

Abstract
Binding interactions between DNA and cationic carriers must be sufficiently strong to prevent nuclease-mediated degradation, yet weak enough to permit transcription. We demonstrate cationic diblock copolymers containing PEG and o-nitrobenzyl moieties that facilitated tailorable DNA complexation and light-activated release. This design unlocks a new approach to advance non-viral gene packaging.
Nonviral gene therapy has the potential to transform treatment for a range of hereditary and acquired diseases without the immunogenic and mutagenic concerns of viral methods.1, 2 Gene carriers such as lipids, peptides, and synthetic polymers can condense and deliver exogenous nucleic acids to dysfunctional cells to manipulate the expression of specific genetic targets and thereby modulate cellular function.3 Cationic polymers are a promising subset of nonviral nucleic acid delivery vehicles due to their synthetic versatility and ability to form electrostatic complexes (polyplexes) with anionic nucleic acids.4 The encapsulation of nucleic acids within polyplexes can prevent enzymatic degradation, reduce serum protein adsorption, and promote cell membrane interactions/endocytosis,5 yet ultimately, nucleic acid release is essential to permit binding by transcription factors (or other protein-based targets) and promote efficient gene expression.6 Balancing these competing requirements is a common challenge in developing successful delivery vehicles.
Weak nucleic acid binding affinity is a substantial and well-documented roadblock to efficient delivery as it leads to polyplex instability, premature nucleic acid release, and nucleic acid degradation.6 For example, Burke and Pun highlighted the importance of polyplex stability in a study of plasmid DNA/polyethylenimine (pDNA/PEI) polyplexes.7 During in vivo delivery to liver tissues, interactions between extracellular matrix components and the pDNA/PEI polyplexes displaced the pDNA from the polyplexes, resulting in reduced cellular internalization of pDNA as well as degradation of the pDNA in the extracellular space. Strategies to improve nucleic acid encapsulation and polyplex stability include increasing the number of cationic polymers in the polyplex, increasing the polycation charge density, increasing the length of the polycation, or incorporating hydrophobic groups within the polycation.8-11 Specifically, Layman et al. showed that increased lengths of poly(2-dimethylaminoethyl methacrylate) shielded pDNA from degradation and hence increased in vitro gene expression by 20-fold as the weight-average molecular weight (Mw) increased from 43,000 g/mol to 915,000 g/mol.9 Rice and coworkers greatly enhanced both polyplex stability and circulatory half-life through the incorporation of hydrophobic acridine groups into polycations; these acridine groups imposed thermodynamic penalties for disassociation and enhanced binding affinities by coupling electrostatic interactions with intercalation between the acridine aromatic rings and the pDNA nucleobases.12 Consideration of the complexation strategies mentioned above has led to improved nanocarrier designs for maintaining pDNA activity during in vivo nucleic acid delivery.13-15
Although several strategies exist to improve polyplex packaging and ultimately enhance stability, high affinity polymers present a challenge within cells as they can impede nucleic acid release.6, 16 Schaffer et al. and Choosakoonkriang et al. showed that increasing polycation length beyond a certain point (with poly(l-lysine) and PEI, respectively) decreased the transfection efficiency in vitro due to a lack of nucleic acid release from the polyplexes.17, 18 Also, Schaffer et al. and Erbacher et al. independently concluded that reduced cationic charge on poly(l-lysine) improved transfection by enhancing polyplex dissociation in solution.17, 19 Additionally, multiple studies have correlated increasing cytotoxicity to increasing cationic charge densities in polyplexes.10, 20, 21 These examples highlight inconsistent extracellular vs. intracellular needs for gene carriers, and suggest that alternative nucleic acid packaging strategies are necessary.
The incorporation of stimuli-responsive functional groups into polymer/nucleic acid complexes can provide complementary functionality for both stable encapsulation and triggered release of nucleic acids.22 For example, Grinstaff and coworkers reported amphiphiles that contained both cationic headgroups to bind to pDNA and terminal esters that were susceptible to enzymatic hydrolysis.23 Gene transfer studies in Chinese Hamster Ovary (CHO) cells showed that lipoplexes susceptible to enzymatic hydrolysis enhanced transfection efficiency 100-fold in comparison to amphiphiles that were stable toward enzymes; the enhancement was attributed to the formation of anionic carboxylates that simultaneously reduced the binding affinity toward pDNA and destabilized endosomal membranes. In a related example, Abbott and coworkers developed ferrocene-containing cationic lipids with reduction/oxidation (redox) sensitive nucleic acid binding capacity.24 The authors showed that chemical or electrochemical oxidation/reduction of the ferrocene groups could reversibly alter the charge of the lipids and thereby produce shifts between lamellar (reduced) and amorphous (oxidized) lipoplex morphologies, which was directly correlated with deactivation/activation of gene transfer.25 Subsequent studies demonstrated the utility of these approaches in cell patterning, as the authors spatially controlled gene transfection in COS-7 cultures by incubating the cells with inactivated lipoplexes and selectively delivering ascorbic acid to a subpopulation of cells isolated by a hollow cylinder (among other methods); the ascorbic acid activated lipoplexes in the subpopulation and facilitated efficient gene transfer.26
