

Summary
In response to many apoptotic stimuli, oligomerization of Bax is essential for mitochondrial outer membrane permeabilization and the ensuing release of cytochrome c. These events are accompanied by mitochondrial fission that appears to require Drp1, a large GTPase of the dynamin superfamily. Loss of Drp1 leads to decreased cytochrome c release by a mechanism that is poorly understood. Here we show that Drp1 stimulates tBid-induced Bax oligomerization and cytochrome c release by promoting tethering and hemifusion of membranes in vitro. This function of Drp1 is independent of its GTPase activity and relies on arginine 247 and the presence of cardiolipin in membranes. In cells, overexpression of Drp1 R247A/E delays Bax oligomerization and cell death. Our findings reveal a novel function of Drp1 and provide a new insight into the mechanism of Bax oligomerization.
Introduction
In mammalian cells undergoing apoptosis, many proteins that are confined to the intermembrane space of mitochondria are released as a result of the mitochondrial outer membrane (MOM) becoming permeable. Permeabilization of the MOM is ensured by pro-apoptotic members of the Bcl-2 family such as Bax or Bak (Kroemer et al., 2007; Schinzel et al., 2004). Whereas Bak is an integral MOM protein, Bax is inactive in the cytosol and soluble, or loosely attached to mitochondria, until it is activated by a diverse array of apoptotic stimuli (Youle and Strasser, 2008). Bax then undergoes conformational changes, translocates and inserts into the mitochondrial outer membrane (MOM) and oligomerizes, inducing MOM permeabilization (MOMP). These conformational rearrangements occur in the MOM and require a tight cooperation between Bax or Bak, BH3-only proteins and the lipid bilayer (Lucken-Ardjomande and Martinou, 2005). The current model postulates that tBid serves as a receptor for Bax, allowing its insertion and oligomerization in the membrane (Lovell et al., 2008). Other BH3-only proteins such as Bim, MAP-1, or Puma could also directly bind and recruit Bax in the MOM as does tBid (Certo et al., 2006; Gallenne et al., 2009; Gavathiotis et al., 2008; Marani et al., 2002; Tan et al., 2005; Walensky et al., 2006). Moreover, components of the TOM complex and Endophilin B1/Bif-1 appear to be important to fine-tune Bax insertion and oligomerization (Bellot et al., 2006; Etxebarria et al., 2008; Ott et al., 2007). Several findings also suggest that components of the mitochondrial fission machinery, including the large GTPase of the dynamin superfamily Drp1 (Heymann and Hinshaw, 2009), may play a role in MOMP and cytochrome c release (Youle and Strasser, 2008).
We previously reported that while tBid appears to be sufficient to trigger Bax oligomerization in synthetic liposomes (Kuwana et al., 2002; Lovell et al., 2008; Lucken-Ardjomande et al., 2008; Terrones et al., 2004), additional proteins are required to induce efficient Bax oligomerization in the MOM (Roucou et al., 2002). We searched for these proteins using a minimal cell-free assay and identified Drp1 as a protein capable of stimulating oligomerization of Bax upon its insertion into liposomes. We provide strong evidence that Drp1 stimulates Bax oligomerization by promoting membrane remodeling.
Results
Drp1 stimulates tBid-dependent Bax oligomerization in liposomes
Our previous work showed that a MOM-associated protein is required to induce efficient tBid-induced Bax oligomerization (Roucou et al., 2002). To identify this protein, we took advantage of a recently described acellular assay to monitor Bax oligomerization, based on the observation that Bax oligomers are partially resistant to trypsin (Goping et al., 1998; Lucken-Ardjomande et al., 2008). In line with our previous data, when incubated with tBid and liposomes containing phosphatidylcholine, phosphatidylethanolamine and cardiolipin (PC/PE/CL liposomes), a fraction of Bax became resistant to trypsin proteolysis (Figure 1A). Addition of high salt-extracted liver mitochondrial proteins stimulated tBid-induced Bax oligomerization, confirming the presence of a Bax activating factor (BAF) in this extract (Figure 1A). In addition, we found that cytosolic extracts from rat liver or brain, or HeLa cells, also stimulated Bax oligomerization (Figure 1A, F). Bax activation required the presence of a functional tBid BH3 domain and was prevented by Bcl-xL (Figure 1A). Proteolysis of the brain cytosolic extract with proteinase K abolished BAF activity. Thus, BAF(s) is/are protein(s) present in the cytosol or weakly attached to mitochondria. Because of its high BAF specific activity, the brain cytosolic extract was selected for biochemical purification of BAF. Cation exchange and size exclusion chromatographies allowed enrichment of BAF specific activity in high molecular weight fractions (P1-P3; Figure 1B). However, purification to homogeneity was not achieved due to a loss of activity during additional purification steps. We therefore decided to identify by mass spectrometry and/or Western blot analysis the proteins present in P1, which displayed the highest specific activity, and to test them individually in the Bax activation assay. Negative results were obtained with a number of different proteins (data not shown, see the list in the legend of Figure 1B), until we identified and tested Drp1 (Figure 1C). Recombinant Drp1 was able to activate Bax in a dose-dependent manner (Figure 1D, E), only in the presence of tBid (data not shown). To test whether Drp1 was responsible for the BAF activity present in the cytosolic extracts, we compared the activity of cytosolic extracts from control HeLa cells (expressing shLuc) and from HeLa cells depleted of Drp1 by RNA interference (shDrp1) (Parone et al., 2006). Drp1 depletion decreased the activity of the cytosol (Figure 1F), implicating it in Bax oligomerization. However, a residual activity was still present in the Drp1-depleted cytosol perhaps because of an incomplete removal of Drp1 or of the existence of additional BAFs.
Figure 1. Drp1 promotes Bax oligomerization in the presence of tBid.
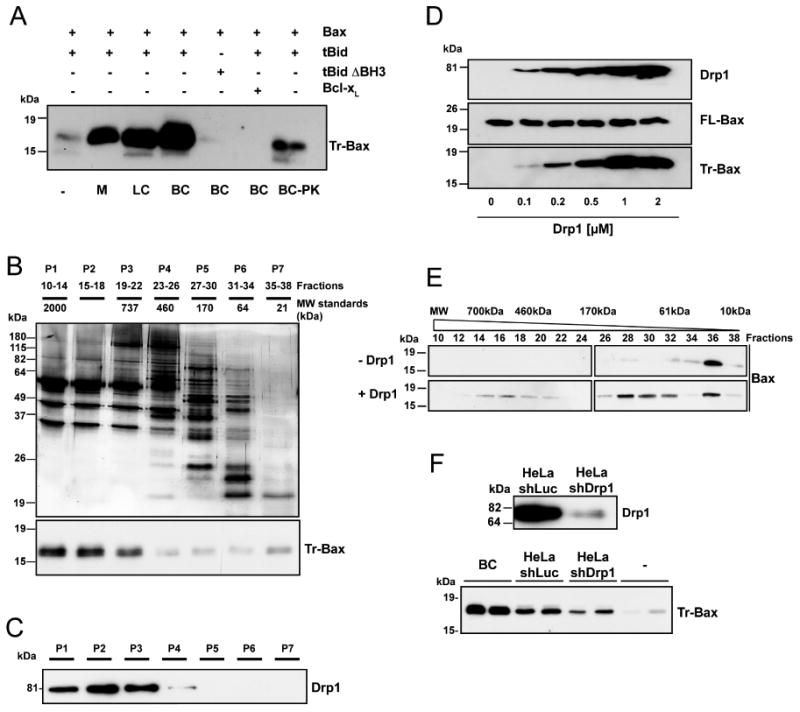
(A) PC/PE/CL (54/20/26 mol%) liposomes were incubated with the indicated proteins at 30°C for 30 min before ultracentrifugation, resuspension in KCl buffer and incubation with trypsin for 2 h at 30°C. Trypsin-resistant Bax (Tr-Bax) was analyzed by immunoblotting. M: salt-extracted liver mitochondrial proteins (400 μg); LC: rat liver cytosolic extract (200 μg); BC: rat brain cytosolic extract (200 μg); BC-PK: BC (200 μg) treated with proteinase K. Bax (50 nM); tBid (10 nM); tBid ΔBH3 (10 nM); Bcl-xL (1 μM). The blot is representative of three independent experiments.
