

Abstract
The hypoxia-sensing transcriptional factor HIF1α is implicated in a variety of hepato-pathological conditions; however, the contribution of hepatocyte-derived HIF1α during progression of alcoholic liver injury is still controversial. HIF1α induces a variety of genes including those involved in apoptosis via p53 activation. Increased hepatocyte apoptosis is critical for progression of liver inflammation, stellate cell activation, and fibrosis. Using hepatocyte-specific HIF1α-deficient mice (ΔHepHIF1α−/−), here we investigated the contribution of HIF1α to ethanol-induced hepatocyte apoptosis and its role in amplification of fibrosis after carbon tetrachloride (CCl4) exposure. Moderate ethanol feeding (11% of kcal) induced accumulation of hypoxia-sensitive pimonidazole adducts and HIF1α expression in the liver within 4 days of ethanol feeding. Chronic CCl4 treatment increased M30-positive cells, a marker of hepatocyte apoptosis in pair-fed control mice. Concomitant ethanol feeding (11% of kcal) amplified CCl4-induced hepatocyte apoptosis in livers of wild-type mice, associated with elevated p53K386 acetylation, PUMA expression, and Ly6c+ cell infiltration. Subsequent to increased apoptosis, ethanol-enhanced induction of profibrotic markers, including stellate cell activation, collagen 1 expression, and extracellular matrix deposition following CCl4 exposure. Ethanol-induced exacerbation of hepatocyte apoptosis, p53K386 acetylation, and PUMA expression following CCl4 exposure was attenuated in livers of ΔHepHIF1α−/− mice. This protection was also associated with a reduction in Ly6c+ cell infiltration and decreased fibrosis in livers of ΔHepHIF1α−/− mice. In summary, these results indicate that moderate ethanol exposure leads to hypoxia/HIF1α-mediated signaling in hepatocytes and induction of p53-dependent apoptosis of hepatocytes, resulting in increased hepatic fibrosis during chronic CCl4 exposure.
Keywords: Apoptosis, carbon tetrachloride, collagen, ethanol, fibrosis, hepatic stellate cells, hypoxia, hypoxia-inducible factor 1α, inflammation, liver
Introduction
The initiation and progression of fibrosis in the liver is tightly regulated by multiple pathways involving different hepatic and immune cell types (Bataller and Brenner 2005; Bataller et al. 2011). While macrophages and hepatic stellate cells (HSC) are considered the primary cell types mediating the inflammatory and profibrotic responses to liver injury (Ramachandran and Iredale 2012a,b), recent evidence indicates that hepatocytes also contribute to development of liver fibrosis (Copple et al. 2009). For example, apoptosis of hepatocytes is a critical factor for progression of liver fibrosis (Canbay et al. 2004). Phagocytosis of apoptotic hepatocytes by macrophages and HSCs activates these cells, resulting in the induction of proinflammatory/profibrogenic responses in the liver (Canbay et al. 2004). Further, hepatocyte-specific deletion of transcription factors, including Snail, a hypoxia-sensitive transcription factor (Rowe et al. 2011), and Smad 7, a regulator of transforming growth factor (TGFβ) expression (Dooley et al. 2008), attenuate hepatic fibrosis, suggesting a role for hepatocyte-generated signals during activation of liver fibrosis. Taken together, studies such as these identify the hepatocyte as a critical player during the propagation of liver fibrosis.
Chronic alcohol consumption is a major risk factor for development of hepatic fibrosis (Bataller et al. 2011); however, the molecular mechanisms of alcohol-driven profibrogenic responses in the liver are still not well understood. Understanding the pathophysiological mechanisms for alcohol's contribution to the development of hepatic fibrosis will aid in the development of pharmacological interventions to both prevent and reverse alcoholic liver disease.
Recent studies have identified several pathways by which ethanol can enhance fibrotic responses. For example, ethanol-induced hepatocyte apoptosis is associated with increased fibrogenesis (Roychowdhury et al. 2012). Furthermore, ethanol metabolism by hepatocytes likely contributes to the development of hepatic fibrosis. The impact of the by-products of alcohol metabolism, acetaldehyde and reactive oxygen species (ROS), has been well studied. Acetaldehyde and ROS activate HSC and induce fibrosis via transcriptional upregulation of profibrotic mediators, including TGFβ1, and thus increase extracellular matrix deposition (Mormone et al. 2011). However, the role for hypoxia, another consequence of ethanol metabolism (Wang et al. 2013), in ethanol-driven amplification of liver fibrosis has not been well studied and is the focus of the current investigation.
Cellular responses to hypoxia are primarily mediated via stabilization of the hypoxia-inducible factor 1α (HIF1α), an oxygen-sensing transcription factor (Nath and Szabo 2012). In response to hypoxia, HIF1α translocates from cytosol to the nucleus and forms dimers with HIF1β, a nuclear protein, and subsequently triggers transcription of a variety of genes, including the proinflammatory/fibrotic genes such as VEGF, iNOS, and COX-2 (Hirota et al. 2009). Hepatocyte-specific HIF1α is critical for a variety of liver pathologies including ischemia/reperfusion injury and drug-induced liver injury (Cursio et al. 2008; Wu et al. 2008; Nath and Szabo 2012). Stabilization of HIF1α in hepatocytes has also been implicated in the progression of liver fibrosis following bile duct ligation (BDL)-induced hepatic injury (Moon et al. 2009).
While it is clear that ethanol feeding induces localized hypoxia in the liver (Nishiyama et al. 2012; Chiang et al. 2013), the role of HIF1α in different stages of alcoholic liver disease is still controversial and not well understood. For example, HIF1α in hepatocytes has been reported to play both a protective (Nishiyama et al. 2012) and injurious (Nath and Szabo 2012) role during ethanol-induced steatosis and inflammation in mouse models of ad libitum ethanol feeding.
While these conflicting results on the role of HIF1α in the early stages of ethanol-induced liver injury demand more detailed studies (Mehal 2012), it is also critical to ascertain the role of HIF1α in the association between chronic ethanol consumption and hepatic fibrosis. Making use of a novel model in which moderate ethanol exposure exacerbates CCl4-induced hepatic fibrosis, we recently identified hepatocyte apoptosis as a critical contributor to ethanol's ability to amplify CCl4-induced fibrosis (Roychowdhury et al. 2012). One potential pathway of ethanol-induced apoptosis in the context of CCl4-induced fibrosis is via HIF1α-dependent pathways. HIF1α-induced apoptotic cell death is largely p53-dependent (Greijer and van der Wall 2004) and p53 is activated during progression of CCl4-induced liver fibrosis (Guo et al. 2013). Therefore, here we hypothesized that ethanol-driven hypoxia/HIFα activation induces p53 activation and apoptosis in hepatocytes, which subsequently leads to amplification of CCl4-induced profibrotic responses in the liver. Making use of hepatocyte-specific HIF1α-deficient mice, here we identified that moderate ethanol feeding in the context of CCl4 exposure activated hypoxia/HIF1α-mediated signaling in hepatocytes, leading to increased acetylation of p53 and hepatocyte apoptosis. These HIF1α-dependent responses in hepatocytes were critical for the exacerbation of CCl4-induced profibrotic responses following moderate ethanol exposure.
