Institutional Animal Use Committee.
AAV
Mouse Wnt3a and Wnt5a-expressing AAV2 virus was prepared as follows. The ORF of Wnt3a and Wnt5a was cloned into the AAV-IRES-hrGFP vector (Stratagene) and virus was created according to the Stratagene helper-free system protocol. In brief, virus plasmid, pHelper, and pAAV-RC were transfected into AAV-293 cells. The cells were lysed by freeze thawing and the virus was concentrated via the ViraBind system (Cell Biolabs). GFP virus was a gift from Dr. W. Bowers (University of Rochester, Rochester NY). Viral genome copy numbers were calculated according to their optical density at 260 and 280 nm (Sommer et al., 2003).
Tissue preparation
Animals were deeply anesthetized and transcardially perfused with 50 ml of room temperature PBS followed by 50 ml of ice-cold 4% formaldehyde in 100 mm PB, pH 7.4. Tissue was dissected from the animals and postfixed in the formaldehyde solution for 4 h at 4°C. Optic nerves were equilibrated with PBS for 12 h before clearing. Spinal cord tissue was prepared for cryostat sectioning by equilibrating the tissue with 30% sucrose in 100 mm PB, pH 7.4, for 48 h. Tissue was frozen in Cryo-Gel embedding media (Instrumedics) in dry ice-cooled 2-methylbutane. Coronal sections 20 μm thick were prepared using standard cryostat procedures.
Optic nerve clearing
Optic nerves were organically desiccated and solvent cleared as described previously (Ertürk et al., 2012). Four percent formaldehyde-fixed optic nerves were incubated in solutions of 50, 80, and 100% tetrahydrofuran in water for 30 min each and then placed in 100% dichloromethane for 20 min. The nerves were placed in a 1:2 solution of benzyl alcohol and benzyl benzoate (BAAB) for 3 min and mounted onto uncoated slides in BAAB solution using a strip of Parafilm as a spacer.
Immunofluorescence staining
Immunofluorescence staining of tissue sections and cell cultures was performed as described previously (Levine et al., 1993). Primary antibodies are listed at the end of the Materials and Methods section. Secondary antibodies were all raised in donkey. Alexa Fluor(AF)-488 anti-chicken, AF-647 anti-rabbit, and AF-647 anti-rat were obtained from Jackson ImmunoResearch, and AF-594 anti-rabbit and AF-594 anti-rat were obtained from Invitrogen.
BrdU labeling
Animals were administered two intraperitoneal injections (10 mg/kg) of BrdU in normal saline 4 and 2 h before being killed and perfused with 4% PFA. Staining was performed using antigen retrieval as described previously (Morgan et al., 1994, 1997; Tang et al., 2007).
In vitro methods
Dissociated cultures of OPCs were prepared as described previously (McCarthy and de Vellis, 1980; Yang et al., 2005). OPCs were isolated from P0–P2 rats and expanded in a mixed glial culture in DMEM + 10% FBS. The cultures were shaken to suspend OPCs, and the cells were subjected to two sessions of differential adhesion for 30 min to enrich for OPCs. OPCs were plated on PLL-coated glass coverslips in DMEM + 10% FBS and permitted to adhere for 4 h before being cultured in defined media. Basic oligodendrocyte differentiation media was DMEM, N2 supplement (Gemini Bio-Products), 0.5% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin (Life Technologies), 40 ng/ml thyroxine (T4), and 30 ng/ml triiodothyronine (T3; Sigma). In different experiments, this media was supplemented with either 10 ng/ml PDGF-AA (PeproTech), Wnt conditioned media as described below, or pure Wnt3a (Millipore). L-cells stably transfected to express Wnt3a and Wnt5a and control L-cells were obtained from ATTC. Conditioned media was prepared by growing the cells to 90% confluence and incubating the cells in DMEM containing 0.5% FBS for 3 d. The media was collected, centrifuged to remove cellular debris, filtered, and stored at 4°C. A human Fc mouse Frizzled-5 cysteine-rich domain fusion protein (Fzl5-CRD-IgG) was purified using protein A chromatography from conditioned media collected from stably transfected 293T cells.
Analytical methods
Imaging.
Confocal imaging was performed on an Olympus FluoView confocal microscope. Wide field images were obtained on a Zeiss Axioskop II with AxioVision (Zeiss) software and were stitched together in Photoshop (Adobe). Stereology was performed using Stereo Investigator and Neurolucida (MicroBrightField) attached to a Zeiss Axioskop II.
SCI.
We counted cells within the penumbra region of the lesions using the optical fractionator probe of Stereo Investigator (MicroBrightField). We define the penumbra as a region 150 μm wide surrounding the lesion core. The lesion core was identified by staining coronal sections of injured spinal cord with anti-GFAP antibodies. GFAP immunoreactivity is lost in the lesion core and upregulated along the borders of the core.
Due to section shrinkage in the z-axis, the average mounting thickness was 16 μm. We used guard intervals at the top and bottom of each section equal to 10% (∼1.6 μm) of the mounted thickness to exclude regions of the tissue disrupted by sectioning. The population estimates were performed using a counting frame of 150 × 150 μm and a sampling grid of 300 × 300 μm, effectively sampling 25% of each cord at nonbiased, systematically arranged, and randomly positioned intervals. We counted cells in five sections spaced 200 μm apart and centered on the injury epicenter (defined by the section having the largest injury area). All biological replicates had a coefficient of error as calculated by the Gundersen method of ≤10% for each marker (Gundersen, 1986; Gundersen et al., 1999). The cell population densities in each region of interest between PDGFrαCreERT2/+, β-cateninf/f, Rosa26YFP/YFP, and control PDGFrαCreERT2/+, Rosa26YFP/YFP mice at each time point were compared using Student's t test with significance defined as p ≤ 0.05.
Optic nerve.
The crush site was identified by YPF expression. Confocal stacks extending through the optic nerve were obtained at the crush site and 1000 μm distal to the crush. We counted the total number of axons that crossed planes drawn perpendicular to the nerve at 25, 50, 150, and 1000 μm from the crush site. The five longest axons were identified by bright-field epifluorescent microscopy and the distance from the edge of the crush was calculated in AxioVision (Zeiss).
Viral injections.
Due to the small area of involvement in the viral overexpression model, subsampling within sections was not performed for BrdU double-labeled cells. Five sections spaced 200 μm apart were selected surrounding the injection site. An area of interest surrounding all GFP+ cells was drawn to inscribe the extent of viral infection. All double-labeled Iba1+/BrdU+ and NG2+/BrdU+ cells were counted within this area of interest. To determine the density of NG2+, Iba1+, and GFAP cells, we counted the total number of cells within a 150 × 150 × 16 μm area about the centroid of the infected area on three sections centered on the injection. A staining intensity ratio was obtained at the injection site by measuring the average intensity of a 150 × 150 μm area at the centroid of the infected area on a single confocal plane and dividing it by the average intensity of a similar area containing equal portions gray and white matter in the contralateral spinal cord. Two measurements were made per animal and averaged.
Statistical analysis
All datasets were obtained from a biological system and assumed to be part of a normal distribution after passing a Shapiro–Wilk's normality test despite n < 10. For comparison between control and β-catenin knock-out (KO) population analysis at 2, 5, and 20 days post injury (dpi), we performed a Student's t test for each time point. To compare multiple groups in the viral overexpression data and in vitro proliferation data, we performed an ANOVA with a post hoc Tukey test. For comparison of axon outgrowth within the optic nerve, we performed a two-way ANOVA with a post hoc Holm–Sidak multiple-comparison test using genotype and distance from lesion edge as independent variables. We assumed p < 0.05 to be significant.
Primary antibodies
Primary antibodies included the following: rat anti-BrdU (Accurate Chemical & Scientific) 1:1000, rabbit anti-GFP (Invitrogen) 1:1000, chicken anti-GFP (Aves) 1:1000, rabbit anti-GFAP (DAKO) 1:2000, rabbit anti-Iba1 (Wako) 1:500, rabbit anti-Axin2 (Abcam) 1:500, rabbit anti-NG2 1:1000, mouse anti-O4 (a gift from R. Bansi, University of Connecticut Health Science Center, Farmington, CT) 1:250, mouse anti-β-catenin (Millipore) 1:250, and mouse anti-β-actin (Sigma) 1:10,000.
Results
To investigate the role of canonical Wnt signaling in the glial reactions to SCI, we developed a genetic model using two strains of transgenic mice: PDGFrαCreERT2/+, β-cateninf/f, Rosa26YFP/YFP and control PDGFrαCreERT2/+, Rosa26YFP/YFP mice. Tamoxifen treatment of mice carrying the PDGFrαCreERT2/+, Rosa26YFP/YFP mice transgenes results in the expression of YFP almost exclusively in OPCs (Rivers et al., 2008). In the triple transgenic mice (PDGFrαCreERT2/+, β-cateninf/f, Rosa26YFP/YFP), tamoxifen treatment also results in the deletion of the first five exons of β-catenin thereby blocking transcription and silencing canonical Wnt signaling (Brault et al., 2001). To validate the cellular specificity and utility of this model, we treated both strains of animals with tamoxifen and counted the number of YFP+ cells in the thoracic spinal cord at intervals after treatment (Fig. 1B). After tamoxifen treatment, equal numbers of YFP+ cells were present throughout the spinal cord of both strains and the number of YFP+ cells approximately doubled between 7 and 25 d post treatment. There were no significant differences in the appearance of the YFP+ cells between the β-catenin KO and control animals (Fig. 1 A). At 7 d post-tamoxifen treatment, >95% of the YFP+ cells expressed the OPC marker NG2 and this percentage declined slightly over the next 18 d, most likely reflecting the normal differentiation of the NG2+ cells into oligodendrocytes (Fig. 1C).