(B) Purification steps of Bax-activating proteins: silver stained SDS-polyacrylamide gel electrophoresis of BC proteins fractionated by size exclusion chromatography (top panel); Bax-activating proteins (assessed as in A) are found in P1-P3 fractions (lower panel). Blots are representative of at least three independent experiments. P1 displayed the highest specific BAF activity and was found to contain GAPDH, Aldolase, Tubulin, Microtubule Associated Protein 1, Dynein, Spectrin, Actin, Gelsolin, Alpha Actinin, Cofilin and Drp1. Except Drp1, all proteins were found to be inactive in the Bax oligomerization assay.
(C) Immunoblot showing that Drp1 is present in P1-P3 fractions.
(D) Dose-response analysis of in vitro tBid-induced Bax oligomerization with increasing concentrations of recombinant Drp1. PC/PE/CL liposomes were incubated with 10 nM tBid and 50 nM Bax and increasing amounts of recombinant Drp1 before trypsin digestion and Bax analysis. Upper immunoblots show levels of Drp1 and Bax before trypsin digestion. The blot is representative of three independent experiments.
(E) Analysis of tBid-induced Bax oligomerization in isolated liposomes in the absence or presence of 1 μM Drp1 by size exclusion chromatography. PC/PE/CL liposomes were incubated with 10 nM tBid, 50 nM Bax and 1 μM Drp1. Liposomes were then lysed in 2% CHAPS, 200 nM NaCl, and proteins fractionated by size exclusion chromatography. The elution profile of Bax was analyzed by immunoblotting.
(F) Upper panel: Western blot analysis of Drp1 in control (shLuc) and Drp1-depleted (shDrp1) HeLa cells. Lower panel: analysis of Bax activating capacity of control and Drp1-depleted HeLa cytosolic extracts (200 μg each). Brain cytosol (BC) was also tested at 200 μg. In each experiment, the same cytosolic extract from HeLa cells was tested in duplicate. The experiment was repeated twice, using the same cell lines but with a different cytosolic extract preparation.
Cardiolipin is essential for Drp1's ability to promote Bax oligomerization
tBid has been previously reported to preferentially bind mitochondrial contact sites, to display a strong affinity for the negatively charged phospholipid CL [(Lutter et al., 2000) and Figure 2B] and to promote Bax oligomerization in CL-containing membranes (Kuwana et al., 2002; Lucken-Ardjomande et al., 2008). The concentration of CL at contact sites has been reported to represent around 25% of the phospholipid content (Ardail et al., 1990). This corresponds to the CL composition of the liposomes used in our study. When the amount of CL was reduced to 10%, a significant effect of Drp1 on tBid-induced Bax oligomerization was also observed, although less important than the effect obtained with liposomes containing higher CL concentrations (Figure 2A). In addition we found that Drp1 had a stronger affinity for CL than for phosphatidylserine (PS), another negatively charged phospholipid (Figure 2B), whereas the integral membrane protein hFis1, used as a control, bound equally well to CL- or PS-containing liposomes (Figure 2B). Since both tBid and Drp1 showed an affinity for CL, the question arose as to whether tBid could alter binding of Drp1 to liposomes or vice versa. Figure S1 shows that, under our experimental conditions, the binding of tBid and Drp1 was not significantly different whether the proteins were added alone or together, in the presence of Bax. Binding of Drp1 to CL could be mediated by electrostatic interactions between negative charges of the phospholipid and positively charged amino acids exposed at the surface of the protein. We therefore mutated a number of these amino acids, including R201, R247, K255 and K256 of the GTPase domain of Drp1 (Figure S2A). Mutation of these residues, except R247, did not alter binding to liposomes (data not shown). Arginine 247 is a residue that is exposed at the surface of Drp1 and is conserved between dynamins 1 and 2 and Drp1 of various species (Figure S2A, B). Mutations of R247 to an alanine or a glutamic acid (Drp1 R247A/E) significantly impaired the ability of Drp1 to bind CL-containing liposomes (Figure 2C), without impairing the GTPase activity of the protein (Figure 2D). In addition, the mutant proteins proved to be less efficient in promoting Bax oligomerization (Figure 2E), indicating that the binding of Drp1 to CL-containing liposomes is required for stimulating tBid-induced Bax oligomerization.
Figure 2. Drp1 acts independently of its GTPase activity, requires ATP, and its binding to cardiolipin is essential for tBid-induced Bax oligomerization.
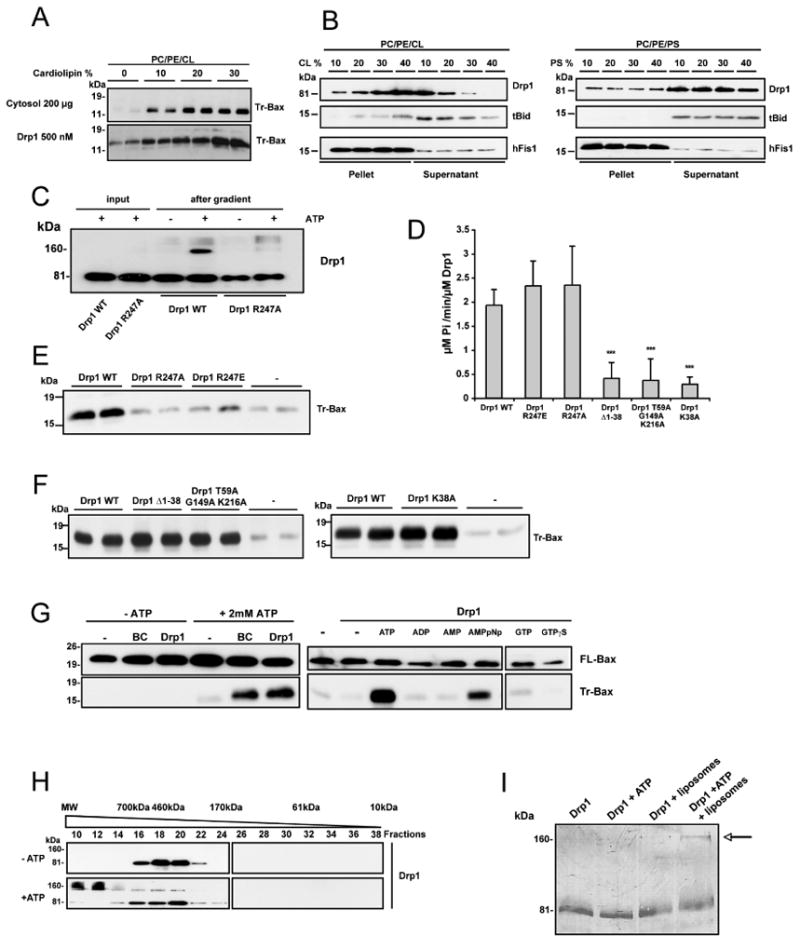
(A) PC/PE/CL liposomes were prepared with increasing concentrations of CL and concomitant reduction of phosphatidylcholine. They were incubated with 10 nM tBid and 50 nM Bax and with 200 μg brain cytosol or 500 nM Drp1 before trypsin digestion and Bax analysis. This blot is representative of three independent experiments.
(B) Preferential binding of Drp1 and tBid to CL. Liposomes with increasing amounts of CL (PC/PE/CL, left panel) or PS (PC/PE/PS, right panel) were incubated in the presence of Drp1 (1 μM) or tBid (10 nM), centrifuged and analyzed by Western blotting for the presence of Drp1 or tBid in the pellet or the supernatant. As a control, liposomes were incubated with the integral membrane protein hFis1. Each panel represents a separate experiment. This blot is representative of two independent experiments. See also Figure S1.
(C) PC/PE/CL (54/20/26) liposomes were incubated with Drp1 R247A or Drp1 WT (500 nM each) alone in the presence or absence of ATP and loaded on a sucrose gradient before ultracentrifugation. Drp1 R247A and Drp1 WT were analyzed by immunoblotting in the suspension before gradient centrifugation and after centrifugation in the floating liposomal suspension. This blot is representative of three independent experiments.