Materials and Methods
Materials
Female C57BL/6J mice (10–12 weeks old) were purchased from Jackson Labs (Bar Harbor, Maine). Lieber-DeCarli high-fat ethanol and control diets were purchased from Dyets (Bethlehem, PA). Antibodies were from the following sources: antipimonidazole (PMD)-adduct (Hypoxyprobe Inc., Burlington, MA), HIF1α (Novus Biologicals, Littleton, CO), collagen 1 (Southern Biotech, Birmingham, AL), α-smooth muscle actin (αSMA, Sigma-Aldrich, St. Louis, MO), CYP2E1 (Abcam, Eugene, OR), HSC70 (Santa Cruz Biotechnology, Inc, Santa Cruz, CA), acetylated p53K386 (Abcam), PUMA (Millipore, Billerica, MA), and M30/anticytokeratin 18 fragments (Roche, Mannheim, Germany). Ly6c-FITC antibody was obtained from AbD Serotec (Raleigh, NC). Direct red was obtained from Sigma-Aldrich. Alexa Fluor 488 and 568-conjugated secondary antibodies were obtained from Invitrogen (Carlsbad, CA).
Generation of hepatocyte-specific HIF1α-deficient mice
A homozygous colony of hepatocyte-specific HIF1α-deficient (ΔHepHIF1α−/−) mice on a C57BL/6 background (Ding et al. 2004) was established at the Cleveland Clinic by serial crossing of HIF1αfl/fl (B6.129-HIF1atm3Rsjo/J) mice with Alb-Cre (B6.Cg-Tg(Alb-cre)21Mgn/J) mice that express the Cre-recombinase under the albumin promoter. Both the parental strains were obtained from Jackson Labs. Mice were genotyped after each generation and checked for hepatocyte-specific conditional deletion of HIF1α allele. Genomic DNA was extracted by digesting mouse tail tissue in proteinase-K solution (1 mg/mL, Roche) at 50°C overnight. Hair and undigested debris were removed by centrifugation and DNA was collected via ethanol precipitation. Real-time polymerase chain reaction analysis was done using Bullseye EvaGreen SYBR qPCR reagent (MidSci, St. Louis, MO) on a Chromo4 Cycler (MJ Research/Bio-Rad, Hercules, CA) using primer sequences as follows: Hif1α, forward primer CGT GTG AGA AAA CTT CTG GAT G; reverse primer AAA AGT ATT GTG TTG GGG CAG T; cre, forward primer TGA TGG ACA TGT TCA GGG ATC; reverse primer CAG CCA CCA GCT TGC ATG A. PCR conditions were detailed as follows: 95°C for 5 min, 95°C for 30 sec, 58°C for 30 sec, 72°C for 30 sec followed by 35 cycles of 95°C for 30 sec, 72°C for 5 min, and 4°C on hold. PCR products were resolved on a 2% agarose gel with ethidium bromide; Hif1a primers yield three potential fragments, a 565-bp fragment for a WT animal, 565 and 615 bp fragments for a heterozygote, and 615-bp fragment for a KO animal. Cre primers yielded a 865-bp fragment. Primer concentration was used as 0.5 μmol/L per primer and dNTP concentration used was 0.2 mmol/L.
Mouse models of moderate ethanol and carbon tetrachloride
In order to study the interaction between ethanol and CCl4-induced liver fibrosis, mice were subjected to chronic CCl4-treatment combined with moderate ethanol (11% of kcal) feeding. The concentration and duration of ethanol feeding used in this model does not induce expression of cytochrome P450 mono-oxygenase (CYP2E1), a key enzyme for ethanol metabolism, as well as bioactivation of CCl4 (Roychowdhury et al. 2012; Chiang et al. 2013) (Fig. S2). The combination of moderate ethanol with CCl4 results in an exacerbation of hepatic fibrosis, characterized by increased activation of HSC and accumulation of extracellular matrix (Roychowdhury et al. 2012; Chiang et al. 2013).
Female mice were housed in shoe-box cages (two animals/cage) with microisolator lids. Standard microisolator handling procedures were used throughout the study. Mice were randomized into ethanol-fed and pair-fed groups and then adapted to control liquid diet for 2 days. The ethanol-fed group was then allowed free access to an ethanol-containing diet with 1% (vol/vol) ethanol for 2 days, then 2% (vol/vol) ethanol for the remainder of the study. The 2% diet provided 11% of calories as ethanol. After 4 days of ethanol feeding, mice were injected with 0.25 μL/g CCl4 (Sigma, St. Louis, MO, Cat. No. 270652), followed by 0.50 μL/g CCl4 3 days later and then administered 1 μL/g body weight of CCl4 (intraperitoneally twice a week) using 100 μL Hamilton syringes fitted with 26G 5/8 inch needles for 5 weeks. Mice continued on either the control or the ethanol diet (11% of calories) during the CCl4 exposure period.
Seventy-two hours after the last CCl4 injection, mice were anesthetized, blood samples taken into nonheparinized syringes from the posterior vena cava, and livers excised. Portions of each liver were then either fixed in formalin or frozen in optimal cutting temperature (OCT) compound (Sakura Finetek U.S.A., Inc., Torrance CA) for histology, frozen in RNAlater (Qiagen, Valencia, CA) or flash frozen in liquid nitrogen and stored at −80°C until further analysis. Blood was transferred to ethylenediamine-N,N,N',N'-tetraacetic acid (EDTA)-containing tubes for the isolation of plasma. Plasma was then stored at −80°C.
All procedures using animals were approved by the Cleveland Clinic Institutional Animal Care and Use Committee.
In order to confirm that each of the parental strains had a similar response to EtOH/CCl4 exposure, Sirius red staining was assessed as a measure of fibrosis. C57BL/6 mice as well as the parental HIF1αfl/fl and Alb-cre mice exhibited comparable increases in ethanol-induced Sirius red staining following chronic CCl4 exposure (see Fig. 4). Therefore, C57BL/6 mice were used as controls for all other studies.
Figure 4.
