

Abstract
Brain-derived neurotrophic factor (BDNF) is produced by developing and mature gastrointestinal (GI) tissues that are heavily innervated by autonomic neurons and may therefore control their development or function. To begin investigating this hypothesis, we compared the morphology, distribution, and density of intraganglionic laminar endings (IGLEs), the predominant vagal GI afferent, in mice with reduced intestinal BDNF (INT-BDNF−/−) and controls. Contrary to expectations of reduced development, IGLE density and longitudinal axon bundle number in the intestine of INT-BDNF−/− mice were increased, but stomach IGLEs were normal. INT-BDNF−/− mice also exhibited increased vagal sensory neuron numbers, suggesting that their survival was enhanced. To determine whether increased intestinal IGLE density or other changes to gut innervation in INT-BDNF−/− mice altered feeding behavior, meal pattern and microstructural analyses were performed. INT-BDNF−/− mice ate meals of much shorter duration than controls, resulting in reduced meal size. Increased suppression of feeding in INT-BDNF−/− mice during the late phase of a scheduled meal suggested that increased satiation signaling contributed to reduced meal duration and size. Furthermore, INT-BDNF−/− mice demonstrated increases in total daily intermeal interval and satiety ratio, suggesting that satiety signaling was augmented. Compensatory responses maintained normal daily food intake and body weight in INT-BDNF−/− mice. These findings suggest a target organ-derived neurotrophin suppresses development of that organ's sensory innervation and sensory neuron survival and demonstrate a role for BDNF produced by peripheral tissues in short-term controls of feeding, likely through its regulation of development or function of gut innervation, possibly including augmented intestinal IGLE innervation.
Keywords: vagus nerve, nodose ganglion, ingestive behavior, meal patterns, growth factor, food intake
Introduction
Brain-derived neurotrophic factor (BDNF) has numerous roles in neural development and function, including potent anorexigenic activity in the CNS (Rios et al., 2001). Several developing and mature peripheral tissues also express BDNF, including those of the gastrointestinal (GI) tract, some at even higher levels than in brain (Lommatzsch et al., 1999; Fox, 2006; Fox and Murphy, 2008; Fox et al., 2013a). However, the anorexigenic potential of GI BDNF has not been explored. The gut is innervated by several pathways that influence feeding directly or indirectly through regulation of GI reflexes, including vagal afferents and efferents, sympathetic efferents, dorsal root afferents, and enteric neurons (Wood et al., 1999). Some of these pathways express BDNF or its high-affinity receptor tyrosine kinase B (trkB), and therefore, changes in GI BDNF levels could alter the development or function of these pathways and consequently modify their effects on feeding (Coulie et al., 2000; Chai et al., 2003; Sclafani et al., 2003; Grider et al., 2006; Boesmans et al., 2008; Fu et al., 2011).
One of these pathways, vagal afferents, expresses trkB during development and maturity (Ernfors et al., 1992; Wetmore and Olson, 1995), and BDNF knock-out (KO) results in substantial loss of vagal afferents and disrupts survival or formation of vagal mechanoreceptors that supply the neonatal stomach wall (Jones et al., 1994; Erickson et al., 1996; Fox and Murphy, 2008; Murphy and Fox, 2010). These findings suggest that some vagal afferents may depend on GI BDNF for survival or other aspects of development. Additionally, BDNF loss from mature GI tissues could alter vagal GI afferents by perturbing their maintenance as demonstrated for vagal baroreceptors or by disrupting their sensory transduction as shown for some cutaneous mechanoreceptors (Carroll et al., 1998; Postigo et al., 2002). Among vagal GI afferents, intraganglionic laminar endings (IGLEs) are the predominant vagal mechanoreceptor innervating GI smooth muscle (SM) and are concentrated in the region of the upper GI tract in which most food collects during consumption of a meal (Fox et al., 2000; Wang and Powley, 2000). They consist of plates of densely packed terminal puncta that innervate myenteric ganglia and are activated by muscle wall tension or stretch and possibly by satiation hormones, such as CCK (Rodrigo et al., 1982; Schwartz and Moran, 1994; Zagorodnyuk et al., 2001).
Here, we have begun to explore the functions of GI BDNF by testing the hypotheses that it supports survival or maintenance of IGLEs and has anorexigenic properties. Mice with reduced BDNF levels restricted primarily to the intestine were used. By decreasing the number of tissues experiencing reduced BDNF, this approach bypassed the perinatal lethality of global BDNF KOs, enabling investigation of IGLEs and feeding in mature animals. Our goals were to characterize the effects of reduced intestinal BDNF levels on structure, distribution, and density of IGLEs and on daily food intake, body weight, meal patterns, and microstructure. We predicted that reduced intestinal BDNF would decrease IGLE survival, and this or other altered intestinal innervation would reduce satiation and thus increase meal size, an effect that could contribute to overeating.
Materials and Methods
Animals
SM22αcre (also referred to as transgelincre;Tg(Tagln-cre)1Her/J; catalog #004746; The Jackson Laboratory; Holtwick et al., 2002) and BDNF+/lox (Rios et al., 2001) mice were maintained at 23°C on a 12 h (14/10 h for breeding) light/dark schedule (lights on at 5:00 A.M.) with ad libitum access to tap water and Laboratory Rodent Diet 5001 (PMI Nutrition). All procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (eighth edition) and American Association for Accreditation of Laboratory Animal Care guidelines and were approved by the Purdue University Animal Care and Use Committee.
Use of SM22αcre driver strain to target BDNF KO to SM.
To study the affect of GI BDNF on the development of IGLEs, the SM22αcre mouse strain was used. This strain was chosen because Cre recombinase expression in SM22αcre mice is primarily restricted to SM, one GI tissue that expresses BDNF. Previous reports showed high recombination efficiency in SM with a Cre-dependent reporter strain (Lepore et al., 2005). In addition, our laboratory and others have used Rosa26 reporter mice to map the spatiotemporal expression of SM22αcre-mediated recombination at several developmental ages. These studies found that SM22αcre mice produced sufficient Cre expression to drive recombination in SM of the GI wall and associated blood vessels at ages when vagal GI afferents enter the gut (Pan et al., 2011; Fox et al., 2013a). This was determined to begin at approximately E13 and continued until at least E17, based on the pattern of β-galactosidase expression in the stomach and intestines. Furthermore, in embryos derived from crosses of SM22αcre and BDNFlox reporter mice, highly efficient Cre-mediated recombination was primarily restricted to SM of the walls of blood vessels supplying GI tract tissues (Fox et al., 2013a). Moreover, in embryos derived from crosses of SM22αcre and NT-3lox reporter mice, recombination was restricted to SM of the intestine wall and a small portion of the stomach wall, as well as to SM of GI blood vessels (Fox et al., 2013b).
We have not previously characterized recombination produced by the SM22αcre transgene in the adult GI tract. Nevertheless, the large degree of overlap between BDNF expression in the adult GI wall, which occurs throughout its SM (Lommatzsch et al., 1999), and the highly efficient SM22αcre-mediated recombination in the SM of the stomach and intestine in adults (Lepore et al., 2005), suggests that floxed BDNF coding sequences would be mostly eliminated from GI SM of adult offspring derived from matings of SM22αcre and BDNFlox mice in the present study. Also, global BDNF KO mice exhibit a fatal respiratory defect and die shortly after birth (Katz, 2005). However, a targeted BDNF KO produced using SM22αcre mice was predicted to be viable, and the mice should exhibit normal brainstem respiratory circuits because the global loss of BDNF that leads to these symptoms will be greatly reduced and primarily restricted to SM.
Generation of mice with BDNF KO targeted to SM.
To target the BDNF KO to SM, three generations of mice were used. First, SM22αcre/+;BDNF+/+ and SM22α+/+;BDNF+/lox mice were crossed to generate SM22αcre/+;BDNF+/lox mice. These SM22αcre/+;BDNF+/lox mice were then mated among themselves to obtain SM22αcre/+;BDNFlox/lox mice, which are homozygous SM-targeted BDNF KO mice. These mice are referred to as “INT-BDNF−/− mice” because the KO was mainly restricted to the intestine (see Results, Assessment of SM-targeted BDNF KO). These double-transgenic mice have a targeted genetic ablation of BDNF from embryonic and possibly mature SM and are distinguished from the triple-transgenic SM22α–BDNFKO mice we made previously in which one BDNF allele was deleted globally and the other one was removed only from SM (SM22αcre/+;BDNFneo/lox; Fox et al., 2013a). The current breeding strategy also resulted in heterozygous SM-targeted BDNF KO mice (INT-BDNF+/−). Four additional genotypes were obtained, including the following: (1) SM22α+/+;BDNF+/+ mice, which are wild-type (WT), with no transgenic elements; (2) SM22αcre/+;BDNF+/+ mice, which have the Cre recombinase transgene but no loxP sites flanking BDNF coding sequences; (3) SM22α+/+;BDNF+/lox mice, which have one floxed BDNF allele but no Cre recombinase transgene; and (4) SM22α+/+;BDNFlox/lox mice, which have both BDNF alleles floxed but have no Cre recombinase transgene. Animals with the Cre transgene only (SM22αcre/+;BDNF+/+) or one copy of the floxed BDNF allele only (SM22α+/+;BDNFlox/+) did not differ from WT mice in any variable tested (data not shown) and were therefore combined with WT mice in some instances to form the control group for more efficient animal use. Offspring genotypes were determined by PCR analysis of DNA extracted from tail tips removed at weaning.
Assessment of SM-targeted BDNF KO
We previously demonstrated efficacy of the SM22αcre transgene at targeting BDNF KO to GI SM of embryos as described above. However, it was unclear whether Cre-mediated recombination also occurred in the adult GI tract. Additionally, SM22αcre-mediated recombination was found to occur in the ventromedial hypothalamus (VMH), the dorsolateral frontal cortex, and the hippocampus (Fox et al., 2013a), brain regions that could influence feeding behavior. Therefore, we compared BDNF mRNA expression in tissues of INT-BDNF−/− mice with those of WTs to examine whether this manipulation reduced BDNF levels in the mature stomach and intestine, as well as the hypothalamus and a portion of the frontal cortex.
RNA extraction and cDNA synthesis.
RNA was extracted from the hypothalamus, prefrontal cortex, ventral stomach, and the first 2 cm of duodenum of adult WT (n = 10) and INT-BDNF−/− (n = 10) mice. Whole stomach and intestine walls were used because it was not possible to ensure total separation of adjacent layers in unfixed tissue. Mice were killed by cervical dislocation at 3–4 months of age to coincide with the anatomical and behavioral experiments in this study. Tissues were immediately dissected and then homogenized in TRIzol (Invitrogen), and RNA was extracted according to the protocol of the manufacturer. Each 1 μg of RNA sample was incubated with DNase1 (Invitrogen) to remove genomic DNA, and first-strand cDNA was synthesized using the Maxima enzyme mix (Thermo Fisher Scientific) in 20 μl PCR reactions according to the instructions of the manufacturer.
qRT-PCR.
To ensure that intact BDNF mRNA of the expected size was obtained, RT-PCR was performed. β-Actin cDNA from each sample was also amplified to assess the integrity of the isolated total RNA. Reverse transcriptase was omitted from duplicate samples after DNase treatment to confirm the removal of genomic DNA. After cDNA synthesis and DNase treatment, 2 μl of cDNA was amplified using the TaqPCRx DNA polymerase kit (Invitrogen), and products were visualized using standard gel electrophoresis. Primer sequences used included the following (Kawakami et al., 2002; Zermeno et al., 2009): BDNF forward, 5′-GAA GAG CTG CTG GAT GAG GAC-3′; BDNF reverse, 5′-TTC AGT TGG CCT TTT GAT ACC-3′; β-actin forward, 5′-TGG TGG GTA TGG GTC AGA AGG ACT C-3′; and β-actin reverse, 5′-CAT GGC TGG GGT GTT GAA GGT CTC A-3′.