(D) GTPase activity of Drp1 WT and Drp1 mutants (1 μM each). Values are means of three independent experiments ± SD. Drp1 GTPase mutants differed from WT with ***: p< 0.001.
(E) Comparison of the effects of Drp1 WT, Drp1 R247A and Drp1 R247E (1 μM each) on tBid-induced Bax oligomerization. PC/PE/CL liposomes were incubated in the presence of 10 nM tBid, 50 nM Bax and the indicated proteins before trypsin digestion and Bax analysis. The blot is representative of three independent experiments. See also Figure S2.
(F) Analysis of Drp1 GTPase mutants (1 μM each) for their capacity to stimulate tBid-induced Bax oligomerization. Blots show duplicates and are representative of three independent experiments.
(G) In vitro analysis of tBid-induced Bax oligomerization in the absence or presence of ATP (left blot) or in the presence of ATP or other nucleotides, each tested at 2.5 mM (right blot). Experiments were performed in the presence or absence of 1 μM Drp1 or 200 μg brain cytosol (BC) (right blot; the two parts of this blot correspond to the same membrane and identical exposure times). Upper blots show Bax binding to liposomes. Blots are representative of at least three independent experiments.
(H) Analysis of Drp1 by size exclusion chromatography. Liposomes were incubated with Drp1 (1 μM), Bax (50 nM) and tBid (10 nM) in the presence or absence of 2.5 mM ATP, solubilized with CHAPS, separated by size exclusion chromatography and the elution profile of Drp1 was analyzed by immunoblotting.
(I) Drp1 (1 μM) was incubated with liposomes and/or 2.5 mM ATP, run on a polyacrylamide gel under reducing conditions and detected by Coomassie staining after transfer to a nitrocellulose membrane. The arrow points to Drp1 dimers.
Drp1 stimulates Bax oligomerization independently of its GTPase activity
Since Drp1 is a large GTPase of the dynamin family, we further tested whether its GTPase activity was necessary to promote tBid-induced Bax oligomerization. Key amino acids in the GTPase domain were mutated to produce GTPase-deficient mutants (Drp1 K38A, Drp1Δ1-38, and Drp1 T59A/G149A/K216A) (Figure 2D). Interestingly, all of these mutants were still capable of stimulating Bax oligomerization (Figure 2F), indicating that the GTPase activity of Drp1 is dispensable for Bax oligomerization.
Supporting this finding, the buffer used to assay BAF in vitro did not contain GTP. However, it contained ATP, the role of which was therefore tested. In the presence of cytosol or Drp1 and tBid, but without ATP, Bax oligomerization dropped to the level obtained in the presence of Bax and tBid alone (Figure 2G, left blot), indicating that ATP was important for Drp1 to stimulate tBid-induced Bax oligomerization. Other nucleotides, including GTP, GTPγS, ADP and AMP could not substitute for ATP (Figure 2G, right blot), unless used at supraphysiological concentrations (>5 mM; data not shown). In contrast, the non-hydrolysable ATP analogue AMPpNp was almost as efficient as ATP, indicating that hydrolysis of ATP was not required for Bax activation (Figure 2G). Accordingly, we excluded the possibility that Drp1 acted as an ATPase (data not shown).
ATP was not required for membrane binding of Drp1 (Figure 2C), but proved to have an impact on its quaternary structure in the presence of liposomes. In agreement with previous data, Drp1 was purified as a tetramer as assessed by size exclusion chromatography (Zhu et al., 2004). In the presence of either ATP alone (data not shown) or Bax, tBid and CL-containing liposomes (Figure 2H), Drp1 remained tetrameric. However, in the presence of ATP, tBid, Bax and liposomes, it was eluted in large molecular weight (MW) fractions, suggesting that the protein formed larger oligomers. A similar elution profile was obtained in the absence of tBid and Bax (data not shown). Drp1 present in the large MW fractions migrated both as a monomer (∼80 kDa) and a dimer (∼160 kDa) on SDS-PAGE, suggesting incomplete disassembly by the SDS present in the buffer (Figure 2H). This dimer was also detected by SDS-PAGE and Coomassie staining upon incubation of 500 nM Drp1 with liposomes and ATP (Figure 2I and see also Figure 2C). Further studies are necessary to determine how ATP promotes formation of high order Drp1 oligomers.
Drp1 promotes tethering and hemifusion of cardiolipin-containing membranes
Interestingly, in the presence of ATP, liposomes clustered in a Drp1 dose-dependent manner, as shown by visual observation (Figure 3A) and by a characteristic rise in the turbidity of the liposome suspension (Nakatogawa et al., 2007) (Figure 3B). These aggregates disappeared after the addition of proteinase K, indicating that Drp1 was responsible for membrane tethering (Figure 3B).
Figure 3. Drp1 triggers membrane tethering.
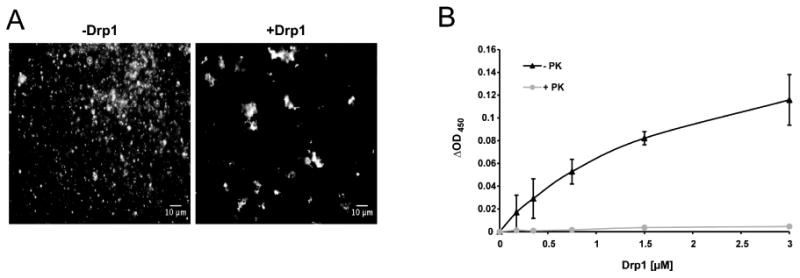
(A) Fluorescence microscopy images of PC/PE/CL liposomes containing 1% NBD-PE in the presence of ATP and the presence or absence of 1 μM Drp1.
(B) Dose-response of liposome aggregation in the presence of increasing concentrations of Drp1 (black line). Proteinase K was added to aggregated liposomes and the turbidity of the liposome suspension was measured 5 min later (grey line). Turbidity of the suspension was measured at 450 nm. ΔOD values represent the difference between OD450 of the liposome suspension incubated in the presence of proteins and OD450 values in the absence of proteins. Results are means of three independent experiments ± SD.
Liposome aggregation could simply represent membrane bridging, but could also represent hemifusion (i.e. fusion of the outer leaflets of adjacent membranes, while inner leaflets remain intact) or complete fusion (i.e. the merger of both inner and outer leaflets) of apposed membranes. In order to test these possibilities, we used a lipid mixing assay, which is based on fluorescence resonance energy transfer from 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4yl) (NBD-PE) to 1,2-dioleoyl-snglycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rho-PE) (Struck et al., 1981). PC/PE/CL large unilamellar vesicles (LUVs) were prepared with or without addition of NBD-PE and Rho-PE. When the two dyes are present at an appropriate concentration in the same liposome, the fluorescence of NBD is quenched by rhodamine. Upon fusion of these LUVs with unlabeled LUVs, the distance between the two dyes increases, resulting in a dequenching of NBD fluorescence. When the two LUV populations were mixed in the presence of Drp1, NBD fluorescence increased in a time and dose-dependent manner, indicating that lipid mixing had occurred (Figure 4A, B, C).
Figure 4. Drp1 potentiates tBid-induced Bax oligomerization by promoting hemifusion of cardiolipin-containing membranes.