The above systems highlight the merits of stimuli-responsive constructs and suggest enormous potential benefits for gene nanocarriers whose properties are sensitive to externally-applied stimuli. These types of materials present additional opportunities for patterned gene expression in cellular subpopulations; for example, substrate-mediated gene delivery and microfluidic devices, among other techniques, have been used to prepare cultures that are applicable to guiding neurite extension and preparing defined interfaces for directed tissue development.27, 28 However, the use of externally-applied stimuli (e.g. light29) offers tremendous potential for generating practical and large scale cultures with patterned, targeted, activated/deactivated gene expression. Light-mediated gene transfer also potentially avoids complications in topical wound repair associated with the off-target expression of growth factors, which has been cited as a key reason for clinical failure of recombinant erythropoietin.30 Photo-responsive polymers are particularly appealing due to the versatility and spatial resolution afforded by light. Photo-responsive systems can undergo isomeric rearrangements or photochemical cleavage reactions that depend on the photoactive functional group utilized.31 In particular, the o-nitrobenzyl (o-NB) ester absorbs UV and near-infrared light and severs to form a carboxylic acid and a nitrosobenzaldehyde.32, 33 The production of the carboxylic acid introduces a negatively charged carboxylate ion in aqueous conditions above pH ≈ 3.32
Recent studies have detailed the incorporation of photo-responsive moieties into various polymeric systems for drug and gene delivery.31, 32, 34-36 In particular, solution assemblies whose hydrophobic portions contained o-NB moieties could disassemble in response to irradiation with light and release dyes or drugs.37-39 In one such example, Johnson et al. synthesized a photocleavable polymer-drug conjugate that tethered doxorubicin (dox) to a poly(norbornene-PEG) bottle-brush copolymer using an o-NB derivative.40 The polymer was noncytotoxic in the absence of UV irradiation and demonstrated successful release of dox in cultured breast cancer cells after UV irradiation. Other researchers have incorporated the o-NB functionality into block copolymer (BCP) systems for nucleic acid delivery. For example, two groups have complexed pDNA into polyplexes by using PEI modified with o-NB–containing crosslinkers.41, 42 In one case, polyplexes were formed and pDNA was subsequently caged within the polyplexes by methacrylamide crosslinking,41 whereas in the other case, PEI was first crosslinked with o-NB urethane linkers and then used to form polyplexes.42 In these examples, the crosslinkers in the polyplexes could be degraded to yield low molecular weight PEI via irradiation with UV light. Transfection by the crosslinked polyplexes was limited in the absence of UV light but became detectable in a portion of cells at levels similar to those induced by standard PEI (e.g. 25 kDa PEI) when cells were irradiated with UV light subsequent to polyplex delivery. However, while the application of UV light may have caused changes in polyplex structure/stability, UV irradiation did not induce pDNA liberation in either example. These results indicate that improved release efficacy in light-activated systems might substantially improve gene transfer potential. Other o-NB-containing systems include photoactive dendrons, metallic nanoparticles, and peptides that facilitated nucleic acid complexation and release.43-45 The above systems present strategies to achieve efficient gene complexation, stimuli-triggered photocleavage, or enhanced gene transfer.
To satisfy the contradictory demands imposed on nucleic acid carriers, our BCP provides an innovative light-mediated charge-reversal design that simultaneously and independently controls both binding and release of nucleic acids. Additionally, this BCP uniquely combines tunable pDNA binding and photo-responsive nucleic acid release with salt stability and resistance toward protein adsorption to satisfy the multiplicity of requirements in nucleic acid transfer. To that end, we demonstrate the controlled and scalable synthesis of this photoactive and cationic diblock copolymer and show its biocompatibilty, stable and tunable pDNA binding, and capacity for light-triggered release.
Critical criteria for new nucleic acid carriers include: (1) complete complexation and condensation of nucleic acids into polyplexes; (2) minimized charge content to reduce cytotoxicity yet retain binding capacity; (3) salt and serum stability; and (4) capability for nucleic acid unpackaging. We incorporated these features into the design of a BCP architecture that utilized PEG as a nonfouling/stealth46 component and a methacrylate-based cationic block to facilitate nucleic acid complexation. We employed a PEG macroinitiator to provide access to controlled radical polymerization (CRP) techniques, i.e. atom-transfer radical polymerization (ATRP), and to reduce the overall charge content in the polymer to moderate cytotoxicity. CRP enables tunable molecular weights, compositions, dispersities, and end-group functionalities. A vital building block in our scheme was the monomer, 5-(3-((tert-butoxycarbonyl)amino)propoxy)-2-nitrobenzyl methacrylate (Boc-APNBMA), which contained a methacrylate and protected amine functionality to facilitate ATRP. Photo-responsive character was introduced through the incorporation of an o-NB ester moiety, which also added the potential for the formation of hydrophobic interactions that could stabilize polyplexes. Cleavage of the Boc-protecting group following ATRP revealed the final cationic and photo-responsive BCP. The monomer, the subsequent protected BCP, and cationic BCP were prepared as depicted in Scheme 1. The placement of the photoactive o-NB ester between the polymer backbone and the ammonium cation is key to the BCP design as applied to nucleic acid delivery. This location leads to charge reversal and the release of hydrophobic aryl units upon irradiation to facilitate the catch and release mechanism desired for efficient nucleic acid delivery. Thus, the design combines critical criteria for efficient nucleic acid delivery, including: nonfouling PEG components, polymers with tailorable composition and molecular weights, cationic and hydrophobic moieties that support tight polyplex formation, and photo-responsive functional groups for controlled spatiotemporal release of the nucleic acid. This combination greatly enhances the potential of these stimuli-responsive BCPs for gene delivery.
