

Abstract
Mouse Double Minute homolog 4 (MDM4) gene upregulation often occurs in human hepatocellular carcinoma (HCC), but the molecular mechanisms responsible for its induction remain poorly understood. Here, we investigated the role of the phosphoinositide-3-kinase/v-akt murine thymoma viral oncogene homolog/mammalian target of Rapamycin (PI3K/AKT/mTOR) axis in the regulation of MDM4 levels in HCC.
The activity of MDM4 and the PI3K/AKT/mTOR pathway was modulated in human HCC cell lines via silencing and overexpression experiments. Expression of main pathway components was analyzed in an AKT mouse model and human HCCs.
MDM4 inhibition resulted in growth restraint of HCC cell lines both in vitro and in vivo. Inhibition of the PI3K-AKT and/or mTOR pathways lowered MDM4 protein levels in HCC cells and reactivated p53-dependent transcription. De-ubiquitination by ubiquitin-specific protease 2a and AKT-mediated phosphorylation protected MDM4 from proteasomal degradation and increased AKT protein stability. The eukaryotic elongation factor 1A2 (EEF1A2) was identified as an upstream inducer of PI3K supporting MDM4 stabilization. Also, we detected MDM4 protein upregulation in an AKT mouse model and a strong correlation between the expression of EEF1A2, activated/phosphorylated AKT, and MDM4 in human HCC (each rho>.8, P<.001). Noticeably, a strong activation of this cascade was associated with shorter patients' survival.
Conclusions
The EEF1A2/PI3K/AKT/mTOR axis promotes the protumorigenic stabilization of the MDM4 protooncogene in human HCC via a post-transcriptional mechanism. The activation level of the EEF1A2/PI3K/AKT/mTOR/MDM4 axis significantly influences the survival probability of HCC patients in vivo and may thus represent a promising molecular target.
Keywords: Liver cancer, oncogene, tumor suppressor gene, functional inactivation
Introduction
The tumor suppressor p53 plays a pivotal role in the regulation of cell cycle, apoptosis, metabolism, and genetic stability in mammalian cells.1 Thus, it is not surprising that inactivation of p53 is one of the most frequent events during human carcinogenesis, with p53 mutations occurring in about 50% of all human cancers.2 In contrast to other cancer types, p53 mutation frequency in hepatocellular carcinoma (HCC) shows a substantial geographical variation. Indeed, HCC display a surprisingly low frequency of p53 mutations in Western countries, ranging between 10 to 20%, whereas up to 50% of HCC samples from Southeast Asia harbor p53 mutations.3-5 Such a different frequency is mainly due to the exposure to certain etiological agents (e.g. aflatoxin B1).6 Nevertheless, although point mutations represent the most common mechanism responsible for p53 inactivation in cancer, additional mechanisms achieving the same effect in the absence of p53 mutations have been described. Functional inactivation of wild-type p53 by post-transcriptional mechanisms such as phosphorylation and proteasomal degradation has been demonstrated in various human cancers, including HCC.4 In mammalian cells, the activity of wild-type p53 is mainly under control of its negative regulator protein, Mouse Double Minute homolog 2 (MDM2), which maintains p53 at low levels, thus allowing the growth of normal cells. MDM2 is able to bind to the transactivation domain of p53 and promotes its proteasomal degradation by functioning as an E3 ligase.7,8 Another p53 binding-protein, Mouse Double Minute 4 (MDM4, MDMX, or HDMX), has been identified in mammalian cells.9,10 Due to its sequence homology to MDM2, MDM4 can also bind to the N-terminal transactivation domain of p53 and act as a repressor of p53 transcriptional activity.11 Noticeably, amplification of the MDM4 gene locus and MDM4 overexpression have been described in a variety of human cancers, mostly those harboring a wild-type p53 gene.12 Recently, we have reported MDM4 gene locus amplification in human HCC, which was associated with its transcriptional upregulation. Functional experiments further emphasized the oncogenic properties of MDM4 in supporting cell growth and proliferation in vitro.13 However, chromosomal gains did not explain the increased MDM4 activity observed in HCC samples with a balanced MDM4 gene locus. A number of protein kinases have been reported to be involved in the regulation of MDM4. Indeed, phosphorylation of MDM4 by Ataxia Teleangiectasia Mutated and Checkpoint Kinase 2 has been shown to promote its destabilization in response to DNA damage in vitro.14,15 Less is known about the positive regulation of MDM4 activity by phosphorylation. In a recent study, Lopez-Pajares and colleagues found that the v-akt murine thymoma viral oncogene homolog (AKT) kinase specifically phosphorylates MDM4 at serine residue 367, leading to its stabilization in vitro. 16 The phosphoinositide-3-kinase (PI3K)/AKT pathway regulates many cellular processes, including cell proliferation, survival, growth, and motility that are critically altered during tumorigenesis. Increasing evidence indicates that activation of AKT signaling is a crucial event in human hepatocarcinogenesis.17-19 Here, we investigated the possible crosstalk between MDM4 and the PI3K/AKT pathway in hepatocarcinogenesis by using in vitro and in vivo approaches. We found that upregulation of the eukaryotic elongation factor 1A2 (EEF1A2) promotes the stabilization of MDM4 in human HCC via a post-transcriptional mechanism involving the AKT/mammalian target of Rapamycin (mTOR) signaling pathway.
Material and Methods
Human Tissue Samples
Five normal livers, 48 HCCs harboring wild-type p53, and corresponding peritumorous non-neoplastic liver tissues (PT) from a previous study were used.20 Patient characteristics are shown in Table 1. Liver tissues were kindly provided by Snorri S. Thorgeirsson (National Cancer Institute, Bethesda, MD). Institutional Review Board approval was obtained from participating hospitals and the National Institutes of Health.
Table 1. Characteristics of the patients in the HCC cohort and results from survival analysis.