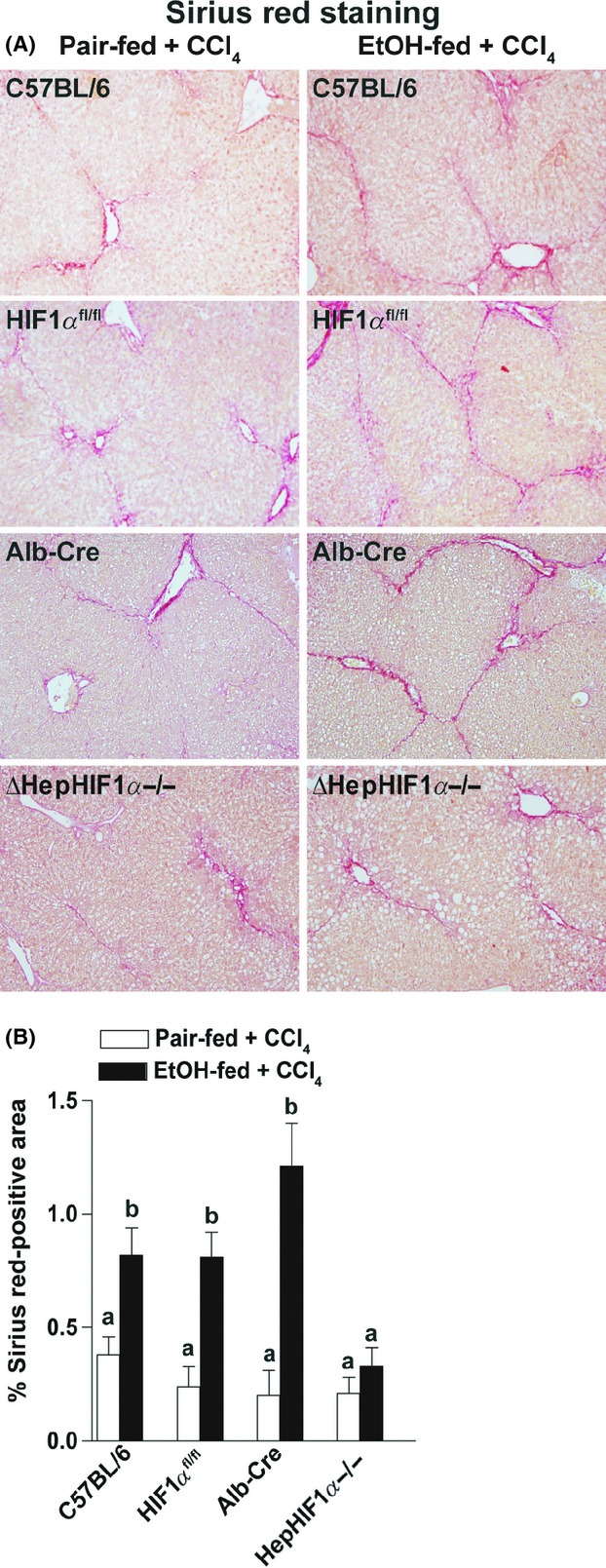
Ethanol-induced exacerbation of CCl4-induced Sirius red staining was reduced in livers of ΔHepHIF1α−/− mice. C57BL/6, HIF1αfl/fl, Alb-cre, and ΔHepHIF1α−/− mice were allowed free access to diets with ethanol- (11% of kcal) or pair-fed controls for 4 days followed by CCl4 injections, as described in Materials and Methods section. (A) Paraffin-embedded liver sections were deparaffinized and stained for Sirius red staining. Images are acquired using 20× objective. (B) Areas positive for Sirius red were quantified using Image Pro-Plus software and analyzed. Values with different alphabetical superscripts were significantly different from each other, P < 0.05 (n = 4–6).
Detection of hypoxia
After 4 days of ethanol feeding mice were injected with the hypoxia-sensing drug, PMD, 1 h before euthanasia (Chiang et al. 2013). Samples were collected as detailed above.
Western blot analysis
Frozen liver tissue (0.5–1.0 g) was homogenized in lysis buffer (10 mL/g tissue) containing 50 mmol/L Tris-HCl, pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mmol/L NaCl, 1 mmol/L EDTA with added protease inhibitors Complete™ (Roche Diagnostics, Mannheim, Germany), 17.5 μg/mL aprotinin, 5 μg/mL bestatin, 10 μg/mL leupeptin, 1 mg/mL bacitracin, and 20 μg/mL E64), and phosphatase inhibitors (1 mmol/L vanadate and 10 mmol/L Na pyrophosphate) using 15 strokes in a glass on glass homogenizer (loose pestle). After 15 min on ice, samples were centrifuged at 16,000g for 15 min to remove insoluble material followed by protein concentration measurement using the bicinchoninic acid (BCA) assay (Pritchard et al. 2007). Liver lysates were then normalized and prepared in Laemmli buffer and boiled for 5 min. Samples were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to membranes for western blotting. Membranes were incubated with rabbit anti-CYP2E1-antibody (Research Diagnostics, Inc., Flanders, NJ, 1:200 dilution) overnight at 4°C, then washed and incubated for 1 h in anti-rabbit secondary antibody (1:25000 dilution) coupled to horseradish peroxidase. Immunoreactive proteins were detected using enhanced chemiluminescence, images were collected, and signal intensities were quantified using Eastman Kodak Co. Image Station 4000R (Eastman Kodak Company, Rochester, NY).
CYP2E1 activity assay
Activity of CYP2E1 was determined by measuring p-nitrophenol hydroxylation in whole liver extract as described earlier (Wu and Cederbaum 2008).
Isolation of RNA and quantitative real-time polymerase chain reaction
Total RNA was isolated and reverse transcribed followed by amplification using qRT-PCR. The relative amount of target mRNA was determined using the comparative threshold (Ct) method by normalizing target mRNA Ct values to those of 18S (Mandal et al. 2010). The reaction mix (25 μL) contained: cDNA, Applied Biosystems SYBR green kit (Life Technologies, Carlsbad, CA) and primers at final concentrations of 200 μmol/L. RT-PCR was performed in the Mx3000P cycler (Agilent, Santa Clara, CA): 95°C for 10 min, 40 cycles of 15 sec at 95°C, 30 sec at 60°C, 30 sec at 72°C followed by 1 min at 95°C, 30 sec at 55°C, and 30 sec at 95°C. The relative amount of target mRNA was estimated using the Ct method by normalizing target mRNA Ct values to those of 18S (Mandal et al. 2010). ΔCt values were used for the statistical analysis of RT-PCR data.
Immunohistochemistry
Formalin-fixed paraffin-embedded liver sections were deparaffinized and stained for PMD adduct, Sirius red, α-SMA and cytokeratin-18 fragments/M30 staining. Frozen liver sections were used for staining HIF1α, collagen 1, and Ly6c (Roychowdhury et al. 2012). All images presented in the results are representative of at least three images per liver and four mice per experimental condition.
Statistical analysis
All values presented represent means ± standard error of mean, with n = 3–6 experimental points. Data were analyzed by analysis of variance using the general linear models procedure (SAS, Carey, NC). Data were log transformed if needed to obtain a normal distribution. Follow-up comparisons were made by least square means testing. Student's t-test was used for comparing values obtained from two groups (used only for Fig. 1).
Figure 1.
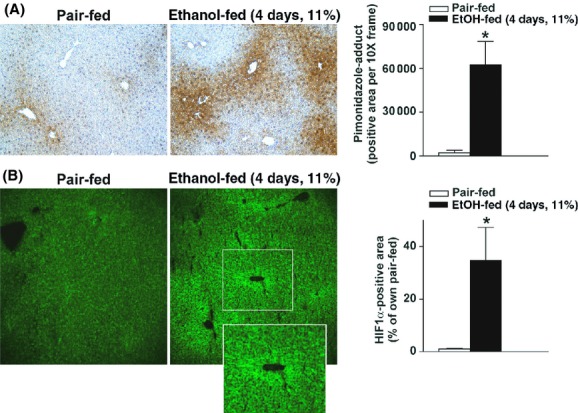
Moderate ethanol feeding induced hypoxia and expression of HIF1α in mouse livers. (A, B) C57BL/6J wild-type mice were allowed free access to ethanol-containing diets with ethanol- (11% of kcal) or pair-fed controls. (A) Animals were injected with PMD 1 h before euthanasia. Paraffin-embedded liver sections were deparaffinized and stained for PMD-adducts. Images were acquired using 10× objective. (B) Frozen liver sections from pair- or ethanol-fed (11% of kcal) mice were stained for HIF1α. Images were acquired using 20× objective. *P < 0.05 pair-fed compared to ethanol-fed mice.