Figure 1.
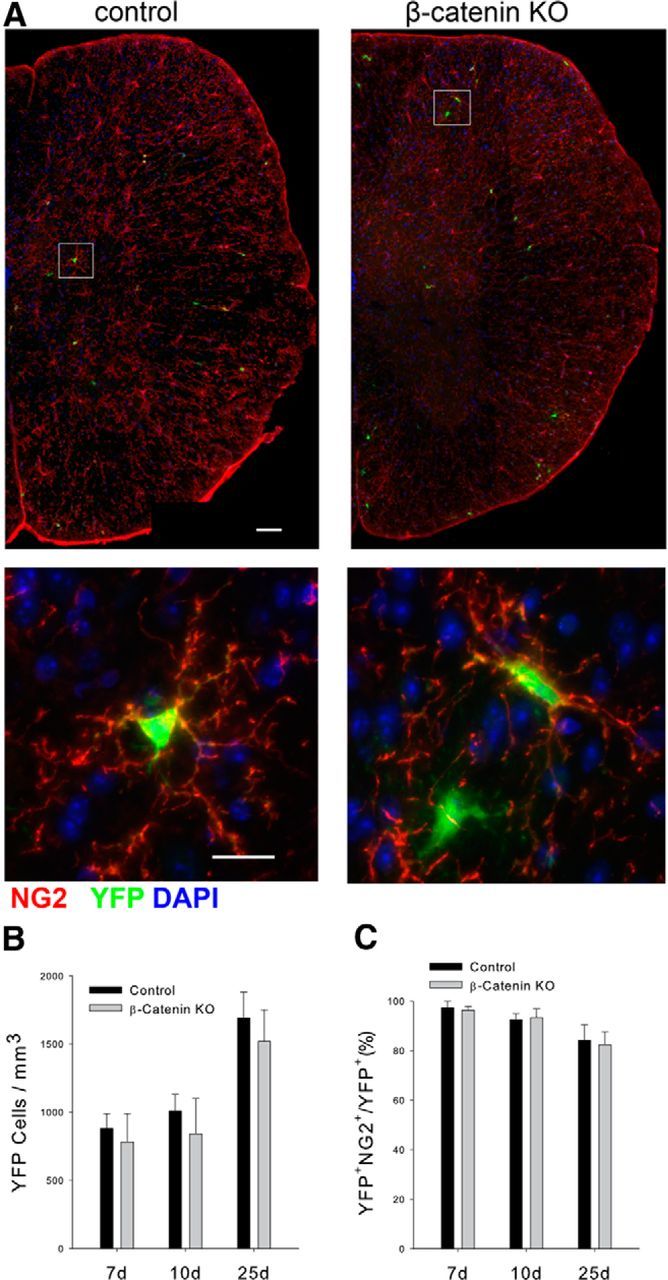
Characterization of the β-catenin KO and control animals. A, The appearance of YFP+ cells in control and β-catenin KO spinal cord 10 d after initiating tamoxifen treatment. Scale bar, 50 μm. Bottom shows individual YFP+, NG2+ OPCs from both genotypes. Scale bar, 10 μm. B, Quantitation of the density of YFP+ cells at the indicated times after initiating tamoxifen treatment. C, The percentage of YFP+ cells colabeled with NG2. There is no statistical difference in the density of OPCs between the two genotypes; n = 3 for all time points and groups.
To monitor ongoing β-catenin-dependent Wnt signaling, we immunofluorescently stained tissue sections for axin2, a direct target for the β-catenin-TCF transcriptional activator complex (Yan et al., 2001; Jho et al., 2002; Lustig et al., 2002). In the uninjured spinal cord, numerous axin2+ cells were present throughout the gray matter and scattered axin2+ cells were present in the lateral white matter (Fig. 2A, left). YFP+ cells in the uninjured control animals were axin2+, whereas YFP+ cells in the KO animals expressed little or no axin2 protein (Fig. 2B). Thus, conditional deletion of β-catenin in adult spinal cord OPCs reduces canonical Wnt signaling but does not alter the normal appearance or short-term development of the cells.
Figure 2.
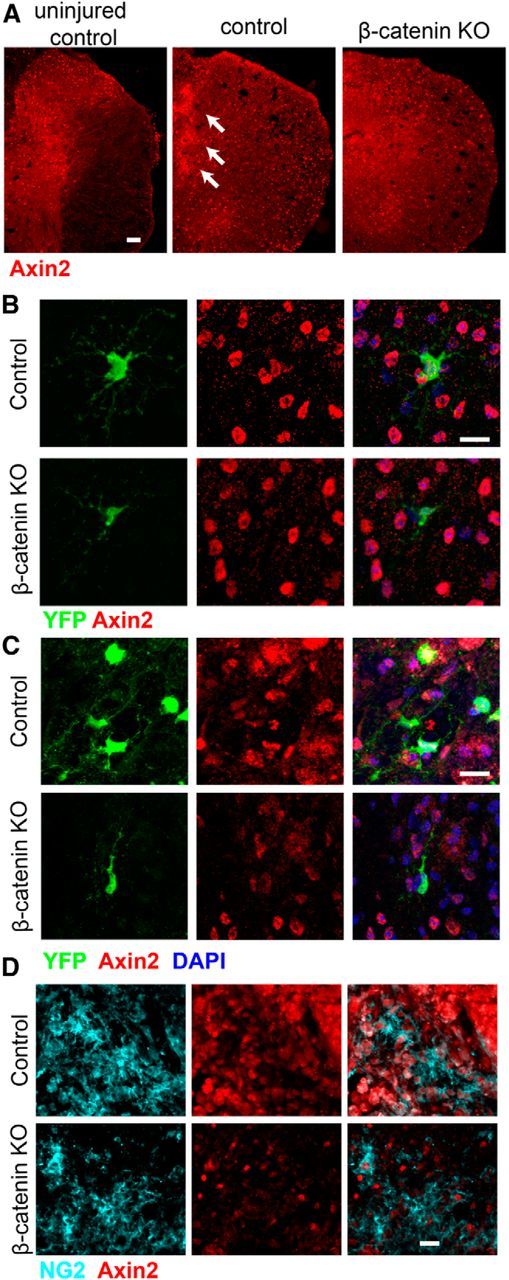
Axin2 immunoreactivity in control and KO mice. A, Anti-axin2 staining of wild-type uninjured thoracic spinal cord, injured control, and injured KO spinal cord at 5 dpl. Scale bar, 50 μm. B, Axin2 immunoreactivity in uninjured control and KO YFP-positive OPCs. Axin2 immunoreactivity is reduced in OPCs in the β-catenin KO mice. Scale bar, 10 μm. C, Axin2 immunoreactivity in YFP+ cells at 5 dpl in injured control and KO animals. Scale bar, 10 μm. D, Anti-NG2 and anti-axin2 staining of 5 dpl injured spinal cord in control and KO mice. Scale bar, 15 μm. Knocking out β-catenin in OPCs reduces axin2 expression throughout the spinal cord and in both the YFP+ and NG2+ cell populations.
To confirm that canonical Wnt signaling increases after SCI (Liu et al., 2008a; Miyashita et al., 2009; Gonzalez et al., 2012), control and KO female mice (6–8 weeks of age) were induced with tamoxifen for 3 and 2 d later, given moderate (45 kdyn) contusion injuries to the dorsal spinal cord at T10. At 5 d after a moderate contusion injury (5 days post lesion, dpl), the number of axin2+ cells in control animals had increased and the cells were present throughout both the gray and white matter (Fig. 2A, middle). Axin2+ cells were particularly dense along the lesion border (Fig. 2A, middle, arrows). Axin2+ cells were present at lower densities in the β-catenin KO animals (Fig. 2, right), although they were still elevated compared with noninjured control animals. YFP+ cells in the control animals expressed moderate to high levels of axin2, whereas in the β-catenin KO animals, axin2 immunoreactivity was barely detectable in individual YFP+ cells (Fig. 2C). Of note, many cells that did not express YFP were axin2+ in both the control and KO animals. To confirm that OPCs were among the cells expressing axin2, we double stained sections from 5 dpl animals with antibodies against axin2 and NG2. As shown in Figure 2D, in the control animals, NG2+ cells in the lesion penumbra were axin2+ whereas NG2-expressing cells in the KO animals did not contain axin2 immunoreactivity. These data confirm that injury induces canonical Wnt signaling in OPCs and other cells and show that β-catenin is necessary for this signaling in OPCs.