| Characteristic | Level | # patients | Median survival (months) |
Univariate survival analysis | Multivariate survival analysis | ||
|---|---|---|---|---|---|---|---|
| p-value | HR (95% CI) | p-value | HR (95% CI) | ||||
| Age | Less than 62 | 12 | 18 | 0.39 | Reference | ||
| 62-66 | 12 | 29 | 0.88 (0.39-2.01) | ||||
| 67-75 | 13 | 12 | 1.71 (0.75-3.92) | ||||
| 76+ | 11 | 39 | 1.09 (0.46-2.57) | ||||
|
| |||||||
| Gender | Male | 43 | 18 | 0.13 | Reference | ||
| Female | 5 | 32 | 0.45 (0.16-1.29) | ||||
|
| |||||||
| Cirrhosis | No | 13 | 32 | 0.39 | Reference | ||
| Yes | 35 | 18 | 0.75 (0.39-1.45) | ||||
|
| |||||||
| Etiology | HBV | 24 | 17 | 0.45 | Reference | ||
| HCV | 17 | 23 | 1.38 (0.72-2.64) | ||||
| alcohol | 6 | 34 | 0.71 (0.29-1.77) | ||||
| Wilson's disease | 1 | 62 | 0.51 (0.07-3.82) | ||||
|
| |||||||
| Tumor size | <3 cm | 9 | 51 | 0.04 | Reference | 0.64 | Reference |
| ≥3 cm | 39 | 15 | 2.20 (1.01-4.79) | 0.81 (0.34-1.93) | |||
|
| |||||||
| Serum AFP | <300 ng/ml | 19 | 44 | 0.50 | Reference | ||
| ≥300 ng/ml | 29 | 15 | 1.23 (0.68-2.23) | ||||
|
| |||||||
| Edmondson / Steiner | II | 14 | 44 | 0.26 | Reference | ||
| III | 21 | 23 | 0.93 (0.46-1.86) | ||||
| IV | 13 | 14 | 1.64 (0.76-3.52) | ||||
|
| |||||||
| EEF1A2 | <311 | 24 | 52 | < 0.0001 | Reference | < 0.0001 | Reference |
| ≥311 | 24 | 10 | 35.1 (7.74-159) | 32.2 (7.01-148) | |||
|
| |||||||
| AKT | <213 | 25 | 18 | 0.11 | Reference | 0.22 | Reference |
| ≥213 | 23 | 23 | 1.67 (0.89-3.12) | 1.48 (0.79-2.79) | |||
|
| |||||||
| pAKT | <235 | 24 | 52 | < 0.0001 | Reference | < 0.0001 | Reference |
| ≥235 | 24 | 10 | 22.1 (6.26-78.1) | 20.3 (5.64-72.8) | |||
|
| |||||||
| MDM4 | <201 | 24 | 52 | < 0.0001 | Reference | < 0.0001 | Reference |
| ≥201 | 24 | 10 | 22.8 (6.46-80.5) | 21.2 (5.87-76.4) | |||
|
| |||||||
| USP2a | <192 | 24 | 50 | < 0.0001 | Reference | 0.0002 | Reference |
| ≥192 | 24 | 10 | 4.17 (2.20-7.88) | 3.80 (1.90-7.59) | |||
|
| |||||||
| p53 | <118 | 24 | 10 | < 0.0001 | Reference | < 0.0001 | Reference |
| ≥118 | 24 | 52 | 0.04 (0.01-0.15) | 0.05 (0.01-0.17) | |||
|
| |||||||
| p21 | <89 | 25 | 10 | < 0.0001 | Reference | < 0.0001 | Reference |
| ≥89 | 23 | 51 | 0.14 (0.06-0.29) | 0.15 (0.07-0.33) | |||
Hydrodynamic Injection and Mouse Treatment
Wild-type FVB/N mice were subjected to hydrodynamic injection as described previously.17 Briefly, 10 μg of the pCMV/SB and pT3-EF1α-HA-myr-AKT constructs in a ratio of 1:25 were diluted in 2 mL of 0.9% NaCl, filtered, and injected into the lateral tail vein of seven week old mice in 7 to 9 seconds. Injected mice were monitored and sacrificed in groups after 12 weeks and 28 weeks. An additional group of AKT-injected mice was subjected, four weeks after hydrodynamic injection, to administration of either vehicle (n=4) or Sirolimus (Rapamune; 5 mg/kg, n=5) by oral administration for 5 days. Liver tissue was harvested 5 hours after the last dose. Sirolimus was obtained from the UCSF Pharmacy. Mice were housed, fed, and treated in accordance with protocols approved by the Committee for Animal Research at the University of California, San Francisco.
Cell lines, transfection, xenograft model, and treatments
Culturing conditions, transfections of human HCC cell lines, xenograft model, and treatment with specific inhibitors were performed as described in Supplementary Materials.
Western Blot Analysis and Immunoprecipitation
Liver tissues were processed as reported in Supplementary Materials. The primary antibodies used are shown in Supplementary Table 1.
Tissue microarrays and immunohistochemistry
The tissue microarray (TMA) and the immunohistochemical analyses are described in the Supplementary Materials.
Quantitative Real-Time Reverse-Transcription Polymerase Chain Reaction
Quantitative Real-Time Reverse-Transcription Polymerase Chain Reaction was performed as reported previously.21 Primer sequences are listed in Supplementary Table 2.
Statistical Analysis
Statistical analyses were performed as reported in the Supplementary Materials.
Results
MDM4 sustains the growth of HCC cells via p53-dependent and independent mechanisms
Previous findings indicated that oncogenic activity of MDM4 is due to its ability to inactivate the transcriptional function of the p53 tumor suppressor gene.11 To test the relevance of MDM4 for HCC cell growth in vitro, HuH6 and HepG2 (harboring wild-type p53) as well as HuH7 and Hep3B cells (harboring mutated or deleted p53) were subjected to MDM4 silencing via siRNA (Fig. 1A-C). Suppression of MDM4 led to growth restraint in all cell lines tested, due to decreased proliferation and increased apoptosis (Fig. 1B/C). The in vivo relevance of these findings could be confirmed using a xenograft mouse model (Fig. 1D). In addition, double inhibition of MDM4 and p53 using gene-specific siRNAs partially rescued the effect of MDM4 knockdown in HepG2 cells, while HuH7 cells remained largely unaffected (Suppl. Fig. 1). Suppression of MDM4 resulted in a strong increase of p53 target genes, including p21 and PUMA, in p53 wild-type cell lines, with little or no changes in expression of the same genes in p53 mutant cell lines (Fig. 1A). Treatment with the MDM4 inhibitor SJ-172550, which disrupts the binding between p53 and MDM4,22 resulted in a dose-dependent growth inhibition and upregulation of p53 target genes only in cell lines with wild-type p53, with no appreciable effects on the same parameters in cell lines with mutant p53 (Fig. 1A, Suppl. Fig. 2-5). On the other hand, transient overexpression of MDM4 in the SNU423 cell line, harboring a mutant p53 gene, resulted in growth acceleration in vitro (Suppl. Fig. 6; P <.01). Altogether, these data indicate that MDM4 promotes the growth of HCC cells in a p53-dependent and -independent manner.
Fig. 1.
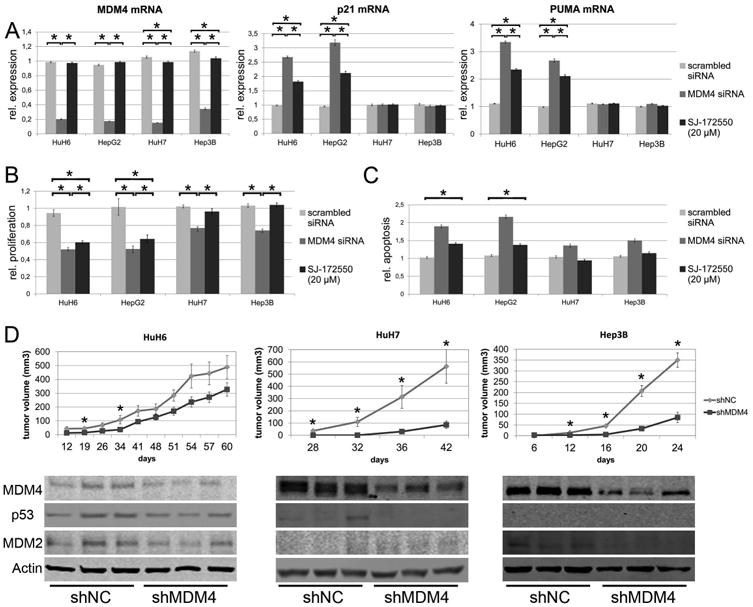
MDM4 exerts protumorigenic effects via p53-dependent and –independent mechanisms. (A) MDM4, p21, and PUMA mRNA levels following siRNA-mediated silencing of MDM4 (dark grey bars) in HuH6 (p53- wildtype), HepG2 (p53-wildtype), Hep3B (p53-deleted), and HuH7 (p53-mutated) cells and after treatment with SJ-172550 (20μM, black bars), a blocker of the MDM4-p53 interaction. Induction of p53 target genes is only observed in HCC cell lines with p53 wild-type alleles. The values shown are normalized against the transfection reagent. (B) Both siRNA-mediated silencing of MDM4 (dark grey bars) and SJ-172550 treatment (black bars) reduces proliferation and (C) increases apoptosis in HCC cells (normalized against oligofectamine-treated cells). (D) 5 × 106 cells transfected with either an shRNA targeting MDM4 (shMDM4) or a neutral control shRNA (shNC) were subcutaneously injected into the flank of NOD/SCID mice (HuH6: n=14; HuH7, Hep3B: n = 12 tumors). Tumor formation and growth was significantly reduced in shMDM4 transfected cells (upper panel; * P < .05). Reduced MDM4 expression is seen in tumor tissues derived from shMDM4 transfected HCC cells (lower panel). In addition, p53 levels are decreased in shMDM4-expressing HuH6 and HuH7 cells, while MDM2 expression is lowered in HuH6 and Hep3B cells.