To quantify the extent of BDNF mRNA loss from GI and brain samples, qPCR was also performed on all mRNA samples. Amplification of 1 μg of cDNA from the first-strand reaction was performed in triplicate using previously established protocols (Unger et al., 2007; Cordeira et al., 2010; Fox et al., 2013a). Based on these protocols, primer sequences used included the following: BDNF forward, 5′-GAA AGT CCC GGT ATC CAA AG-3′; BDNF reverse, 5′-CCA GCC AAT TCT CTT TTT-3′; β-actin forward, 5′-GGC TGT ATT CCCC TCC ATC G-3′; and β-actin reverse, 5′-CCA GTT GGT AAC AAT GCC ATG T-3′. All primers were optimized such that the correlation coefficient was 0.99–1.0 and the PCR efficiency was 95–100%. Real-time PCR amplification was performed using an iCycler and the iQ SYBR Green Supermix (Bio-Rad).
Anterograde labeling of vagal GI afferents
Injections of wheat-germ agglutinin–horseradish conjugate into the nodose ganglion.
To visualize vagal afferent axons and terminals in the GI tract of control, INT-BDNF+/−, and INT-BDNF−/− mice, the nerve tracer horseradish peroxidase conjugated to wheat-germ agglutinin (WGA–HRP) was injected into the left nodose ganglion (controls, n = 15; INT-BDNF+/−, n = 11; and INT-BDNF−/−, n = 12). This method was used because, to date, it has been the most successful for labeling consistent, large numbers of vagal sensory elements supplying the GI wall and, therefore, has been the most useful for quantification of vagal sensory axons and their terminal endings, as well as for making quantitative comparisons of these elements between groups of mutant and control animals (Fox et al., 2000, 2001a,b, 2002; Wang and Powley, 2000). The left nodose was chosen for injections because it was known to provide much greater innervation to the proximal intestine compared with the right nodose ganglion in mice and rats (Fox et al., 2000, 2001b; Wang and Powley, 2000). In the study in which this laterality was quantified in mouse, this difference was >10-fold for the left nodose compared with the right (∼550 vs 40 IGLEs, respectively; Fox et al., 2001b, their Fig. 9A). In contrast, the ventral and dorsal stomach halves each receive a similar density of innervation from the left and right nodose ganglia, respectively. Therefore, we focused on the left nodose ganglion, ventral stomach, and proximal intestine to assess sufficiently large populations of IGLEs in both stomach and intestine of the same mice. Whole mounts were not included in the analyses if vagal afferent innervation could not be adequately observed. For example, large portions of the stomach or intestine with unlabeled fibers indicated an incomplete injection. Also, sometimes vagal afferent fibers could not be visualized because of dense artifactual staining that can occur with this method, and these instances were also dropped from the analysis (Mesulam, 1978). Based on these criteria, data from three controls, two INT-BDNF+/−, and three INT-BDNF−/− mice that had been injected with WGA–HRP were not included in the analysis. Therefore, in this study, 79% of the animals injected were included in the final analysis (12 controls: 4 males, 8 females; 9 INT-BDNF+/−: 6 males, 3 females; and 9 INT-BDNF−/−: 5 males, 4 females).
Mice were anesthetized with intraperitoneal injections of a ketamine hydrochloride (75 mg/kg; Ketaset; Fort Dodge) and xylazine (50 mg/kg; Anased; Lloyd Laboratories) mixture. The left nodose ganglion was exposed, and WGA–HRP (0.5 μl, 4%; Vector Laboratories) was pressure injected (40 psi, 4 ms; PicoSpritzer III; General Valve Corporation) into the ganglion using a glass micropipette (inner diameter, 25 μm).
Tissue processing.
Twenty to 22 h after nodose ganglion injection, mice were anesthetized using Brevital sodium (120 mg/kg; JHP Pharmaceuticals) or the same ketamine–xylazine mixture described above. Mice were perfused for 5–10 min with 0.9% saline (35°C, pH 7.4) until the liver cleared, and then the stomach was expanded with 0.9% saline (35°C, pH 7.4). Next, the mice were fixed with chilled 3% paraformaldehyde (PFA)/0.75% glutaraldehyde at 4 ml/min for 30 min. The ventral half of the stomach and the first 8 cm of the intestine opened along the mesentery were processed as whole mounts as described previously (Fox et al., 2000). The SM was separated from the mucosa of the GI tract wall by sharp dissection using Dumont #7 forceps, processed using tetramethylbenzidine according to the protocol by Mesulam (1978), and mounted on gelatin-coated slides. Next, the tissues were flattened by placing weights on a slide covering the tissue for 30 min. They were then air dried overnight and cleared with xylene the following day. The slides were then coverslipped using DPX mounting medium (Sigma-Aldrich).
Quantification of vagal elements.
HRP-labeled vagal sensory elements, including IGLEs, free axons, and axon bundles present in the ventral stomach and first 8 cm of duodenum in INT-BDNF−/− and INT-BDNF+/− mutants and controls, were quantified using a previously characterized method (Fox et al., 2000). The ventral half of the stomach was tested in this study because the left nodose ganglion, which was the side injected with WGA–HRP in all mice, projects mainly to the ventral stomach in the mouse (Fox et al., 2000). Briefly, a sampling grid was used to normalize the varying size and shape of each stomach and then used to locate comparable sampling sites in which vagal elements were quantified. The sampling grid used for the stomach consisted of six rows and eight columns, resulting in 48 sampling sites, which accounted for 13% of the total stomach area. A 1 cm2 counting grid that consists of 100 1-mm2 squares placed in the ocular of the microscope was then used to quantify vagal elements at each sampling site. In contrast, the proximal 8 cm of the small intestine was sampled by aligning the counting grid with one of the edges of the longitudinal cut through the intestine wall and with the anterior edge of the tissue block where it was separated from the pylorus. The counting grid was moved sequentially along the entire width of the intestine, and vagal elements were counted at each of the sampling sites. This was repeated at 5 mm intervals moving caudally along the small intestine until the proximal 8 cm of its length had been sampled. This resulted in sampling at ∼80 different sites, covering 20% of the total area of this region of the small intestine.
Quantification of IGLEs.
Criteria for IGLE identification were as determined previously: an IGLE must consist of (1) a laminar aggregate of (2) fine terminal puncta (3) within the neuropil of a myenteric ganglion, and (4) must cover all or part of the myenteric ganglion (Rodrigo et al., 1975, 1982; Neuhuber, 1987; Fox et al., 2000). Counting of IGLEs was conducted at 100× magnification and verified at 200× when necessary. The number of squares of the counting grid that contained IGLEs were counted rather than attempting to count individual IGLE leaves, because overlapping IGLE leaves were difficult to identify. In particular, in the present study, because some intestinal IGLEs in mutants appeared larger than normal and were accompanied by what appeared to be increased numbers of axons, it was not possible to determine whether these larger IGLEs were composed of increased numbers of terminal puncta originating from a single axon or of multiple, overlapping IGLE leaves, arising from multiple axons. Thus, “IGLE density” as used here represents the number of counting grid squares within the sampled area, totaled and represented in number per square millimeter, that contained IGLE terminal puncta rather than the number of IGLEs per unit area. Density of IGLEs was analyzed for the entire ventral stomach wall and by stomach compartment. Based on the rows and columns calculated for the sampling grid, IGLEs in the three most proximal columns represented the forestomach, the middle three columns represented the corpus, and the two most distal columns represented the antrum.
Quantification of intestinal vagal sensory axon bundles and individual axons.
To aid interpretation of the mechanisms underlying increased IGLE innervation of the small intestine (see Results), individual intestinal fibers and fiber bundles running in either circular or longitudinal orientations in the intestine of controls (n = 9) and INT-BDNF−/− mice (n = 9) were quantified. Three controls were dropped from analysis because their fiber bundles were not labeled well enough to obtain meaningful counts. Note that dropping these animals did not significantly change the mean IGLE numbers for this group. Moreover, this reduced the magnitude of the difference in fiber bundle counts between mutants and controls. A single axon fiber was defined as an axon that was not contained within a vagal fiber bundle and was not closely apposed to other axons. A bundle was defined as two or more axons coursing together in parallel and in close apposition. Each time a circular or longitudinal axon fiber or axon bundle crossed the top or left sides of the squares of the counting grid at each sampling site, it was counted.
Quantification of axon bundles grouped by diameter.
The diameters of intestinal fiber bundles in circular or longitudinal orientations of INT-BDNF−/− mice and controls were measured. Every axon bundle that crossed the top and left sides of each square of the counting grid was counted at each sampling site, and its diameter was measured. Bundle diameters were measured to the nearest 0.5 μm, because this was the smallest observable diameter bundle using the WGA–HRP nerve tracer. Therefore, measured bundles were grouped according to 0.5 μm increases in diameter, and those within each size range were quantified. Axon bundle diameters were measured by placing a micrometer scale in the ocular of the microscope, aligning it perpendicular to the long axis of each axon bundle and directly reading its width.
Nodose ganglion cell counts
After cardiac perfusion with chilled 4% PFA fixative, the right and left nodose ganglia were removed from WT (n = 9) and INT-BDNF−/− (n = 14) mice. Ganglia were postfixed in 4% PFA for 48 h and then transferred to 10% buffered Formalin for a minimum of 7 d. The ganglia were then embedded in paraffin, sectioned at a thickness of 10 μm, and stained with cresyl violet (0.5% cresyl violet acetate). The first section that contained a substantial number of stained neurons was counted, followed by every subsequent 10th section containing stained neurons that spanned the entire ganglion. Counting was done at 200× magnification to ensure that only neurons with a clear nucleus, stained nucleolus, and complete neuronal profile were counted. The average number of cells per section were obtained and then multiplied by the total number of sections per ganglion to estimate the total number of neurons per ganglion.
Body weight
Body weight measurements of all genotypes generated were initiated at weaning, when the animals were 3 weeks of age, and continued until meal pattern collection began at 3–4 months of age. Male and female mice were weighed once a week on the same day each week, except during the 22 d of the meal pattern experiments when mice were weighed daily. Sample sizes included in the long-term body weight measurements were as follows: for males, WT (n = 7), SM22αcre/+;BDNF+/+ (n = 10), SM22α+/+;BDNFlox/+ (n = 4), SM22α+/+;BDNFlox/lox (n = 6), INT-BDNF+/− (n = 10), and INT-BDNF−/− (n = 10); and for females, WT (n = 7), SM22αcre/+;BDNF+/+ (n = 10), SM22α+/+;BDNFlox/+ (n = 10), SM22α+/+;BDNFlox/lox (n = 2), INT-BDNF+/− (n = 10), and INT-BDNF−/− (n = 10). Seven male and six female WT mice, seven male and seven female INT-BDNF+/−, and seven male and eight female INT-BDNF−/− mice included in these body weight measurements were also used in the body composition and meal pattern analyses.
Body composition
Body composition was measured on the day before meal pattern testing was initiated, before fasting, when the animals were 3–4 months of age. All of the mice used in the body composition analysis were the same mice used for the meal pattern analysis experiment. Precise measurements of body composition parameters, including total body fat, lean mass, body fluids, and total body water were determined in WT (n = 13; 7 males, 6 females), INT-BDNF+/− (n = 14; 7 males, 7 females), and INT-BDNF−/− (n = 15; 7 males, 8 females) mice using an echo MRI whole-body composition analyzer (EchoMRI-900; Echo Medical Systems) in live mice without anesthesia (Taicher et al., 2003; Tinsley et al., 2004). Fat and lean mass were calculated as total grams and also as a percentage of total body mass.
Meal pattern analysis
A balanced diet (20 mg of dustless 380 precision pellets; Bio-Serv) was used in automated feeding machines used to examine meal patterns. The macronutrient composition of this diet was 22% protein, 66% carbohydrate, and 12% fat, with a caloric density of 3.623 kcal/g. This is comparable with the chow diet that was composed of 28% protein, 60% carbohydrate, and 12% fat, with a caloric density of 3.04 kcal/g. WT (n = 13), INT-BDNF+/− (n = 14), and INT-BDNF−/− (n = 15) mice at 3–4 months of age were housed individually in plastic cages equipped with automated pellet dispensers (Coulbourn Instruments) programmed and operated using Graphic State software (version 2.0; Coulbourn Instruments) that was also used for collection and analysis of eating patterns as described previously (Fox and Byerly, 2004; Biddinger and Fox, 2010). Each automated pellet dispenser was equipped with an infrared photobeam and sensor that checked for changes in photobeam state every 100 ms. A pellet sitting in the dispenser interrupted the beam. Each time the mouse removed a pellet from the dispenser, the photobeam was detected and another pellet was dispensed. Mice were adapted to the test room and cages for a minimum of 1 week before the start of meal pattern collection. During that week, animals received three limited preexposures to the test diet, each consisting of 10 of the Bio-Serv precision pellets, to prevent neophobia at the start of testing. All animals used in the experiment ate all 10 pellets during each preexposure. Intake patterns were monitored 18 h each day, and animals were fasted the remaining 6 h, during which time cage and automated feeder maintenance was performed and mice were weighed. Each daily session began at the start of the dark phase of the light cycle (lights out at 5:00 P.M.) and extended 6 h into the light phase, and meal pattern data were collected for 22 consecutive days. Mice of each genotype were tested in parallel to control for any inadvertent variations in the testing conditions.