OPCs proliferate after contusive SCI and accumulate in the penumbral region of the injury site (McTigue et al., 2001; Lytle and Wrathall, 2007; Lytle et al., 2009). To determine whether canonical Wnt signaling is required for this accumulation, we counted the number of YFP+ cells in the area surrounding the injury site using nonbiased stereology as described previously (see Materials and Methods). As shown in Figure 3, A and B, YFP+ cells accumulated in the lesion penumbra in control animals, but this accumulation was much reduced in the KO animals. In control animals, the number of YFP+ cells increased 10-fold at 5 dpl (1010 ± 120/mm3 in uninjured cases, n = 4, 10,017 ± 2001/mm3 at 5 dpi, n = 4) and continued to increase more slowly thereafter. In contrast, the density of YFP+ cells in the KO animals did not increase significantly between 2 and 5 dpl and was almost threefold less than that of control animals at 20 dpl (Fig. 3B). Consistent with the almost exclusive expression of YFP in NG2+ OPCs, the density of all NG2+ cells in the injury penumbra was also reduced at 2, 5 and 20 dpl in the KO animals compared with control animals (Fig. 3C,E). YFP+, NG2+ cells in the control animals underwent a morphological transformation typical of the injury response. At 5 dpl, they had shortened processes and enlarged cell bodies (Fig. 3D). In contrast, YFP+, NG2+ cells in the KO animals retained their stellate appearance and their long multibranched processes (Fig. 3D). The reduced density of the NG2+ and YFP+ cells in the KO animals arises, at least in part, from a failure of the β-catenin KO cells to proliferate as shown by reduced BrdU incorporation at 2 and 5 dpl compared with control YFP+ cells (Fig. 3F,G). BrdU incorporation into the total NG2+ cell population was reduced in the KO animals to an extent similar to that of the YFP+ cells (Fig. 3H). As the glial scar matured between 5 and 20 dpl, the number of YFP+ cells in both genotypes increased and the number of NG2+ cells decreased, most likely reflecting the continuing proliferation and differentiation of YFP+ cells into either NG2-oligodendrocytes or Schwann cells (McTigue et al., 2001; Zawadzka et al., 2010). Thus, abrogating β-catenin signaling in OPCs reduces their proliferation and accumulation in the tissue surrounding SCI.
Figure 3.

OPC proliferation and accumulation after injury is reduced in the β-catenin KO mice. A, Appearance of YFP-positive cells surrounding the lesion site at 5dpi. The dotted line outlines the contusion site. B, Quantitation of the density of YFP+ cells at the indicated postinjury times, n = 4. C, The appearance of YFP+, NG2+ OPCs at the indicated postinjury times. In control animals, OPCs accumulate around the lesion site, retract their processes, and have increased immunoreactivity for NG2. In the KO animals, OPCs retain their ramified appearance at all postinjury survival times. D, High-power views of YFP+,NG2+ cells at 5 dpl. In control animals, the cells have swollen cell bodies and short processes whereas KO NG2+,YFP+ retain their long multibranched processes. E, Quantitation of the density of NG2+ OPCs at the indicated postinjury times, n = 4. F, Quantitation of BrdU incorporation into YFP+ cells at the indicated postinjury times, n = 4. G, An example of a BrdU+, YFP+, and NG2+ cell in control animal 5 dpi. H, Quantitation of BrdU incorporation into NG2+ cells at the indicated postinjury times, n = 4. B, E, F, and H, *p < 0.02, **p < 0.005. Scale bars: A, C, 50 μm; D, 15 μm; G, 10 μm.
SCI results in astrocyte hypertrophy and microglial proliferation and accumulation (Dahl and Bignami, 1974; Giulian et al., 1989). Whether the initial reactions of OPCs to injury play a role in the subsequent reactions of these glial cell types is unknown. We therefore next asked whether reducing the OPC reaction to SCI alters the injury-induced responses of microglia, macrophages, and astrocytes. Reactive microglia and macrophages were identified using a polyclonal antibody against Iba1 that identifies both of these two cell types. As shown in Figure 4, A and B, the accumulation of microglia/macrophages around the injury site was reduced in the β-catenin KO animals compared with control animals, particularly at 5 dpl when the microglia/macrophage reaction is at its maximum. The insets show that the Iba1+ cells present in the KO animals had ramified processes compared with cells in the control animals, which had the amoeboid appearance typical of activated cells. The accumulation of microglia/macrophages after SCI arises from both cell migration and local cell division. Surprisingly, there were no differences in the ratio of Iba1+ cells incorporating BrdU (Fig. 4C) at 2 and 5 dpl, suggesting that the reduced cell accumulation in the KO animals is due to a failure of inflammatory cell mobilization and migration rather than reduced proliferation.
Figure 4.

Injury-induced reactions of microglia are reduced in the β-catenin KO animals. A, The appearance of microglia/ macrophages in control and KO animals at the indicated postinjury survival times. B, Quantitation of the density of Iba1+ cells in the injury penumbra at the indicated postinjury survival times; **p < 0.001. n = 4. C, BrdU incorporation in Iba1+ cells at the indicated postinjury times. There is no statistically significant difference in the percentage of Iba1+ cells that take up BrdU in the two genotypes. Scale bars: A, 100 μm; insets, 10 μm.
The astrocyte hypertrophy that follows SCI forms a barrier to cell invasion and restores the boundary between CNS and non-CNS tissue (Bush et al., 1999). Astrocytic reactions to contusive injury were also reduced in the KO animals and overall levels of anti-GFAP immunoreactivity were reduced at 5 dpl in the β-catenin KO animals (Fig. 5). This reduction was most apparent at the single cell level, and astrocytes in the KO animals lacked the dense bundles of GFAP-containing intermediate filaments seen in the control cells (Fig. 5, insets). At 20 dpi in control animals, the injury site resolves into a fluid- and cell-filled granuloma encapsulated by the processes of reactive astrocytes (Fig. 5). Although reactive astrocytes were present around the granuloma that forms in the KO animals, the astrocytic borders of this granuloma were poorly defined compared with the sharp edges seen in the control animals. Thus, when the OPC reaction to injury is suppressed, the reactions of microglia and astrocytes are also muted. This suggests that OPCs, which are among the first cells to react to several different types of CNS injury (Levine et al., 2001), may orchestrate the reactive responses of other glial cells.
Figure 5.
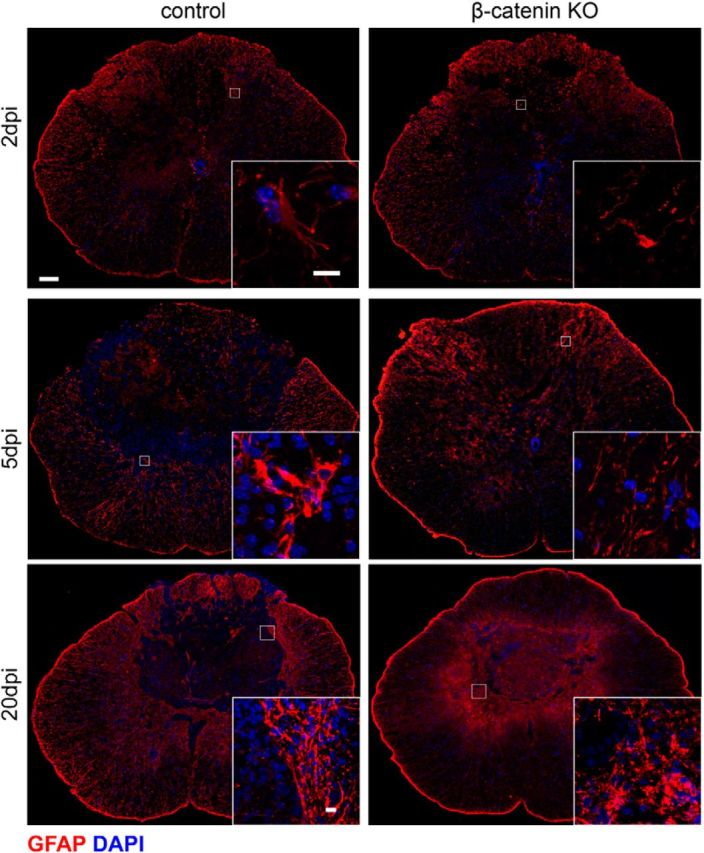
Astrocyte hypertrophy is reduced in the KO animals. GFAP-immunoreactivity at the indicated postinjury survival times in control and β-catenin KO animals. Insets show higher magnification views of the boxed areas. Astrocytes are hypertrophic at 5 dpi in the control animals but appear unchanged in the KO animals. At 20 dpi, astrocytes form a compact border around at the injury site; KO astrocytes are irregular in appearance and do not form a compact border. Scale bars: A, 50 μm; insets, 10 μm.
Despite these differences in the accumulation of OPCs and microglia after injuries, lesion size did not vary significantly between the control and β-catenin KO animals at 2 and 5 dpl. There was a slight trend toward smaller lesion sizes at 20 dpl (lesion size was 15% smaller in the KO animals), but this did not reach significance (p = 0.06, Student's t test).