Scheme 1.

Synthesis of Boc-APNBMA, mPEG-b-P(Boc-APNBMA)n, and mPEG-b-P(APNBMA•HCl)n.
Developing structure-property relationships is critical to the design and optimization of polymeric nanocarriers and necessitates the synthesis of polymers with well-defined characteristics. Thus, ATRP of Boc-APNBMA from a mPEG-Br macroinitiator (5,300 g/mol, Đ = 1.05) was used to generate well-defined mPEG-b-P(Boc-APNBMA) BCPs. Boc-APNBMA content and overall copolymer molecular weight were tuned by manipulating the monomer to macroinitiator ratio.47 The size exclusion chromatography (SEC) traces in Fig. 1 demonstrate the shift in elution volume upon a change in the monomer to macroinitiator ratio. Narrow molecular weight distributions (Đ ≤ 1.16) for mPEG-b-P(Boc-APNBMA)n (in which n represents the calculated degree of polymerization of the Boc-APNBMA block) supported the controlled nature of the polymerization during Boc-APNBMA chain extension.
Fig. 1.

Size exclusion chromatograms of mPEG-b-P(Boc-APNBMA)n.
Analysis of 1H NMR spectroscopy data confirmed the chemical structures of the monomer, protected BCP, and cationic diblock copolymer (Fig. 2). After ATRP, the appearance of mPEG-associated resonances at ≈ 3.6 ppm, the elimination of resonances from the methacrylate C=C bond between 5.7-6.3 ppm, and the broadened resonances that correlate to the repeat unit functional groups supported the successful synthesis of mPEG-b-P(Boc-APNBMA)n. The disappearance of the resonances at 1.4 ppm following acid treatment indicated complete conversion of mPEG-b-P(Boc-APNBMA)n to mPEG-b-P(APNBMA•HCl)n. The charged nature of mPEG-b-P(APNBMA•HCl)n necessitated analysis in a polar solvent (DMSO-d6) and provided further evidence for the deprotection. Comparing the integrations of the terminal methoxy (3.4 ppm), PEG methylene (3.6 ppm), and Boc methylene (1.4 ppm) resonances enabled the calculation of the number-average molecular weights (Mn)s for mPEG-b-P(Boc-APNBMA)n, which were 8,400 g/mol and 14,600 g/mol for the two polymers depicted in Fig. 1. Using a range of monomer to initiator ratios yielded a series of BCPs, and the two discussed herein illustrate the ability to tune and control molecular weight and dispersity. The integrations from the remaining resonances did not change following Boc deprotection, indicating that no cleavage of the pendant ester residues occurred during synthesis or workup. Table 1 summarizes the molecular weights obtained from 1H NMR spectroscopy and the dispersities obtained from SEC for the two protected polymers and their deprotected counterparts.
Fig. 2.
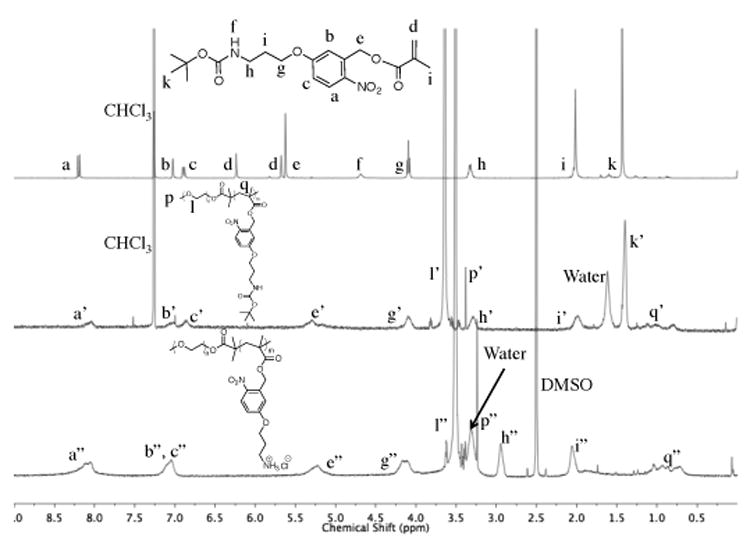
1H NMR spectra of Boc-APNBMA monomer (top) in CDC13, mPEG-b-P(Boc-APNBMA)n (middle) in CDC13, and mPEG-b-P(APNBMA•HCl)n (bottom) in DMSO-d6. Letters with a single prime (′) indicate resonances from the protected BCP and letters with a double prime (″) indicate resonances from mPEG-b-P(APNBMA•HCl)n.
Table 1.