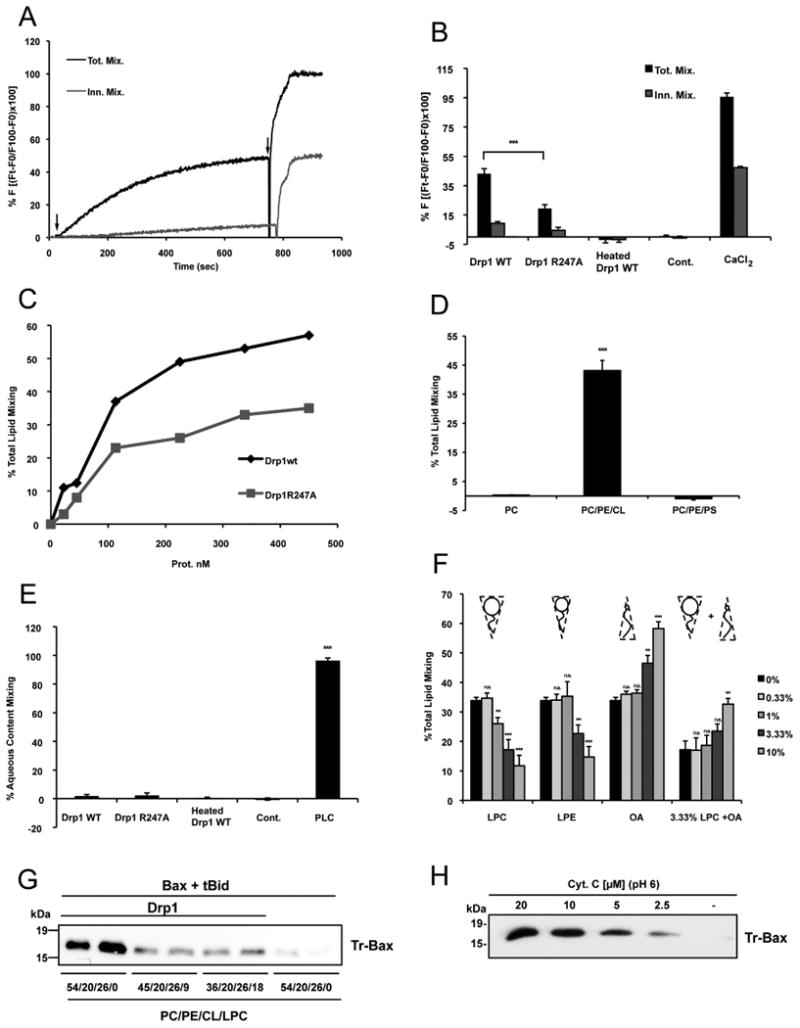
(A) Representative time courses of total lipid mixing (Tot. Mix.) and inner monolayer lipid mixing (Inn. Mix.) induced by 225 nM Drp1 WT in PC/PE/CL LUVs. Lipid mixing was monitored by the NBD/Rhodamine lipid dilution assay. For inner monolayer lipid mixing NBD + Rho-liposomes were treated with appropriate amounts of sodium dithionite to quench NBD fluorescence of the outer leaflet. Unlabeled and NBD + Rho-Labeled liposomes were mixed and incubated for 5 min before protein addition. The extent of lipid mixing was quantified on a percentage basis according to the equation: (Ft − F0/F100 − F0) × 100 where Ft is the measured fluorescence of protein-treated LUVs at time t, F0 is the initial fluorescence of the LUV suspension before protein addition, and F100 is the fluorescence value after complete disruption of LUVs by addition of 10 mM β-octylglucoside (OG). First arrow shows time of addition of Drp1 WT to the liposome suspension and the second arrow denotes addition of OG.
(B) Comparison of the degree of total lipid mixing and inner monolayer lipid mixing elicited by Drp1 WT, Drp1 R247A and heat-denatured Drp1 WT (Heated Drp1 WT) and 10 mM CaCl2. As a control, Drp1 WT was incubated with NBD/Rho containing LUVs but in the absence of unlabeled LUVs (Cont.). 100% total lipid mixing value corresponds to LUVs treated with OG. Mean values ± S.E. are shown for three to seven independent experiments. Drp1 WT differed from Drp1 R247A with: *** p < 0.001. See also Figure S3.
(C) Dose response of Drp1 WT and Drp1 R247A on total lipid mixing.
(D) Lipid mixing elicited by Drp1WT in LUVs containing either PC (100), PC/PE/CL (54/20/26) or PC/PE/PS (28/20/52). Mean values ± S.E. are shown for three independent experiments. PC and PC/PE/PS differed from PC/PE/CL with *** p < 0.001.
(E) Comparison of the degree of aqueous content mixing elicited by Drp1 WT, Drp1 R247A, heat-denatured Drp1 WT (Heated Drp1 WT) and 0.1 nM Phospholipase C from Bacillus cereus (PLC). Content mixing was monitored by ANTS/DPX aqueous content mixing assay; 100% of content mixing was determined using LUVs containing both 12.5 mM ANTS and 45 mM DPX. Drp1 WT was incubated with ANTS containing LUVs as a control (Cont.). Mean values ± S.E. are shown for three independent experiments. Drp1 WT, Drp1 R247A, and heated Drp1 WT differed from PLC with: *** p < 0.001.
(F) Total lipid mixing induced by 158 nM Drp1WT in PC/PE/CL vesicles upon external addition of indicated concentrations of LPC, LPE and OA. Mean values ± S.E. are shown for three to seven independent experiments. Each LPC, LPE, OA concentration differed from 0% with **: p < 0.01. ***: p < 0.001 or was not significantly different (ns).
(G) PC/PE/CL liposomes containing 9 or 18 mol% LPC were used in in vitro Bax oligomerization assays. Drp1 was used at 1 μM. Blot shows duplicates and is representative of three independent experiments.
(H) PC/PE/CL liposomes were incubated with tBid and Bax and increasing concentrations of cytochrome c at pH 6 before assaying Bax oligomerization. The blot is representative of three independent experiments.
The level of NBD fluorescence induced by Drp1 was about 50% of that obtained upon addition of detergent, which provides the maximum level of fluorescence (100%). As expected, the extent of lipid mixing obtained with Drp1 R247A was significantly reduced compared to Drp1 WT (Figure 4B, C). On the other hand, no increase in NBD fluorescence was measured with heat-denatured Drp1 or when Drp1 was incubated in the presence of labeled LUVs alone (Figure 4B). Finally, consistent with the preferential binding of Drp1 to CL, replacing this phospholipid with PS, maintaining the net charge of the vesicles, completely abolished the ability of Drp1 to trigger an increase in NBD fluorescence (Figure 4D). Thus, CL is critical for Drp1-induced lipid mixing. A similar conclusion was reached when lipid mixing was monitored by FACS analysis (Figure S3A, B). We then investigated whether Drp1-induced lipid mixing resulted from complete membrane fusion or membrane hemifusion. We prepared asymmetrically labeled liposomes by adding the membrane-impermeable reductant sodium dithionite to the liposome suspension to selectively quench the fluorescence of NBD in the outer leaflet. The validity of the assay was checked with calcium which is known to trigger complete fusion (Ortiz et al., 1999). In the presence of 10 mM calcium, a significant increase in NBD fluorescence was still measured upon incubation of dithionite-treated LUVs with unlabeled LUVs, indicating that fusion of the inner leaflets of apposed membranes, i.e. complete fusion of vesicles, had occurred (Figure 4B). In contrast to calcium, under these conditions, Drp1 WT or Drp1 R247A had a small impact on NBD fluorescence, indicating that Drp1 mainly triggers fusion of the outer leaflet with minimal inner leaflet mixing (Figure 4A, B). Consistent with these results, no fusion pore formation and content mixing occurred in the presence of Drp1. This result was established using the content-mixing fusion assay where liposomes filled with 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS) are mixed with liposomes filled with p-xylylenebis(pyridinium bromide) (DPX) (Ellens et al., 1985). Vesicle fusion results in quenching of ANTS fluorescence. As a positive control, we used phospholipase C (PLC) from Bacillus cereus that had previously been reported to induce membrane fusion (Basanez et al., 1996) (Figure 4E). In contrast to PLC, neither Drp1 WT nor Drp1 R247A induced aqueous content mixing, indicating that Drp1 does not trigger lipid pore formation and complete membrane fusion (Figure 4E).
According to the widely accepted stalk-pore fusion model (Chernomordik and Kozlov, 2008), hemifusion is thought to start with the formation of a stalk, a local connection between the contacting monolayers of two membranes. The stalk then extends connecting the facing monolayers (hemifusion) before pore formation (fusion) occurs. The model predicts that addition of inverted cone shaped lipids (i.e. positive curvature-inducing lipids) such as lyso-phosphatidylcholine (LPC) or lyso-phosphatidylethanolamine (LPE) to contacting membrane leaflets should prevent formation of hemifused intermediates (Chernomordik et al., 1995), whereas cone shaped lipids such as oleic acid (OA), which induce negative curvatures, should promote formation of hemifusion intermediates. Therefore, to confirm that Drp1 induced lipid mixing through formation of hemifusion intermediates, we added sub-lytic concentrations of LPC or LPE (Chernomordik et al., 1993) or OA to the vesicles (Figure 4F; see also Figure S3C). Addition of LPC or, to a lesser degree, LPE that possesses a less positive intrinsic curvature than LPC, significantly decreased total lipid mixing induced by Drp1 in a dose-dependent manner. On the other hand, addition of OA slightly promoted Drp1-induced lipid mixing. When OA and LPC were added together, OA was able to counteract the inhibitory effect of LPC on lipid mixing. These data strongly argue that Drp1-induced lipid mixing is mediated by formation of membrane hemifusion intermediates.