PI3K-AKT signaling is involved in the regulation of the MDM4 protein levels in HCC
Since it has been shown that the AKT serine/threonine kinase can stabilize MDM4 in various cancer cell lines,16 we assessed the role of the PI3K-AKT pathway in the regulation of MDM4 in human HCC cells lines. For this purpose, we performed a time-course experiment using the PI3K inhibitor LY294002 in the same cell lines. MDM4 protein decrease started 4 h after treatment and MDM4 protein levels remained low compared to DMSO treated (control) cells for at least 24 h. This effect was observed in all analyzed cell lines independently of the p53 gene status and was paralleled by a decrease in MDM2 protein in HepG2 and HuH7 cells (Fig. 2A, Suppl. Fig. 7). Gene specific siRNA-mediated targeting of AKT1 and/or AKT2 isoforms (Fig. 2C, Suppl. Fig. 8/9) confirmed the involvement of the PI3K-AKT axis in the regulation of the MDM4 protein levels. LY294002 treatment did not affect MDM4 mRNA levels (Fig. 2B, P >.05), suggesting that the PI3K pathway regulates MDM4 expression through a post-transcriptional mechanism. Importantly, the decrease in MDM4 protein levels was associated with the reactivation of p53-dependent transcriptional activity, as determined by quantitative RT-PCR of several p53 target genes in HepG2 cells (p53 wild-type; Fig. 2B). Indeed, a strong and significant increase of p21 mRNA was recorded in LY294002 treated cells as early as 4 h after treatment (2.27 ± 0.57 vs. 1.0 ± 0.02 in DMSO-treated cells; P <.05). Similar effects were observed for the BCL2-associated × protein (BAX; at 8 h: 1.26 ± 0.07 vs. 1.0 ± 0.02, respectively; P <.01), the BCL2 binding component 3 (PUMA; at 4h: 1.44 ± 0.02 vs. 1.0 ± 0.07, respectively; P <.01), and the MDM2 (at 8h: 1.55 ± 0.10 vs. 1.0 ± 0.03, respectively; P <.01).
Fig. 2.
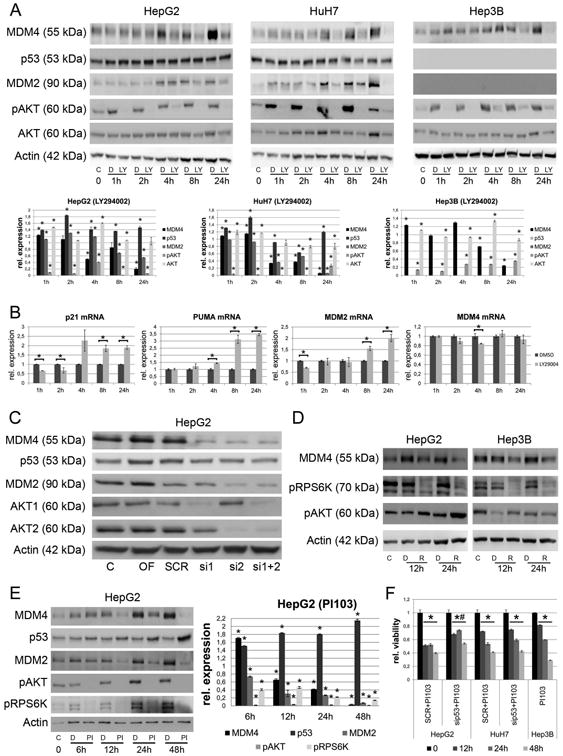
AKT and mTOR signaling are involved in the regulation of MDM4 protein expression. (A) The PI3KInhibitor LY294002 reduces the phosphorylation of AKT and the MDM4 protein levels in all three HCC cell lines in a time-dependent manner. This effect is associated with reduced MDM2 and increased p53 expression in HepG2 and HuH7 cells. Note, the p53-deficient Hep3B cells do not express the p53-target gene MDM2. The lower panel shows the results from densitometric analysis. * P < .05 (B) LY294002 treatment induces the mRNA-expression of p53 target genes (p21, PUMA, MDM2, P < .05), while the transcription of MDM4 is not significantly affected (P > .05). Dark grey bars represent DMSO treated and light grey bars represent LY294002-treated HepG2 cells. (C) Validation of the influence of AKT signaling on MDM4 protein levels by treating HepG2 cells using isoform specific siRNAs against AKT1 and AKT2. (D) The mTORC1 inhibitor Rapamycin reduces the phosphorylation of RPS6K and the MDM4 protein levels in HepG2 cells. (E) PI103 inhibits activation of both AKT- and mTOR signaling, abolishes the expression of MDM4 and MDM2 and increases the p53 level in HepG2 cells in a time-dependent manner. (F) PI103 treatment significantly reduces the cell viability in HepG2, HuH7 and Hep3B cells compared to DMSO treated cells, respectively (* P < .05). In HepG2 cells siRNA-mediated inhibition of p53 significantly reduces the effect of PI103 treatment on cell viability (# P < .05). Abbreviations: C, untreated control; D, DMSO; LY, LY294002; OF, oligofectamine control; SCR, scrambled siRNA; R, rapamycin; PI, PI103; si1, siRNA targeting AKT1; si2, siRNA targeting AKT2.
The mTOR pathway cooperates with PI3K/AKT signaling to stabilize MDM4
Since AKT activation can lead to the induction of the mTOR signaling,19 we next analyzed whether the mTOR pathway contributes to the regulation of MDM4. Treatment with the mTOR complex 1 (mTORC1) inhibitor, Rapamycin, resulted in a significant reduction of MDM4 protein levels in HCC cells. The specificity of the Rapamycin effect was confirmed by the decreased phosphorylation of the downstream mTORC1 target, ribosomal protein S6 kinase (RPS6K) (Fig. 2D, Suppl. Fig. 10). In addition, treatment with PI103 (a PI3K-, mTORC1-, and mTORC2-multikinase inhibitor) efficiently knocked down the MDM4 expression in HCC cell lines for at least 48 h (Fig. 2E, Suppl. Fig. 11) and significantly reduced the cell viability of HepG2 and Hep3B cells after 24 h (40% and 42% reduction compared to DMSO-treated controls, respectively; P <.01) and 48 h (62% and 71% reduction compared to DMSO-treated controls, respectively; P <.01; Fig. 2F) following treatment. The influence of both branches of the mTOR pathway (mTORC1 and mTORC2) on the regulation of the MDM4 protein level was further confirmed by siRNA-mediated silencing of regulatory associated protein of mTORC1 (RAPTOR) and RAPTOR independent companion of mTORC2 (RICTOR; Suppl. Fig. 12).