Results
Moderate ethanol feeding induced hypoxia in mouse liver
Chronic ethanol feeding induces hypoxia in mouse liver (Nishiyama et al. 2012; Chiang et al. 2013). Using immunohistochemical staining, accumulation of hypoxia-sensing PMD adducts was detected in mouse liver as early as 4 days after moderate ethanol feeding (4d, 11%, Fig. 1A). PMD adducts were primarily visualized in hepatocytes around the central veins (Fig. 1A). Moderate ethanol feeding for 4 days also increased HIF1α protein expression in mouse liver (4d, 11%, Fig. 1B), primarily localized around the central veins. HIF1α protein accumulation was not detected in livers of ΔHepHIF1α−/− mice (Fig. S1A).
Moderate ethanol feeding combined with CCl4 exposure also increased HIF1α protein accumulation in liver of wild-type, but not ΔHepHIF1α−/− mice (Fig. S1B). HIF1α protein accumulated in both the nucleus and cytosol and did not exhibit any zonal distribution.
Hepatocyte HIF1α deficiency reduced the numbers of apoptotic hepatocytes in mouse liver
Hepatocyte apoptosis is an important contributor to the progression of hepatic fibrosis (Canbay et al. 2004; Roychowdhury et al. 2012). Hypoxia induces apoptosis in a variety of cell types, including hepatocytes. Caspase-mediated cytokeratin-18 fragmentation, detected by M30 staining, is a specific indicator of hepatocyte apoptosis. While higher doses of ethanol induces hepatocytes apoptosis, moderate ethanol feeding alone does not result in hepatocyte apoptosis (Cohen et al. 2010). In contrast, CCl4 exposure resulted in a modest degree of hepatocyte apoptosis, as detected by M30-positive hepatocytes. This response was exacerbated by concomitant ethanol feeding (Fig. 2A and B). While hepatocyte-specific deletion of HIF1α had no effect on M30-positive hepatocytes in pair-fed mice, the absence of HIF1α in hepatocytes ameliorated the exacerbation of M30-positive hepatocytes in ethanol-fed mice (Fig. 2A and B).
Figure 2.
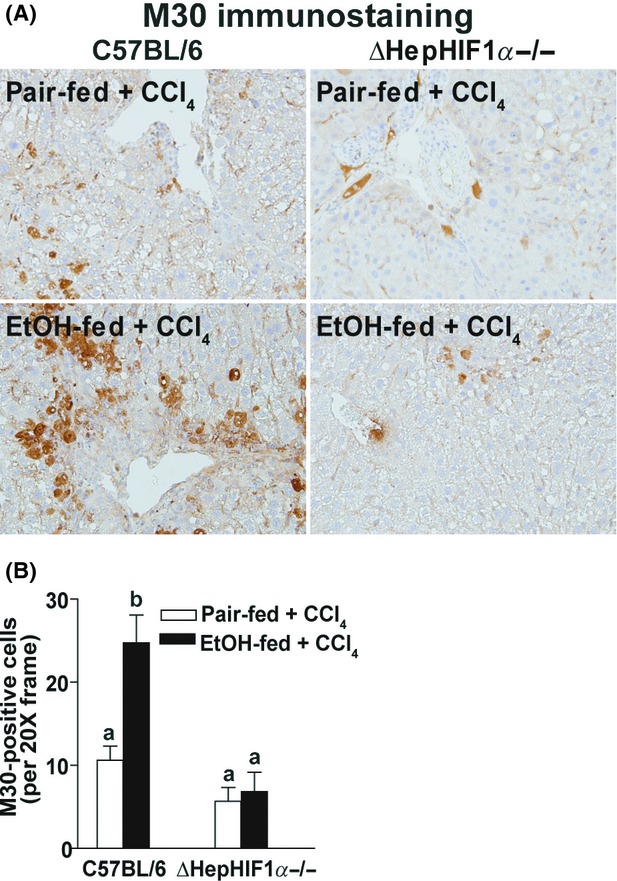
Exacerbation of CCl4-induced hepatocyte apoptosis by ethanol was attenuated in livers of ΔHepHIF1α−/− mice. C57BL/6 wild-type (WT) and ΔHepHIF1α−/− mice were allowed free access to diets with ethanol- (11% of kcal) or pair-fed controls for 4 days followed by CCl4 injections, as described earlier. (A) Paraffin-embedded liver sections were deparaffinized and stained for M30/CK18 fragments, a marker for hepatocyte apoptosis. Images were acquired using a 20× objective. (B) Total numbers of M30+ cells were counted. Values with different alphabetical superscripts were significantly different from each other, P < 0.05 (n = 4–6).
CYP2E1 expression, assessed by Western blot and enzymatic activity (Fig. S2), the concentration of alanine aminotransferase (ALT) in the plasma, and triglycerides in the liver were not affected by diet or genotype (Table S1).
Deficiency of HIF1α in hepatocytes reduced acetyl-p53K386 and PUMA expression in hepatocytes
In response to hypoxia, HIF1α induces apoptosis in a p53-dependent manner (Sermeus and Michiels 2011). p53 activity is regulated via both transcriptional and posttranslational mechanisms. In particular, acetylation of p53 is essential for its activation and DNA-binding activity (Tang et al. 2008). Under the conditions of our study, neither CCl4 exposure nor ethanol feeding affected expression of p53 mRNA in the liver (data not shown). In contrast, the immunoreactive quantity of acetylated p53 (Ac-p53K386) was increased in response to CCl4 exposure (Fig. 3A). Further, concomitant ethanol feeding increased the numbers of Ac-p53K386-stained cells (Fig. 3A, inset). Costaining with Ac-p53K386 and collagen 1 revealed that while a major population of Ac-p53K386-positive cells were localized in close proximity of the fibrotic septa, some of the Ac-p53K386-positive cells were also visualized farther from the collagen fibers (Fig. 3B). Deletion of HIF1α in hepatocytes reduced the numbers of Ac-p53K386-positive cells following ethanol/CCl4 exposure (Fig. 3A).
Figure 3.

Exacerbation of CCl4-induced p53 acetylation and PUMA expression by ethanol were attenuated in livers of ΔHepHIF1α−/− mice. C57BL/6 wild-type (WT) and ΔHepHIF1α−/− mice were allowed free access to diets with ethanol- (11% of kcal) or pair-fed controls for 4 days followed by CCl4 injections, as described earlier. (A) Ac-p53/K386, (B) Ac-p53/K386 (green) and collagen 1 (red) costaining, or (C) PUMA. Images were acquired using a 40× objective. PUMA (green) was costained with the nuclear stain DAPI (blue). (D) Total number of PUMA+ cells was counted. Values with different alphabetical superscripts were significantly different from each other, P < 0.05 (n = 4–6).
PUMA, a p53-transcription-dependent effector molecule, is critical for p53-driven proapoptotic activities (Tang et al. 2008). PUMA expression was detected in both hepatocytes and nonparenchymal cells (NPCs) in livers of CCl4-exposed mice, with higher numbers observed after ethanol feeding (Fig. 3B). Hepatocyte-specific HIF1α-deficiency reduced the numbers of PUMA-positive cells following ethanol/CCl4 exposure (Fig. 3C). However, the number of PUMA-positive cells following CCl4 exposure in pair-fed mice was not different between livers of ΔHepHIF1α−/− compared to wild-type mice (Fig. 3D).