Membrane hemifusion is sufficient to stimulate tBid-induced Bax oligomerization
Whereas LPC significantly reduced membrane hemifusion induced by Drp1, it also blocked the effect of Drp1 on Bax oligomerization (Figure 4G), suggesting that the capacity of Drp1 to facilitate tBid-induced Bax oligomerization is related to its capacity to promote membrane hemifusion.
Cytochrome c at pH 6 has previously been reported to stimulate membrane hemifusion (Kawai et al., 2005). To demonstrate further the role of membrane hemifusion as the mechanism mediating the effect of Drp1 on tBid-induced Bax oligomerization, we tested whether hemifusion triggered by cytochrome c at pH 6 (Figure S3D) would also promote Bax oligomerization. We found that tBid-induced Bax oligomerization was enhanced by increasing concentrations of cytochrome c at pH 6 (Figure 4H), indicating that rather than Drp1 per se, it is the membrane hemifusion process that is directly responsible for potentiating tBid-induced Bax oligomerization.
Overexpression of Drp1 R247A or Drp1 R247E delays apoptosis
We tested the physiological relevance of part of our findings in cellulo. We first analyzed the migration pattern of Drp1 from HeLa cells undergoing actinomycin D (ActD)-induced apoptosis by SDS-PAGE. In agreement with previous data (Breckenridge et al., 2003; Frank et al., 2001; Wasiak et al., 2007), we observed that Drp1 was recruited to mitochondria during apoptosis (Figure 5A). Moreover, consistent with the data obtained with liposomes, we found that a proportion of Drp1 was detected as a ∼ 160 kDa band on SDS-PAGE, suggesting that Drp1 oligomerizes during apoptosis. We then expressed Drp1 R247A/E mutants in HeLa cells and tested their impact on mitochondrial morphology, Bax oligomerization and cytochrome c release during apoptosis (Figures 5B-D, 6A-E). Drp1 R247 A/E was found to co-immunoprecipitate with Drp1 WT, indicating that they likely formed hetero-oligomers (Figure S4). Importantly, in 80% of cells expressing mutant Drp1 R247E, mitochondria formed very elongated tubules, which showed a number of vesicular dilatations (Figures 5B-D). These morphological alterations of mitochondria suggest that, despite a normal GTPase activity, Drp1 R247A/E has an impaired fission activity and exerts a dominant negative effect over endogenous Drp1. Taken together with the weaker binding of Drp1 R247A to CL-containing liposomes, these findings suggest that CL could play a role in the mitochondrial recruitment and/or activity of Drp1, in a manner similar to what has been previously shown for the assembly of the dynamin related protein Mgm1 in the inner mitochondrial membrane (DeVay et al., 2009).
Figure 5. Drp1 oligomerization during apoptosis and impact of Drp1 247A/E mutants on mitochondrial morphology.
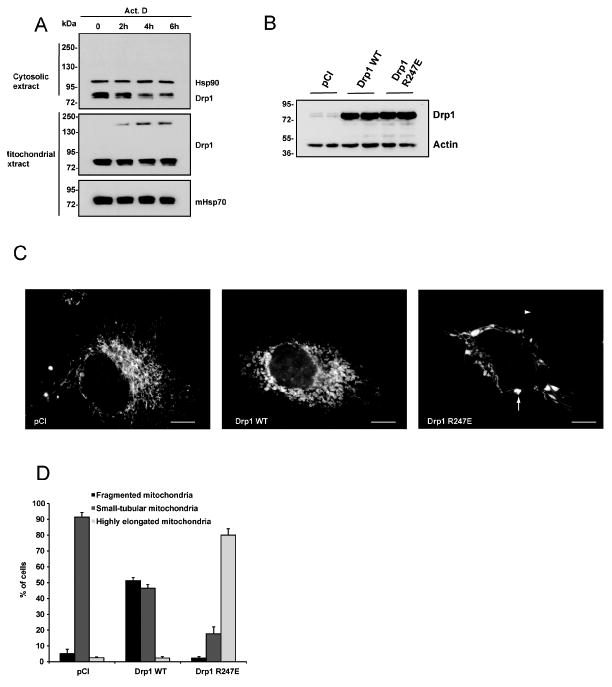
(A) Mitochondrial and cytosolic extracts were prepared from HeLa cells cultured in the absence (time 0) or in the presence of ActD (3 μM) for 2, 4 and 6 h. Cell extracts were separated by SDS-PAGE in the absence of DTT and analyzed for Drp1 by Western blotting. Hsp90 and mHsp70 were used as loading controls for cytosolic and mitochondrial extracts respectively. The blots are representative of four independent experiments.
(B-D): HeLa cells were transfected with an empty DNA vector (pCI) or with plasmids encoding Drp1 WT or Drp1 R247E together with a mitochondria-targeted YFP. 48h later Drp1 expression levels were quantified in total cell extracts by Western blot using an antibody to Drp1. Actin was used as a loading control. In parallel, mitochondrial morphology was analyzed by fluorescence microscopy. (B) Immunoblot shows that cells transfected with plasmids encoding Drp1 WT or Drp1 R247E expressed equivalent amounts of Drp1 WT and Drp1 R247E proteins. Actin was used as a loading control. See also Figure S4. (C) Morphology of mitochondria in HeLa cells observed by fluorescence microscopy. White arrow denotes the presence of vesicular dilatations while arrowhead denotes highly elongated mitochondria. Bar is 5 μm (D) Quantification of mitochondrial morphology. Mitochondria were divided into three classes: small tubular, which corresponds to cells mainly filled with small filaments of ∼2 μm or less; fragmented that corresponds to punctiform mitochondria; highly elongated, which corresponds to cells mainly filled with mitochondria > 5μm.
Figure 6. Expression of Drp1 R247A/E mutants decreases cytochrome c release and Bax oligomerization in response to apoptotic stimuli.
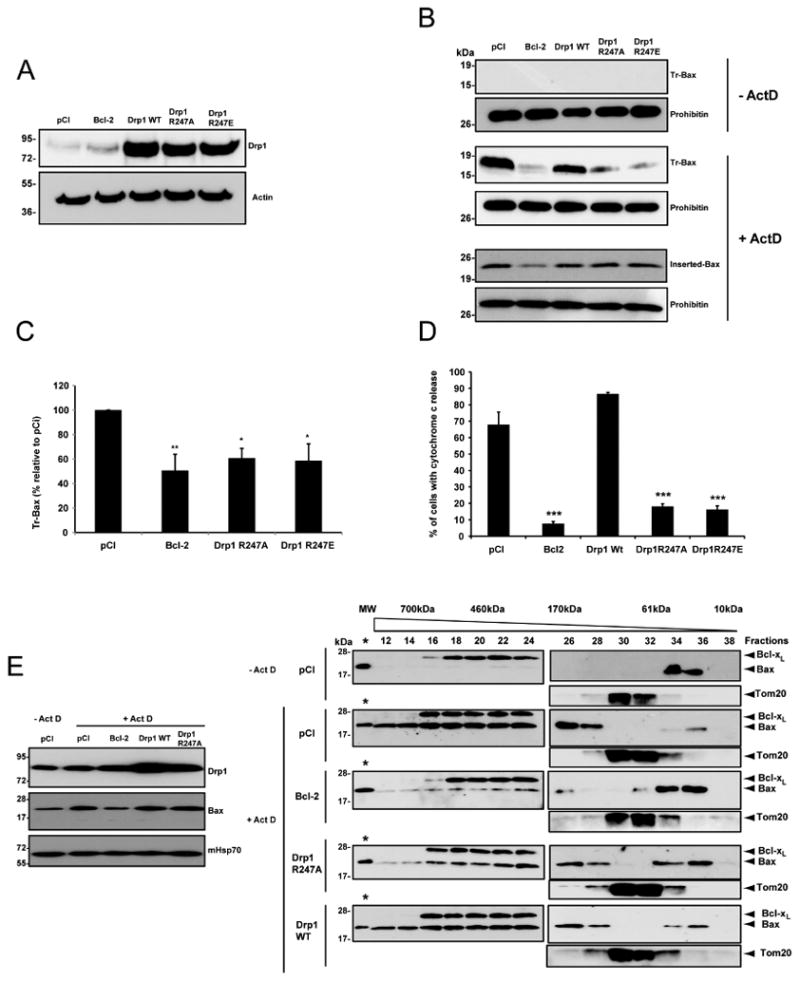
(A) In all experiments performed to assess cytochrome c release, Bax oligomerization, and cell death (Figure 7), HeLa cells were transfected with an empty vector (pCI) or with plasmids encoding Bcl-2, Drp1 WT or Drp1 R247A/E. The efficacy of transfection varied between 60 to 80%. Drp1 WT and Drp1 R247A/E were overexpressed at equivalent levels as shown by the immunoblot perfomed with an antibody to Drp1. Actin was used as a loading control.