The EEF1A2 oncogene induces MDM4 upregulation via AKT activation
Recently, we have found that EEF1A2 is frequently upregulated concomitant with MDM4 in human HCC and exerts oncogenic functions.13 Since EEF1A2 has been described to promote its protumorigenic effect at least partly through activation of the AKT protooncogene,23 we assessed whether the same applies to HCC. Noticeably, siRNA-mediated silencing of EEF1A2 in human HCC cells resulted in decreased phosphorylation of AKT and RPS6K. Additionally, MDM4 levels were markedly decreased (Fig. 3A). As a consequence, transcription of p53 target genes was significantly upregulated (p21: 2.9-fold; BAX: 1.9-fold; PUMA: 1.5-fold, and MDM2: 2.8-fold compared to scrambled-treated cells, each P <.05), while the MDM4 mRNA level was even lowered (Fig. 3B). Conversely, forced overexpression of EEF1A2 in HuH6 cells by transient transfection led to growth acceleration and reduction of apoptosis (Fig. 3D/E; P <.01), which were paralleled by activation of AKT and upregulation of MDM4 (Fig. 3C). The changes induced by EEF1A2 overexpression were almost completely reverted by the treatment with the AKT1/2 inhibitor (Fig. 3C-E; P <.01). Altogether, the present data indicate that EEF1A2 promotes MDM4 upregulation via AKT-dependent mechanisms in HCC cells.
Fig. 3.
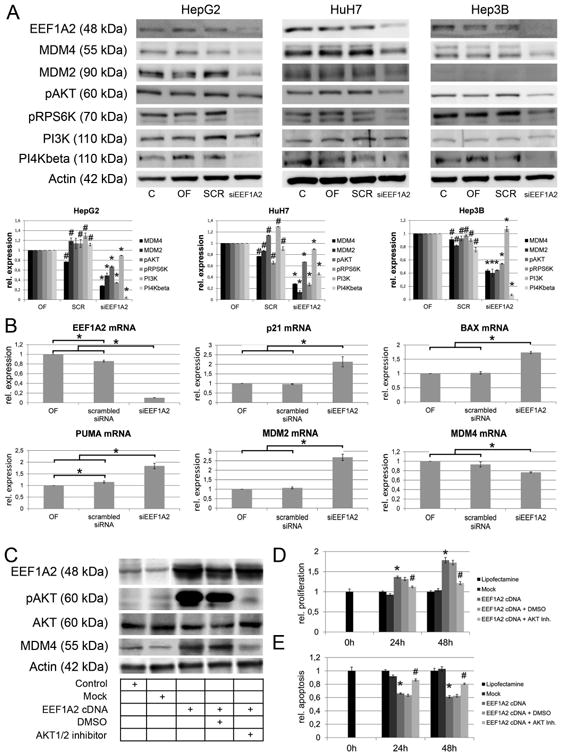
EEF1A2 is involved in the regulation of MDM4 protein expression. (A) siRNA-mediated knockdown of EEF1A2 expression diminishes the activation of both AKT- and mTOR signaling via reduction of PI4KB expression, which leads to reduced MDM4 protein expression. Upper panel: representative Western blots; lower panel: relative protein expression according to densitometric analysis of Western blots normalized against β-Actin expression and oligofectamine control. *siEEF1A2 vs. OF and SCR, P < .05; #OF vs. SCR, P < .05. (B) Reactivation of p53 function following siRNA-mediated silencing of EEF1A2 is indicated by activation of p21, BAX, puma, and MDM2 mRNA expression (P < .05). (C) Western immunoblotting following EEF1A2 cDNA transfection in HuH6 cells. The EEF1A2 induced upregulation of MDM4 can be rescued by pharmacological inhibition of AKT1/2. (D/E) Transfection of EEF1A2 increases the proliferation (D) and reduces the apoptotic rate (E), which can be partially prevented by AKT1/2 inhibition. * EEF1A2 cDNA vs. Mock, P < .05; # EEF1A2 cDNA vs. EEF1A2 cDNA + AKT1/2 inhibitor, P < .05. Abbreviations: C, untreated control; OF, oligofectamine control; SCR, scrambled siRNA; PI4KB, phosphatidylinositol 4-kinase beta; siEEF1A2, siRNA targeting EEF1A2; AKT Inh., AKT1/2 inhibitor.
AKT stabilizes MDM4 and prevents its degradation
To explore the molecular mechanism responsible for MDM4 downregulation, we analyzed its protein stability following inhibition of PI3K (by using the PI3K inhibitor LY294002), with and without simultaneous inhibition of protein biosynthesis using cycloheximide. A significant decrease in MDM4 protein levels was detected in HepG2 cells after combined treatment with LY294002 and cycloheximide, when compared to the administration of either LY294002 or cycloheximide alone after 8 and 24 hours, respectively (P <.05; Fig. 4A, Suppl. Fig. 13), thus confirming a profound impact of the PI3K-AKT pathway on the stability of the MDM4 protein in human HCC cells. To assess whether active AKT signaling may protect MDM4 from proteasomal degradation, we pharmacologically inhibited the proteasome activity using MG132 while treating HepG2, Hep3B, or HuH7 cells with LY294002. Interestingly, MG132 treatment rescued the decrease of the MDM4 protein level observed following LY294002 single treatment (Fig. 4B). This protective effect was observed in all cell lines analyzed, indicating an underlying p53- and MDM2-independent molecular mechanism.
Fig. 4.
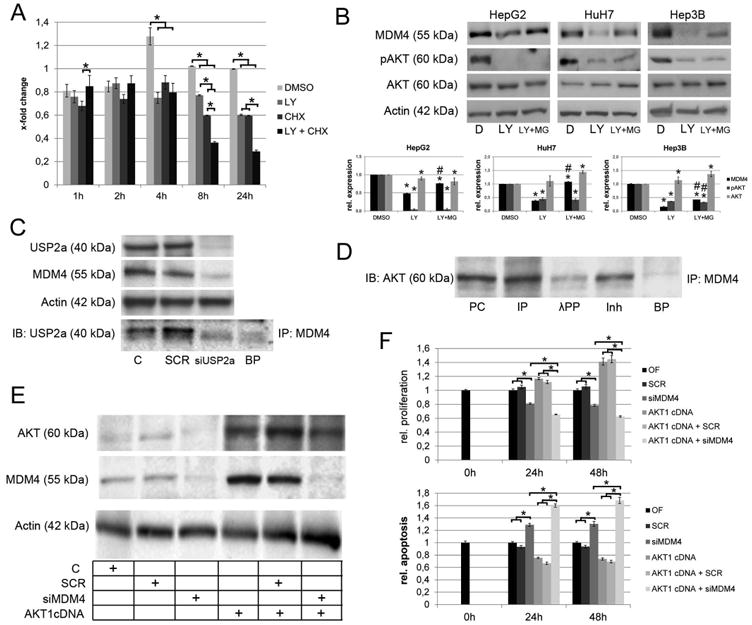
Mechanisms involved in the post-transcriptional regulation of MDM4. (A) Densitometric analysis of the immunoblots following LY294002 and/or CHX treatment. AKT signaling promotes stabilization of the MDM4 protein as indicated by significantly reduced MDM4 protein levels following LY294002 treatment of HCC cells with CHX-induced termination of protein biosynthesis (8h: 0.60 ± 0.001 compared to 0.36 ± 0.001 in CHXtreated controls, P<.05; 24 h: 0.60 ± 0.01 vs. 0.29 ± 0.01, respectively, P<.05). (B) Inhibition of proteasomal degradation rescues the MDM4 protein levels after LY294002 treatment of HCC cells. Lower panel: densitometric analysis of Western blots. *DMSO vs. LY294002 or LY294002 + MG132, P < .05; #LY294002 vs. LY294002 + MG132, P < .05 (C) USP2a physically interacts with MDM4 and siRNA-mediated silencing of USP2a reduces the MDM4 protein levels indicating that USP2a-mediated deubiquitination is involved in the stabilization of the MDM4 protein. (D) Co-immunoprecipitation demonstrating a physical interaction between AKT and MDM4 in HCC cells. Preincubation with lambda protein phosphatase reduces the AKT-MDM4 interaction. (E) Western immunobloting following transfection of in HLE cells using an AKT1 cDNA with/without simultaneous siRNA inhibition of MDM4. (F) Transfection of AKT1 increase the proliferation (upper panel) and decreases the apoptotic rate (lower panel), which can be rescued by siRNAmediated knockdown of MDM4. Abbreviations: BP, blocking peptide; C, untreated control; CHX, cycloheximide; D, DMSO; IB, immunoblot; Inh, inhibitor; incubation with lambda protein phosphatase and phosphatase inhibitor; IP, immunoprecipitation; λPP, lambda protein phosphatase; LY, LY294002; MG, MG132; SCR, scrambled siRNA; PC, positive control; siUSP2a, siRNA targeting USP2a.