Hepatocyte-specific deletion of HIF1α attenuated CCl4-induced fibrosis in mouse liver
Apoptosis is critical for progression of fibrosis in mouse liver. Since apoptosis of hepatocytes following moderate ethanol/CCl4 exposure was HIF1α-dependent, it would be expected to contribute in the exacerbation of fibrosis by ethanol. Moderate ethanol feeding (11% of kcal) during CCl4 exposure increased liver fibrosis in C57BL/6 mice, resulting in enhanced bridging of Sirius red staining, compared to CCl4-treated pair-fed mice (Fig. 4A and B). Like C57BL/6 mice, the parental HIF1αfl/fl and Alb-cre mice also exhibited ethanol-driven increases in Sirius red staining following chronic CCl4 exposure (Fig. 4A and B).
If hepatocyte-HIF1α contributes to ethanol-induced exacerbation of CCl4-mediated fibrosis, deficiency of HIF1α in hepatocytes should ameliorate this increase in fibrosis. Hepatocyte-specific deletion of HIF1α attenuated ethanol-induced exacerbation of CCl4-induced extracellular matrix (ECM) deposition, as detected by Sirius red staining (Fig. 4A and B). However, in pair-fed control mice, CCl4-induced hepatic ECM deposition was not influenced by the expression of HIF1α in hepatocytes (Fig. 4A and B).
Hepatic expression of collagen 1A1 mRNA (Fig. 5C) and collagen 1 protein (Fig. 5A and B) was also increased in the livers of C57BL/6 mice following combined exposure to ethanol and CCl4. Hepatocyte-specific HIF1α-deletion attenuated the increase in CCl4-induced collagen 1A1 mRNA and collagen 1 protein expression in ethanol-fed mice but not in pair-fed control mice (Fig. 5A–C).
Figure 5.

Ethanol-induced exacerbation of CCl4-induced collagen 1 and smooth muscle actin expression were reduced in livers of ΔHepHIF1α−/− mice. C57BL/6 wild-type and ΔHepHIF1α−/− mice were allowed free access to diets with ethanol- (11% of kcal) or pair-fed controls for 4 days followed by CCl4 injections, as described in Materials and Methods section. (A) Frozen liver sections were used for collagen 1 staining. Images are acquired using 20× objective. (B) Areas positive for collagen 1 were quantified using Image Pro-Plus software and analyzed. (C) mRNA expression of Col1A1 was detected in mouse livers using qRT-PCR measurement. (D) Paraffin-embedded liver sections were deparaffinized and stained for αSMA staining. Images are acquired using 10× objective. (E) Areas positive for αSMA were quantified using Image Pro-Plus software and analyzed. (F) mRNA expression of αSMA was detected in mouse livers using qRT-PCR measurement. Values with different alphabetical superscripts were significantly different from each other, P < 0.05 (n = 4–6).
Activation of HSC is a hallmark of liver fibrosis. In response to profibrogenic stimuli, quiescent HSCs are transformed to their activated phenotype and produce collagen fibers in the liver. If hepatocyte-derived HIF1α is involved in the activation of HSC during ethanol/CCl4 exposure, then the ΔHepHIF1α−/− mice should be protected from this ethanol-induced amplification of HSC activation. Both mRNA (Fig. 5F) and protein expression of αSMA (Fig. 5D and E), a marker of HSC activation, increased in the liver of C57BL/6 mice following combined exposure to ethanol and CCl4 compared to mice pair-fed control diets during CCl4 treatment. Deletion of HIF1α specifically in hepatocytes attenuated this ethanol-induced increase in αSMA expression (Fig. 5D–F).
HIF1α deficiency in hepatocytes reduced infiltration of monocytes/macrophages in mouse liver
During early phases of liver fibrosis circulating innate immune cells, including Ly6c+ monocyte/macrophages, migrate to the site of injury to engulf the damaged cells. If attenuation of fibrosis following ethanol/CCl4-combined exposure in the liver of ΔHepHIF1α−/− mice was associated with reduced monocyte/macrophages infiltration, recruitment of Ly6c+ cells to the liver should also be reduced in the livers of ΔHepHIF1α−/− mice. Ly6c+ cells were increased following ethanol/CCl4-combined exposure compared to CCl4 in pair-fed control mice (Fig. 6A and B). The majority of Ly6c+ cells were localized in close vicinity of collagen fibers (Fig. 6C). This increase in Ly6c+ cells was reduced in CCl4-treated livers of ethanol-fed ΔHepHIF1α−/− mice compared to the C57BL/6 mice (Fig. 6A and B). The number of Ly6c+ cells was not different between ΔHepHIF1α−/− and wild-type pair-fed control mice exposed to CCl4.
Figure 6.
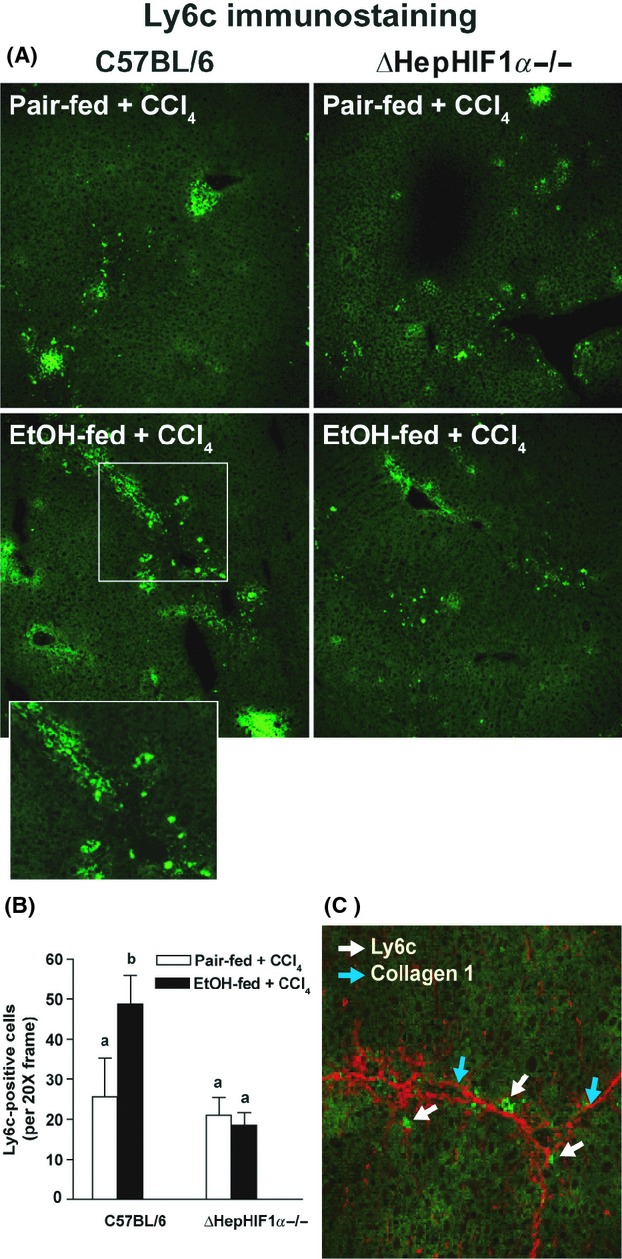
Exacerbation of CCl4-induced Ly6c+ cell infiltration by ethanol were attenuated in livers of ΔHepHIF1α−/− mice. C57BL/6 wild-type (WT) and ΔHepHIF1α−/− mice were allowed free access to diets with ethanol- (11% of kcal) or pair-fed controls for 4 days followed by CCl4 injections, as described earlier. (A) Frozen liver sections were used for Ly6c staining. Images were acquired using a 20× objective. (B) Total numbers of Ly6c+ cells were counted. Values with different alphabetical superscripts were significantly different from each other, P < 0.05 (n = 4–6). (C) Ly6C+ (green) and collagen 1 (red) were costained in frozen liver sections from C57BL/6 mice exposed to both ethanol and CCl4. White arrows indicate collagen 1-stained fibers and blue arrows indicate Ly6C+-stained cells.