Meal criteria.
Meal initiation was defined as a minimum of seven pellet removals with a minimum of 20 min between responses. Once a meal was initiated, meal termination was defined as the onset of a 20 min interval with no food intake. These criteria were determined as described previously (Fox and Byerly, 2004; Biddinger and Fox, 2010). These criteria were applied to the raw data using the Graphic State software to identify the times of onset and termination of each meal, which were used to calculate several meal parameters. These were considered to be good estimates based on the observation that mice consumed all or almost all of each pellet, as evidenced by the minute amount of spillage present on cage floors.
Meal microstructure.
The first meal each day (defined as spontaneous food intake during the first 30 min after mice gained access to the food at the start of the daily test session) was subjected to microstructural analysis. This involves characterization of changes in food intake rate over the course of this meal (Davis, 1998; Fox and Byerly, 2004). Analysis of meal microstructure allows for partial deconstruction of oropharyngeal positive feedback and GI negative feedback contributions to food intake, which combine to regulate short-term or within-meal ingestive behavior. The initial rate of food intake is influenced almost exclusively by oropharyngeal positive feedback that drives food intake. In contrast, the subsequent decay in the rate of feeding has two components. The first involves a rapid decay in feeding rate that is regulated by both oropharyngeal positive and GI negative feedback. The second component is represented by a low and relatively stable rate of intake that typically declines to end the meal and is influenced mainly by GI negative feedback, or satiation, signaling activated by the accumulation of food in the GI tract. Initial intake rate was estimated by the amount of food consumed during minute 1, and changes in this rate across the 30 min feeding session were estimated by determining the amount consumed during each of the subsequent 29 min of food intake.
Statistical analysis and graphical display of data
Changes in BDNF mRNA levels between controls and INT-BDNF−/− mice were determined by comparing changes in the constitutively expressed housekeeping gene β-actin and BDNF. This was done using the Livak and Schmittgen method (Livak and Schmittgen, 2001). Briefly, the difference in threshold cycle values (typically referred to as CT values) between BDNF and β-actin in each tissue was calculated, and this difference was compared in WT and mutant mice as determined previously (Fox et al., 2013a). All counting of neural elements in this study was performed by the same experimenter (J.E.B.) and done blind to genotype. One-way ANOVA was used to test for differences between genotypes in IGLE density, number of fibers, number of axon bundles, numbers of nodose ganglion neurons, body weight, and body composition. Meal pattern variables were tested in WT, INT-BDNF+/−, and INT-BDNF−/− mice using pairwise repeated-measures ANOVA across days 5–22. Meal microstructure data were analyzed using Tongue Twister software (version 1.45). Food intake between WT and INT-BDNF−/− mice during meal microstructure analysis was tested using one-way ANOVA for minute 1 and repeated-measures ANOVA over multiple time points. Statistics were tested using Statistica software (version 6.0; StatSoft). p values <0.05 were considered significant. Values are presented as means ± SEMs. Graphs were constructed with GraphPad Prism (version 4.0; GraphPad Software). Figure layouts were organized using Photoshop (version 6.0; Adobe Systems). Photoshop was also used to adjust brightness and contrast on photomicrographs and to apply scale bars and text to images.
Results
Assessment of the SM-targeted BDNF KO
We demonstrated previously in embryos derived from crosses of BDNFlox reporter mice and SM22αcre mice that highly efficient Cre-mediated recombination was primarily restricted to SM of the walls of blood vessels supplying GI tract tissues (Fox et al., 2013a). Similarly, in embryos derived from crosses of NT-3lox reporter mice and SM22αcre mice, recombination was restricted to SM of the intestine wall and a small portion of the stomach wall, as well as to SM of GI blood vessels (Fox et al., 2013b). Previous characterization of SM22αcre and BDNF expression in mature tissues suggests that Cre-mediated recombination would have been efficient and primarily restricted to SM of adult INT-BDNF−/− mice in the present study (Lommatzsch et al., 1999; Lepore et al., 2005). To determine the extent to which BDNF mRNA was decreased in the stomach and intestine of adult INT-BDNF−/− mice compared with controls, qPCR was performed. Additionally, BDNF mRNA in the hypothalamus and frontal cortex was examined to ensure that it was not significantly altered at these sites. First, nonquantitative RT-PCR was performed to verify mRNA integrity and BDNF mRNA size. Gel electrophoresis of RT-PCR products from the hypothalamus, frontal cortex, ventral stomach, and proximal small intestine showed bands of BDNF and β-actin reverse-transcribed cDNA at the expected sizes of 332 and 266 bp, respectively (Kawakami et al., 2002; Zermeno et al., 2009). There were no bands visible in PCR products made from mRNA that had not been reverse transcribed, suggesting that treatment of samples with DNase was effective at eliminating genomic DNA. qPCR showed that a significant decrease in BDNF mRNA levels only occurred in the proximal small intestine (86%; t(18) = 1.77, p < 0.05) in INT-BDNF−/− mice compared with controls (Fig. 1). Smaller nonsignificant reductions in BDNF mRNA levels in INT-BDNF−/− mice compared with controls occurred in the ventral stomach (54%; p = 0.076), hypothalamus (24%; p = 0.28), and frontal cortex (20%; p = 0.44). This suggested that the KO was primarily successful at reducing BDNF expression in the small intestine, while leaving BDNF expression in the brain mostly intact as relative levels of BDNF mRNA remained high in the CNS. However, we were not able to test mRNA levels in separate layers of the GI wall because the intimate relationship of the layers to one another makes it nearly impossible with unfixed tissue to cleanly separate adjacent tissue layers, especially in the instance of the myenteric plexus and adjacent SM layers. Consequently, in adults, we could not determine whether the BDNF KO was restricted to SM or whether BDNF expression that occurs in epithelial cells and myenteric neurons of the GI tract was altered (Lommatzsch et al., 1999; Grider et al., 2006; Boesmans et al., 2008), nor could we assess the degree to which BDNF mRNA remaining in these epithelial cells and myenteric neurons contributed to the incomplete reductions of BDNF message in the stomach and intestine. Furthermore, it is rare for a Cre-mediated conditional gene KO to achieve 100% loss of protein. Therefore, in young adult INT-BDNF−/− mice, we only claim that BDNF expression in the whole intestine wall is reduced (not eliminated) and suggest that the intestine was the primary site of Cre-mediated recombination. This is consistent with the more complete SM22α-driven Cre-mediated recombination in the intestine compared with the stomach observed previously in embryos (Fox et al., 2013a,b). The differential Cre expression in gastric and intestinal SM may have occurred because, in the initial creation of the SM22αcre transgenic mice, all of the gene regulatory elements required for gastric expression were not included in the transgene or regulatory elements situated near the site where the transgene inserted into the chromosome may have suppressed gastric SM expression. Levels of BDNF mRNA normalized to β-actin mRNA in controls were set at 100% (control hypothalamus, 100 ± 38%; frontal cortex, 100 ± 83.5%; ventral stomach, 100 ± 44.6%; 0–2 cm duodenum, 100 ± 48.5%).
Figure 1.

BDNF mRNA levels in tissues of young adult mice. Bars represent relative percentage BDNF ± SEM mRNA expression in CNS and GI tissues of INT-BDNF−/− mice (n = 10) compared with controls (n = 10) normalized to β-actin mRNA. Control values were set at 100%. The only tissue tested that showed a significant decrease in BDNF mRNA levels was the proximal small intestine of INT-BDNF−/− mice compared with WT mice (86%; p < 0.05), suggesting that this was the primary site of Cre-mediated recombination in INT-BDNF−/− mice. In contrast, smaller nonsignificant decreases in BDNF mRNA levels were observed in the stomach (54%), hypothalamus (24%), and frontal cortex (20%) of INT-BDNF−/− mice compared with WTs.
IGLE innervation of the stomach and intestine
Vagal afferent innervation, including IGLEs, were labeled in the stomach and intestine of control, INT-BDNF+/−, and INT-BDNF−/− mice to determine the role of intestinal BDNF in the development of these vagal elements that supply the SM. Found throughout most of the GI tract, IGLEs typically form networks of sensory nerve terminals in the myenteric plexus. They consist of laminar aggregates of fine terminal puncta that are closely associated with myenteric ganglia.
IGLE morphology and density in the stomach
Vagal afferent innervation observed in the stomachs of both controls and mutants displayed this characteristic lattice pattern of innervation, in which vagal afferent axons and fiber bundles extended axons that terminated in single IGLEs or small groups of IGLEs (Fig. 2A,C). Morphology of IGLEs in the stomach of control, INT-BDNF+/−, and INT-BDNF−/− mice appeared to be similar, because the vagal afferent axons could be seen giving rise to IGLEs that had a distinct leaf-like appearance and punctate endings interdigitating with elements of both the neurons and the surrounding capsule of myenteric ganglia (Fig. 2A,C). IGLEs were quantified as total stomach IGLE density and also separately for each of its major compartments, including the forestomach, corpus, and antrum, and determined to be evenly distributed across these compartments, which is consistent with other studies and in other strains of mice (Fig. 2E; Fox et al., 2000, 2001a,b, 2002). No differences were detected in total stomach IGLE density between any of the genotypes (controls vs INT-BDNF+/−, p = 0.201; controls vs INT-BDNF−/−, p = 0.409; INT-BDNF+/− vs INT-BDNF−/−, p = 0.885) or in any of the stomach compartments between any of the genotypes (p values are listed in Table 1). In adults, BDNF was not significantly reduced in the stomach, consistent with the intestinal bias of the SM22αcre-mediated BDNF KO in embryos. This could have been the reason gastric IGLE density was not altered in INT-BDNF−/− mice. Alternatively, it is possible that BDNF was significantly reduced in adult gastric SM, but this decrease was masked by high levels of BDNF in other gastric tissues, including the myenteric plexus and mucosa. If this did occur, it would suggest that BDNF expressed in SM is not essential for normal development or maintenance of gastric IGLEs.
Figure 2.

The morphology and density of IGLEs in the stomach (A, C, E) and intestine (B, D, F). The morphology of IGLEs in the stomach was normal in INT-BDNF−/− mice. IGLEs observed in the stomachs of both controls (A) and mutants (C) displayed the characteristic pattern of IGLE innervation, which can be seen as vagal afferent axons giving rise to the leaf-like IGLEs composed of numerous, densely packed, punctate endings. There were no differences in IGLE density between any of the genotypes or between any of the stomach compartments (INT-BDNF−/−, n = 9; INT-BDNF+/−, n = 9; control, n = 12; E). In the intestine, although the general innervation pattern and IGLE structure was similar in controls (B) and INT-BDNF−/− mice (D), the IGLE terminals appeared larger and more numerous in mutants. Furthermore, fiber bundles often appeared to contain more fibers or be larger in diameter in mutants. Quantification of IGLE density in the first 8 cm of the small intestine demonstrated a significant 40% increase in IGLE density in INT-BDNF−/− mice compared with controls (p < 0.05; group sizes were the same as for the stomach; F). This increase in IGLE density was evenly distributed across the 0–4 and 4–8 cm segments of the proximal small intestine. Arrows in A–D indicate IGLEs (double arrow in D indicates a tightly packed group of small IGLEs). Arrowheads in B and D indicate vagal sensory axon bundles. Open arrowhead in D points to a single vagal afferent axon. Scale bars: (in C) A, C, 150 μm; (in D) B, D, 50 μm.