The data presented above show that abrogating canonical Wnt signaling in OPCs after SCI reduces OPC accumulation, inflammation, and reactive astrogliosis. Because OPCs and astrocytes produce large amounts of growth-inhibitory chondroitin sulfate proteoglycans (CSPGs) among other growth-inhibitory molecules, we next asked whether there was a reduced accumulation of CSPGs in the KO animals. As shown in Figure 6, CSPG immunoreactivity was reduced at 20 dpi in the KO animals compared with the control injured animals. To determine whether glial scar reduction affected the ability of damaged axons to regenerate, we used a minimally invasive infraorbital optic nerve crush model (Fig. 7). After an infraorbital crush, damaged RGC axons sprout but fail to grow into the glial environment of the damaged distal segment unless the growth state of the retinal ganglion neurons is experimentally manipulated (Benowitz and Yin, 2010). We crushed the left optic nerve of control and KO mice and measured axon regeneration 3 weeks later in whole-mount preparations of solvent-cleared optic nerves (Ertürk et al., 2012). As shown in Figure 7, A and B, YFP+ cells accumulate at the crush site in control animals and this accumulation is reduced in the KO animals. To measure axon regeneration, we counted the total number of axons that crossed planes drawn perpendicular to the nerve at 25, 50, 150, and 1000 μm from the crush site (Fig. 7C). Approximately three times more axons grew past the proximal edge of the crush site in KO optic nerves compared with control optic nerves. We also measured the length of the five longest axons in each control and KO animal (Fig. 7D). The mean length was 3.6-fold longer in the KO mice (1322 ± 27.3 μm in the KO vs 365 ± 66 μm in control animals, mean ± SE). In the KO animals, many labeled axons had arrowhead-shaped growth cone-like endings (Fig. 7F) whereas in the control animals, labeled axons had dystrophic end bulbs (Fig. 7E). Thus, reducing glial scarring and associated inflammation creates an environment that is permissive for axon regeneration.
Figure 6.

CSPG immunoreactivity is reduced in the KO spinal cords. Monoclonal antibody CS-56 stain of control and KO spinal cords at 20 dpl. Scale bar, 100 μm.
Figure 7.

Retinal ganglion cell (RGC) axon regeneration in control and KO animals. A, B, Whole-mount preparations of optic nerves 3 weeks after an infraorbital crush. CTB-labeled RGC axons are red. The chevrons indicate the crush site. YFP+ cells are abundant in control animals and form a glial scar that is greatly reduced in the KO animals. CTB-labeled axonal profiles extend past the crush site in the KO animals. Scale bar, 50 μm. C, Mean axon number at the indicated distances from the proximal edge of the injury. Control, n = 4; β-catenin KO, n = 6. D, The lengths of the five longest axons in each of the control and KO animals represented in shades of red and green, respectively. Horizontal line indicates the median for the longest axons in control and KO animals. E, Confocal image of a dystrophic growth cone from a control animal. F, A labeled growth cone in a KO animal. Note the elongated form and the thin filopodium; **p < 0.001, *p < 0.05. Scale bar E and F, 10 μm.
These results suggest that β-catenin-dependent canonical Wnt signaling is necessary for the glial reactions to injury. Because β-catenin also participates in multiple cellular processes (Valenta et al., 2012), we used a gain-of-function approach to determine whether increasing Wnt expression and subsequent β-catenin-dependent signaling is sufficient to provoke an injury-like response in glial cells in uninjured animals. We injected AAV viruses expressing GFP alone, Wnt3a and GFP, or Wnt5a and GFP into the dorsal columns of 6- to 8-week-old female C57BL/6J mice and examined the region surrounding the injection site for reactive-like glial changes 10 d later. To ensure that the injected viruses were activating canonical Wnt signaling, we performed two control experiments. First, we examined axin2 reactivity and, as shown in Figure 8A, there was a large increase in the expression of axin2 surrounding the Wnt3a virus injection site. Axin2 was detected in both OPCs and microglia (data not shown). Second, we injected the Wnt3a-expressing virus into control and β-catenin KO animals and examined YFP expression. As shown in Figure 8B, YFP+ cells were abundant in the control animals but much reduced in the KO animals. Thus, injecting Wnt3a-expressing AAV induced canonical signaling around the injection site and a β-catenin-dependent increase in the density of YFP+ OPCs.
Figure 8.
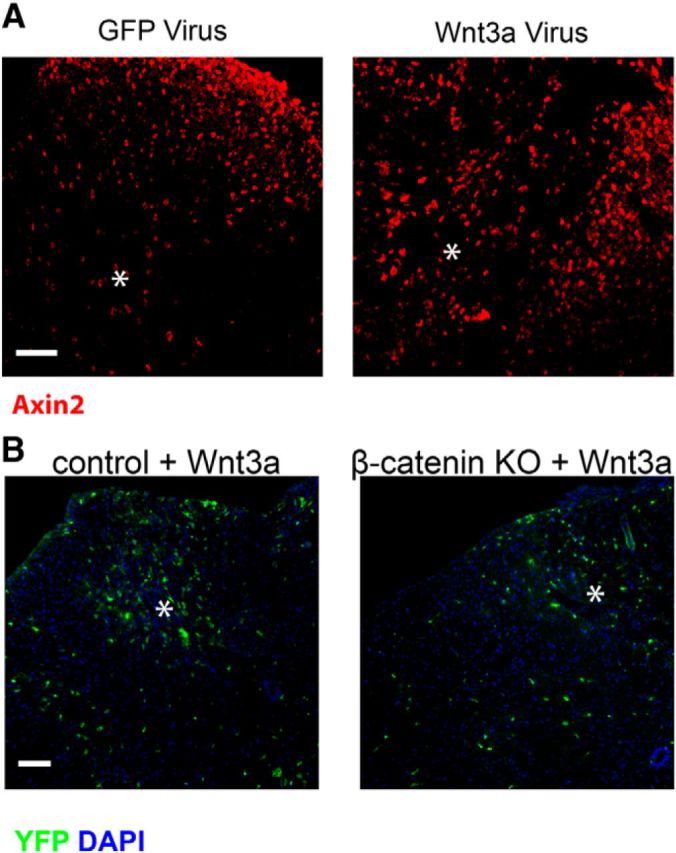
Exogenous Wnt3a activates β-catenin-dependent canonical signaling. A, Axin2 immunoreactivity surrounding virus injection sites (asterisk). B, YFP-positive cells after AAV-Wnt3a virus injection in control or KO mice. Scale bars: A, B, 50 μm.
Injecting AAV-Wnt3a increased the density of NG2+ OPCs in the vicinity of the injection site (Fig. 9A–C) whereas after injection of either AAV-GFP or AAV-Wnt5a, the density of OPCs did not change significantly. After injection of AAV-Wnt3a, OPCs at the injection site were tightly clustered and had enlarged cell bodies and increased immunoreactivity to the NG2 CSPG (Fig. 9B). Injection of AAV-Wnt5a also altered the morphology of OPCs; they had fewer but longer processes. This may reflect activated planar polarity pathways. The Wnt3a-mediated increase in cell density was due, at least in part, to increased cell division as the incorporation of BrdU into OPCs after a pulse label was increased fourfold in the Wnt3a-injected animals compared with AAV-eGFP-injected animals (Fig. 9C). The BrdU-positive cells were clustered around the injection site (data not shown). These results show that the initiation of canonical Wnt signaling in adult OPCs stimulates their proliferation and accumulation similar to what occurs after acute SCI.
Figure 9.
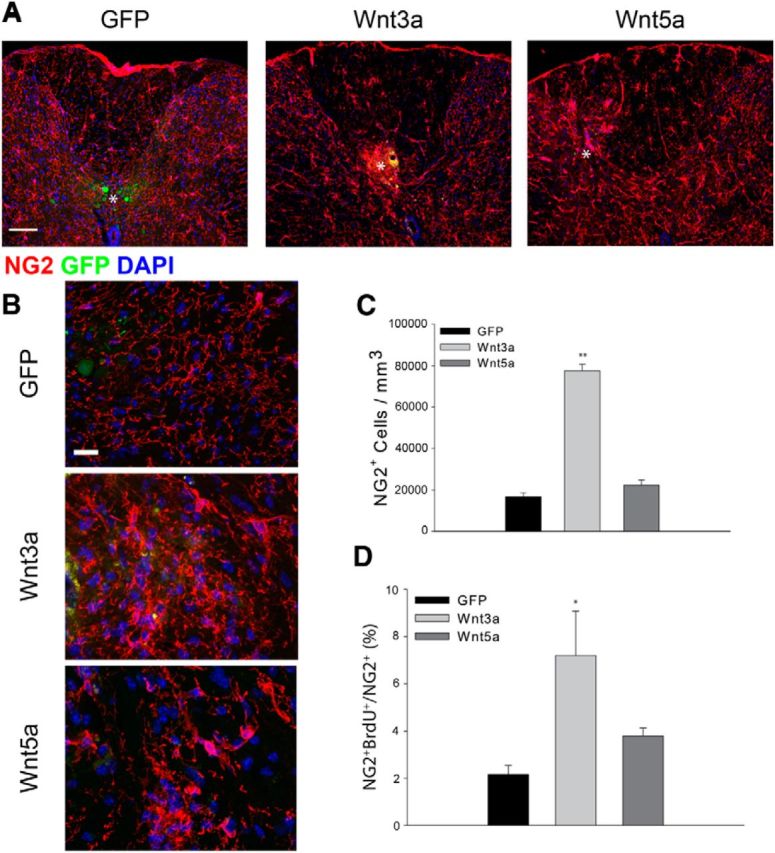
Exogenous Wnt3a increases OPC density. A, The density of NG2+ (red) OPCs increases in Wnt3a virus-injected animals but does not change significantly after injection of either a GFP virus or a Wnt5a-expressing virus. White asterisk indicates centroid of the injection site. B, High-power views of NG2 immunoreactivity after injection of the indicated AAV viruses. C, Quantitation of cell density 10 d post virus injection. D, BrdU incorporation into NG2+ cells after a pulse label. Wnt3a, n = 8; Wnt5a, n = 4; GFP, n = 6; **p = 0.001, *p = 0.01. Scale bars: A, 50 μm; B, 10 μm.