Since ubiquitination is a crucial step in flagging proteins for proteasomal degradation and the ubiquitin-specific protease 2a (USP2A), which acts as a de-ubiquitinase, was recently reported to be induced by AKT in human HCC,17 and to protect MDM4 from proteolysis,24 we explored a possible protective effect of USP2a on the MDM4 protein levels. We not only detected decreased MDM4 protein levels after siRNA-mediated silencing of USP2a in the Focus cell line (expressing high levels of USP2a),17 but we were also able to co-immunoprecipate USP2a and MDM4 indicating their interaction, which was impaired following USP2a knockdown (Fig. 4C). Also, since a direct interaction between MDM4 and AKT in various tumor cell lines was documented in vitro,16 we evaluated whether the same occurs in human HCC cell lines. Immunoprecipitation analyses confirmed a physical interaction between AKT and MDM4 in human HCC cells indicating that MDM4 is a substrate of AKT kinase activity in human HCC. Of note, the interaction between MDM4 and AKT was significantly decreased when the protein lysates were incubated with lambda protein phosphatase (which removes phosphate groups from phosphorylated serine, threonine, and tyrosine residues in proteins) prior to immunoprecipitation (Fig. 4D). The latter observation indicates that MDM4 requires to be phosphorylated in order to bind to AKT.
The crosstalk between AKT and MDM4 was further investigated in the human HLE cell line stably overexpressing AKT1.17 The latter cell line showed increased levels of AKT, pAKT, and MDM4 when compared with the non-transfected counterpart (Fig. 4E). Of note, suppression of MDM4 by siRNA induced a much stronger growth inhibition in HLE cells stably-transfected with AKT1 when compared with untransfected cells (Fig. 4F, P <.01). These results indicate that the cell growth properties of AKT on HCC cells depend, at least partly, on MDM4.
Activation of the EEF1A2-AKT-MDM4 cascade has functional relevance in vivo and is associated with shorter patients' survival
To investigate whether activated AKT signaling stabilizes MDM4 also in vivo, we took advantage of the recently generated AKT mouse model of hepatocarcinogenesis, which has been shown to express elevated protein levels of the de-ubiquitinase USP2a.17 Increased protein levels of activated/phosphorylated AKT, USP2a, and MDM4 were detected by Western blotting in 12 and 28 weeks old AKT mice compared to wild-type littermates (Fig. 5 A,B; each P <.001). Also, a strong correlation between AKT phosphorylation and USP2a expression was observed (Spearman's rho=0.88, P <.001), pAKT and MDM4 protein expression (Spearman's rho=0.93, P <.001), and USP2a and MDM4 protein expression (Spearman's rho=0.91, P <.001), while the mRNA levels of MDM4 were not altered in AKT mice compared to the wild-type controls (3.3 ± 0.6 respectively 2.9 ± 0.7 vs. 3.2 ± 0.5; each P >.05; Fig. 5C). Immunohistological analyses revealed an upregulation of MDM4 in pAKT-expressing neoplastic lesions in AKT mice (Fig. 5D). Of note, treatment of AKT mice with the mTORC1 inhibitor Rapamycin resulted in downregulation of p-AKT, USP2a, and MDM4, as assessed by Western blot analysis (Fig. 5E).
Fig. 5.
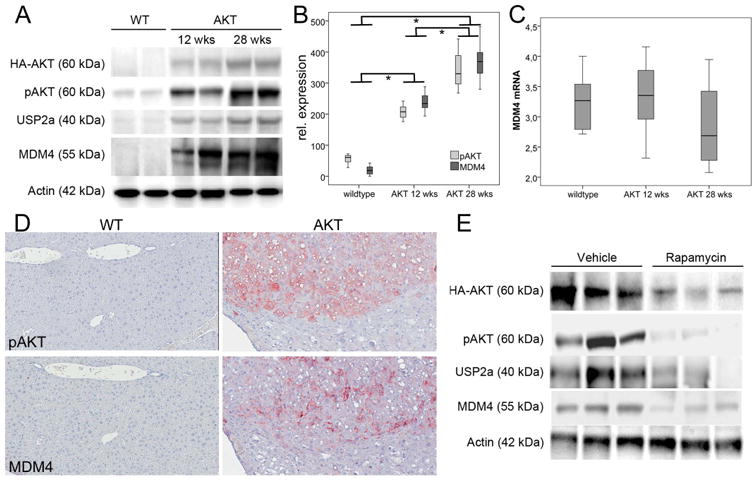
Upregulation of MDM4 in AKT mice. (A) Representative Western blots in wild-type and mice (12 and 28 weeks old). (B) The densitometric analyses reveal a co-regulation of pAKT, USP2a, and MDM4, while the MDM4 transcription (C) is not altered between wild-type and animals. (D) Representative immunohistochemicial stainings of wild-type (left panel) and AKT (right panel) for pAKT (upper panel) and MDM4 (lower panel). Original magnification: 20×. (E) Representative Westernblots in AKT transgenic mice treated with vehicle or Rapamycin. Abbreviation: wks, weeks; WT, wildtype.
Finally, to investigate whether the EEF1A2-PI3K-AKT-mTOR-MDM4 cascade is relevant in human liver tumors, we analyzed the expression of the main signaling components in a collection of human HCCs (n=48). Using Western blotting, we observed a significant increase of EEF1A2 protein expression in human HCCs compared to normal liver (NL) and peritumorous, non-neoplastic liver tissue (PT; 66 ± 2 (NL) vs. 82 ± 2 (PT) vs. 285 ± 16 (HCC); P <.001). This upregulation was paralleled by an upregulation of pAKT (46 ± 4 (NL) vs. 83 ± 1 (PT) vs. 254 ± 15 (HCC); P <.001; Fig. 6A/B), USP2a (36 ± 8 (NL) vs. 44 ± 2 (PT) vs. 190 ± 10 (HCC); P <.001; Fig. 6A/B), and MDM4 (69 ± 5 (NL) vs. 72 ± 2 (PT) vs. 226 ± 13 (HCC); NL/PT vs. HCC: P <.001; Fig. 6A/B), while p21 protein levels were significantly reduced in human HCCs (255 ± 10 (NL) vs. 242 ± 4 (PT) vs. 110 ± 8 (HCC); NL/PT vs. HCC: P <.001; Fig. 6A/B). Survival analysis revealed that high levels of EEF1A2, pAKT, USP2a, and MDM4 associate with a shorter survival of HCC patients after liver resection (each p<.001; Fig. 6C). In univariate analyses, tumor size was the only clinico-pathological parameter that showed an association with patients' survival in the investigated cohort. Indeed, patients with tumors larger than 3 cm showed a median survival time of 15 months, compared to 51 months for patients diagnosed with smaller tumors (HR=2.20, p-value=0.04). In multivariate analysis high level of EEF1A2, pAKT, USP2 and MDM4 were clearly associated with a shorter patients' survival (Table 1). Importantly, correlation analyses revealed a strong positive association between EEF1A2 and pAKT (rho: 0.87, P <.001), EEF1A2 and USP2a (rho: 0.76, P <.001), EEF1A2 and MDM4 (rho: 0.88, P <.001), pAKT and USP2a (rho: 0.74, P <.001), pAKT and MDM4 (rho: 0.94, P < .001), USP2a and MDM4 (rho: 0.80, P <.001), while a strong negative association was recorded between EEF1A2 and p21 (rho: -0.75, P <.001), pAKT and p21 (rho: -0.75, P <.001), USP2a and p21 (rho: -0.66, P <.001), and MDM4 and p21 (rho: -0.79, P <.001). These data were further substantiated by immunohistological analysis of an independent HCC cohort (n=76; Supplementary Materials), which revealed a positive association of EEF1A2 and pAKT immunostaining (rho: 0.27, P <.05), EEF1A2 expression and nuclear MDM4 expression (rho: 0.41, P <.001), nuclear MDM4 and p53 (rho: 0.23, P <.05), and p53 and p21 immunosignals (rho: 0.76, P <.001), while expression of MDM4 and p21 revealed a significant inverse association (rho: -0.79, P <.001; Fig. 6D).