Table 1.
p values for IGLE density comparisons in Figure 2E
| Control versus INT-BDNF+/− | Control versus INT-BDNF−/− | INT-BDNF+/− versus INT-BDNF−/− | |
|---|---|---|---|
| Total stomach | 0.201 | 0.409 | 0.885 |
| Forestomach | 0.367 | 0.443 | 0.954 |
| Corpus | 0.074 | 0.227 | 0.443 |
| Antrum | 0.578 | 0.794 | 0.814 |
IGLE density was quantified in each compartment of the stomach, and these values were combined to yield IGLE density for the entire stomach. Values represent p values for pairwise comparisons between each genotype. IGLE density did not differ between genotypes for the total stomach or for its forestomach, corpus, or antrum compartments.
IGLE morphology and density in the small intestine
As in the stomach, vagal afferent axon bundles and individual fibers also give rise to IGLE nerve terminal endings throughout the small intestine. The innervation patterns of sensory vagal axon bundles in the intestine are typically found in a lattice pattern formed by the perpendicular branching of bundles that orient parallel to either circular or longitudinal SM fibers as they follow the organization of the myenteric plexus (Fox et al., 2000; Wang and Powley, 2000). This innervation pattern was seen in both controls and mutants (Figs. 2B,D, 3). Surprisingly, however, the IGLE terminals appeared to be larger and more numerous in INT-BDNF−/− mice compared with controls. Quantitatively, this resulted in a 40% increase in IGLE density in INT-BDNF−/− mice compared with controls in the first 8 cm of the small intestine (Fig. 2F; F(1,20) = 4.52, p < 0.05). This finding was unexpected, because our hypothesis, based in part on the finding that global BDNF KO results in the loss of a large proportion of vagal afferents, predicted that KO of BDNF from the GI tract would lead to a reduced density of vagal GI afferents. The increase in IGLE density in the INT-BDNF−/− mice appeared to be consistent across the 0–4 and 4–8 cm portions of the intestine, although individually these differences were not significant (0–4 cm, p = 0.053; 4–8 cm, p = 0.071; Fig. 3F). As noted in Materials and Methods, the HRP labeling method we used did not permit us to distinguish whether this increase in IGLE density was attributable an increase in IGLE number or to an increase in branching of their axons or terminals. There were no differences in IGLE density between controls and INT-BDNF+/− mice or between INT-BDNF+/− and INT-BDNF−/− mice across any length of duodenum measured [0–4 cm duodenum: controls vs INT-BDNF+/−, p = 0.331; INT-BDNF+/− vs INT-BDNF−/−, p = 0.310; 4–8 cm duodenum: controls vs INT-BDNF+/−, p = 0.348; INT-BDNF+/− vs INT-BDNF−/−, p = 0.743; total (0–8 cm) duodenum: controls vs INT-BDNF+/−, p = 0.268; INT-BDNF+/− vs INT-BDNF−/−, p = 0.426].
Figure 3.

Vagal afferent innervation pattern in the proximal small intestine. Low-magnification photomontages of vagal afferent innervation of the same region of the duodenum are shown in control (A) and INT-BDNF−/− (B) mice. These images illustrate the lattice pattern composed of circular and longitudinal axon bundles running in near-perpendicular orientations and the increased IGLE density and axon bundle diameter in INT-BDNF−/− mice compared with controls. Arrows denote IGLEs, and arrowheads indicate axon bundles. Scale bar: (in B) A, B, 2.5 μm.
There were no genotype differences in the total area of the stomach or the small intestine, the width of the small intestine at the pyloric sphincter, the width of the small intestine at its junction with the cecum, or the length of the small intestine (Table 2). Therefore, the increase in IGLE density in the intestine of INT-BDNF−/− mice was almost certainly not attributable to a change in the area of the intestine, and the failure to observe a genotype difference in gastric IGLE density was probably not attributable to altered area of the stomach.
Table 2.
Length, width, and area of selected GI organ whole-mount regions
| Control | INT-BDNF+/− | INT-BDNF−− | Control versus INT-BDNF+/− | Control versus INT-BDNF−/− | INT-BDNF+/− versus INT-BDNF−/− | |
|---|---|---|---|---|---|---|
| Stomach area | 19.16 ± 1.66 | 21.07 ± 1.86 | 23.17 ± 2.03 | 0.947 | 0.950 | 0.905 |
| Intestine area | 37.5 ± 12.81 | 32.04 ± 4.96 | 30.31 ± 3.36 | 0.239 | 0.322 | 0.773 |
| Intestine width at pylorus | 1.0 ± 0.12 | 0.8 ± 0.16 | 0.83 ± 0.11 | 0.145 | 0.216 | 0.941 |
| Intestine width at cecum | 0.7 ± 0.38 | 0.6 ± 0.12 | 0.55 ± 0.04 | 0.675 | 0.213 | 0.687 |
| Intestine length | 46.6 ± 4.75 | 46.25 ± 1.69 | 44.3 ± 2.84 | 0.803 | 0.369 | 0.600 |
Several parameters of GI organ size were measured in mice of each genotype. Values are group means ± SEMs (first 3 columns) and p values for pairwise comparisons between each genotype (last 3 columns). These values were determined in early adulthood, when the mice were processed for WGA–HRP staining. Stomach and intestine area (square centimeters), intestine width at pylorus and cecum (centimeters), and intestine length from pylorus to cecum (centimeters) are listed.
Nodose ganglion cell counts
The two most probable mechanisms underlying the increased intestinal IGLE density in INT-BDNF−/− mice are increased survival of vagal sensory neurons that innervate the intestine, providing it with increased numbers of IGLE axons, or increased branching of the normal number of intestinal vagal sensory axons or terminals. To aid determination of whether increased IGLE survival occurred, vagal sensory neuron number was quantified in control and INT-BDNF−/− mice. Consistent with the increased survival of vagal GI afferents in INT-BDNF−/− mice, there was a significant 47% increase in total nodose ganglion neuron number in INT-BDNF−/− mice compared with controls (F(1,22) = 5.81, p = < 0.05; Fig. 4C). Interestingly, the increase in neuron number in INT-BDNF−/− mice compared with controls, when tested separately for the right and left nodose ganglia, was only significant on the right (left nodose: 25% increase, p = 0.129; right nodose: 62% increase, F(1,22) = 8.92, p < 0.05; Fig. 4C). Although there was no statistically significant difference in neuron number in the left nodose ganglion, labeling of vagal afferents by injection of WGA–HRP into the left ganglion led to detection of a significant increase in IGLE terminal density in the intestine of mutants. This suggests that reduced intestinal BDNF may have altered other aspects of IGLE development instead of, or in addition to, survival that contributed to increased intestinal IGLE density. In particular, reduced BDNF may have had effects on IGLE axon or terminal branching. Thus, it is possible that increased IGLE axon branching accounted for the increase in intestinal IGLE density in INT-BDNF−/− mice or that this increased axon branching combined with the surplus of axons arising from the nonsignificant 25% increase in left nodose neuron number to produce the significant increase in intestinal IGLE density. As described in detail in Materials and Methods (Anterograde labeling of vagal GI afferents), the left nodose was chosen for injections because it provides much greater innervation to the proximal intestine compared with the right nodose ganglion in mice and rats (Fox et al., 2000, 2001b; Wang and Powley, 2000). Thus, if the right nodose ganglion had been injected and if the increased number of nodose neurons contributed to augmented intestinal IGLE density in INT-BDNF−/− mice, then this increase in IGLE density when computed as a percentage would probably have been larger than that computed after left nodose ganglion injections. However, the absolute increase in density of IGLEs labeled by right nodose ganglion injection would have been much smaller than the increase in those labeled by left nodose ganglion injections as a consequence of the dramatically smaller number of IGLEs arising from the right nodose ganglion. Another potential consequence of the right nodose ganglion showing a larger and significant increase in neuron number compared with the left that we cannot rule out is that it is possible that, if we had injected the right nodose with WGA–HRP and examined the dorsal stomach, we might have found an increase in IGLE density in INT-BDNF−/− mice.
Figure 4.
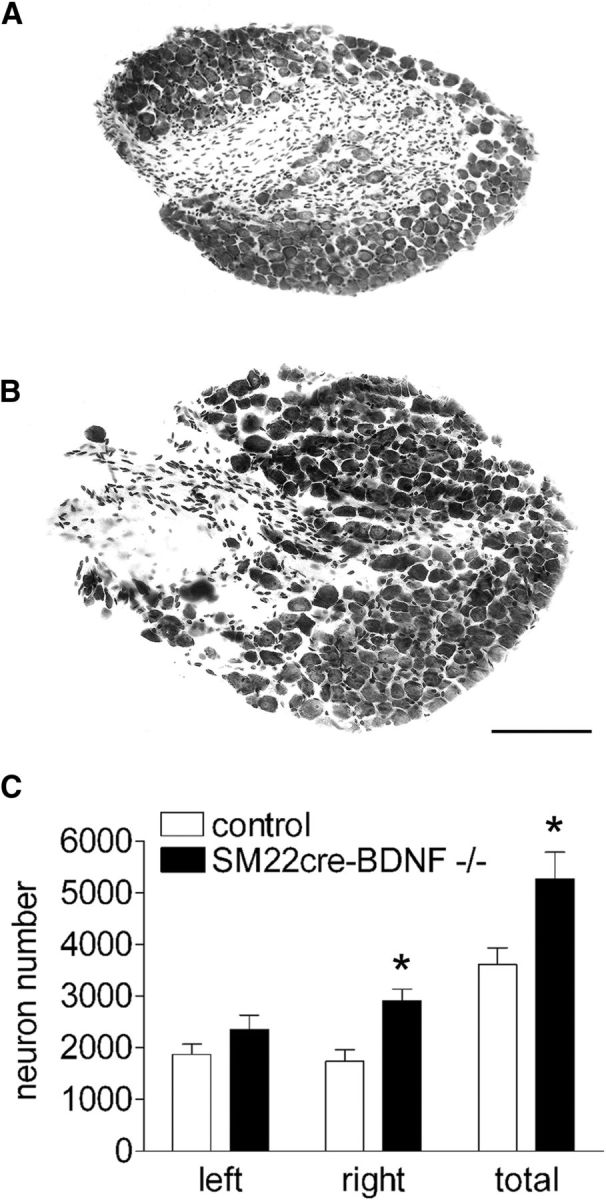
Comparison of neuron numbers in nodose ganglia of control and INT-BDNF−/− mice. Representative images of the right nodose ganglion are shown in a control (A) and an INT-BDNF−/− (B) mouse. The total (left and right nodose ganglia combined) and the right nodose demonstrated significant increases in vagal sensory neuron number in INT-BDNF−/− mice (n = 14) compared with controls (n = 9, 47% and 62% increases, respectively; both p < 0.05; C), whereas the left nodose ganglia showed a nonsignificant 25% increase in neuron number in INT-BDNF−/− mice. Scale bar: (in B) A, B, 150 μm.
Quantification of intestinal vagal sensory axons and axon bundles
To investigate whether the increased intestinal IGLE innervation was reflected in increased density of individual longitudinal or circular axon fibers or axon bundles, these elements were quantified in control and INT-BDNF−/− mice (Fig. 5A,B). There were no statistically significant differences in the density of any of the vagal elements quantified between INT-BDNF−/− mice and controls.
Figure 5.

Comparisons of densities of single vagal sensory axons, axon bundles, and numbers of axon bundles of specific diameter ranges in the proximal 8 cm of the small intestine in INT-BDNF−/− and control mice. The numbers of single afferent axon fibers (A) and afferent axon bundles (B) were similar in controls (n = 9) and INT-BDNF−/− mice (n = 9). In contrast, INT-BDNF−/− mice showed an increase in the number of longitudinal bundles in the 1.5–2.0 (p < 0.05) and 3.5–4.0 (p < 0.01) μm diameter ranges compared with controls (C). There were no differences in circular axon bundle number between mutants and controls within any diameter range (D).