We also examined the effects of Wnt overexpression on microglia and astrocytes. Consistent with previous studies showing that microglia respond to exogenous Wnts (Halleskog et al., 2011; Kilander et al., 2011), injection of AVV-Wnt3a increased the density of Iba1+ microglia fivefold; injection of AAV-Wnt5a also increased microglial cell density, albeit to a lesser degree (Fig. 10A,B). There were no differences in the percentage of Iba1+ cells incorporating BrdU after a 2 h pulse between the control and Wnt3a-injected or Wnt5a-injected animals (data not shown). Neither Wnt3A nor Wnt5A increased the density of GFAP-positive astrocytes (Fig. 10A,B), but both Wnts lead to significant increases in GFAP expression (Fig. 10C). The effects of Wnt5a on GFAP expression were more modest than that of Wnt3a. These results demonstrate that the increased expression of Wnt3a provokes an injury-like response in glial cells. While this suggests a key role of canonical Wnt signaling in regulating the glial responses to SCI, we cannot rule out a contribution of noncanonical signaling.
Figure 10.
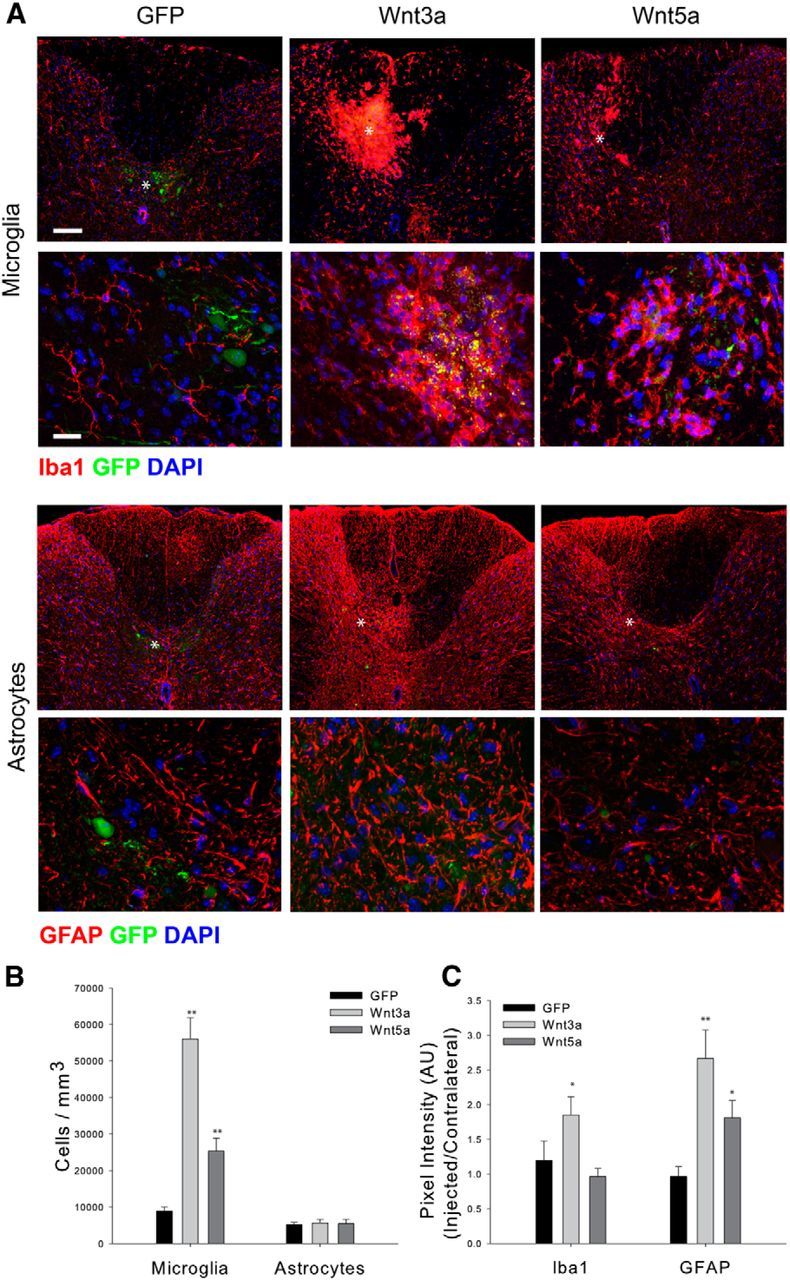
Exogenous Wnts activate microglia/macrophages and induce astrocyte hypertrophy. A, The appearance of Iba1-positive (red) microglia/macrophages and GFAP-positive astrocytes after AAV virus injection. The second and fourth row show higher power views of the cells. B, Quantitation of cell density after virus injection. Microglia but not astrocytes are increased in density after Wnt3a virus injection. C, Pixel intensity for the indicated antigens. Wnt3a induces increases in the expression of Iba1 and GFAP. The effects of Wnt5a are more modest than those of Wnt3a. Wnt3a, n = 8; Wnt5a, n = 4; GFP, n = 6; **p = 0.001, *p = 0.01. Scale bars: A, top, 50 μm; A, second row, 10 μm. White asterisk indicates centroid of the injection site.
Last, to confirm that canonical Wnt signaling is mitogenic to OPCs, we grew neonatal rat OPCs in culture, treated the cells with media conditioned by L-cells secreting either Wnt3a or Wnt5a, and measured BrdU incorporation into the cells. As shown in Figure 11, exposure to Wnt3a-conditioned media but not Wnt5a-conditioned media stimulated BrdU incorporation into NG2+ OPCs. Purified soluble Wnt3a was also an effective mitogen and its effects were blocked by soluble Frizzled5-CRD-hFc, which blocks the binding of Wnts to their LRP receptors (Ishikawa et al., 2001; Yamamoto et al., 2006; DeAlmeida et al., 2007). Neither Wnt3a nor Wnt5a stimulated BrdU incorporation into O4+ pre-oligodendrocytes.
Figure 11.
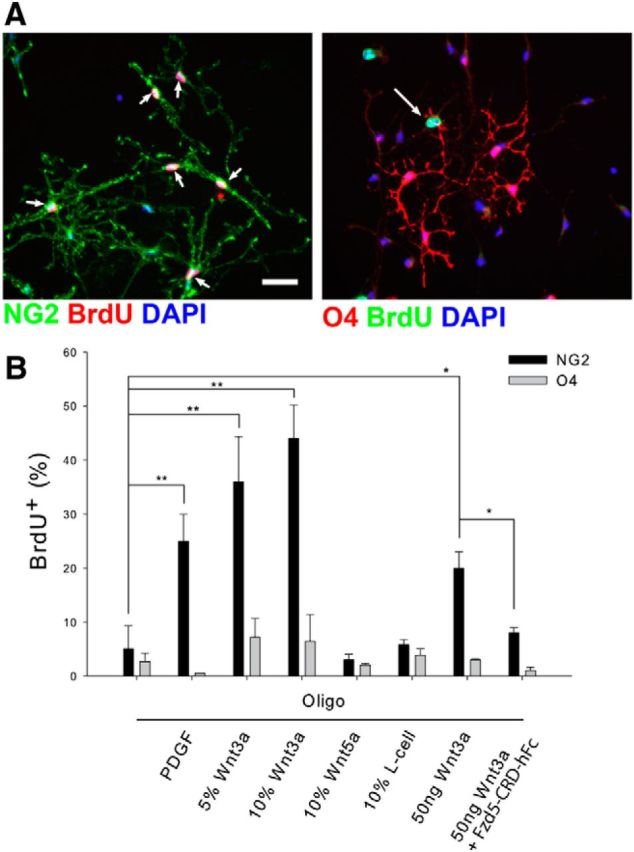
Wnt3a is mitogenic for OPCs. A, NG2+ OPCs but not 04+ pre-oligodendrocytes incorporate BrdU after exposure to canonical Wnt3a. Arrows point to double-labeled cells. Scale bar, 50 μm. B, Quantitation of BrdU incorporation for OPCs grown in oligodendrocyte media containing the indicated additives. Addition of an IgG-Frizzled-CRD fusion protein inhibits the effect of Wnt3a; *p = 0.02, **p < 0.001, where not indicated, results were not significant, n ≥ 4 in all cases. A minimum of 150 cells were counted for each condition in each experiment.