Discussion
Ethanol consumption induces hypoxia in the liver, particularly around the central veins (Nishiyama et al. 2012; Chiang et al. 2013). Hypoxia induces expression of multiple genes, regulated by the hypoxia-sensing transcription factors, HIF1α and HIF2α, in a cell-type-dependent manner (Nath and Szabo 2012). However, the contribution of hypoxia to development of ethanol-induced fibrosis has not yet been studied. Since chronic ethanol feeding, even at high ethanol concentrations, does not result in significant liver fibrosis in mice, we have recently adopted an alternative approach by exposing the mice to chronic CCl4 in presence of a moderate ethanol diet (Roychowdhury et al. 2012; Chiang et al. 2013). Using this model of ethanol-induced exacerbation of apoptosis and fibrosis, we have identified the importance of ethanol-induced hypoxia/HIF1α signaling in mediating the response, associated with increased p53 acetylation, PUMA expression, and hepatocyte apoptosis in livers from CCl4-exposed mice exposed to moderate ethanol. Importantly, hepatocyte-derived HIF1α also contributed to ethanol-induced amplification of profibrotic responses following chronic exposure to CCl4.
HIF1α, an important mediator of cellular and systemic responses to hypoxia, regulates the expression of a variety of genes associated with different hepato-pathologies. Moderate ethanol exposure increased HIF1α protein accumulation was detected in both nucleus and cytosol. Accumulation of HIF1α in the cytosol could be a consequence of either a temporary saturation of the nuclear transport machinery for HIF1α or a continued stabilization of HIF1α prior to nuclear transport. Here we find that hepatocyte HIF1α is an essential mediator of hepatocyte apoptosis in response to combined exposure to moderate ethanol and CCl4, but not in response to CCl4 alone. Although, moderate ethanol exposure alone is insufficient to trigger apoptosis in hepatocytes, our data indicate that ethanol-induced HIF1α accumulation acted to prime hepatocytes to a second injurious signal, for example, CCl4, contributing to an exacerbated proapoptotic response.
This specific contribution of hepatocyte HIF1α during moderate ethanol exposure is most likely a consequence of ethanol metabolism and the generation of a localized hypoxic environment. Metabolism of ethanol dysregulates energy metabolism, histone deacetylation, and apoptosis by impairing mitochondrial bioenergetics and NADH/NAD+ ratio in hepatocytes (Zakhari 2006). HIF1α can also impair cellular redox balance by increasing glucose metabolism and reducing mitochondrial function (Vengellur et al. 2003). Therefore, it is likely that ethanol-induced hypoxia/HIF1α expression sensitizes hepatocytes to a second hit, such as CCl4, leading to the increased proapoptotic environment in hepatocytes.
HIF1α-driven apoptosis is largely p53-dependent (Sermeus and Michiels 2011). While p53 expression is increased in some models of chronic ethanol feeding and CCl4 exposure (Guo et al. 2013), posttranslational modifications including acetylation, phosphorylation, methylation, sumoylation, and ubiquitination mediate activation of p53 transcriptional activity. Phosphorylation prevents the binding of p53 with Mdm2, a negative regulator of p53 activation, while acetylation of p53 is central to its DNA-binding activity and upregulation of the downstream mediators, such as PUMA, NOXA, and Bax, responsible for its proapoptotic activity (Tang et al. 2008). Recently, increased p53 acetylation has been implicated to hepatocyte apoptosis in a model of nonalcoholic fatty liver disease (Castro et al. 2013). Importantly, ethanol, both in vivo and in cultured cells, increases the acetylation of multiple proteins (Shepard and Tuma 2009). The most well-studied example is the ethanol-induced increase in histone acetylation that is mediated via decreased activity of sirtuin 1 (SIRT1), an NAD+-dependent protein deacetylase (Zakhari 2006). Increased NADH/NAD+ ratios generated during ethanol metabolism serve to decrease the activity of SIRT1 in hepatocytes (Zakhari 2006; Elliott and Jirousek 2008). Here, we used immunoreactive Ac-p53K386 as an indicator of p53 acetylation and detected increased Ac-p53K386 in both hepatocytes and NPCs near the fibrotic septa after CCl4 exposure. Concomitant ethanol feeding and CCl4 increased the numbers of Ac-p53K386-positive cells compared to pair-fed controls. Importantly, acetylation of p53 in response to ethanol and CCl4 was HIF1α-dependent. While we have not yet characterized the mechanism for increased acetylation of p53 under these conditions, it is known that acetylation of p53 is regulated via SIRT1-dependent deacetylation (Shepard and Tuma 2009), consistent with a likely interaction between ethanol and SIRT1-dependent deacetylation in the context of ethanol and CCl4-induced injury.
As further evidence for an increase in p53-dependent transcriptional activity in response to ethanol/CCl4, expression of PUMA, a downstream target of p53 activation (Qiu et al. 2011) was also increased in a HIF1α-dependent manner in hepatocytes and NPCs. The reduction in PUMA-positive hepatocytes in livers of ΔHepHIF1α−/− mice is consistent with protection of hepatocytes from apoptosis in these mice.
Phagocytosis of dying hepatocytes induces a variety of proinflammatory cytokines/chemokines resulting in infiltration of monocytes and macrophages in the liver. Under the combined insult of moderate ethanol and CCl4, there was an increased recruitment of Ly6c+ cell populations compared to mice treated with only CCl4. Deficiency of HIF1α in hepatocytes reduced the numbers of hepatic Ly6c+ cells following combined exposure to ethanol and CCl4. These data suggest that hepatocyte-derived HIF1α contributes to increased hepatic infiltration of the proinflammatory/fibrotic populations of monocytes and macrophages following ethanol feeding, which in turn can modulate profibrotic responses following exposure to CCl4. Reduced numbers of Ly6c+ could be the result of altered chemokine release from HIF1α-deficient hepatocytes and/or a indirect result of decreased HSC activation and fibrosis. The exact mechanistic interaction between HIF1α in hepatocytes and the recruitment of Ly6c+ cells from the circulation will require further investigation.
HIF1α regulates the expression of a variety of genes associated with different hepato-pathologies, including SREBP (Nishiyama et al. 2012), a critical mediator of ethanol-induced hepatic steatosis, as well as VEGF and iNOS, key genes for progression of liver fibrosis and angiogenesis (Keith et al. 2012). In response to hypoxia, hepatocytes also release a variety of profibrotic molecules including PDGF-A, PDGF-B, and plasminogen activator inhibitor (PAI-1) in a HIF1α-dependent manner (Copple et al. 2009). Because ethanol metabolism results in a localized hypoxic environment within the liver, we hypothesized that HIF1α in hepatocytes is critical for the amplification of fibrosis by even moderate concentrations of ethanol. Indeed, we find that ΔHepHIF1α-deficient mice are protected from the exacerbation of CCl4-induced fibrosis by moderate ethanol.