In addition to free axon or axon bundle density as assessments of whether an increase in axon numbers occurred in mutants, it is possible that a subset of fiber bundles within a specific range of diameters would have been augmented. To examine this, diameters of axon bundles running in both the longitudinal and circular orientations measured to the nearest 0.5 μm in control and INT-BDNF−/− mice and the number of bundles within each diameter range are plotted in Figure 5C and D, respectively. The largest-diameter bundles, 4.5–5.0 μm diameter, were rarely observed in either the longitudinal or circular orientations in either genotype. In control mice, the numbers of both longitudinal and circular axon bundles progressively decreased from the smallest to the largest diameter range examined. This same pattern was generally observed in INT-BDNF−/− mice, except for the longitudinal and circular bundles in the 1.5–2.0 μm diameter range, which were greatest in number. In the smallest diameter longitudinal bundles observed, the 0.5–1.0 μm diameter range, there were no differences between control and INT-BDNF−/− mice (14.8% increase in mutants; p = 0.688; Fig. 5C). However, at the 1.5–2.0 μm diameter range, INT-BDNF−/− mice showed a greater number of longitudinal bundles compared with control mice (118% increase; F(1,17) = 5.02, p < 0.05). At the 2.5–3.0 μm diameter range, there was also an increase in number of longitudinal axon bundles for INT-BDNF−/− mice compared with controls, but it was not significant (150%; p = 0.071). At the 3.5–4.0 μm diameter range, INT-BDNF−/− mice showed a significant increase in the number of longitudinal bundles compared with control mice (267% increase; F(1,17) = 9.64, p < 0.01). The magnitude of this latter difference should be interpreted cautiously because there were small numbers of fiber bundles in each group. The finding of increased numbers of longitudinally oriented axon bundles at some diameter ranges is consistent with the nonsignificant increase in number of vagal sensory ganglion neurons in the nodose ganglion contributing to the augmented intestinal IGLE density. However, it could also be consistent with increased axon branching, if this branching occurred proximal to the intestine (between the nodose ganglion and the intestine). The increase in number of axon bundles within specific diameter ranges observed in INT-BDNF−/− mice compared with controls was specific to those oriented longitudinally, because there were no differences observed in the number of bundles at any diameter in the circular orientation between INT-BDNF−/− mice and controls (Fig. 5D).
Note that we have used counts of HRP-labeled axon bundles of specified diameters as a proxy for actual axon counts. Ensuring that axon bundles of a given diameter contain similar numbers of axons would require use of another method, such as counts of axons imaged with an electron microscope.
Analysis of body weight, body composition, and feeding behavior
Feeding behavior and body weight of mice with reduced BDNF expression in the intestine were examined to assess the anorexigenic potential of BDNF, possibly mediated by the increase in intestinal IGLE density or alterations that may have occurred in development or function of other autonomic neurons that innervate the intestine. If GI BDNF has similar effects on feeding as in the CNS, we hypothesized that reduced BDNF in the intestine would result in hyperphagia and increased weight gain compared with controls with normal levels of GI BDNF.
Body weight, body composition, and daily food intake
Growth curves derived from weekly body weight measures of males and females of all genotypes generated by the breeding strategy used to produce INT-BDNF−/− mice from 3 weeks (weaning) to 4 months of age (young adulthood) were similar (Fig. 6). Additionally, during the 22 d of meal pattern experiments at 3–4 months of age, daily body weights of the three genotypes tested (WT, INT-BDNF+/−, and INT-BDNF−/−) were nearly identical (Fig. 7A). Body composition of these three groups determined the day before meal pattern collection, before fasting, also revealed no differences in either fat mass or lean mass, in either males or females, here calculated as percentage of body weight with mice of each group consisting of ∼10% fat (controls vs INT-BDNF+/−, p = 0.896; INT-BDNF+/− vs INT-BDNF−/−, p = 0.725; controls vs INT-BDNF−/−, p = 0.888; Fig. 7B). Also during the meal pattern experiments, total food intake of the Bio-Serv balanced pellet diet was calculated each day. There were also no genotype group differences in average daily food intake (controls vs INT-BDNF+/−, p = 0.694; INT-BDNF+/− vs INT-BDNF−/−, p = 0.228; controls vs INT-BDNF−/−, p = 0.162; Fig. 7C). Moreover, the average daily food intake, which was ∼3.0 g/d, was similar to the amount of balanced diet consumed by various WT mice reported by other investigators and in our previous studies (Kernie et al., 2000; Fox et al., 2001b; Biddinger and Fox, 2010; Krashes et al., 2011). The failure to find genotype group differences in body weight, body composition, or daily food consumption suggests, counter to our hypothesis, that neither the loss of BDNF from the intestine wall nor the gene manipulations used to produce this targeted BDNF KO altered long-term regulation of food intake and body weight.
Figure 6.
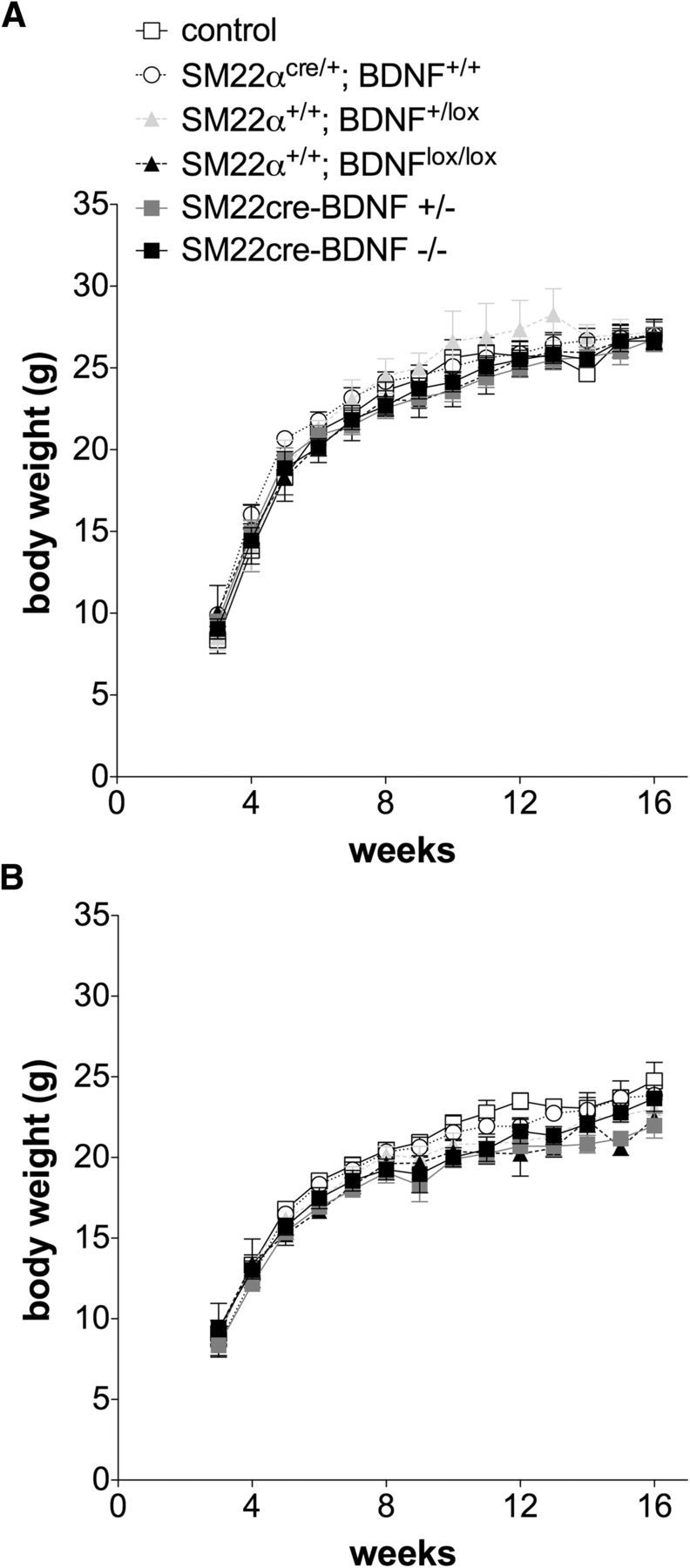
Body weights of male and female mice of all genotypes generated. There were no differences in body weight between any of the six genotypes generated by the breeding strategy used in this study from weaning at 3 weeks of age to young adulthood at 16 weeks of age in either males (A) or females (B). Males: WT (n = 7), SM22αcre/+;BDNF+/+ (n = 10), SM22α+/+;BDNFlox/+ (n = 4), SM22α+/+;BDNFlox/lox (n = 6), INT-BDNF+/− (n = 10), and INT-BDNF−/− (n = 10). Females: WT (n = 7), SM22αcre/+;BDNF+/+ (n = 10), SM22α+/+;BDNFlox/+ (n = 10), SM22α+/+;BDNFlox/lox (n = 2), INT-BDNF+/− (n = 10), and INT-BDNF−/− (n = 10).
Figure 7.
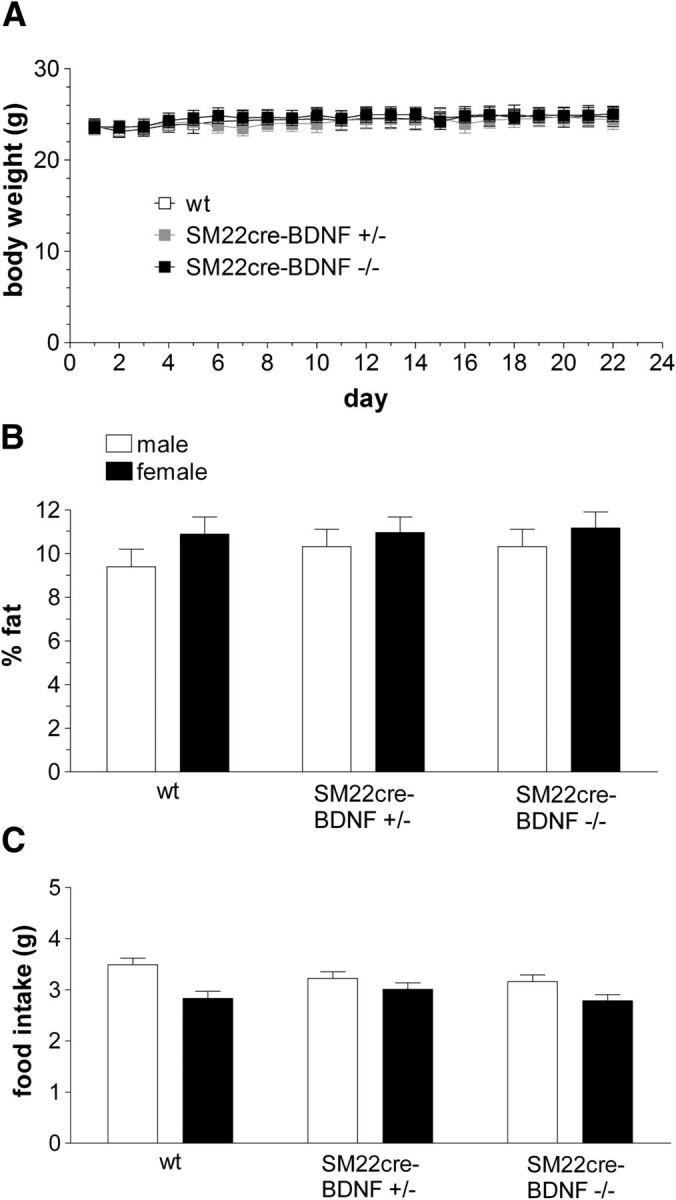
Body weight, body fat, and daily food intake of mice used in the meal pattern experiment. During the 22 d of meal pattern testing, when mice were 3–4 months of age, no differences in body weight emerged between WT (n = 13; 7 males, 6 females), INT-BDNF+/− (n = 14; 7 males, 7 females), and INT-BDNF−/− (n = 15; 7 males, 8 females) mice (A). In addition, there were no differences in body fat, here calculated as percentage of body weight, between any of the three genotypes or between males and females (B). Average total daily food intake during the meal pattern collection remained normal in INT-BDNF+/− and INT-BDNF−/− mice in both males and females (C).
Analysis of meal patterns
Because long-term food intake and body weight did not appear to be altered in INT-BDNF−/− mice and any changes to innervation of the gut in these mice, including the increased intestinal IGLE density, could have altered their short-term feeding, several meal pattern parameters were characterized. Eating patterns of WT, INT-BDNF+/−, and INT-BDNF−/− mice stabilized by day 5, and therefore, data obtained from days 5–22 were analyzed. This adaptation should have minimized any possible differences in learning ability between mutants and controls that might have occurred if BDNF levels in the hippocampus were decreased (Fox et al., 2013a). The means ± SEMs for body weight averaged across days 5–22 of testing for each group are listed in Table 3.