Discussion
Glial cells react to injury of the brain or spinal cord by forming a complex scar tissue at the injury site. This scar is both beneficial and detrimental to wound healing and damage repair (Rolls et al., 2009). In the early stages of wound healing, astrocytes form a barrier that keeps invading cells, mostly macrophages and leukocytes, out of the undamaged areas (Faulkner et al., 2004; Hermann et al., 2008). They are also important factors in the re-establishment of the blood–brain barrier. At later stages, however, astrocytes, together with degenerating oligodendrocytes, OPCs, and meningeal cells, create a biochemical environment that is nonpermissive for axon regeneration (Fitch and Silver, 2008). Although this suggests that preventing scar formation might promote axonal regeneration (Fawcett et al., 2012), ablating reactive astrocytes or suppressing their reaction to injury leads paradoxically to more extensive damage and reduced repair, most likely because of an increased early inflammatory response (Faulkner et al., 2004; Hermann et al., 2008). Here we have shown that preventing canonical Wnt signaling in OPCs not only suppresses their reactions to the injury but also reduces the reactions of microglia and astrocytes. Together these reduced glial reactions create an environment that is supportive rather than repressive to axonal regeneration and repair.
Wnts are secreted morphogens known mostly for their role as regulators of pattern formation and cell fate during early development (Moon et al., 1997). They also have complex effects on developing oligodendrocyte precursor cells. Canonical Wnt signaling delays the differentiation of OPCs into myelinating oligodendrocytes (Shimizu et al., 2005; Fancy et al., 2009; Feigenson et al., 2009), but it also upregulates the expression of proteolipid protein in mouse oligodendrocytes (Tawk et al., 2011). In the uninjured adult CNS, Wnts regulate adult neurogenesis in the dentate gyrus (Lie et al., 2005) and multiple aspects of synaptic transmission (Inestrosa and Arenas, 2010). Canonical Wnt signaling is also mitogenic to those adult neural stem cells that specifically generate oligodendrocytes (Ortega et al., 2013). Although there is ongoing β-catenin-dependent Wnt signaling in adult OPCs (White et al., 2010; Fig. 2), reducing this signaling in OPCs by deleting β-catenin did not alter either the density or appearance of OPCs at least over a 25 d time course. Thus β-catenin-dependent signaling is not necessary for the normal slow proliferation of adult OPCs. The binding of platelet-derived growth factor, a major mitogen for OPCs, to its α-receptor in developing OPCs activates both Wnt-β-catenin-dependent and PI3K-mTor-dependent signaling pathways, both of which can drive cell proliferation (Hill et al., 2013). Thus, the Wnt pathway may be dispensable for basal proliferation. In contrast, β-catenin signaling is necessary for the injury-induced rapid proliferation of OPCs and increasing Wnt3a expression in the uninjured spinal cord is sufficient to initiate proliferation and a reactive response in OPCs. Thus increases in the expression of Wnts and their receptors after SCI (Liu et al., 2008b; Miyashita et al., 2009; Fernández-Martos et al., 2011; Gonzalez et al., 2012) are likely responsible, at least in part, for the rapid proliferation and mobilization of OPCs after injury.
Eliminating β-catenin-dependent signaling in OPCs had striking and unexpected consequences for the reactions to injury of other glial cells. Both microglia/macrophage accumulation and astrocyte hypertrophy were significantly reduced. This suggests that the OPC reactive response regulates the responses of microglia/macrophages. We can only speculate as to the mechanisms underlying these cell nonautonomous effects on microglia. First, CSPGs can directly activate microglia in vitro (Rolls et al., 2008). Reactive OPCs and astrocytes are major sources of secreted and cell-associated CSPGs after injury (Levine, 1994; Asher et al., 2000) and the expression of CSPGs is reduced in the β-catenin KO animals. Consistent with the notion that reducing CSPGs can reduce microglial reactions to injury is the finding that there is reduced inflammation after lysolecithin-induced demyelination in the spinal cords of NG2-null mice (Kucharova et al., 2011). Second, canonical Wnt signaling activates microglia and increases the expression of pro-inflammatory cytokines in vitro (Halleskog et al., 2011). As demonstrated here (Fig. 10), Wnt3a causes a robust activation and accumulation of microglia in vivo. Oligodendrocyte lineage cells express high levels of Wnt3 mRNA (Ortega et al., 2013) and Wnt mRNA is detected in NG2-positive cells after SCI (Liu et al., 2008a). This suggests that by reducing the number of reactive OPCs, we may have reduced Wnt tone and availability to other cells, including microglia, in the injured area. The reduced number of axin2+ cells throughout the injured spinal cord (Fig. 2) is consistent with this idea. Third, because the same percentage of microglia incorporated BrdU in both the control and KO animals, β-catenin deletion in OPCs is likely to affect the mobilization and migration of microglia and macrophages into the glial scar rather than their proliferation. Reactive OPCs secrete MMP9, which can open the blood–brain barrier (Seo et al., 2013), and they are a potential source of chemokines and chemoattractants for inflammatory cells. Reducing any of these functions would attenuate the inflammatory response. Regardless of the exact mechanism, it is clear that there are complex interactions between activated OPCs and activated microglia/macrophages (Wu et al., 2002). Cytokines produced by activated microglia and platelets induce reactive changes in OPCs (Rhodes et al., 2006) and these reactive changes, together with an increase in OPC number, are necessary for the accumulation and activation of microglia. Deleting β-catenin-dependent signaling in OPCs appears to disrupt these complex interactions leading to both a reduced accumulation of OPCs and a reduced inflammatory response to SCI.
Since astrocyte hypertrophy may be induced by activated microglia (Emmetsberger and Tsirka, 2012), the attenuation of microglia activation in the KO animals could lead to the reduced astrocyte hypertrophy and reduced expression of GFAP. In the injured normal spinal cord, the barrier formed by reactive astrocytes in the early stages of wound healing confines microglia/macrophages to the injured areas (Faulkner et al., 2004). In the β-catenin KO animals, although the barrier between the injured spinal cord and non-neural tissue and cells is reduced and there are fewer hypertrophic astrocytes (Fig. 5), inflammation is also reduced. Thus manipulating the OPC reaction to injury has dramatic effects on the overall development and characteristics of the glial scar. The activation of OPCs is a common feature of several different types of CNS injury (Levine et al., 2001). Perturbing this activation by reducing canonical Wnt signaling has revealed an essential role for these cells in orchestrating the responses of several different cell types to injury.
One technical question that arises is how can a relatively small number of induced cells have such a striking effect on the terrain of the damaged spinal cord. Recombination efficiency at any specific locus is influenced by many factors including the local concentration of tamoxifen, the kinetics of nuclear import of cre, the distance between LoxP sites, and local chromatin structure (Vooijs et al., 2001). Although only ∼12–15% of the NG2-positive OPCs in the uninjured spinal cord express YFP after tamoxifen treatment, this is likely to be an underestimation of the true number of cells that have either undergone recombination at the β-catenin locus or failed to activate Wnt-dependent intracellular signaling pathways. First, overall axin2 immunoreactivity, a direct readout of Wnt signaling, is greatly reduced in the KO spinal cords compared with control animals (Fig. 2). Second, we estimate that ∼63% of the NG2+ cells at 5 dpl in control animals express axin2 whereas only 40% of the NG2+ cells in the KO animals are axin2+. Considering that injured control animals have three times more NG2+ cells in the penumbral region at 5 dpl than do the β-catenin-KO animals, we calculate that there are ∼4-fold fewer Wnt responding OPCs per unit volume surrounding the injury site in the KO animals relative to control animals. This reduction is sufficient to reduce the extent of glial scarring. Third, when we compared the percentage of YFP-positive cells labeled with BrdU after a pulse at 2 and 5 d post injury to the percentage of all the NG2-positive cells (both YFP positive and YFP negative) that had incorporated BrdU, the numbers were identical. Thus the total NG2-positive cell population behaved identically to a smaller YFP-positive population. These considerations suggest that recombination at the Rosa26 locus is less efficient than recombination at the β-catenin locus and we had, in fact, deleted β-catenin from large numbers of OPCs. It is important to note that by reducing overall Wnt tone (see above), we may have also reduced the injury responses of those OPCs that did not undergo recombination at the β-catenin locus.
The glial scar is considered both a physical and chemical barrier to successful axon regrowth (Fitch and Silver, 2008). Numerous factors contribute to this barrier function including the secretion of CSPGs by astrocytes and reactive OPCs (Asher et al., 2000; Tan et al., 2006), inhibitory molecules contained within myelin debris (Lee and Zheng, 2012), repulsive axon guidance molecules secreted by invading meningeal cells (De Winter et al., 2002), and contact repulsion mediated by invading macrophages (Horn et al., 2008). By eliminating β-catenin-dependent signaling in OPCs, we created an environment that is permissive to axon regeneration even in the absence of manipulating the growth state of the retinal ganglion cells. This is consistent with the notion that adult neurons are not intrinsically incapable of regeneration, rather the environment of the damaged CNS prevents regeneration (David and Aguayo, 1981). Targeting Wnt signaling either alone or combined with other approaches (Lu et al., 2012) may be a basis for designing new therapies for encouraging axonal repair and regeneration.
Footnotes
This work was supported by an NMSS Collaborative Research Center Grant to J.M.L., National Institutes of Health (NIH) Grant HL107493 to K.-I.T., and an NIH predoctoral fellowship to J.P.R.