Characterization of mPEG-b-P(Boc-APNBMA)n and mPEG-b-P(APNBMA•HCl)n using 1H NMR spectroscopy and SEC.
| Mna(g/mol) | Đb | |
|---|---|---|
| mPEG-Br | 5,300 | 1.05 |
| mPEG-b-P(Boc-APNBMA)7.9 | 8,400 | 1.16 |
| mPEG-b-P(APNBMA•HCl)7.9 | 7,900 | - |
| mPEG-b-P(Boc-APNBMA)23.6 | 14,600 | 1.11 |
| mPEG-b-P(APNBMA•HCl)23.6 | 13,100 | - |
Determined using 1H NMR spectroscopy and subsequently used to calculate the degree of polymerization for each polymer (7.9 and 23.6).
Determined using SEC.
Two assays were employed to analyze polyplex formation in this work. The first assay, an ethidium bromide exclusion assay, probes the extent of nucleic acid compaction into polyplexes; ethidium bromide fluoresces upon intercalation between nucleic acid base pairs to reveal the location of the nucleic acids migrating down an electrophoresis gel. A tightly formed polyplex excludes the fluorescent dye, which reduces or eliminates fluorescence. The mPEG-b-P(APNBMA•HCl)7.9 complexed pDNA into polyplexes at N/P ≥ 2, with tight polyplex formation occurring at N/P ≥ 5 as shown by the complete exclusion of ethidium bromide (Fig. 3a). The mPEG-b-P(APNBMA•HCl)23.6 formed tight polyplexes at N/P ≥ 1 and completely excluded ethidium bromide at N/P ≥ 4 (Fig. S1a). The lower N/P ratio necessary to form tight polyplexes with the longer polycation length indicated an enhanced binding affinity between pDNA and the longer polycation, which highlights the benefits of utilizing tailorable BCPs for gene complexation. The satisfactory binding of both of the mPEG-b-P(APNBMA•HCl)n polymers initiated further binding studies to probe polyplex formation. Using mPEG-b-P(APNBMA•HCl)7.9 with pDNA at N/P = 5 as a model system, a YOYO-1 iodide fluorescence quenching assay was used to quantitatively determine the level of fluorescence quenching induced by polyplex formation. Specifically, YOYO-1 iodide also labels pDNA through intercalation binding interactions, and pDNA packaging into polyplexes can position the bound YOYO-1 iodide fluorophores in sufficient proximity for self-quenching.48 pDNA complexation with mPEG-b-P(APNBMA•HC1)7.9 produced decreased fluorescence as tighter polyplexes formed at higher N/P ratios (Fig. 3b). A plateau in fluorescence occurred near N/P ≈ 5, supporting the ethidium bromide exclusion data in Fig. 3a.
Fig. 3.

Characterization of mPEG-b-P(APNBMA•HCl)7.9/pDNA polyplex size and stability. (a) Gel electrophoresis of polyplexes formed at varying N/P ratios. (b) YOYO-1 fluorescence quenching assay. (c) DH of polyplexes formed at various N/P ratios in HEPES buffer. (d) DH of polyplexes at N/P = 5 after a 60 min incubation in various salt solutions. Error bars in (c) and (d) represent the standard deviation from the mean of three independent measurements of polyplex DH. An asterisk (*) indicates a statistically significant difference between the indicated samples (p < 0.05).
Polyplexes were further analyzed using dynamic light scattering (DLS) to investigate polyplex size. DLS analyses identified hydrodynamic diameters (DH) of ≈ 150 nm for mPEG-b-P(APNBMA•HCl)7.9/pDNA polyplexes at N/P ≥ 4 (Fig. 3c) and ≈ 150 nm for mPEG-b-P(APNBMA•HCl)23.6/pDNA polyplexes at N/P ≥ 3 (Fig. S1b). These data suggest potential compatibility with the size limits for endocytic internalization into cells.49 Increasing the N/P ratio led to slight decreases in DH, which was consistent with the tighter polyplex structures determined by the fluorescence quenching assay. These complementary results suggested tight polyplex formation at N/P ≥ 5 for both polymers. Thus, polyplexes were formed at N/P = 5 in all subsequent analyses.
Polyplex stability within high salt and/or protein-rich environments is an additional key parameter for predicting the in vivo performance of gene delivery vehicles. High salt environments can induce polyplex aggregation and precipitation, and serum proteins can adsorb on the surface of polyplexes and significantly alter the size as well as induce immune clearance.50 As described earlier, we expected that the inclusion of a nonfouling PEG coating would enhance the stability in solution. Hence, we tested the nonfouling capacity by performing DLS experiments in the presence of salt or serum with the mPEG-b-P(APNBMA•HCl)7.9/pDNA polyplexes. The polyplexes exhibited no change in size following incubation in mild buffered solution (HEPES buffer) or water over a period of 60 min at 23 °C. Incubation in protein-supplemented medium (Opti-MEM) or cell-culture grade PBS solutions containing physiological saline (150 mM NaCl) only modestly increased polyplex DH by ≈ 30 nm (1.2×), which suggested improved polyplex stability in high salt media relative to other nucleic acid carriers in the literature.48 In comparison, Johnson et al. reported an increase of approximately 1 μrn (10-fold) for the DH of PEI/pDNA and poly(l-lysine)/pDNA polyplexes over 15 min in PBS.48 In the present work, the nearly constant polyplex sizes suggested greater stability in the high salt conditions commonly encountered in vitro and in vivo (Fig. 4d) and indicated a potential for extension to cell-based applications.
Fig. 4.