(B, C) Analysis of Bax insertion in the outer mitochondrial membrane by alkali treatment and Bax oligomerization by the trypsin digestion assay. HeLa cells were transfected as indicated in (A). 72 h after transfection, apoptosis was induced with 3 μM ActD (+ ActD). 4 h later, mitochondria were isolated and analyzed for Bax insertion by alkali treatment (inserted Bax) or Bax oligomerization by trypsin digestion (Tr-Bax). Tr-Bax was also analyzed in untreated HeLa cells (-ActD). Prohibitin was used as a mitochondrial loading control. Note that although Bax insertion was similar in cells transfected with pCI, Drp1 WT or Drp1 R247A/E vectors, Tr-Bax levels were significantly lower in cells expressing Drp1 R247A/E or Bcl-2 as shown in (C) which is a compilation of results from four different experiments performed for pCI, Bcl-2, Drp1 R247A and Drp1 R247E. Results represent a quantification of the immunoblots for trypsin-resistant Bax. Data are mean ± SE (n = 6); *p < 0.05, ** p < 0.01.
(D) HeLa cells were transfected as indicated in (A). 72 h later cells were treated with ActD for 4 h in the presence of 100 μM z-VAD to avoid their detachment from the culture dish, and immunostained for cytochrome c. Cells were analyzed by fluorescence microscopy and the number of cells with diffused cytochrome c staining was counted. Histograms display the means of three independent experiments ± S.E; ***: p < 0.001
(E) Analysis of Bax oligomerization by size exclusion chromatography. HeLa cells were transfected as indicated in (A). 72 h after transfection, cells were treated with 3 μM ActD to induce apoptosis. 6h later mitochondria were isolated from cells undergoing apoptosis and from pCI transfected cells that were not exposed to ActD, as a control. Proteins were extracted from membranes with 2% CHAPS in 200 mM NaCl and fractionated by size exclusion chromatography. Proteins in the eluted fractions were precipitated with tricholoroacetic acid and separated by SDS-PAGE before Bax immunoblotting (Right panel). 1 ng recombinant Bax was loaded on every blot (labeled with an asterisk) to control that every blot was exposed similarly to X ray film during the chemiluminescence detection of proteins, allowing direct comparison of all immunoblots. In addition, Bcl-xL and Tom 20 were used as loading controls. The left panel shows levels of Bax that were associated with mitochondria before extraction. Results confirm those shown in (B), lower panel). Drp1 and mHsp70 (used as a loading control) were also blotted. See also Figure S5.
Bax oligomerization, monitored by the trypsin digestion assay (Figures 6B, C), by size exclusion chromatography (Figure 6E) or cross-linking (Figure S5), and MOMP, analysed through the release of cytochrome c (Figure 6D), were significantly decreased in both Drp1 R247A/E and Bcl-2 expressing cells compared to cells transfected with an empty vector or to cells overexpressing Drp1 WT. Thus, expression of Drp1 R247A/E significantly decreased Bax oligomerization and MOMP in vivo. As a consequence, cells expressing the Drp1 mutants were more resistant to apoptosis than cells expressing equivalent amounts of Drp1 WT or than cells transfected with an empty plasmid when exposed to 3 μM ActD or UV irradiation (Figures 7A, B). Although the protection was significant at 6 hours after exposure to the stress stimulus, no protection was observed after 24 hours (data not shown), consistent with previously published results (Parone et al., 2006; Sheridan et al., 2008). This indicates that other mechanisms can substitute for Drp1 to trigger Bax oligomerization.
Figure 7. Expression of Drp1 R247A/E delays cell death.
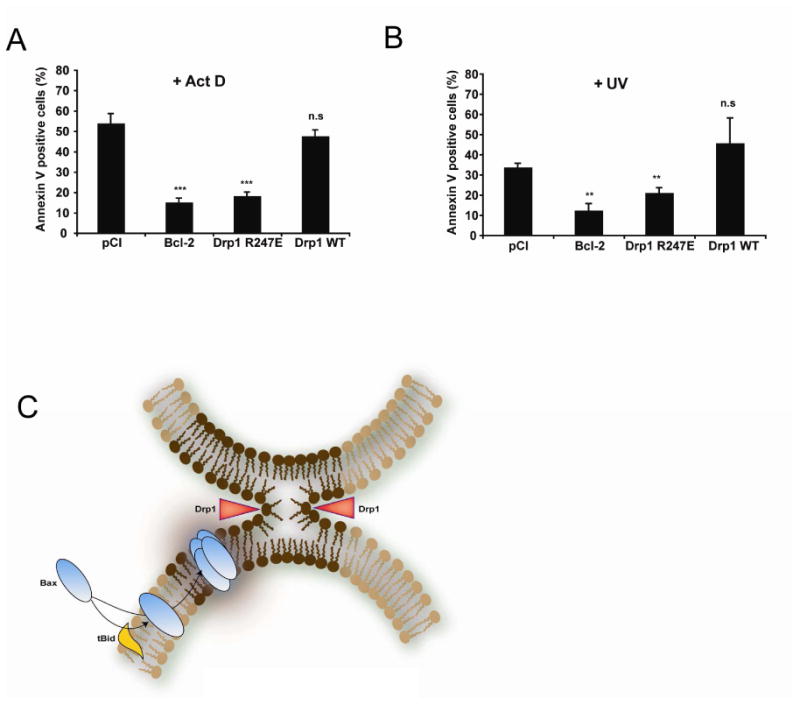
(A, B) HeLa cells were transfected with an empty vector (pCI) or with plasmids encoding Bcl-2, Drp1 WT or Drp1 R247E. 72 h later, cell cultures were treated with 3 μM Act D (A) or UV- irradiated (60 mJ/cm2) (B) and apoptosis quantified 6 h later by Annexin V staining and FACS analysis. Values are the average of six independent experiments ± S.E. **: p < 0.01; ***: p < 0.001; (n.s.), not significant.
(C) Model explaining the role of mitochondrial membrane hemifission or hemifusion intermediates in Bax oligomerization. During apoptosis, Bax is recruited to the outer mitochondrial membrane by tBid (or other BH3 only proteins) where it inserts. At the same time, Drp1 constricts the organelle as indicated by red arrowheads, triggering the formation of a hemifission intermediate. This membrane remodeling (dark brown part of the membrane) promotes Bax oligomerization. We speculate that contact sites between the inner and outer membranes, that are enriched in CL, could also be privileged sites for the formation of hemifusion intermediates, independently of Drp1. Contact sites could therefore represent additional sites in which Bax oligomerization would occur.