The authors declare no competing financial interests.
References
- Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE, Levine JM, Margolis RU, Rogers JH, Fawcett JW. Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J Neurosci. 2000;20:2427–2438. doi: 10.1523/JNEUROSCI.20-07-02427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz LI, Yin Y. Optic nerve regeneration. Arch Ophthalmol. 2010;128:1059–1064. doi: 10.1001/archophthalmol.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/S0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Cadigan KM, Liu YI. Wnt signaling: complexity at the surface. J Cell Sci. 2006;119:395–402. doi: 10.1242/jcs.02826. [DOI] [PubMed] [Google Scholar]
- Dahl D, Bignami A. Heterogeneity of the glial fibrillary acidic protein in gliosed human brains. J Neurol Sci. 1974;23:551–563. doi: 10.1016/0022-510X(74)90027-6. [DOI] [PubMed] [Google Scholar]
- David S, Aguayo AJ. Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Science. 1981;214:931–933. doi: 10.1126/science.6171034. [DOI] [PubMed] [Google Scholar]
- DeAlmeida VI, Miao L, Ernst JA, Koeppen H, Polakis P, Rubinfeld B. The soluble wnt receptor Frizzled8CRD-hFc inhibits the growth of teratocarcinomas in vivo. Cancer Res. 2007;67:5371–5379. doi: 10.1158/0008-5472.CAN-07-0266. [DOI] [PubMed] [Google Scholar]
- De Winter F, Oudega M, Lankhorst AJ, Hamers FP, Blits B, Ruitenberg MJ, Pasterkamp RJ, Gispen WH, Verhaagen J. Injury-induced class 3 semaphorin expression in the rat spinal cord. Exp Neurol. 2002;175:61–75. doi: 10.1006/exnr.2002.7884. [DOI] [PubMed] [Google Scholar]
- Donnelly EM, Madigan NN, Rooney GE, Knight A, Chen B, Ball B, Kinnavane L, Garcia Y, Dockery P, Fraher J, Strappe PM, Windebank AJ, O'Brien T, McMahon SS. Lentiviral vector delivery of short hairpin RNA to NG2 and neurotrophin-3 promotes locomotor recovery in injured rat spinal cord. Cytotherapy. 2012;14:1235–1244. doi: 10.3109/14653249.2012.714865. [DOI] [PubMed] [Google Scholar]
- Emmetsberger J, Tsirka SE. Microglial inhibitory factor (MIF/TKP) mitigates secondary damage following spinal cord injury. Neurobiol Dis. 2012;47:295–309. doi: 10.1016/j.nbd.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertürk A, Mauch CP, Hellal F, Förstner F, Keck T, Becker K, Jährling N, Steffens H, Richter M, Hübener M, Kramer E, Kirchhoff F, Dodt HU, Bradke F. Three-dimensional imaging of the unsectioned adult spinal cord to assess axon regeneration and glial responses after injury. Nat Med. 2012;18:166–171. doi: 10.1038/nm.2600. [DOI] [PubMed] [Google Scholar]
- Fancy SP, Baranzini SE, Zhao C, Yuk DI, Irvine KA, Kaing S, Sanai N, Franklin RJ, Rowitch DH. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009;23:1571–1585. doi: 10.1101/gad.1806309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JW, Schwab ME, Montani L, Brazda N, Müller HW. Defeating inhibition of regeneration by scar and myelin components. Handb Clin Neurol. 2012;109:503–522. doi: 10.1016/B978-0-444-52137-8.00031-0. [DOI] [PubMed] [Google Scholar]
- Feigenson K, Reid M, See J, Crenshaw EB, 3rd, Grinspan JB. Wnt signaling is sufficient to perturb oligodendrocyte maturation. Mol Cell Neurosci. 2009;42:255–265. doi: 10.1016/j.mcn.2009.07.010. [DOI] [PubMed] [Google Scholar]
- Fernández-Martos CM, González-Fernández C, González P, Maqueda A, Arenas E, Rodríguez FJ. Differential expression of Wnts after spinal cord contusion injury in adult rats. PLoS One. 2011;6:e27000. doi: 10.1371/journal.pone.0027000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch MT, Silver J. CNS injury, glial scars, and inflammation: inhibitory extracellular matrices and regeneration failure. Exp Neurol. 2008;209:294–301. doi: 10.1016/j.expneurol.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Chen J, Ingeman JE, George JK, Noponen M. The role of mononuclear phagocytes in wound healing after traumatic injury to adult mammalian brain. J Neurosci. 1989;9:4416–4429. doi: 10.1523/JNEUROSCI.09-12-04416.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez P, Fernandez-Martos CM, Gonzalez-Fernandez C, Arenas E, Rodriguez FJ. Spatio-temporal expression pattern of frizzled receptors after contusive spinal cord injury in adult rats. PLoS One. 2012;7:e50793. doi: 10.1371/journal.pone.0050793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen HJ. Stereology of arbitrary particles. A review of unbiased number and size estimators and the presentation of some new ones, in memory of William R. Thompson. J Microsc. 1986;143:3–45. doi: 10.1111/j.1365-2818.1986.tb02764.x. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB, Kiêu K, Nielsen J. The efficiency of systematic sampling in stereology–reconsidered. J Microsc. 1999;193:199–211. doi: 10.1046/j.1365-2818.1999.00457.x. [DOI] [PubMed] [Google Scholar]
- Halleskog C, Mulder J, Dahlström J, Mackie K, Hortobágyi T, Tanila H, Kumar Puli L, Färber K, Harkany T, Schulte G. WNT signaling in activated microglia is proinflammatory. Glia. 2011;59:119–131. doi: 10.1002/glia.21081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann PM, Nicol JJ, Bulloch AG, Wildering WC. RGD-dependent mechanisms in the endoneurial phagocyte response and axonal regeneration in the nervous system of the snail Lymnaea stagnalis. J Exp Biol. 2008;211:491–501. doi: 10.1242/jeb.013102. [DOI] [PubMed] [Google Scholar]
- Hill RA, Patel KD, Medved J, Reiss AM, Nishiyama A. NG2 cells in white matter but not gray matter proliferate in response to PDGF. J Neurosci. 2013;33:14558–14566. doi: 10.1523/JNEUROSCI.2001-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn KP, Busch SA, Hawthorne AL, van Rooijen N, Silver J. Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci. 2008;28:9330–9341. doi: 10.1523/JNEUROSCI.2488-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inestrosa NC, Arenas E. Emerging roles of Wnts in the adult nervous system. Nat Rev Neurosci. 2010;11:77–86. doi: 10.1038/nrn2755. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Tamai Y, Zorn AM, Yoshida H, Seldin MF, Nishikawa S, Taketo MM. Mouse Wnt receptor gene Fzd5 is essential for yolk sac and placental angiogenesis. Development. 2001;128:25–33. doi: 10.1242/dev.128.1.25. [DOI] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilander MB, Halleskog C, Schulte G. Recombinant WNTs differentially activate beta-catenin-dependent and -independent signalling in mouse microglia-like cells. Acta Physiol. 2011;203:363–372. doi: 10.1111/j.1748-1716.2011.02324.x. [DOI] [PubMed] [Google Scholar]
- Kimelman D, Xu W. Beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene. 2006;25:7482–7491. doi: 10.1038/sj.onc.1210055. [DOI] [PubMed] [Google Scholar]
- Kucharova K, Chang Y, Boor A, Yong VW, Stallcup WB. Reduced inflammation accompanies diminished myelin damage and repair in the NG2 null mouse spinal cord. J Neuroinflammation. 2011;8:158. doi: 10.1186/1742-2094-8-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimoto T, Yin Y, Omura K, Gilbert HY, Kim D, Cen LP, Moko L, Kügler S, Benowitz LI. Long-distance axon regeneration in the mature optic nerve: contributions of oncomodulin, cAMP, and pten gene deletion. J Neurosci. 2010;30:15654–15663. doi: 10.1523/JNEUROSCI.4340-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Zheng B. Role of myelin-associated inhibitors in axonal repair after spinal cord injury. Exp Neurol. 2012;235:33–42. doi: 10.1016/j.expneurol.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon S, Yin Y, Nguyen J, Irwin N, Benowitz LI. Lens injury stimulates axon regeneration in the mature rat optic nerve. J Neurosci. 2000;20:4615–4626. doi: 10.1523/JNEUROSCI.20-12-04615.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JM. Increased expression of the NG2 chondroitin-sulfate proteoglycan after brain injury. J Neurosci. 1994;14:4716–4730. doi: 10.1523/JNEUROSCI.14-08-04716.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JM, Stincone F, Lee YS. Development and differentiation of glial precursor cells in the rat cerebellum. Glia. 1993;7:307–321. doi: 10.1002/glia.440070406. [DOI] [PubMed] [Google Scholar]
- Levine JM, Reynolds R, Fawcett JW. The oligodendrocyte precursor cell in health and disease. Trends Neurosci. 2001;24:39–47. doi: 10.1016/S0166-2236(00)01691-X. [DOI] [PubMed] [Google Scholar]