Table 3.
Additional meal pattern data
| WT | INT-BDNF+/− | INT-BDNF−/− | WT versus INT-BDNF+/− | WT versus INT-BDNF−/− | INT-BDNF+/− versus INT-BDNF−/− | |
|---|---|---|---|---|---|---|
| Body weight | 24.36 ± 1.01 | 24.16 ± 0.92 | 24.61 ± 0.80 | 0.947 | 0.950 | 0.905 |
| Meal number | 9.53 ± 0.46 | 9.79 ± 0.45 | 10.81 ± 0.43 | 0.239 | 0.322 | 0.773 |
| Total meal duration | 174.83 ± 23.7 | 158.85 ± 22.86 | 93.44 ± 22.08* | |||
| IMI | 62.50 ± 2.77 | 67.16 ± 2.77 | 68.55 ± 2.58 | 0.145 | 0.216 | 0.941 |
Additional meal pattern data (first 3 columns, mean ± SEM; last 3 columns, p values associated with genotype comparisons for parameters that yielded no significant differences). Each measure was averaged daily. The values are based on the average of these daily values over days 5–22 of behavioral testing. Body weight (grams), total meal duration (minutes), and IMI (minutes) are listed.
*p < 0.05, significantly different from WT and INT-BDNF+/− groups.
INT-BDNF−/− mice exhibited changes in several meal pattern parameters compared with WT mice. Most notably, INT-BDNF−/− mice had a large decrease in average meal duration compared with controls (60%; F(1,17) = 7.22, p < 0.05; Fig. 8A), which over the course of each day accumulated to produce a decrease in average total meal duration (total time spent eating during each daily test session; 47%; F(1,17) = 7.32, p < 0.05; Table 3). This reduction in meal duration in INT-BDNF−/− mice was compensated for through an increase in average eating rate (F(1,17) = 6.14, p < 0.05; Fig. 8B). However, this compensation was only partial because the decrease in meal duration for the INT-BDNF−/− mice still contributed to a modest but significant 20% decrease in meal size compared with WT mice (F(1,17) = 8.27, p < 0.01; Fig. 8C). The decreases in both meal duration and meal size suggest that there was an increase in satiation in INT-BDNF−/− mice compared with WTs. Moreover, the 20% reduction in the average meal size of INT-BDNF−/− mice was compensated for by a small, nonsignificant increase of ∼1.3 meals each day by INT-BDNF−/− compared with WT mice (Table 3). Consequently, the decrease in meal size did not result in a decrease in total food intake by INT-BDNF−/− mice compared with controls. There were no differences between controls and INT-BDNF+/− mice on any meal parameter tested. However, INT-BDNF−/− mice showed decreased total (F(1,17) = 5.84, p < 0.05) and average (F(1,17) = 6.12, p < 0.05) meal duration and decreased meal size (F(1,17) = 7.23, p < 0.05) compared with INT-BDNF+/− mice (Fig. 8A,C; Table 3). These meal parameter differences between homozygous and heterozygous targeted BDNF KO mice suggest that the presence of one intact BDNF allele in the intestine wall was sufficient to mostly compensate for at least some of the effects of the reduction in BDNF levels caused by the targeted allele.
Figure 8.

Meal pattern and microstructure parameters associated with satiation in INT-BDNF−/−, INT-BDNF+/−, and WT mice. INT-BDNF−/− mice (n = 15) displayed a large decrease in average meal duration compared with WT (n = 13) and INT-BDNF+/− (n = 14) mice (both p < 0.05; A). The reduced meal duration observed in INT-BDNF−/− mice was partially compensated for through an increase in average eating rate in INT-BDNF−/− mice compared with WTs (p < 0.05; B). Because this compensation was only partial, INT-BDNF−/− mice also demonstrated a modest decrease in meal size compared with INT-BDNF+/− mice (p < 0.05) and WTs (p < 0.01) (C). These decreases in meal duration and meal size suggested that satiation was increased in INT-BDNF−/− mice compared with WTs and INT-BDNF+/− mice. INT-BDNF−/− mice displayed a trend toward increased average IMI compared with WT mice (Table 3) that accrued to a significant increase in total daily IMI compared with WT controls (p < 0.05; D). This trend toward an increase in average IMI also contributed to a significant increase in satiety ratio in INT-BDNF−/− mice compared with WTs (p < 0.01; E). These increases in total daily IMI and satiety ratio in INT-BDNF−/− mice suggested that they had an increase in satiety, which indicates that a given amount of food produced greater satiety for them compared with controls. In terms of the meal microstructure analysis, both WT and INT-BDNF−/− mice showed a similar high initial food intake rate during minute 1 (F). Because the first minute of feeding provides an estimate of initial intake rate, which is mainly regulated by oropharyngeal positive feedback, this suggested that the contribution of this feedback to food intake was not altered in INT-BDNF−/− mice. Mutants and WTs also exhibited a similar rapid decay of food intake rate during minutes 2–5 (F), a process controlled by both oropharyngeal positive and GI negative feedback. However, starting at minute 6, INT-BDNF−/− mice exhibited a greater decrease in their food intake rate compared with WTs (G), suggesting that the mutants had increased GI negative feedback signaling. These findings are consistent with the interpretation that satiation signaling was augmented in INT-BDNF−/− mice.
In addition to meal pattern changes in INT-BDNF−/− mice that are associated with increased satiation or within-meal effects that lead to meal termination, there were changes in the feeding behavior of the mutants that could imply they also had increased satiety or between meal effects that delay initiation of a subsequent meal. In particular, the average intermeal interval (IMI) showed a small increase in INT-BDNF−/− mice compared with WT mice that, although not in itself significant (p = 0.09; Table 3), accumulated over the course of each test day to yield a significant 20% increase in average total IMI in INT-BDNF−/− mice compared with WT (F(1,17) = 6.14, p < 0.05; Fig. 8D). Furthermore, this small, nonsignificant increase in average IMI contributed to a 27% increase in satiety ratio for INT-BDNF−/− compared with control mice (Fig. 8E; F(1,17) = 12.02, p < 0.01). Satiety ratio is the ratio of IMI to the preceding meal size, and therefore, this increase suggests that a given amount of food produced greater satiety in INT-BDNF−/− mice than it did in WTs. There were no differences in average IMI, total IMI, or satiety ratio that suggested that changes in satiety occurred between controls and INT-BDNF+/− mice or between INT-BDNF+/− and INT-BDNF−/− mice (Fig. 8D,E; Table 3).
Analysis of meal microstructure
Changes in either oropharyngeal positive feedback or GI negative feedback could contribute to altered satiation. Importantly, the bulk of the GI negative feedback signals are transmitted by vagal afferents (Smith, 1996; Schwartz et al., 2000). To provide additional evidence in support of the interpretation that augmentation of negative feedback signaling from gut-to-brain contributed to the decreased meal duration and meal size of INT-BDNF−/− mice, microstructure of the first 30 min of food intake on days 5–22 of meal pattern data collection was examined. Three microstructural parameters have been used to characterize changes in the rate of eating over the course of a meal (Davis, 1998; Fox and Byerly, 2004): (1) initial consumption rate, mainly influenced by oropharyngeal stimulation; (2) early component of decay of eating rate, influenced by both oropharyngeal positive feedback and GI negative feedback signaling; and (3) late component of decay of eating rate, mainly influenced by GI negative feedback. Therefore, augmented GI negative feedback in INT-BDNF−/− mice would be most apparent as an increase in the late decay of eating rate. In contrast, reduced oropharyngeal positive feedback would be most evident as a decrease in intake rate during the first minute of food access. Both WT and INT-BDNF−/− mice showed a similar, high initial intake rate during minute 1, and in fact, this rate was slightly, albeit nonsignificantly, higher in INT-BDNF−/− mice than in WTs, suggesting that altered oropharyngeal input did not contribute to their reduced meal duration and meal size (p = 0.289; Fig. 8F). Mutants and controls also exhibited a similar rapid decay of food intake rate during minutes 2–5 (p = 0.390; Fig. 8F). However, starting at minute 6, the INT-BDNF−/− mice began to show an increased rate of decay of food intake compared with the controls, represented as an average 40% decrease in their sustained intake rate across minutes 6–30 (F(1,24) = 7.36, p < 0.05; Fig. 8G). Moreover, consistent with reduced average meal duration in INT-BDNF−/− mice compared with WTs, during the first meal, most mutants stopped eating, albeit briefly, at approximately minute 8, whereas most WT mice did not stop eating until approximately minute 26. These findings are consistent with augmented GI negative feedback signaling, or satiation, in INT-BDNF−/− mice and could further imply that the increase in IGLE innervation of the small intestine or changes to other autonomic innervation of the intestine in these mice were sufficient in magnitude to alter short-term controls of food intake. Furthermore, the lack of genotype group differences in body weight, body composition, and daily food intake in INT-BDNF−/− mice described above suggests that the alterations to meal pattern and microstructure parameters that were detected were likely to have been primary effects of reduced intestinal BDNF levels on short-term feeding behavior rather than having been secondary to changes in daily food intake, body weight, or body composition.
Discussion
To determine whether BDNF produced by the intestine regulates development or maintenance of vagal GI afferents, food intake, body weight, or body composition, mice with a targeted BDNF KO restricted mainly to the intestine were generated. This manipulation surprisingly led to increased IGLE density in the intestine, but not in the stomach, and to increased numbers of vagal sensory neurons and longitudinal intestinal axon bundles. Furthermore, INT-BDNF−/− mice displayed a large decrease in meal duration that resulted in reduced meal size. Increased suppression of feeding during the later phase of the first meal suggested that this was attributable to augmented satiation signaling. Additionally, INT-BDNF−/− mice demonstrated increased total daily IMI and satiety ratios, implying that satiety signaling was increased. INT-BDNF−/− mice compensated for augmentation of satiation and satiety to maintain normal daily food intake and body weight through a significant increase in average eating rate and a small nonsignificant increase in meal number. These findings are the first to suggest that a target organ-derived neurotrophin acts to reduce sensory innervation density and neuron number. They are also the first to demonstrate a role for BDNF produced by a peripheral tissue in short-term controls of feeding, likely through its regulation of development or function of autonomic innervation of the gut, possibly including augmented intestinal IGLE innervation.
The current findings implicate intestinal BDNF in suppression of intestinal IGLE survival, or possibly axon growth, guidance, or branching of their axons, functions that run counter to expectations for a neurotrophic factor. Neurons are overproduced during development and are thought to compete for limiting amounts of target-derived neurotrophic factors, which support their survival by preventing apoptosis (Oppenheim, 1991). Although the effects of global BDNF KOs have typically been consistent with this framework, leading to decreased innervation in other sensory systems, augmented innervation of skin by mechanoreceptors and autonomic fibers has been observed (Fundin et al., 1997; Rice et al., 1998).
Possible mechanisms of increased IGLE density
During development, the BDNF KO in the GI tract occurred mainly in vascular SM (Fox et al., 2013a). This could imply that loss of BDNF secretion from GI blood vessels during development promoted IGLE survival, or growth, guidance, or branching of IGLE axons that extend alongside these vessels to reach the intestine. Additionally, development of mucosal vagal afferents and other autonomic fibers that grow along these vessels as they develop could have been altered. In the adult, the KO probably occurred throughout the SM of the intestinal wall in INT-BDNF−/− mice. However, because technical considerations impeded our ability to confirm that BDNF loss from the intestine wall was restricted to SM, it is possible that BDNF was reduced in myenteric neurons or mucosa instead of, or in addition to, SM. If BDNF loss from adult intestinal SM occurred, it could have enhanced the maintenance, terminal branching, or sensory transduction of IGLEs and altered other intestinal innervation. Consistent with increased neuron survival, positive correlations between sensory neuron number and density of peripheral nerve fibers and terminals have been observed in numerous studies involving manipulations of growth factors in the skin, tongue, and gut (Albers et al., 1996; Fox et al., 2001b; Krimm, 2007). Alternatively, BDNF has been shown to promote growth, guidance, and branching of axons in several sensory systems, including vagal afferents (Cohen-Cory and Fraser, 1995; Lentz et al., 1999; Tucker, 2002; Hellard et al., 2004). If GI BDNF regulates IGLE axon growth, guidance, or branching but in the opposite direction—i.e., by suppressing these processes—the loss of this suppression in INT-BDNF−/− mice could have increased growth or branching of intestinal IGLE axons or terminals and thus contributed to the increase in intestinal IGLE density.