Discussion
Inhibition of Drp1 has previously been reported to delay cytochrome c release and apoptosis in vitro (Brooks et al., 2007; Frank et al., 2001; Germain et al., 2005; Ishihara et al., 2009; Lee et al., 2004; Cassidy-Stone et al., 2008). Moreover, Drp1-null mouse embryos fail to undergo developmentally regulated apoptosis during neural tube formation in vivo (Wakabayashi et al., 2009). Our results now explain that Drp1 participates in apoptosis by stimulating Bax oligomerization, thereby enhancing MOMP. By promoting Bax oligomerization and massive cytochrome c efflux, Drp1 could ensure that mitochondrial function is irreversibly altered during apoptosis, and that a sufficient amount of cytochrome c is released to activate APAF1. This would prevent cells from recovering after MOMP, a scenario that has been shown in cells expressing low caspase activity and high levels of GAPDH (Colell et al., 2007). Drp1 may therefore correspond to the previously described macromolecular cytosolic factor termed PEF (permeability enhancing factor), which was defined as a proteinaceous macromolecule able to enhance MOMP by tBid and Bax (Kluck et al., 1999).
Furthermore, we report that, in vitro, Drp1 promotes Bax oligomerization by triggering membrane tethering and hemifusion, independently of its GTPase activity. Rather than a direct interaction between Bax and Drp1, which we have not been able to detect (data not shown), we provide evidence that formation of a Drp1-induced membrane hemifusion intermediate promotes tBid-induced Bax oligomerization. This membrane structure promoted by Drp1 in a reconstituted system may recapitulate the membrane remodeling that occurs at mitochondrial fission sites in cells undergoing apoptosis. Indeed, analogous to membrane fusion, membrane fission also proceeds via a pathway of intermediate structures. It has been shown that at membrane constriction sites, dynamins would pinch the membrane and bring into contact the inner leaflets of the membrane, allowing the formation of what has been called a hemifission intermediate (Kozlovsky and Kozlov, 2003). Thus, during mitochondrial fission, Drp1 could constrict the MOM and trigger the formation of a hemifission intermediate at fission sites (See Figure 7 C for a model). The occurrence of this mitochondrial membrane remodeling during apoptosis may provide the appropriate membrane curvature, lipid composition and/or lateral pressure profile that are considered to be essential determinants for integral membrane protein oligomerization (Basañez et al., 2002; Lucken-Ardjomande et al., 2008; van den Brink-van der Laan et al., 2004) and may explain the preferential localization of Bax at mitochondrial fission sites in apoptotic cells (Karbowski et al., 2002).
Our data also suggest that any mechanism that would lead to formation of membrane hemifusion intermediates during apoptosis may promote Bax oligomerization. Indeed, in the liposome assay, cytochrome c, which is able to trigger membrane hemifusion at pH 6, triggered tBid-induced Bax oligomerization as well as Drp1. Privileged sites where such a membrane remodeling could occur, are contact sites between the inner and outer mitochondrial membranes, which are higly enriched in CL. We postulate that oligomerization of Bax in, or nearby, hemifusion or hemifission intermediates would increase membrane tension and damage these very fragile structures, triggering leakage. Therefore, hemifusion/hemifission intermediates could be thought of as an Achille's heel targeted by Bax to damage the outer mitochondrial membrane and trigger cytochrome c release.
Experimental Procedures
Liposome preparation
In most experiments, liposomes were prepared as described by Lucken-Ardjomande et al. (2008). For lipid mixing assays, lipid mixtures at the indicated ratios were co-dissolved in chloroform/methanol (2:1). Organic solvents were removed by evaporation under nitrogen stream followed by incubation under vacuum for 2 h. Dried lipid films were resuspended in the required buffers. LUVs were formed using 10 freeze/thaw cycles and two polycarbonate membranes of 0.2-μm pore size for extrusion (Nucleopore, San Diego, CA). Unless otherwise stated, liposome composition was PC/PE/CL (54:20:26, mol/mol). Egg yolk PC, bovine heart CL, bovine brain PE and bovine brain LPC were from Sigma or Avanti.
Lipid mixing and aqueous content mixing assays in large unilamellar vesicles
The resonance energy transfer assay of Struck et al. (1981) was used to monitor membrane lipid mixing. Briefly PC/PE/CL (54: 20: 26) LUVs containing 2 mol% of NBD-PE and 2 mol% Rh-PE were mixed with 9 fold excess of unlabelled LUVs. NBD-PE emission was monitored at 530 nm with the excitation wavelength set at 465 nm. A 515 nm cut-off filter was placed between the sample and the emission monochromator to avoid scattering interference. Inner monolayer lipid mixing was measured using asymmetrically labeled membrane vesicles produced by the quenching of the outer leaflet NBD-PE fluorescence upon addition of sodium dithionite. Dithionite was removed by gel filtration in Sephadex G-25, using 125 mM KCl as elution buffer.
Content mixing was monitored by the ANTS/DPX assay performed by Ellens et al. (1985). LUVs containing either a) 25 mM ANTS, 40 mM NaCl, and 10 mM HEPES, b) 90 mM DPX and 10 mM HEPES, or c) 12.5 mM ANTS, 45 mM DPX, 20 mM NaCl, and 10 mM HEPES were obtained by separating the non encapsulated material by gel filtration on a Sephadex G-25 column eluted with 10 mM Hepes, 100 mM NaCl (pH 7.4). Osmolarity of all buffers was adjusted to 200 mOsm. ANTS emission was monitored at 530 nm with the excitation wavelength set at 360 nm. A 470 nm cut-off filter was placed between the sample and the emission monochromator to avoid scattering interference. The 0% vesicle content mixing was set by using a 1:1 mixture of ANTS and DPX liposomes. The 100% mixing of contents corresponded to the fluorescence of the vesicles containing co-encapsulated ANTS and DPX.
Unless otherwise stated, LUVs (60 μM lipid) were incubated with 1mM ATP and 1mM MgCl2 prior to treatment with Drp1. Experiments were performed in an SLM-2 Aminco-Bowman luminescence spectrometer (Spectronic Instruments, Rochester, NY) in a thermostated 1-cm path length cuvette with constant stirring at 37 °C.
Supplementary Material
Acknowledgments
The authors thank P. Chiesa and D. Caille for excellent technical assistance, Dr R. Cartoni for help with electron microscopy, Dr D. Tondera for some exploratory work on Bax oligomerization, Dr L. Chernomordik and Dr M. Schick for helpful discussions on the process of membrane hemifusion and fusion pore formation, Dr T. Halazonetis for help with the 3D structure of Drp1 and Dr. M. Koppen and Alexis Jourdain for careful reading of the manuscript. PM is supported by grants from the Swiss National Science Foundation (310000-109402), the Juvenile Diabetes Foundation International (1-2007-158), and the European Union (FP7 BETAIMAGE 222980). DM is supported by DFG grant MA 1081/7-2. EBW is supported by NIH grants RO1 EY016164, RO1 NS0474556, RO1 NS055193 (EBW) and EU grants MCEXT-033534, MCIRG-046536 (RS). This work was funded by the Swiss National Science Foundation (subsidy 31003A-124968/1 to JCM), the European union (UE6-ONCODEATH-037278), OncoSuisse and the Geneva Department of Education. OT was supported by a post-doctoral fellow from Ministerio de Ciencia e Innovación, Gobierno de España.