Cell proliferation assays following treatment with (a) varying concentrations of free polymers [mPEG-b-P(APNBMA•HCl)n or PEI], or (b) mPEG-b-P(APNBMA•HCl)7.9/pDNA polyplexes, UV irradiation, or UV irradiation following polyplex incubation. Asterisks (*) indicate statistically significant differences between the indicated samples (p < 0.05).
Cell viability assays probed the cytotoxicities of the two BCPs and revealed a slightly reduced cytotoxicity for mPEG-b-P(APNBMA•HCl)7.9 as compared with mPEG-b-P(APNBMA•HCl)23.6 (Fig. 4a). The IC50 values for the BCPs were 1.05 μM (7.9 repeat units) and 0.77 μM (23.6 repeat units), both of which were significantly higher than the reported IC50 value for PEI (0.28 μM in NIH/3T3 cells48). The slightly increased toxicity for the higher molecular weight polymer was consistent with polymer toxicity studies in the literature, which show that increasing polycation molecular weight correlates with increasing cytotoxicity.9 To determine whether the polymers would maintain low cytotoxicity under the conditions used for transfection, additional viability assays were performed using mPEG-b-P(APNBMA•HCl)7.9 polyplexes alone or in combination with UV treatment. Neither the polyplexes alone nor the polyplexes combined with UV irradiation induced significant cell losses, indicating that both the treatment conditions and products of the cleavage reaction were not harmful to cells (Fig. 4b). Moreover, the biocompatibility of o-NB-based systems and their photocleavage byproducts is well-documented for photodegradable hydrogels in vitro,51, 52 beads decorated with photolabile peptides for cell adhesion and release,53, 54 and in vivo delivery of fluorescent dyes, and o-NB-based systems are also postulated to extend to clinical trials.56 These literature precedents, together with the biocompatibility of UV-irradiated mPEG-b-P(APNBMA•HCl)7.9/pDNA polyplexes demonstrated in vitro (see Fig. 4b), suggest that these materials may be suitable for extension to further in vitro and in vivo studies.
As mPEG-b-P(APNBMA•HCl)7.9 exhibited favorable properties for nucleic acid packaging and cell treatment, additional UV irradiation studies were performed to assess the polymer's efficacy for light-triggered pDNA release. We irradiated mPEG-b-P(APNBMA•HC1)7.9 with 365 nm light at an intensity of 200 W/m2 and monitored the relative characteristic absorbance of the o-NB ester at 316 nm to determine the extent of photocleavage.57 The peak at 316 nm displayed dramatic decreases up to 20 min, small changes from 20 to 40 min, and no change between 40 min and 60 min (Fig. 5a), and the decrease in the characteristic absorbance of o-NB (316 nm) was accompanied by the formation of a new absorbance peak at higher wavelength that increased in intensity as irradiation time increased, consistent with the literature cited above.57 Photolysis was confirmed using 1H NMR spectroscopy, which revealed a decreased intensity for resonances from the benzylic –CH2– and the formation of the nitrosobenzaldehyde salt (Fig. S2). The decreasing absorbance at 316 nm as a function of irradiation time followed an exponential decay, and fitting the decay enabled the determination of an exponential decay constant (τ = 5.7 min for n = 7.9 and τ = 3.7 min for n = 23.6) (Fig. S3). The subsequent irradiation of mPEG-b-P(APNBMA•HCl)7.9/pDNA polyplexes yielded a similar decrease in UV-Vis absorbance at 316 nm up to 20 min of irradiation (Fig. 5b), when compared to the free polymer (Fig. 5a), consistent with cleavage in the polyplexes. The exponential decay constant for the mPEG-b-P(APNBMA•HCl)7.9/pDNA polyplexes was 2.7 min under the same irradiation conditions used for photolysis of the polymer samples (Fig. S3).
Fig. 5.

UV-visible spectroscopy of (a) mPEG-b-P(APNBMA•HCl)7.9 following exposure to 365 nm light at 200 W/m2 for 0 (black), 2.5 (red), 5 (blue), 10 (green), 20 (orange), 40 (purple), and 60 (teal) min; and (b) pDNA/mPEG-b-P(APNBMA•HCl)7.9 polyplexes following exposure to 200 W/m2 for 0 (black), 2.5 (red), 5 (blue), 10 (green), 20 (orange), 40 (purple), and 60 (teal) min. Gel electrophoresis of pDNA/mPEG-b-P(APNBMA•HCl)7.9 polyplexes following exposure to (c) UV at varying irradiation times, and (d) a combination of SDS surfactant at a sulfate/phosphate (S/P) ratio of 20 and subsequent UV irradiation. The arrows in (c) and (d) indicate the location of higher intensity fluorescence bands, which potentially correlate to supercoiled DNA or open circular DNA that is interacting with photocleavage fragments and hence prohibited from migrating as free pDNA. The free pDNA (“Free Plasmid”) in (c) and (d) was irradiated with UV light to demonstrate the lack of UV-mediated crosslinking.