Interestingly, deficiency of hepatocyte-HIF1α did not reduce the profibrotic parameters in pair-fed mice exposed to CCl4 alone, further indicating that hypoxia due to ethanol metabolism may be a unique contributor to hepatic fibrosis in the context of CCl4. Induction of fibrosis in response to CCl4, a potent hepatotoxin, is initiated by hepato-cellular necrosis followed by a surge of hepatic inflammation. Moderate ethanol feeding has no direct impact on necrotic injury of hepatocytes by CCl4, as indicated by ALT concentrations in the plasma (14, Table S1). In turn the data suggest that increased apoptosis of hepatocytes during ethanol exposure drives increased activation of HSC and subsequent ECM production.
It is interesting to note that, in contrast to the hepatocyte HIF1α-independent progression of fibrosis in control mice exposed to CCl4, liver fibrosis following BDL is reduced in livers of ΔHepHIF1α−/− mice (Moon et al. 2009). The differential contribution of HIF1α in BDL- versus CCl4-driven fibrosis could be a consequence of pathophysiological differences between these two models. In contrast to the hepatocyte apoptosis and necrosis induced by CCl4, development of fibrosis following BDL proceeds with less severe hepatocyte injury, instead triggered by increased portal inflammation and proliferation of biliary epithelia and oval cells (Starkel and Leclercq 2011). Similarly, a differential role for PAI-1 in CCl4 compared to BDL-induced fibrosis has been observed; PAI-1 is profibrotic in CCl4-induced fibrosis, but antifibrotic during BDL-induced fibrosis (von Montfort et al. 2010).
In summary, we found that expression of HIF1α in hepatocytes following moderate ethanol exposure is critical for amplification of CCl4-induced proapoptotic, as well as profibrogenic, responses in mouse liver. Moreover, ethanol-induced fibrosis was associated with increased hepatic Ly6c+ cells, suggesting that HIF1α expression in hepatocytes also contributes to infiltration of monocyte/macrophage in the liver. Taken together, we have identified a critical link between activation of hepatocyte apoptosis, dependent on HIF1α/p53 axis, and development of hepatic fibrosis using a mouse model of ethanol-induced liver fibrosis. The association between ethanol and increased acetylation of p53 is of potential clinical relevance, as a variety of drugs to enhance deacetylase activity are currently under investigation (Elliott and Jirousek 2008; Shepard and Tuma 2010).
Acknowledgments
We are grateful to Colleen Croniger and Dave DeSantis for their help with the genotyping of ΔHepHIF1α−/− mice. The authors would also like to thank Manoa Hui for her assistance in preparing figure graphics and Jazmine Danner for her help in caring and breeding all animals used in this study.
Glossary
- BDL
bile duct ligation
- HIF 1α
hypoxia-inducible factor 1α
- HSC
hepatic stellate cells
- NPC
nonparenchymal cells
- OCT
optimal cutting temperature
- PMD
pimonidazole
- ROS
reactive oxygen species
- TGFβ
transforming growth factor
- WT
wild type
- αSMA
α-smooth muscle actin
Disclosure
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Ethanol-induced HIF1α expression in wild-type and ΔHepHIF1α−/− mouse livers. C57BL/6J wild-type mice were allowed free access to diets with ethanol- (11% of kcal) or pair-fed controls for 4 days (A) followed by CCl4 injections twice a week for 5 weeks and maintained on ethanol- or control diets (B). Frozen liver sections were stained for HIF1α. Images were acquired using 40× objective (n = 3–6).
Figure S2. CYP2E1 expression in wild-type and ΔHepHIF1α−/− mouse livers. C57BL/6 wild-type (WT) and ΔHepHIF1α−/− mice were allowed free access to diets with ethanol (11% of kcal) or pair-fed controls for 4 days. Mice were then injected with CCl4 twice a week for 5 weeks and maintained on ethanol- or control diets. (A) CYP2E1 protein expression was measured in liver lysates using Western blot. Images are representative of at least 3–6 mice per group and values under the image are means and SEM. P, Pair-fed; E, EtOH-fed. (B) CYP2E1 activity was measured in liver homogenates. n = 3–6.
Table S1. Body weight, food intake, plasma levels of liver enzyme and hepatic triglyceride after moderate ethanol feeding (32d, 11%) in presence of chronic CCl4 exposure.
Table S2. Gene-specific primer sequences for RT-PCR.
References
- Bataller R, Brenner DA. Liver fibrosis. J Clin Investig. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataller R, Rombouts K, Altamirano J, Marra F. Fibrosis in alcoholic and nonalcoholic steatohepatitis. Best Pract Res Clin Gastroenterol. 2011;25:231–244. doi: 10.1016/j.bpg.2011.02.010. [DOI] [PubMed] [Google Scholar]
- Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39:273–278. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- Castro RE, Ferreira DM, Afonso MB, Borralho PM, Machado MV, Cortez-Pinto H, et al. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J Hepatol. 2013;58:119–125. doi: 10.1016/j.jhep.2012.08.008. [DOI] [PubMed] [Google Scholar]
- Chiang DJ, Roychowdhury S, Bush K, McMullen MR, Pisano S, Niese K, et al. Adenosine 2A receptor antagonist prevented and reversed liver fibrosis in a mouse model of ethanol-exacerbated liver fibrosis. PLoS ONE. 2013;8:e69114. doi: 10.1371/journal.pone.0069114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JI, Roychowdhury S, McMullen MR, Stavitsky AB, Nagy LE. Complement and alcoholic liver disease: role of C1q in the pathogenesis of ethanol-induced liver injury in mice. Gastroenterology. 2010;139:664–674. doi: 10.1053/j.gastro.2010.04.041. 674.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copple BL, Bustamante JJ, Welch TP, Kim ND, Moon JO. Hypoxia-inducible factor-dependent production of profibrotic mediators by hypoxic hepatocytes. Liver Int. 2009;29:1010–1021. doi: 10.1111/j.1478-3231.2009.02015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cursio R, Miele C, Filippa N, Van Obberghen E, Gugenheim J. Liver HIF-1 alpha induction precedes apoptosis following normothermic ischemia-reperfusion in rats. Transpl Proc. 2008;40:2042–2045. doi: 10.1016/j.transproceed.2008.05.037. [DOI] [PubMed] [Google Scholar]
- Ding WX, Ni HM, DiFrancesca D, Stolz DB, Yin XM. Bid-dependent generation of oxygen radicals promotes death receptor activation-induced apoptosis in murine hepatocytes. Hepatology. 2004;40:403–413. doi: 10.1002/hep.20310. [DOI] [PubMed] [Google Scholar]
- Dooley S, Hamzavi J, Ciuclan L, Godoy P, Ilkavets I, Ehnert S, et al. Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology. 2008;135:642–659. doi: 10.1053/j.gastro.2008.04.038. [DOI] [PubMed] [Google Scholar]
- Elliott PJ, Jirousek M. Sirtuins: novel targets for metabolic disease. Curr Opin Investig Drugs. 2008;9:371–378. [PubMed] [Google Scholar]