Footnotes
Supplemental Data: Supplemental Data include Supplemental Experimental Procedures and five figures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ardail D, Privat JP, Egret-Charlier M, Levrat C, Lerme F, Louisot P. Mitochondrial contact sites. Lipid composition and dynamics. J Biol Chem. 1990;265:18797–18802. [PubMed] [Google Scholar]
- Arnoult D, Rismanchi N, Grodet A, Roberts RG, Seeburg DP, Estaquier J, Sheng M, Blackstone C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol. 2005;15:2112–2118. doi: 10.1016/j.cub.2005.10.041. [DOI] [PubMed] [Google Scholar]
- Basanez G, Fidelio GD, Goni FM, Maggio B, Alonso A. Dual inhibitory effect of gangliosides on phospholipase C-promoted fusion of lipidic vesicles. Biochemistry. 1996;35:7506–7513. doi: 10.1021/bi953084a. [DOI] [PubMed] [Google Scholar]
- Basanez G, Sharpe JC, Galanis J, Brandt TB, Hardwick JM, Zimmerberg J. Bax-type apoptotic proteins porate pure lipid bilayers through a mechanism sensitive to intrinsic monolayer curvature. J Biol Chem. 2002;277:49360–49365. doi: 10.1074/jbc.M206069200. [DOI] [PubMed] [Google Scholar]
- Bellot G, Cartron PF, Er E, Oliver L, Juin P, Armstrong LC, Bornstein P, Mihara K, Manon S, Vallette FM. TOM22, a core component of the mitochondria outer membrane protein translocation pore, is a mitochondrial receptor for the proapoptotic protein Bax. Cell Death Differ. 2006;14:785–794. doi: 10.1038/sj.cdd.4402055. [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol. 2003;160:1115–1127. doi: 10.1083/jcb.200212059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, Xie ZJ, Dong Z. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci USA. 2007;104:11649–11654. doi: 10.1073/pnas.0703976104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Chernomordik L, Kozlov MM, Zimmerberg J. Lipids in biological membrane fusion. J Membr Biol. 1995;146:1–14. doi: 10.1007/BF00232676. [DOI] [PubMed] [Google Scholar]
- Chernomordik LV, Kozlov MM. Mechanics of membrane fusion. Nature structural & molecular biology. 2008;15:675–683. doi: 10.1038/nsmb.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik LV, Vogel SS, Sokoloff A, Onaran HO, Leikina EA, Zimmerberg J. Lysolipids reversibly inhibit Ca(2+)-, GTP- and pH-dependent fusion of biological membranes. FEBS Lett. 1993;318:71–76. doi: 10.1016/0014-5793(93)81330-3. [DOI] [PubMed] [Google Scholar]
- Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, Fitzgerald P, Guio-Carrion A, Waterhouse NJ, Li CW, et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell. 2007;129:983–997. doi: 10.1016/j.cell.2007.03.045. [DOI] [PubMed] [Google Scholar]
- DeVay RM, Dominguez-Ramirez L, Lackner LL, Hoppins S, Stahlberg H, Nunnari J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J Cell Biol. 2009;186:793–803. doi: 10.1083/jcb.200906098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellens H, Bentz J, Szoka FC. H+- and Ca2+-induced fusion and destabilization of liposomes. Biochemistry. 1985;24:3099–3106. doi: 10.1021/bi00334a005. [DOI] [PubMed] [Google Scholar]
- Estaquier J, Arnoult D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007;14:1086–1094. doi: 10.1038/sj.cdd.4402107. [DOI] [PubMed] [Google Scholar]
- Etxebarria A, Terrones O, Yamaguchi H, Landajuela A, Landeta O, Antonsson B, Wang HG, Basanez G. Endophilin B1/Bif-1 stimulates BAX activation independently from its capacity to produce large-scale membrane morphological rearrangements. J Biol Chem. 2008 doi: 10.1074/jbc.M808050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Gallenne T, Gautier F, Oliver L, Hervouet E, Noel B, Hickman JA, Geneste O, Cartron PF, Vallette FM, Manon S, et al. Bax activation by the BH3-only protein Puma promotes cell dependence on antiapoptotic Bcl-2 family members. J Cell Biol. 2009;185:279–290. doi: 10.1083/jcb.200809153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, et al. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain M, Mathai JP, McBride HM, Shore GC. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. Embo J. 2005;24:1546–1556. doi: 10.1038/sj.emboj.7600592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goping IS, Gross A, Lavoie JN, Nguyen M, Jemmerson R, Roth K, Korsmeyer SJ, Shore GC. Regulated targeting of BAX to mitochondria. J Cell Biol. 1998;143:207–215. doi: 10.1083/jcb.143.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymann JA, Hinshaw JE. Dynamins at a glance. J Cell Sci. 2009;122:3427–3431. doi: 10.1242/jcs.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai C, Prado FM, Nunes GL, Di Mascio P, Carmona-Ribeiro AM, Nantes IL. pH-Dependent interaction of cytochrome c with mitochondrial mimetic membranes: the role of an array of positively charged amino acids. J Biol Chem. 2005;280:34709–34717. doi: 10.1074/jbc.M412532200. [DOI] [PubMed] [Google Scholar]
- Kluck RM, Esposti MD, Perkins G, Renken C, Kuwana T, Bossy-Wetzel E, Goldberg M, Allen T, Barber MJ, Green DR, et al. The pro-apoptotic proteins, Bid and Bax, cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. J Cell Biol. 1999;147:809–822. doi: 10.1083/jcb.147.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlovsky Y, Kozlov MM. Membrane fission: model for intermediate structures. Biophysical journal. 2003;85:85–96. doi: 10.1016/S0006-3495(03)74457-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell. 2008;135:1074–1084. doi: 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Lucken-Ardjomande S, Martinou JC. Newcomers in the process of mitochondrial permeabilization. J Cell Sci. 2005;118:473–483. doi: 10.1242/jcs.01654. [DOI] [PubMed] [Google Scholar]
- Lucken-Ardjomande S, Montessuit S, Martinou JC. Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell Death Differ. 2008;15:484–493. doi: 10.1038/sj.cdd.4402280. [DOI] [PubMed] [Google Scholar]
- Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol. 2000;2:754–761. doi: 10.1038/35036395. [DOI] [PubMed] [Google Scholar]
- Marani M, Tenev T, Hancock D, Downward J, Lemoine NR. Identification of novel isoforms of the BH3 domain protein Bim which directly activate Bax to trigger apoptosis. Mol Cell Biol. 2002;22:3577–3589. doi: 10.1128/MCB.22.11.3577-3589.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130:165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Ortiz A, Killian JA, Verkleij AJ, Wilschut J. Membrane fusion and the lamellar-to-inverted-hexagonal phase transition in cardiolipin vesicle systems induced by divalent cations. Biophysical journal. 1999;77:2003–2014. doi: 10.1016/S0006-3495(99)77041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M, Norberg E, Walter KM, Schreiner P, Kemper C, Rapaport D, Zhivotovsky B, Orrenius S. The mitochondrial TOM complex is required for tBid/Bax-induced cytochrome c release. J Biol Chem. 2007;282:27633–27639. doi: 10.1074/jbc.M703155200. [DOI] [PubMed] [Google Scholar]
- Parone PA, James DI, Da Cruz S, Mattenberger Y, Donze O, Barja F, Martinou JC. Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol Cell Biol. 2006;26:7397–7408. doi: 10.1128/MCB.02282-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roucou X, Montessuit S, Antonsson B, Martinou JC. Bax oligomerization in mitochondrial membranes requires tBid (caspase-8-cleaved Bid) and a mitochondrial protein. Biochem J. 2002;368:915–921. doi: 10.1042/BJ20020972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinzel A, Kaufmann T, Borner C. Bcl-2 family members: integrators of survival and death signals in physiology and pathology [corrected] Biochim Biophys Acta. 2004;1644:95–105. doi: 10.1016/j.bbamcr.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Sheridan C, Delivani P, Cullen SP, Martin SJ. Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol Cell. 2008;31:570–585. doi: 10.1016/j.molcel.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Tan KO, Fu NY, Sukumaran SK, Chan SL, Kang JH, Poon KL, Chen BS, Yu VC. MAP-1 is a mitochondrial effector of Bax. Proc Natl Acad Sci U S A. 2005;102:14623–14628. doi: 10.1073/pnas.0503524102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrones O, Antonsson B, Yamaguchi H, Wang HG, Liu Y, Lee RM, Herrmann A, Basanez Gb. Lipidic pore formation by the concerted action of pro-apoptotic BAX and tBID. J Biol Chem. 2004;279:30081–30091. doi: 10.1074/jbc.M313420200. [DOI] [PubMed] [Google Scholar]
- van den Brink-van der Laan E, Killian JA, de Kruijff B. Nonbilayer lipids affect peripheral and integral membrane proteins via changes in the lateral pressure profile. Biochim Biophys Acta. 2004;1666:275–288. doi: 10.1016/j.bbamem.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. A stapled BID BH3 helix directly binds and activates BAX. Mol Cell. 2006;24:199–210. doi: 10.1016/j.molcel.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–450. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Zhu PP, Patterson A, Stadler J, Seeburg DP, Sheng M, Blackstone C. Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J Biol Chem. 2004;279:35967–35974. doi: 10.1074/jbc.M404105200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.