The UV-Vis absorption findings suggested complete cleavage of the o-NB ester in both the polymer and polyplex, as well as negligible screening of UV light by the pDNA. Therefore, polyplex destabilization and unpackaging were visualized using gel electrophoresis to confirm the reduced electrostatic interactions following longer irradiation times (Fig. 5c).58 The pDNA migrated through the gel at UV irradiation times ≥ 20 min, which suggested a reduced polyvalency for the BCP, polyplex destabilization, and pDNA release. This mild release demonstrated a loosening of the polyplex structure, which could enable improved transcriptional access to the pDNA in a cellular environment. To further simulate the anionic lipid-rich environment within cells, polyplexes were incubated with sodium dodecyl sulfate (SDS) prior to UV irradiation (Fig. 5d). Addition of SDS at a sulfate/phosphate ratio of 20 without UV irradiation did not induce pDNA release, whereas addition of SDS combined with subsequent UV irradiation caused significant pDNA migration out of the loading well. The arrows in Fig. 5c and Fig. 5d indicate the location of higher intensity fluorescence bands within the smear, which could potentially correlate to supercoiled DNA or open circular DNA that is interacting with photocleavage fragments and hence prohibited from migrating as free pDNA. The reduced migration of the liberated pDNA in the gel in comparison to free pDNA indicated that the photocleavage reaction products either inhibited the mobility of the pDNA through direct interactions or changed the gel characteristics such that migration was limited. Cowman and coworkers reported several experimental parameters that influenced the mobility of hyaluronan in agarose gels, including buffer composition, buffer salt concentration, agarose percentage (i.e. gel pore size), etc.59 We hypothesize that the cleaved fragments could interact with the pDNA and screen the electric field, which would increase the apparent molecular weight, and the varying number of interacting fragments would influence the molecular weight distribution. Further investigations into the dissociated polyplex structure, size, and dispersity are ongoing.
Our findings differ from earlier studies, described above, employing PEI-based o-NB-containing nucleic acid delivery systems, which displayed a complete lack of pDNA release following UV irradiation.41, 42 These systems relied on o-NB-based crosslinkers, and retained relatively high molecular weight polycations (∼2 kDa) even after irradiation with UV light. In contrast, mPEG-b-P(APNBMA•HCl)7.9 undergoes a charge reversal after irradiation with UV light, which could account for the release and migration of pDNA in our system. Regardless of the reduced pDNA mobility, the supposition of photo-triggered polyplex disassembly was supported by the clear shifts in pDNA migration behavior following light irradiation. These promising findings suggested that the mPEG-b-P(APNBMA•HCl)n design could address a key paradox for biocompatible gene carrier designs through the simultaneous incorporation of ammonium cations and hydrophobic residues for strong and tailorable binding and an externally-triggered mechanism to allow on-demand and spatially-resolved release.
Conclusions
In summary, we synthesized a new photo-responsive, methacrylate-based monomer and used ATRP to generate a pair of BCPs with tunable block lengths and narrow molecular weight distributions. These mPEG-b-P(APNBMA•HCl)n BCPs effectively complexed pDNA into salt-stable polyplexes with sizes appropriate for cellular internalization. The longer polycation length reduced the N/P ratio necessary for efficient and tight polyplex formation; however, the longer polycation also was slightly more cytotoxic. Irradiation of mPEG-b-P(APNBMA•HCl)7.9/pDNA with UV light cleaved the o-NB ester and facilitated pDNA release. Considering the favorable complexation and release of pDNA, we are currently investigating the spatiotemporal efficacy and control of these photocleavable BCPs as nucleic acid delivery vehicles, and the influence of cationic block length on nucleic acid encapsulation and release. We anticipate that this novel BCP platform will enable the utility of photoactive polymer assemblies for applications that extend beyond gene delivery to charge reversal membranes and coatings, coacervates, and cellular encapsulation.
Supplementary Material
Acknowledgments
The authors thank the National Institute of General Medical Sciences of the National Institutes of Health (NIH) for financial support through an Institutional Development Award (IDeA) under grant number P20GM103541 and the Delaware Biotechnology Institute (DBI) and Delaware Economic Development Office (DEDO) for financial support through the Bioscience Center for Advanced Technology (Bioscience CAT) award (12A00448). 1H NMR spectra were collected on instrumentation supported by the National Science Foundation (NSF) Chemistry Research Instrumentation and Facilities (CRIF) program grant: MU CEH 0840401. We acknowledge the Center for Molecular and Engineering Thermodynamics at the University of Delaware for providing access to the DLS instrument.
Footnotes
Electronic Supplementary Information (ESI) available: synthetic protocols, gel electrophoresis and DLS of mPEG-b-P(APNBMA•HCl)7.9/pDNA polyplexes, photocleavage profiles: 1H NMR spectroscopy of photocleavage and fits to the exponential decay. See DOI: 10.1039/c000000x/
Contributor Information
Thomas H. Epps, III, Email: thepps@udel.edu.
Millicent O. Sullivan, Email: msullivan@udel.edu.