- Lie DC, Colamarino SA, Song HJ, Désiré L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437:1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- Liu C, Wang Y, Smallwood PM, Nathans J. An essential role for Frizzled5 in neuronal survival in the parafascicular nucleus of the thalamus. J Neurosci. 2008a;28:5641–5653. doi: 10.1523/JNEUROSCI.1056-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang X, Lu CC, Kerman R, Steward O, Xu XM, Zou Y. Repulsive Wnt signaling inhibits axon regeneration after CNS injury. J Neurosci. 2008b;28:8376–8382. doi: 10.1523/JNEUROSCI.1939-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Lin C, King TD, Chen H, Reynolds RC, Li Y. Silibinin inhibits Wnt/beta-catenin signaling by suppressing Wnt co-receptor LRP6 expression in human prostate and breast cancer cells. Cell Signal. 2012;24:2291–2296. doi: 10.1016/j.cellsig.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, van de Wetering M, Clevers H, Schlag PM, Birchmeier W, Behrens J. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol. 2002;22:1184–1193. doi: 10.1128/MCB.22.4.1184-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytle JM, Wrathall JR. Glial cell loss, proliferation and replacement in the contused murine spinal cord. Eur J Neurosci. 2007;25:1711–1724. doi: 10.1111/j.1460-9568.2007.05390.x. [DOI] [PubMed] [Google Scholar]
- Lytle JM, Chittajallu R, Wrathall JR, Gallo V. NG2 cell response in the CNP-EGFP mouse after contusive spinal cord injury. Glia. 2009;57:270–285. doi: 10.1002/glia.20755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue DM, Wei P, Stokes BT. Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord. J Neurosci. 2001;21:3392–3400. doi: 10.1523/JNEUROSCI.21-10-03392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer RL, Miotke J. Rapid initiation of neurite outgrowth onto laminin from explants of adult mouse retina induced by optic nerve crush. Exp Neurol. 1990;107:214–221. doi: 10.1016/0014-4886(90)90138-I. [DOI] [PubMed] [Google Scholar]
- Meyer RL, Miotke JA, Benowitz LI. Injury induced expression of growth-associated protein-43 in adult mouse retinal ganglion cells in vitro. Neuroscience. 1994;63:591–602. doi: 10.1016/0306-4522(94)90552-5. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Koda M, Kitajo K, Yamazaki M, Takahashi K, Kikuchi A, Yamashita T. Wnt-Ryk signaling mediates axon growth inhibition and limits functional recovery after spinal cord injury. J Neurotrauma. 2009;26:955–964. doi: 10.1089/neu.2008.0776. [DOI] [PubMed] [Google Scholar]
- Moon RT, Brown JD, Torres M. WNTs modulate cell fate and behavior during vertebrate development. Trends Genet. 1997;13:157–162. doi: 10.1016/S0168-9525(97)01093-7. [DOI] [PubMed] [Google Scholar]
- Morgan JM, Navabi H, Schmid KW, Jasani B. Possible role of tissue-bound calcium ions in citrate-mediated high-temperature antigen retrieval. J Pathol. 1994;174:301–307. doi: 10.1002/path.1711740410. [DOI] [PubMed] [Google Scholar]
- Morgan JM, Navabi H, Jasani B. Role of calcium chelation in high-temperature antigen retrieval at different pH values. J Pathol. 1997;182:233–237. doi: 10.1002/(SICI)1096-9896(199706)182:2<233::AID-PATH827>3.0.CO%3B2-Y. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. 2009;10:9–22. doi: 10.1038/nrn2495. [DOI] [PubMed] [Google Scholar]
- Ortega F, Gascón S, Masserdotti G, Deshpande A, Simon C, Fischer J, Dimou L, Chichung Lie D, Schroeder T, Berninger B. Oligodendrogliogenic and neurogenic adult subependymal zone neural stem cells constitute distinct lineages and exhibit differential responsiveness to Wnt signalling. Nat Cell Biol. 2013;15:602–613. doi: 10.1038/ncb2736. [DOI] [PubMed] [Google Scholar]
- Paukert M, Bergles DE. Synaptic communication between neurons and NG2+ cells. Curr Opin Neurobiol. 2006;16:515–521. doi: 10.1016/j.conb.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Petrosyan HA, Hunanyan AS, Alessi V, Schnell L, Levine J, Arvanian VL. Neutralization of inhibitory molecule NG2 improves synaptic transmission, retrograde transport, and locomotor function after spinal cord injury in adult rats. J Neurosci. 2013;33:4032–4043. doi: 10.1523/JNEUROSCI.4702-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes KE, Raivich G, Fawcett JW. The injury response of oligodendrocyte precursor cells is induced by platelets, macrophages and inflammation-associated cytokines. Neuroscience. 2006;140:87–100. doi: 10.1016/j.neuroscience.2006.01.055. [DOI] [PubMed] [Google Scholar]
- Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls A, Shechter R, London A, Segev Y, Jacob-Hirsch J, Amariglio N, Rechavi G, Schwartz M. Two faces of chondroitin sulfate proteoglycan in spinal cord repair: a role in microglia/macrophage activation. PLoS Med. 2008;5:e171. doi: 10.1371/journal.pmed.0050171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls A, Shechter R, Schwartz M. The bright side of the glial scar in CNS repair. Nat Rev Neurosci. 2009;10:235–241. doi: 10.1038/nrn2591. [DOI] [PubMed] [Google Scholar]
- Seo JH, Miyamoto N, Hayakawa K, Pham LD, Maki T, Ayata C, Kim KW, Lo EH, Arai K. Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J Clin Invest. 2013;123:782–786. doi: 10.1172/JCI65863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Kagawa T, Wada T, Muroyama Y, Takada S, Ikenaka K. Wnt signaling controls the timing of oligodendrocyte development in the spinal cord. Dev Biol. 2005;282:397–410. doi: 10.1016/j.ydbio.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Simon C, Götz M, Dimou L. Progenitors in the adult cerebral cortex: cell cycle properties and regulation by physiological stimuli and injury. Glia. 2011;59:869–881. doi: 10.1002/glia.21156. [DOI] [PubMed] [Google Scholar]
- Sommer JM, Smith PH, Parthasarathy S, Isaacs J, Vijay S, Kieran J, Powell SK, McClelland A, Wright JF. Quantification of adeno-associated virus particles and empty capsids by optical density measurement. Mol Ther. 2003;7:122–128. doi: 10.1016/S1525-0016(02)00019-9. [DOI] [PubMed] [Google Scholar]
- Tan AM, Colletti M, Rorai AT, Skene JH, Levine JM. Antibodies against the NG2 proteoglycan promote the regeneration of sensory axons within the dorsal columns of the spinal cord. J Neurosci. 2006;26:4729–4739. doi: 10.1523/JNEUROSCI.3900-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Falls DL, Li X, Lane T, Luskin MB. Antigen-retrieval procedure for bromodeoxyuridine immunolabeling with concurrent labeling of nuclear DNA and antigens damaged by HCl pretreatment. J Neurosci. 2007;27:5837–5844. doi: 10.1523/JNEUROSCI.5048-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawk M, Makoukji J, Belle M, Fonte C, Trousson A, Hawkins T, Li H, Ghandour S, Schumacher M, Massaad C. Wnt/beta-catenin signaling is an essential and direct driver of myelin gene expression and myelinogenesis. J Neurosci. 2011;31:3729–3742. doi: 10.1523/JNEUROSCI.4270-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenta T, Hausmann G, Basler K. The many faces and functions of beta-catenin. EMBO J. 2012;31:2714–2736. doi: 10.1038/emboj.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vooijs M, Jonkers J, Berns A. A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO Rep. 2001;2:292–297. doi: 10.1093/embo-reports/kve064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RE, McTigue DM, Jakeman LB. Regional heterogeneity in astrocyte responses following contusive spinal cord injury in mice. J Comp Neurol. 2010;518:1370–1390. doi: 10.1002/cne.22282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Komekado H, Kikuchi A. Caveolin is necessary for Wnt-3a-dependent internalization of LRP6 and accumulation of beta-catenin. Dev Cell. 2006;11:213–223. doi: 10.1016/j.devcel.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Yan D, Wiesmann M, Rohan M, Chan V, Jefferson AB, Guo L, Sakamoto D, Caothien RH, Fuller JH, Reinhard C, Garcia PD, Randazzo FM, Escobedo J, Fantl WJ, Williams LT. Elevated expression of axin2 and hnkd mRNA provides evidence that Wnt/beta-catenin signaling is activated in human colon tumors. Proc Natl Acad Sci U S A. 2001;98:14973–14978. doi: 10.1073/pnas.261574498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Watanabe M, Nishiyama A. Optimization of oligodendrocyte progenitor cell culture method for enhanced survival. J Neurosci Methods. 2005;149:50–56. doi: 10.1016/j.jneumeth.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Young KM, Psachoulia K, Tripathi RB, Dunn SJ, Cossell L, Attwell D, Tohyama K, Richardson WD. Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron. 2013;77:873–885. doi: 10.1016/j.neuron.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawadzka M, Rivers LE, Fancy SP, Zhao C, Tripathi R, Jamen F, Young K, Goncharevich A, Pohl H, Rizzi M, Rowitch DH, Kessaris N, Suter U, Richardson WD, Franklin RJ. CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell. 2010;6:578–590. doi: 10.1016/j.stem.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]