Fig. 6.
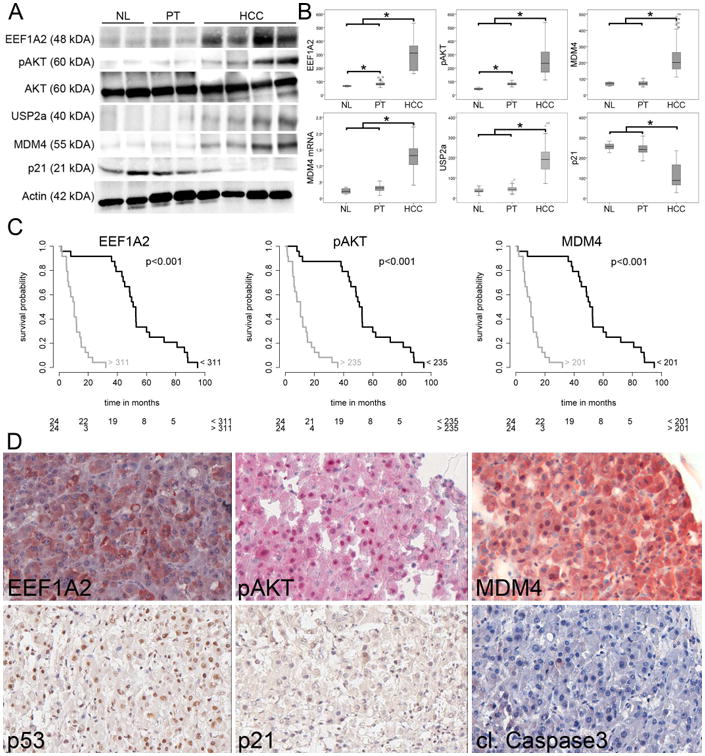
Dysregulation of the EEF1A2-AKT-MDM4 axis in human HCC. (A) Representative Western blots of lysates prepared from NL, PT, and HCC and immunoblotted with the indicated antibodies. (B) Western blot optical densities were normalized to β-actin values and expressed in arbitrary units. (C) Survival probability of human HCC patients depends on the expression level of EEF1A2, pAKT, and MDM4. The number of patients at risk is indicated in the lower part of the survival plots. (D) Representative immunohistochemical stainings of the tissue microarray. High EEF1A2 expression is associated with phosphorylation and thus activation of AKT, which stabilizes MDM4. Despite nuclear p53, there is neither upregulation of p21 nor apoptosis induction (cleaved Caspase 3). Original magnification: 40×. Abbreviations: NL, normal liver, PT, peritumorous non-neoplastic liver.
Thus, our data clearly show that the activation of the EEF1A2-AKT-MDM4 cascade has not only functional relevance in vivo, but its activation level has strong impact on the survival time of HCC patients.
Discussion
MDM4 is one of the main negative regulators of p53 that blocks its transcriptional activity upon binding to its N-terminal transactivation domain.11 Recently, MDM4 has been identified as a candidate oncogene in human liver cancer due to its frequent upregulation in HCC specimens.13 MDM4 inhibition resulted in growth restraint of HCC cell lines both in vitro and in vivo. However, the finding that targeting MDM4 in p53-wildtype HuH6 xenografts only resulted in a moderate reduction of tumor growth compared to p53-deficient cells was unexpected. Besides less efficient engrafting of p53-wildtype cells, we observed a significant reduction of shMDM4 expressing cells as determined by reduced GFP levels in shMDM4- compared to shNC-treated cells and pre-injection controls indicating that the MDM4 knockdown resulted in selection of shMDM4-negative cells (Suppl. Fig. 14). As this finding may affect the translational potential of MDM4 targeting therapies and data on p53-independent functions of MDM4 are limited,25 future studies are needed to dissect p53-dependent and –independent functions of MDM4.
Although amplification of the MDM4 locus is an important mechanism leading to MDM4 upregulation, elevated protein levels of MDM4 are also present in HCC samples without MDM4 amplification.13 In accordance with the latter finding, emerging evidence indicates that additional molecular mechanisms, such as phosphorylation or ubiquitination can modulate the activity and the protein turnover of MDM4 as well.14,15,24 Here, we demonstrate for the first time that activation of the PI3K-AKT-mTOR pathway promotes the upregulation of MDM4 in human HCCs. Pharmacological targeting of the AKT (LY294002) and the mTOR (Rapamycin) pathway alone or in combination (PI103) not only blocked the downstream signaling cascade, but also resulted in a marked downregulation of the MDM4 protein expression. The strongest effect was observed when PI3K, mTORC1, and mTORC2 activities were silenced simultaneously. The relevance of the AKT-mTOR axis for the protumorigenic activity of MDM4 was further demonstrated in a mouse model overexpressing a constitutively active form of AKT in the liver. In this model, we found that active AKT alone is sufficient to induce MDM4 protein levels in vivo. Furthermore, the treatment of AKT mice with Rapamycin prevented MDM4 overexpression, likely due to reduced MDM4 biosynthesis. Accordingly, Rapamycin treatment resulted in reduced activity of the eukaryotic translation initiation factor 4E in colon carcinoma cells, which impaired the translation activity and resulted in decreased MDM2 protein levels.26 A similar mechanism may be involved in the mTOR-mediated regulation of MDM4 in human and murine HCC. Since the MDM4 transcription was neither significantly altered following inhibition of PI3K-AKT-mTOR signaling in vitro nor in the AKT mice, a post-transcriptional mechanism is presumably responsible for the downregulation of the MDM4 protein expression. Indeed, co-administration of the proteasomal inhibitor MG132 rescued the reduced MDM4 protein levels after LY294002 treatment supporting a post-translational mechanism. In particular, the half-life of the MDM4 protein was significantly decreased after silencing of the PI3K-AKT pathway in vitro, suggesting an important role of the AKT kinase in protecting MDM4 from proteasomal degradation. We were able to identify two independent mechanisms that increase the stability of the MDM4 protein, namely the protection of MDM4 from degradation and its phosphorylation by AKT. Indeed, we showed that USP2a physically interacts with MDM4 in human HCC cells and that downregulation of USP2a reduces the MDM4 protein levels, suggesting that the USP2a-mediated de-ubiquitination of MDM4 inhibits its proteolysis. In addition, we were able to demonstrate a direct interaction between phosphorylated MDM4 and AKT in vitro, supporting the idea that MDM4 might be phosphorylated by AKT in human HCC cells, as it has been reported for several other tumor types.16 A schematic model of the EEF1A2-mediated posttranslational stabilization of MDM4 is shown in Fig. 7. Interestingly, recent studies indicated that the phosphorylation of serine 367 by AKT results in the stabilization of MDM4, while phosphorylation of this residue by CHK2 promotes its degradation, implying an important role of the molecular/cellular context in the regulation of MDM4 protein levels.16,27 Since there was no correlation between MDM4 phosphorylation at serine 367 and MDM4 protein levels both in human and AKT mouse tissues (Calvisi et al., data not shown), other, so far undefined serine residues may regulate MDM4 stability in HCC.