- Greijer AE, van der Wall E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J Clin Pathol. 2004;57:1009–1014. doi: 10.1136/jcp.2003.015032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo XL, Liang B, Wang XW, Fan FG, Jin J, Lan R, et al. Glycyrrhizic acid attenuates CCl(4)-induced hepatocyte apoptosis in rats via a p53-mediated pathway. World J Gastroenterol. 2013;19:3781–3791. doi: 10.3748/wjg.v19.i24.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota SA, Beck PL, MacDonald JA. Targeting hypoxia-inducible factor-1 (HIF-1) signaling in therapeutics: implications for the treatment of inflammatory bowel disease. Recent Pat Inflamm Allergy Drug Discov. 2009;3:1–16. doi: 10.2174/187221309787158434. [DOI] [PubMed] [Google Scholar]
- Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P, Roychowdhury S, Park PH, Pratt BT, Roger T, Nagy LE. Adiponectin and heme oxygenase-1 suppress TLR4/MyD88-independent signaling in rat Kupffer cells and in mice after chronic ethanol exposure. J Immunol. 2010;185:4928–4937. doi: 10.4049/jimmunol.1002060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehal WZ. HIF-1alpha is a major and complex player in alcohol induced liver diseases. J Hepatol. 2012;56:311–312. doi: 10.1016/j.jhep.2011.09.009. [DOI] [PubMed] [Google Scholar]
- von Montfort C, Beier JI, Kaiser JP, Guo L, Joshi-Barve S, Pritchard MT, et al. PAI-1 plays a protective role in CCl4-induced hepatic fibrosis in mice: role of hepatocyte division. Am J Physiol. 2010;298:G657–G666. doi: 10.1152/ajpgi.00107.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JO, Welch TP, Gonzalez FJ, Copple BL. Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am J Physiol. 2009;296:G582–G592. doi: 10.1152/ajpgi.90368.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormone E, George J, Nieto N. Molecular pathogenesis of hepatic fibrosis and current therapeutic approaches. Chem Biol Interact. 2011;193:225–231. doi: 10.1016/j.cbi.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath B, Szabo G. Hypoxia and hypoxia inducible factors: diverse roles in liver diseases. Hepatology. 2012;55:622–633. doi: 10.1002/hep.25497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama Y, Goda N, Kanai M, Niwa D, Osanai K, Yamamoto Y, et al. HIF-1alpha induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J Hepatol. 2012;56:441–447. doi: 10.1016/j.jhep.2011.07.024. [DOI] [PubMed] [Google Scholar]
- Pritchard MT, Roychowdhury S, McMullen MR, Guo L, Arteel GE, Nagy LE. Early growth response-1 contributes to galactosamine/lipopolysaccharide-induced acute liver injury in mice. Am J Physiol. 2007;293:G1124–G1133. doi: 10.1152/ajpgi.00325.2007. [DOI] [PubMed] [Google Scholar]
- Qiu W, Wang X, Leibowitz B, Yang W, Zhang L, Yu J. PUMA-mediated apoptosis drives chemical hepatocarcinogenesis in mice. Hepatology. 2011;54:1249–1258. doi: 10.1002/hep.24516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran P, Iredale JP. Liver fibrosis: a bidirectional model of fibrogenesis and resolution. QJM. 2012a;105:813–817. doi: 10.1093/qjmed/hcs069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran P, Iredale JP. Macrophages: central regulators of hepatic fibrogenesis and fibrosis resolution. J Hepatol. 2012b;56:1417–1419. doi: 10.1016/j.jhep.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Rowe RG, Lin Y, Shimizu-Hirota R, Hanada S, Neilson EG, Greenson JK, et al. Hepatocyte-derived Snail1 propagates liver fibrosis progression. Mol Cell Biol. 2011;31:2392–2403. doi: 10.1128/MCB.01218-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychowdhury S, Chiang DJ, Mandal P, McMullen MR, Liu X, Cohen JI, et al. Inhibition of apoptosis protects mice from ethanol-mediated acceleration of early markers of CCl(4) -induced fibrosis but not steatosis or inflammation. Alcohol Clin Exp Res. 2012;36:1139–1147. doi: 10.1111/j.1530-0277.2011.01720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sermeus A, Michiels C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011;2:e164. doi: 10.1038/cddis.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard BD, Tuma PL. Alcohol-induced protein hyperacetylation: mechanisms and consequences. World J Gastroenterol. 2009;15:1219–1230. doi: 10.3748/wjg.15.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard BD, Tuma PL. Alcohol-induced alterations of the hepatocyte cytoskeleton. World J Gastroenterol. 2010;16:1358–1365. doi: 10.3748/wjg.v16.i11.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkel P, Leclercq IA. Animal models for the study of hepatic fibrosis. Best Pract Res Clin Gastroenterol. 2011;25:319–333. doi: 10.1016/j.bpg.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vengellur A, Woods BG, Ryan HE, Johnson RS, LaPres JJ. Gene expression profiling of the hypoxia signaling pathway in hypoxia-inducible factor 1alpha null mouse embryonic fibroblasts. Gene Expr. 2003;11:181–197. doi: 10.3727/000000003108749062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wu D, Yang L, Gan L, Cederbaum AI. Cytochrome P450 2E1 potentiates ethanol induction of hypoxia and HIF-1alpha in vivo. Free Radical Biol Med. 2013;63:175–186. doi: 10.1016/j.freeradbiomed.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Cederbaum A. Cytochrome P4502E1 sensitizes to tumor necrosis factor alpha-induced liver injury through activation of mitogen-activated protein kinases in mice. Hepatology. 2008;47:1005–1017. doi: 10.1002/hep.22087. [DOI] [PubMed] [Google Scholar]
- Wu YL, Piao DM, Han XH, Nan JX. Protective effects of salidroside against acetaminophen-induced toxicity in mice. Biol Pharm Bull. 2008;31:1523–1529. doi: 10.1248/bpb.31.1523. [DOI] [PubMed] [Google Scholar]
- Zakhari S. Overview: how is alcohol metabolized by the body? Alcohol Res Health. 2006;29:245–254. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Ethanol-induced HIF1α expression in wild-type and ΔHepHIF1α−/− mouse livers. C57BL/6J wild-type mice were allowed free access to diets with ethanol- (11% of kcal) or pair-fed controls for 4 days (A) followed by CCl4 injections twice a week for 5 weeks and maintained on ethanol- or control diets (B). Frozen liver sections were stained for HIF1α. Images were acquired using 40× objective (n = 3–6).
Figure S2. CYP2E1 expression in wild-type and ΔHepHIF1α−/− mouse livers. C57BL/6 wild-type (WT) and ΔHepHIF1α−/− mice were allowed free access to diets with ethanol (11% of kcal) or pair-fed controls for 4 days. Mice were then injected with CCl4 twice a week for 5 weeks and maintained on ethanol- or control diets. (A) CYP2E1 protein expression was measured in liver lysates using Western blot. Images are representative of at least 3–6 mice per group and values under the image are means and SEM. P, Pair-fed; E, EtOH-fed. (B) CYP2E1 activity was measured in liver homogenates. n = 3–6.
Table S1. Body weight, food intake, plasma levels of liver enzyme and hepatic triglyceride after moderate ethanol feeding (32d, 11%) in presence of chronic CCl4 exposure.
Table S2. Gene-specific primer sequences for RT-PCR.