References
- 1.Auguste DT, Furman K, Wong A, Fuller J, Armes SP, Deming TJ, Langer R. Journal of Controlled Release. 2008;130:266–274. doi: 10.1016/j.jconrel.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Rorke S, Keeney M, Pandit A. Progress in Polymer Science. 2010;35:441–458. [Google Scholar]
- 3.Burke PA, Pun SH, Reineke TM. ACS Macro Letters. 2013;2:928–934. doi: 10.1021/mz400418j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo X, Huang L. Accounts of Chemical Research. 2011;45:971–979. doi: 10.1021/ar200151m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Namgung R, Kim J, Singha K, Kim CH, Kim WJ. Molecular Pharmaceutics. 2009;6:1826–1835. doi: 10.1021/mp900096u. [DOI] [PubMed] [Google Scholar]
- 6.Pack DW, Hoffman AS, Pun S, Stayton PS. Nature Reviews Drug Discovery. Nature Publishing Group; 2005. pp. 581–593. Editon edn. [DOI] [PubMed] [Google Scholar]
- 7.Burke RS, Pun SH. Bioconjugate Chemistry. 2008;19:693–704. doi: 10.1021/bc700388u. [DOI] [PubMed] [Google Scholar]
- 8.Abdelhady HG, Allen S, Davies MC, Roberts CJ, Tendler SJB, Williams PM. Nucleic Acids Research. 2003;31:4001–4005. doi: 10.1093/nar/gkg462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Layman JM, Ramirez SM, Green MD, Long TE. Biomacromolecules. 2009;10:1244–1252. doi: 10.1021/bm9000124. [DOI] [PubMed] [Google Scholar]
- 10.Reineke TM, Davis ME. Bioconjugate Chemistry. 2003;14:247–254. doi: 10.1021/bc025592k. [DOI] [PubMed] [Google Scholar]
- 11.Allen MH, Green MD, Getaneh HK, Miller KM, Long TE. Biomacromolecules. 2011;12:2243–2250. doi: 10.1021/bm2003303. [DOI] [PubMed] [Google Scholar]
- 12.Kizzire K, Khargharia S, Rice KG. Gene Therapy. 2013;20:407–416. doi: 10.1038/gt.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao Y, Yin Q, Chen L, Zhang Z, Li Y. Bioconjugate Chemistry. 2011;22:1153–1161. doi: 10.1021/bc200008j. [DOI] [PubMed] [Google Scholar]
- 14.Fortune JA, Novobrantseva TI, Klibanov AM. Journal of Drug Delivery. 2011;2011 doi: 10.1155/2011/204058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Y, Haynes MT, Wang Y, Liu F, Huang L. ACS Nano. 2013;7:5376–5384. doi: 10.1021/nn4012384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green JJ, Langer R, Anderson DG. Acc Chem Res. 2008;41:749–759. doi: 10.1021/ar7002336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaffer DV, Fidelman NA, Dan N, Lauffenburger DA. Biotechnology and Bioengineering. 2000;67:598–606. doi: 10.1002/(sici)1097-0290(20000305)67:5<598::aid-bit10>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 18.Choosakoonkriang S, Lobo BA, Koe GS, Koe JG, Middaugh CR. Journal of Pharmaceutical Sciences. 2003;92:1710–1722. doi: 10.1002/jps.10437. [DOI] [PubMed] [Google Scholar]
- 19.Erbacher P, Roche AC, Monsigny M, Midoux P. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1997;1324:27–36. doi: 10.1016/s0005-2736(96)00204-0. [DOI] [PubMed] [Google Scholar]
- 20.Florea B, Meaney C, Junginger H, Borchard G. AAPS PharmSci. 2002;4:1–11. doi: 10.1208/ps040312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang SJ, Bellocq NC, Davis ME. Bioconjugate Chemistry. 2001;12:280–290. doi: 10.1021/bc0001084. [DOI] [PubMed] [Google Scholar]
- 22.Kelley EG, Albert JNL, Sullivan MO, Epps TH. Chemical Society Reviews. 2013;42:7057–7071. doi: 10.1039/c3cs35512h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prata CAH, Zhao Y, Barthelemy P, Li Y, Luo D, McIntosh TJ, Lee SJ, Grinstaff MW. Journal of the American Chemical Society. 2004;126:12196–12197. doi: 10.1021/ja0474906. [DOI] [PubMed] [Google Scholar]
- 24.Abbott NL, Jewell CM, Hays ME, Kondo Y, Lynn DM. Journal of the American Chemical Society. 2005;127:11576–11577. doi: 10.1021/ja054038t. [DOI] [PubMed] [Google Scholar]
- 25.Aytar BS, Muller JPE, Golan S, Hata S, Takahashi H, Kondo Y, Talmon Y, Abbott NL, Lynn DM. Journal of Controlled Release. 2012;157:249–259. doi: 10.1016/j.jconrel.2011.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aytar BS, Muller JPE, Kondo Y, Abbott NL, Lynn DM. ACS Applied Materials & Interfaces. 2013;5:8283–8288. doi: 10.1021/am402594z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Houchin-Ray T, Huang A, West ER, Zelivyanskaya M, Shea LD. Journal of Neuroscience Research. 2009;87:844–856. doi: 10.1002/jnr.21908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Gazit Z, Pelled G, Gazit D, Vunjak-Novakovic G. Integrative Biology. 2011;3:39–47. doi: 10.1039/c0ib00053a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Müller K, Zurbriggen MD, Weber W. Nature Protocols. 2014;9:622–632. doi: 10.1038/nprot.2014.038. [DOI] [PubMed] [Google Scholar]