Fig. 7.
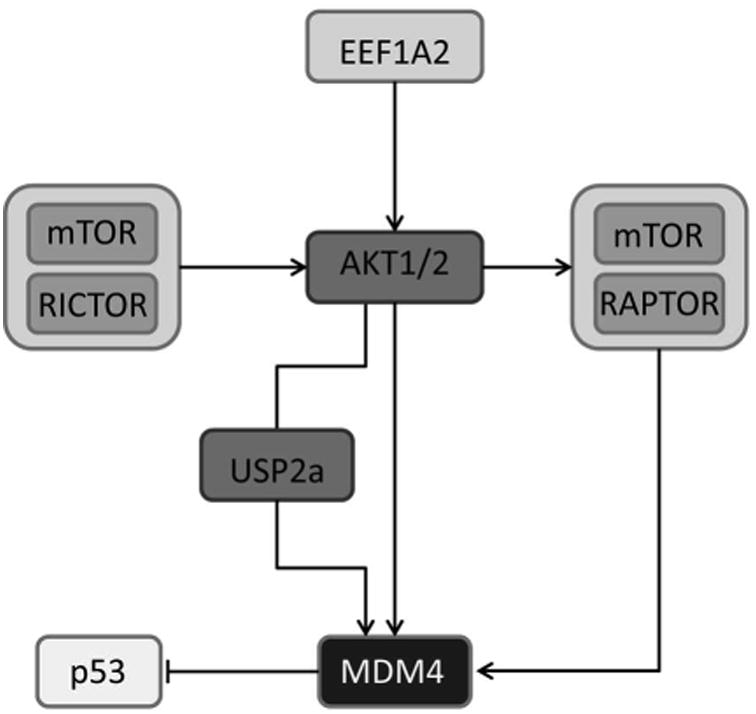
Schematic drawing of the EEF1A2-mediated activation of PI3/AKT/mTOR-signalling that results in posttranslational stabilization of MDM4 via AKT-mediated phosphorylation and USP2a-mediated deubiquitination.
Using array-based comparative genomic hybridization and the definition of minimal overlapping regions, we have previously identified EEF1A2 as a candidate oncogene in human hepatocarcinogenesis.13 Recent data indicated that EEF1A2 overexpression is a hallmark of prostate and ovarian cancer and plays an important role in mammary carcinogenesis.28-30 In particular, EEF1A2 has been reported to induce the activation of AKT protooncogene in breast cancer and rat cell lines.23 Here, we demonstrate that the siRNA-mediated silencing of EEF1A2 results in reduced levels of pAKT and pRPS6K. Also, we found that EEF1A2 overexpression was strongly associated with upregulation of pAKT and MDM4 protein levels in human HCC samples. These data imply that the protumorigenic upregulation of EEF1A2 in human HCCs activates AKT and mTOR signaling, which in turn promote the functional inactivation of p53 via stabilization of the MDM4 protein. Importantly, a strong activation of the EEF1A/PI3K/AKT/mTOR-MDM4 axis was associated with shorter survival of HCC patients. Since PI3K, AKT, and mTOR inhibitors that are well tolerated in tumor patients (including HCC) have been developed,31-33 human HCC patients, whose tumors show activation of AKT or mTOR signaling, should benefit most from targeting these pathways. In these patients suppression of the AKT/mTOR cascade may result in both, the inhibition of the protumorigenic effects driven by AKT/mTOR as well as the induction of tumor-suppressive effects following p53 reactivation. In order to substantiate this intriguing hypothesis and to analyze the p53-dependency of targeting this pathway, HCC patients treated with inhibitors of PI3K-AKT-mTOR signaling should be the tested for p53 mutation and surrogate markers of AKT/mTOR pathway activation, such as phosphorylation of AKT and RPS6K in tumor tissue. Biopsy-based HCC diagnosis prior to respective future clinical trials, as routinely performed in other human cancers, may support predictive testing and rational inclusion of patients likely to benefit more form PI3K-AKT-mTOR inhibition.
In summary, by using in vitro and in vivo approaches we provide evidence that the EEF1A2/PI3K/AKT/mTOR cascade supports the protumorigenic upregulation of MDM4 in human HCC through a post-transcriptional mechanism involving AKT-mediated phosphorylation of MDM4 and USP2a de-ubiquitination. The degree of activation of the EEF1A2/PI3K/AKT/mTOR/MDM4 axis has impact on the survival probability of HCC patients in vivo and may thus represent a promising molecular target.
Supplementary Material
Acknowledgments
We are grateful to Verena Kautz, Marianne Hartmann, Susanne Bösser, Veronika Geissler, Sara Messard, and Eva Eiteneuer for excellent technical assistance. This work was supported by the tissue bank of the National Center for Tumor Diseases Heidelberg.
Financial Support: This work was supported by the Deutsche Forschungsgemeinschaft DFG SFB/TRR77 (subprojects B4 to L.Z., B5 to R.P, B.R., P.S., T.L., Z2 to J.L.B.), Graduiertenkolleg 1172 to J. W. and M. Z., Ev168/2-1 to M.E, and Do622/2-1 to F.D.; National Institutes of Health grants R21CA131625 and R01CA136606 to X.C.; P30DK026743 for UCSF Liver Center; Siemens/DAAD Post Graduate Programme to V. K.; LOEWE inititative Oncogenic Signaling Franfurt to M. Z. (funded by the Hessian Ministry of Higher Education, Research and the Arts; funding reference number III L 4-518/55.004 (2009)) and institutional funds of the Georg-Speyer-Haus by the German Federal Ministry of Health (BMG) and the Ministry of Higher Education, Research and the Arts of the state of Hessen (HMWK) to M.Z.
Abbreviations
- AKT
v-akt murine thymoma viral oncogene homolog
- BAX
BCL2-associated × protein
- cDNA
complementary DNA
- CHX
cycloheximide
- EEF1A2
eukaryotic elongation factor 1A2
- HBV
Hepatitis B virus
- HCV
Hepatitis C virus
- HCC
hepatocellular carcinoma
- MDM2
Mouse Double Minute homolog 2
- MDM4
Mouse Double Minute homolog 4
- mTOR
mammalian target of Rapamycin
- mTORC
mammalian target of Rapamycin complex
- p21
cyclin-dependent kinase inhibitor 1A (CDKN1A, Cip1)
- p53
tumor protein p53
- PI3K
phosphoinositide-3-kinase
- PUMA
BCL2 binding component 3
- RAPTOR
regulatory associated protein of mTOR, complex 1
- RICTOR
RAPTOR independent companion of mTOR, complex 2
- RPS6K
ribosomal protein S6 kinase
- SCR
scrambled siRNA
- shMDM4
shRNA targeting MDM4
- shNC
neutral control shRNA
- siMDM4
siRNA targeting MDM4
- USP2a
ubiquitin-specific protease 2a
Footnotes
Authors' Disclosures of Potential Conflicts of Interest: The authors indicated no potential conflicts of interest.