A single mechanism that could account for increased survival, or axon growth or branching of intestinal IGLEs in INT-BDNF−/− mice is reduced signaling by proBDNF (BDNF precursor) through p75, the low-affinity neurotrophin receptor. This signaling typically has effects antagonistic to those of BDNF activation of trkB and include stimulating apoptosis and inhibiting axon growth and branching (Ichim et al., 2012; Suetterlin et al., 2012). For example, BDNF or p75 KO led to increased axon branching that probably contributed to increases in density of sympathetic innervation of the pineal and submandibular salivary glands and to increased neuron numbers in neonatal superior cervical ganglia (Glebova and Ginty, 2005; Jahed and Kawaja, 2005). Other possible mechanisms mediating increased IGLE density include compensatory increases in expression of other growth factors that support this increase (Erickson et al., 1996; McAllister et al., 1997; Kolbeck et al., 1999; Murphy and Fox, 2010), neurons with low trkB expression favoring suppression by BDNF, and BDNF activation of trkB in some neurons stimulating second-messenger pathways that inhibit development (Rice et al., 1998).
Reduced intestinal BDNF and satiation
The meal pattern and microstructure findings of the present study suggest that satiation is increased in INT-BDNF−/− mice. Because vagal afferents provide the bulk of negative feedback signaling that influences satiation (Smith, 1996; Schwartz et al., 2000) and IGLEs are tension receptors that can sense muscle stretch (Zagorodnyuk et al., 2001), increased intestinal IGLE density is a candidate for mediating this effect. However, establishing this relationship will require additional research because the status of other vagal GI afferents in particular, including intramuscular arrays (IMAs) and mucosal afferents, as well as other pathways innervating the intestine in INT-BDNF−/− mice is not known.
Several experimental approaches have provided convergent evidence for a prominent role of vagal GI afferents, and possibly for IGLEs, in satiation. Disruption of vagal afferents supplying the gut by sensory-selective surgical vagotomy or capsaicin treatment results in increased meal size attributable to increased eating rate or meal duration, effects consistent with reduced vagal negative feedback signaling (Walls et al., 1995; Chavez et al., 1997; Schwartz et al., 1999). Studies using mutations of genes for growth factors or their receptors to manipulate vagal GI afferents implicated intestinal IGLEs in signaling satiation. NT-4 KO mice, which had a 90% decrease in IGLE density in the duodenum, showed increased meal duration and size, whereas NT-4 knockin mice, which appeared to have increased intestinal IGLE density, exhibited a decrease in meal size and increased sensitivity to CCK (Fox et al., 2001b; Chi et al., 2004). Furthermore, c-Kit and steel mutant mice had a severe reduction of forestomach IMAs, yet they exhibited normal satiation (Fox et al., 2002; Chi and Powley, 2003). This is consistent with IGLEs contributing to satiation because they were intact and are the only other major vagal SM mechanoreceptor. However, the contribution of IGLEs in these studies remains provisional because it is not known whether other GI innervation was modified by the gene mutations used, although IMA structure and density were normal in NT-4 KO mice.
It is unlikely that effects of reduced intestinal BDNF levels on vagal efferents contributed to increased satiation because there has been no evidence of disruption of motor neurons, including vagal efferents, in BDNF KO mice (Jones et al., 1994; Murphy and Fox, 2010). If their function was disrupted, similar to perturbation of the enteric nervous system, the digestion of food would be slowed, which could produce symptoms resembling increased satiation. Although disruption of sympathetic innervation of the intestine produced a small decrease in meal size, it was not determined whether this involved reduced meal duration (Fu et al., 2011). Moreover, the pattern of changes in other meal parameters measured was different from that observed in the present study. Both IMI and satiety ratios were decreased in the experiment by Fu et al., whereas in the present study, they were increased. These discrepancies suggest that, although disruption of some aspects of sympathetic intestinal innervation in INT-BDNF−/− mice could have contributed to increased satiation, the relationship is not straightforward. Although it is not known what effects changes to dorsal root afferents might have on meal patterns, their disruption would likely have similar effects on sympathetic perturbation as they drive sympathetic reflexes.
SM22αcre-mediated recombination in the adult brain
We recently reported evidence suggesting that SM22αcre-mediated recombination occurred unexpectedly in three adult brain regions: (1) the anterolateral cerebral cortex; (2) the hippocampus; and (3) the VMH (Fox et al., 2013a). The nonsignificant reductions of BDNF mRNA in the hypothalamus and cortex in the present study probably resulted from recombination of floxed BDNF alleles in some cells within these regions. If this occurred, these BDNF reductions could have contributed to the altered feeding behavior of INT-BDNF−/− mice. However, it is unlikely that they account entirely for these effects. Loss of BDNF from the VMH region results in increased food intake and body weight, which did not occur in INT-BDNF−/− mice (Unger et al., 2007). It is improbable that reduced hippocampal BDNF levels played a significant role in the altered feeding of INT-BDNF−/− mice. Rodents with hippocampal lesions eat short, small meals at two to three times the normal frequency (Clifton et al., 1998), whereas INT-BDNF−/− mice showed no significant increase in meal number, although they ate shorter, smaller meals. Regarding the loss of BDNF from the cortex, structures in this region including the dorsolateral prefrontal and primary somatosensory cortices, which are activated by food-related stimuli, are involved in inhibitory processes, such as satiety (Boubaker et al., 2012). Therefore, reduced function of these cortical areas associated with decreased BDNF expression might be expected to reduce satiety, whereas we found evidence of increased satiety.
Footnotes
This research was supported by National Institutes of Health Grant R01 NS046716 (E.A.F.). We thank Terry Powley, Kim Kinzig, and Ed Bartlett for their helpful comments on a previous draft of the manuscript and Guoping Fan for mice with a floxed BDNF allele (BDNF+/lox) and the associated genotyping protocol. Parts of this work have been published previously in abstract form at the annual meetings of the Society for the Study of Ingestive Behavior in 2012 and the Society for Neuroscience in 2012 and 2013. This manuscript was based in part on a dissertation submitted by J.E.B. in partial fulfillment of the requirements of the PhD degree.
The authors declare no competing financial interests.
References
- Albers KM, Perrone TN, Goodness TP, Jones ME, Green MA, Davis BM. Cutaneous overexpression of NT-3 increases sensory and sympathetic neuron number and enhances touch dome and hair follicle innervation. J Cell Biol. 1996;134:487–497. doi: 10.1083/jcb.134.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddinger JE, Fox EA. Meal parameters and vagal gastrointestinal afferents in mice that experienced early postnatal overnutrition. Physiol Behav. 2010;101:184–191. doi: 10.1016/j.physbeh.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boesmans W, Gomes P, Janssens J, Tack J, Vanden Berghe P. Brain-derived neurotrophic factor amplifies neurotransmitter responses and promotes synaptic communication in the enteric nervous system. Gut. 2008;57:314–322. doi: 10.1136/gut.2007.131839. [DOI] [PubMed] [Google Scholar]
- Boubaker J, Val-Laillet D, Guérin S, Malbert CH. Brain processing of duodenal and portal glucose sensing. J Neuroendocrinol. 2012;24:1096–1105. doi: 10.1111/j.1365-2826.2012.02326.x. [DOI] [PubMed] [Google Scholar]
- Carroll P, Lewin GR, Koltzenburg M, Toyka KV, Thoenen H. A role for BDNF in mechanosensation. Nat Neurosci. 1998;1:42–46. doi: 10.1038/242. [DOI] [PubMed] [Google Scholar]
- Chai NL, Dong L, Li ZF, Du KX, Wang JH, Yan LK, Dong XL. Effects of neurotrophins on gastrointestinal myoelectric activities of rats. World J Gastroenterol. 2003;9:1874–1877. doi: 10.3748/wjg.v9.i8.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez M, Kelly L, York DA, Berthoud HR. Chemical lesion of visceral afferents causes transient overconsumption of unfamiliar high-fat diets in rats. Am J Physiol. 1997;272:R1657–R1663. doi: 10.1152/ajpregu.1997.272.5.R1657. [DOI] [PubMed] [Google Scholar]
- Chi MM, Powley TL. c-Kit mutant mouse behavioral phenotype: altered meal patterns and CCK sensitivity but normal daily food intake and body weight. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1170–R1183. doi: 10.1152/ajpregu.00015.2003. [DOI] [PubMed] [Google Scholar]
- Chi MM, Fan G, Fox EA. Increased short-term food satiation and sensitivity to cholecystokinin in neurotrophin-4 knock-in mice. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1044–R1053. doi: 10.1152/ajpregu.00420.2004. [DOI] [PubMed] [Google Scholar]
- Clifton PG, Vickers SP, Somerville EM. Little and often: ingestive behavior patterns following hippocampal lesions in rats. Behav Neurosci. 1998;112:502–511. doi: 10.1037/0735-7044.112.3.502. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S, Fraser SE. Effects of brain-derived neurotrophic factor on optic axon branching and remodelling in vivo. Nature. 1995;378:192–196. doi: 10.1038/378192a0. [DOI] [PubMed] [Google Scholar]
- Cordeira JW, Frank L, Sena-Esteves M, Pothos EN, Rios M. Brain-derived neurotrophic factor regulates hedonic feeding by acting on the mesolimbic dopamine system. J Neurosci. 2010;30:2533–2541. doi: 10.1523/JNEUROSCI.5768-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulie B, Szarka LA, Camilleri M, Burton DD, McKinzie S, Stambler N, Cedarbaum JM. Recombinant human neurotrophic factors accelerate colonic transit and relieve constipation in humans. Gastroenterology. 2000;119:41–50. doi: 10.1053/gast.2000.8553. [DOI] [PubMed] [Google Scholar]
- Davis JD. A model for the control of ingestion—20 years later. In: Morrison AR, Fluharty SJ, editors. Progress in psychobiology and physiological psychology. San Diego: Academic; 1998. pp. 127–173. [Google Scholar]
- Erickson JT, Conover JC, Borday V, Champagnat J, Barbacid M, Yancopoulos G, Katz DM. Mice lacking brain-derived neurotrophic factor exhibit visceral sensory neuron losses distinct from mice lacking NT4 and display a severe developmental deficit in control of breathing. J Neurosci. 1996;16:5361–5371. doi: 10.1523/JNEUROSCI.16-17-05361.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernfors P, Merlio JP, Persson H. Cells expressing messenger-RNA for neurotrophins and their receptors during embryonic rat development. Eur J Neurosci. 1992;4:1140–1158. doi: 10.1111/j.1460-9568.1992.tb00141.x. [DOI] [PubMed] [Google Scholar]
- Fox EA. A genetic approach for investigating vagal sensory roles in regulation of gastrointestinal function and food intake. Auton Neurosci. 2006;126–127:9–29. doi: 10.1016/j.autneu.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Fox EA, Byerly MS. A mechanism underlying mature-onset obesity: evidence from the hyperphagic phenotype of brain-derived neurotrophic factor mutants. Am J Physiol Regul Integr Comp Physiol. 2004;286:R994–R1004. doi: 10.1152/ajpregu.00727.2003. [DOI] [PubMed] [Google Scholar]