- 30.Miller CP, Blau CA. Gene Therapy. 2008;15:759–764. doi: 10.1038/gt.2008.43. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Y. Macromolecules. 2012;45:3647–3657. [Google Scholar]
- 32.Zhao H, Sterner ES, Coughlin EB, Theato P. Macromolecules. 2012;45:1723–1736. [Google Scholar]
- 33.Il'ichev YV, Schwörer MA, Wirz J. Journal of the American Chemical Society. 2004;126:4581–4595. doi: 10.1021/ja039071z. [DOI] [PubMed] [Google Scholar]
- 34.Thomas SW. Macromolecular Chemistry and Physics. 2012;213:2443–2449. [Google Scholar]
- 35.Kim MS, Diamond SL. Bioorganic & Medicinal Chemistry Letters. 2006;16:4007–4010. doi: 10.1016/j.bmcl.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 36.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Science. 2009;324:59–63. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang J, Tong X, Morris D, Zhao Y. Macromolecules. 2006;39:4633–4640. [Google Scholar]
- 38.Kumar S, Allard JF, Morris D, Dory YL, Lepage M, Zhao Y. Journal of Materials Chemistry. 2012;22:7252–7257. [Google Scholar]
- 39.Liu G, Dong CM. Biomacromolecules. 2012;13:1573–1583. doi: 10.1021/bm300304t. [DOI] [PubMed] [Google Scholar]
- 40.Johnson JA, Lu YY, Burts AO, Lim YH, Finn MG, Koberstein JT, Turro NJ, Tirrell DA, Grubbs RH. Journal of the American Chemical Society. 2010;133:559–566. doi: 10.1021/ja108441d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim MS, Gruneich J, Jing H, Diamond SL. Journal of Materials Chemistry. 2010;20:3396–3403. [Google Scholar]
- 42.Lee H, Kim Y, Schweickert PG, Konieczny SF, Won YY. Biomaterials. 2014;35:1040–1049. doi: 10.1016/j.biomaterials.2013.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kostiainen MA, Smith DK, Ikkala O. Angewandte Chemie. 2007;119:7744–7748. doi: 10.1002/anie.200701200. [DOI] [PubMed] [Google Scholar]
- 44.Han G, You CC, Kim Bj, Turingan RS, Forbes NS, Martin CT, Rotello VM. Angewandte Chemie International Edition. 2006;45:3165–3169. doi: 10.1002/anie.200600214. [DOI] [PubMed] [Google Scholar]
- 45.Yin L, Tang H, Kim KH, Zheng N, Song Z, Gabrielson NP, Lu H, Cheng J. Angewandte Chemie International Edition. 2013;52:9182–9186. doi: 10.1002/anie.201302820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dimitriou MD, Zhou Z, Yoo HS, Killops KL, Finlay JA, Cone G, Sundaram HS, Lynd NA, Barteau KP, Campos LM, Fischer DA, Callow ME, Callow JA, Ober CK, Hawker CJ, Kramer EJ. Langmuir. 2011;27:13762–13772. doi: 10.1021/la202509m. [DOI] [PubMed] [Google Scholar]
- 47.Reaction time was held constant at 24 h.
- 48.Johnson RN, Chu DSH, Shi J, Schellinger JG, Carlson PM, Pun SH. Journal of Controlled Release. 2011;155:303–311. doi: 10.1016/j.jconrel.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ogris M, Steinlein P, Carotta S, Brunner S, Wagner E. AAPS PharmSci. 2001;3:43–53. doi: 10.1208/ps030321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Millili PG, Yin DH, Fan H, Naik UP, Sullivan MO. Bioconjugate Chemistry. 2010;21:445–455. doi: 10.1021/bc900328j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kloxin AM, Benton JA, Anseth KS. Biomaterials. 2010;31:1–8. doi: 10.1016/j.biomaterials.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Siltanen C, Shin DS, Sutcliffe J, Revzin A. Angewandte Chemie. 2013;125:9394–9398. doi: 10.1002/anie.201303965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fomina N, Sankaranarayanan J, Almutairi A. Advanced Drug Delivery Reviews. 2012;64:1005–1020. doi: 10.1016/j.addr.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shin DS, Hyun Seo J, Sutcliffe JL, Revzin A. Chemical Communications. 2011;47:11942–11944. doi: 10.1039/c1cc15046d. [DOI] [PubMed] [Google Scholar]
- 55.Cao J, Huang S, Chen Y, Li S, Li X, Deng D, Qian Z, Tang L, Gu Y. Biomaterials. 2013;34:6272–6283. doi: 10.1016/j.biomaterials.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 56.Liu G, Liu W, Dong CM. Polymer Chemistry. 2013;4:3431–3443. [Google Scholar]
- 57.Aujard I, Benbrahim C, Gouget M, Ruel O, Baudin JB, Neveu P, Jullien L. Chemistry – A European Journal. 2006;12:6865–6879. doi: 10.1002/chem.200501393. [DOI] [PubMed] [Google Scholar]
- 58.The free pDNA (“Free Plasmid”) in Fig. 5c and Fig. 5d was irradiated with 365 nm light at 200 W/m2 to address concerns regarding any potential deleterious effects of UV light on pDNA.
- 59.Lee HG, Cowman MK. Analytical Biochemistry. 1994;219:278–287. doi: 10.1006/abio.1994.1267. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.