References
- 1.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 2.May P, May E. Twenty years of p53 research: structural and functional aspects of the p53 protein. Oncogene. 1999;18:7621–7636. doi: 10.1038/sj.onc.1203285. [DOI] [PubMed] [Google Scholar]
- 3.Kubicka S, Trautwein C, Schrem H, Tillmann H, Manns M. Low incidence of p53 mutations in European hepatocellular carcinomas with heterogeneous mutation as a rare event. J Hepatol. 1995;23:412–419. doi: 10.1016/0168-8278(95)80199-5. [DOI] [PubMed] [Google Scholar]
- 4.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hussain SP, Harris CC. Molecular epidemiology of human cancer. Recent Results Cancer Res. 1998;154:22–36. doi: 10.1007/978-3-642-46870-4_2. [DOI] [PubMed] [Google Scholar]
- 6.Seemann S, Maurici D, Olivier M, Caron de Fromentel C, Hainaut P. The tumor suppressor gene TP53: implications for cancer management and therapy. Crit Rev Clin Lab Sci. 2004;41:551–583. doi: 10.1080/10408360490504952. [DOI] [PubMed] [Google Scholar]
- 7.Momand J, Zambetti GP. Mdm-2: “big brother” of p53. J Cell Biochem. 1997;64:343–352. [PubMed] [Google Scholar]
- 8.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 9.Shvarts A, Bazuine M, Dekker P, Ramos YF, Steegenga WT, Merckx G, van Ham RC, et al. Isolation and identification of the human homolog of a new p53-binding protein, Mdmx. Genomics. 1997;43:34–42. doi: 10.1006/geno.1997.4775. [DOI] [PubMed] [Google Scholar]
- 10.Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, van Ham RC, et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. Embo J. 1996;15:5349–5357. [PMC free article] [PubMed] [Google Scholar]
- 11.Marine JC, Jochemsen AG. Mdmx as an essential regulator of p53 activity. Biochem Biophys Res Commun. 2005;331:750–760. doi: 10.1016/j.bbrc.2005.03.151. [DOI] [PubMed] [Google Scholar]
- 12.Riemenschneider MJ, Buschges R, Wolter M, Reifenberger J, Bostrom J, Kraus JA, Schlegel U, et al. Amplification and overexpression of the MDM4 (MDMX) gene from 1q32 in a subset of malignant gliomas without TP53 mutation or MDM2 amplification. Cancer Res. 1999;59:6091–6096. [PubMed] [Google Scholar]
- 13.Schlaeger C, Longerich T, Schiller C, Bewerunge P, Mehrabi A, Toedt G, Kleeff J, et al. Etiology-dependent molecular mechanisms in human hepatocarcinogenesis. Hepatology. 2008;47:511–520. doi: 10.1002/hep.22033. [DOI] [PubMed] [Google Scholar]
- 14.Pereg Y, Shkedy D, de Graaf P, Meulmeester E, Edelson-Averbukh M, Salek M, Biton S, et al. Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc Natl Acad Sci U S A. 2005;102:5056–5061. doi: 10.1073/pnas.0408595102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 2005;24:3411–3422. doi: 10.1038/sj.emboj.7600812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez-Pajares V, Kim MM, Yuan ZM. Phosphorylation of MDMX Mediated by Akt Leads to Stabilization and Induces 14-3-3 Binding. J Biol Chem. 2008;283:13707–13713. doi: 10.1074/jbc.M710030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, Destefanis G, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology. 2011;140:1071–1083. doi: 10.1053/j.gastro.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C, Tovar V, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135:1972–1983. e1971–1911. doi: 10.1053/j.gastro.2008.08.008. 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ho C, Wang C, Mattu S, Destefanis G, Ladu S, Delogu S, Armbruster J, et al. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology. 2012;55:833–845. doi: 10.1002/hep.24736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calvisi DF, Donninger H, Vos MD, Birrer MJ, Gordon L, Leaner V, Clark GJ. NORE1A tumor suppressor candidate modulates p21CIP1 via p53. Cancer Res. 2009;69:4629–4637. doi: 10.1158/0008-5472.CAN-08-3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pellegrino R, Calvisi DF, Ladu S, Ehemann V, Staniscia T, Evert M, Dombrowski F, et al. Oncogenic and tumor suppressive roles of polo-like kinases in human hepatocellular carcinoma. Hepatology. 2010;51:857–868. doi: 10.1002/hep.23467. [DOI] [PubMed] [Google Scholar]
- 22.Reed D, Shen Y, Shelat AA, Arnold LA, Ferreira AM, Zhu F, Mills N, et al. Identification and characterization of the first small molecule inhibitor of MDMX. J Biol Chem. 2010;285:10786–10796. doi: 10.1074/jbc.M109.056747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amiri A, Noei F, Jeganathan S, Kulkarni G, Pinke DE, Lee JM. eEF1A2 activates Akt and stimulates Akt-dependent actin remodeling, invasion and migration. Oncogene. 2007;26:3027–3040. doi: 10.1038/sj.onc.1210101. [DOI] [PubMed] [Google Scholar]
- 24.Allende-Vega N, Sparks A, Lane DP, Saville MK. MdmX is a substrate for the deubiquitinating enzyme USP2a. Oncogene. 2010;29:432–441. doi: 10.1038/onc.2009.330. [DOI] [PubMed] [Google Scholar]
- 25.Matijasevic Z, Steinman HA, Hoover K, Jones SN. MdmX promotes bipolar mitosis to suppress transformation and tumorigenesis in p53-deficient cells and mice. Mol Cell Biol. 2008;28:1265–1273. doi: 10.1128/MCB.01108-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kao CL, Hsu HS, Chen HW, Cheng TH. Rapamycin increases the p53/MDM2 protein ratio and p53-dependent apoptosis by translational inhibition of mdm2 in cancer cells. Cancer Lett. 2009;286:250–259. doi: 10.1016/j.canlet.2009.05.031. [DOI] [PubMed] [Google Scholar]
- 27.LeBron C, Chen L, Gilkes DM, Chen J. Regulation of MDMX nuclear import and degradation by Chk2 and 14-3-3. Embo J. 2006;25:1196–1206. doi: 10.1038/sj.emboj.7601032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scaggiante B, Dapas B, Bonin S, Grassi M, Zennaro C, Farra R, Cristiano L, et al. Dissecting the expression of EEF1A1/2 genes in human prostate cancer cells: the potential of EEF1A2 as a hallmark for prostate transformation and progression. Br J Cancer. 2012;106:166–173. doi: 10.1038/bjc.2011.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pinke DE, Lee JM. The lipid kinase PI4KIIIbeta and the eEF1A2 oncogene co-operate to disrupt three-dimensional in vitro acinar morphogenesis. Exp Cell Res. 2011;317:2503–2511. doi: 10.1016/j.yexcr.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Pinke DE, Kalloger SE, Francetic T, Huntsman DG, Lee JM. The prognostic significance of elongation factor eEF1A2 in ovarian cancer. Gynecol Oncol. 2008;108:561–568. doi: 10.1016/j.ygyno.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 31.Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, Demanse D, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30:282–290. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 32.Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, Baird RD, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. 2011;29:4688–4695. doi: 10.1200/JCO.2011.35.5263. [DOI] [PubMed] [Google Scholar]
- 33.Zhu AX, Abrams TA, Miksad R, Blaszkowsky LS, Meyerhardt JA, Zheng H, Muzikansky A, et al. Phase 1/2 study of everolimus in advanced hepatocellular carcinoma. Cancer. 2011;117:5094–5102. doi: 10.1002/cncr.26165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.