- Fox EA, Murphy MC. Factors regulating vagal sensory development: potential role in obesities of developmental origin. Physiol Behav. 2008;94:90–104. doi: 10.1016/j.physbeh.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox EA, Phillips RJ, Martinson FA, Baronowsky EA, Powley TL. Vagal afferent innervation of smooth muscle in the stomach and duodenum of the mouse: morphology and topography. J Comp Neurol. 2000;428:558–576. doi: 10.1002/1096-9861(20001218)428:3<558::AID-CNE11>3.0.CO%3B2-M. [DOI] [PubMed] [Google Scholar]
- Fox EA, Phillips RJ, Martinson FA, Baronowsky EA, Powley TL. C-Kit mutant mice have a selective loss of vagal intramuscular mechanoreceptors that innervate gastric smooth muscle. Anat Embryol. 2001a;204:11–26. doi: 10.1007/s004290100184. [DOI] [PubMed] [Google Scholar]
- Fox EA, Phillips RJ, Baronowsky EA, Byerly MS, Jones S, Powley TL. Neurotrophin-4 deficient mice have a loss of vagal intraganglionic mechanoreceptors from the small intestine and a disruption of short-term satiety. J Neurosci. 2001b;21:8602–8615. doi: 10.1523/JNEUROSCI.21-21-08602.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox EA, Phillips RJ, Byerly MS, Baronowsky EA, Chi MM, Powley TL. Selective loss of vagal intramuscular mechanoreceptors in mice mutant for steel factor, the c-Kit receptor ligand. Anat Embryol. 2002;205:325–342. doi: 10.1007/s00429-002-0261-x. [DOI] [PubMed] [Google Scholar]
- Fox EA, Biddinger JE, Jones KR, McAdams J, Worman A. Mechanism of hyperphagia contributing to obesity in brain-derived neurotrophic factor knockout mice. Neuroscience. 2013a;229:176–199. doi: 10.1016/j.neuroscience.2012.09.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox EA, Biddinger JE, Baquet ZC, Jones KR, McAdams J. Loss of neurotrophin-3 from smooth muscle disrupts vagal gastrointestinal afferent signaling and satiation. Am J Physiol Regul Integr Comp Physiol. 2013b;305:R1307–R1322. doi: 10.1152/ajpregu.00337.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Dipatrizio NV, Guijarro A, Schwartz GJ, Li X, Gaetani S, Astarita G, Piomelli D. Sympathetic activity controls fat-induced oleoylethanolamide signaling in small intestine. J Neurosci. 2011;31:5730–5736. doi: 10.1523/JNEUROSCI.5668-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fundin BT, Silos-Santiago I, Ernfors P, Fagan AM, Aldskogius H, DeChiara TM, Phillips HS, Barbacid M, Yancopoulos GD, Rice FL. Differential dependency of cutaneous mechanoreceptors on neurotrophins, trk receptors, and P75 LNGFR. Dev Biol. 1997;190:94–116. doi: 10.1006/dbio.1997.8658. [DOI] [PubMed] [Google Scholar]
- Glebova NO, Ginty DD. Growth and survival signals controlling sympathetic nervous system development. Annu Rev Neurosci. 2005;28:191–222. doi: 10.1146/annurev.neuro.28.061604.135659. [DOI] [PubMed] [Google Scholar]
- Grider JR, Piland BE, Gulick MA, Qiao LY. Brain-derived neurotrophic factor augments peristalsis by augmenting 5-HT and calcitonin gene-related peptide release. Gastroenterology. 2006;130:771–780. doi: 10.1053/j.gastro.2005.12.026. [DOI] [PubMed] [Google Scholar]
- Hellard D, Brosenitsch T, Fritzsch B, Katz DM. Cranial sensory neuron development in the absence of brain-derived neurotrophic factor in BDNF/Bax double null mice. Dev Biol. 2004;275:34–43. doi: 10.1016/j.ydbio.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, Hammer RE, Herz J, Kuhn M. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc Natl Acad Sci U S A. 2002;99:7142–7147. doi: 10.1073/pnas.102650499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichim G, Tauszig-Delamasure S, Mehlen P. Neurotrophins and cell death. Exp Cell Res. 2012;318:1221–1228. doi: 10.1016/j.yexcr.2012.03.006. [DOI] [PubMed] [Google Scholar]
- Jahed A, Kawaja MD. The influences of p75 neurotrophin receptor and brain-derived neurotrophic factor in the sympathetic innervation of target tissues during murine postnatal development. Auton Neurosci. 2005;118:32–42. doi: 10.1016/j.autneu.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Jones KR, Fariñas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz DM. Regulation of respiratory neuron development by neurotrophic and transcriptional signaling mechanisms. Respir Physiol Neurobiol. 2005;149:99–109. doi: 10.1016/j.resp.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Kawakami T, Wakabayashi Y, Isono T, Aimi Y, Okada Y. Expression of neurotrophin messenger RNAs during rat urinary bladder development. Neurosci Lett. 2002;329:77–80. doi: 10.1016/S0304-3940(02)00598-0. [DOI] [PubMed] [Google Scholar]
- Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000;19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolbeck R, Bartke I, Eberle W, Barde YA. Brain-derived neurotrophic factor levels in the nervous system of wild-type and neurotrophin gene mutant mice. J Neurochem. 1999;72:1930–1938. doi: 10.1046/j.1471-4159.1999.0721930.x. [DOI] [PubMed] [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, Lowell BB. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest. 2011;121:1424–1428. doi: 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krimm RF. Factors that regulate embryonic gustatory development. BMC Neurosci. 2007;8(Suppl 3):S4. doi: 10.1186/1471-2202-8-S3-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz SI, Knudson CM, Korsmeyer SJ, Snider WD. Neurotrophins support the development of diverse sensory axon morphologies. J Neurosci. 1999;19:1038–1048. doi: 10.1523/JNEUROSCI.19-03-01038.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepore JJ, Cheng L, Min Lu M, Mericko PA, Morrisey EE, Parmacek MS. High-efficiency somatic mutagenesis in smooth muscle cells and cardiac myocytes in SM22alpha-Cre transgenic mice. Genesis. 2005;41:179–184. doi: 10.1002/gene.20112. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lommatzsch M, Braun A, Mannsfeldt A, Botchkarev VA, Botchkareva NV, Paus R, Fischer A, Lewin GR, Renz H. Abundant production of brain-derived neurotrophic factor by adult visceral epithelia. Implications for paracrine and target-derived neurotrophic functions. Am J Pathol. 1999;155:1183–1193. doi: 10.1016/S0002-9440(10)65221-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron. 1997;18:767–778. doi: 10.1016/S0896-6273(00)80316-5. [DOI] [PubMed] [Google Scholar]
- Mesulam MM. Tetramethyl benzidine for horseradish peroxidase neurohistochemistry: a non-carcinogenic blue reaction product with superior sensitivity for visualizing neural afferents and efferents. J Histochem Cytochem. 1978;26:106–117. doi: 10.1177/26.2.24068. [DOI] [PubMed] [Google Scholar]
- Murphy MC, Fox EA. Mice deficient in brain-derived neurotrophic factor have altered development of gastric vagal sensory innervation. J Comp Neurol. 2010;518:2934–2951. doi: 10.1002/cne.22372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhuber WL. Sensory vagal innervation of the rat esophagus and cardia: a light and electron microscopic anterograde tracing study. J Auton Nerv Syst. 1987;20:243–255. doi: 10.1016/0165-1838(87)90153-6. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Pan Y, Balazs L, Tigyi G, Yue J. Conditional deletion of Dicer in vascular smooth muscle cells leads to the developmental delay and embryonic mortality. Biochem Biophys Res Commun. 2011;408:369–374. doi: 10.1016/j.bbrc.2011.02.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo A, Calella AM, Fritzsch B, Knipper M, Katz D, Eilers A, Schimmang T, Lewin GR, Klein R, Minichiello L. Distinct requirements for TrkB and TrkC signaling in target innervation by sensory neurons. Genes Dev. 2002;16:633–645. doi: 10.1101/gad.217902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice FL, Albers KM, Davis BM, Silos-Santiago I, Wilkinson GA, LeMaster AM, Ernfors P, Smeyne RJ, Aldskogius H, Phillips HS, Barbacid M, DeChiara TM, Yancopoulos GD, Dunne CE, Fundin BT. Differential dependency of unmyelinated and A delta epidermal and upper dermal innervation on neurotrophins, trk receptors, and p75LNGFR. Dev Biol. 1998;198:57–81. doi: 10.1006/dbio.1998.8861. [DOI] [PubMed] [Google Scholar]
- Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, Jaenisch R. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- Rodrigo J, Hernandez J, Vidal MA, Pedrosa JA. Vegetative innervation of the esophagus. II. Intraganglionic laminar endings. Acta Anat. 1975;92:79–100. doi: 10.1159/000144431. [DOI] [PubMed] [Google Scholar]
- Rodrigo J, de Felipe J, Robles-Chillida EM, Pérez Antón JA, Mayo I, Gómez A. Sensory vagal nature and anatomical access paths to esophagus laminar nerve endings in myenteric ganglia. Determination by surgical degeneration methods. Acta Anat. 1982;112:47–57. doi: 10.1159/000145496. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, Moran TH. CCK elicits and modulates vagal afferent activity arising from gastric and duodenal sites. Ann N Y Acad Sci. 1994;713:121–128. doi: 10.1111/j.1749-6632.1994.tb44058.x. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, Salorio CF, Skoglund C, Moran TH. Gut vagal afferent lesions increase meal size but do not block gastric preload-induced feeding suppression. Am J Physiol. 1999;276:R1623–R1629. doi: 10.1152/ajpregu.1999.276.6.R1623. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- Sclafani A, Ackroff K, Schwartz GJ. Selective effects of vagal deafferentation and celiac-superior mesenteric ganglionectomy on the reinforcing and satiating action of intestinal nutrients. Physiol Behav. 2003;78:285–294. doi: 10.1016/S0031-9384(02)00968-X. [DOI] [PubMed] [Google Scholar]
- Smith GP. The direct and indirect controls of meal size. Neurosci Biobehav Rev. 1996;20:41–46. doi: 10.1016/0149-7634(95)00038-G. [DOI] [PubMed] [Google Scholar]
- Suetterlin P, Marler KM, Drescher U. Axonal ephrinA/EphA interactions, and the emergence of order in topographic projections. Semin Cell Dev Biol. 2012;23:1–6. doi: 10.1016/j.semcdb.2011.10.015. [DOI] [PubMed] [Google Scholar]
- Taicher GZ, Tinsley FC, Reiderman A, Heiman ML. Quantitative magnetic resonance (QMR) method for bone and whole-body-composition analysis. Anal Bioanal Chem. 2003;377:990–1002. doi: 10.1007/s00216-003-2224-3. [DOI] [PubMed] [Google Scholar]
- Tinsley FC, Taicher GZ, Heiman ML. Evaluation of a quantitative magnetic resonance method for mouse whole body composition analysis. Obes Res. 2004;12:150–160. doi: 10.1038/oby.2004.20. [DOI] [PubMed] [Google Scholar]
- Tucker KL. Neurotrophins and the control of axonal outgrowth. Panminerva Med. 2002;44:325–333. [PubMed] [Google Scholar]
- Unger TJ, Calderon GA, Bradley LC, Sena-Esteves M, Rios M. Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J Neurosci. 2007;27:14265–14274. doi: 10.1523/JNEUROSCI.3308-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls EK, Phillips RJ, Wang FB, Holst MC, Powley TL. Suppression of meal size by intestinal nutrients is eliminated by celiac vagal deafferentation. Am J Physiol. 1995;269:R1410–R1419. doi: 10.1152/ajpregu.1995.269.6.R1410. [DOI] [PubMed] [Google Scholar]
- Wang FB, Powley TL. Topographic inventories of vagal afferents in gastrointestinal muscle. J Comp Neurol. 2000;421:302–324. doi: 10.1002/(SICI)1096-9861(20000605)421:3<302::AID-CNE2>3.0.CO%3B2-N. [DOI] [PubMed] [Google Scholar]
- Wetmore C, Olson L. Neuronal and nonneuronal expression of neurotrophins and their receptors in sensory and sympathetic ganglia suggest new intercellular trophic interactions. J Comp Neurol. 1995;353:143–159. doi: 10.1002/cne.903530113. [DOI] [PubMed] [Google Scholar]
- Wood JD, Alpers DH, Andrews PL. Fundamentals of neurogastroenterology. Gut. 1999;45:II6–II16. doi: 10.1136/gut.45.2008.ii6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagorodnyuk VP, Chen BN, Brookes SJH. Intraganglionic laminar endings are mechanotransduction sites of vagal tension receptors in the guinea-pig stomach. J Physiol. 2001;534:255–268. doi: 10.1111/j.1469-7793.2001.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zermeño V, Espindola S, Mendoza E, Hernández-Echeagaray E. Differential expression of neurotrophins in postnatal C57BL/6 mice striatum. Int J Biol Sci. 2009;5:118–127. doi: 10.7150/ijbs.5.118. [DOI] [PMC free article] [PubMed] [Google Scholar]