

Significance
Connections are crucial to brain function and a variety of molecular systems direct axonal growth during development and regeneration. An important system involves Celsr2, Celsr3, and Fzd3, membrane proteins that also regulate epithelial planar cell polarity (PCP). Here, we show genetically that Celsr2 and Celsr3 guide axons redundantly, in collaboration with Fzd3 in the same cell populations. However, unlike in epithelial PCP, their action is Vangl1 and Vangl2 independent. Furthermore, expression of Celsr2-3 and Fzd3 in thalamocortical axons and cortical cells is required for the fine mapping of cortical areas. Our findings that Celsr2, Celsr3, and Fzd3 regulate axonal guidance using mechanisms different than epithelial PCP have implications for brain wiring during normal development and regeneration.
Keywords: Cre, anterior commissure, internal capsule, cortical barrels
Abstract
Celsr3 and Fzd3, members of “core planar cell polarity” (PCP) genes, were shown previously to control forebrain axon guidance and wiring by acting in axons and/or guidepost cells. Here, we show that Celsr2 acts redundantly with Celsr3, and that their combined mutation mimics that of Fzd3. The phenotypes generated upon inactivation of Fzd3 in different forebrain compartments are similar to those in conditional Celsr2-3 mutants, indicating that Fzd3 and Celsr2-3 act in the same population of cells. Inactivation of Celsr2-3 or Fzd3 in thalamus does not affect forebrain wiring, and joint inactivation in cortex and thalamus adds little to cortical inactivation alone in terms of thalamocortical projections. On the other hand, joint inactivation perturbs strongly the formation of the barrel field, which is unaffected upon single cortical or thalamic inactivation, indicating a role for interactions between thalamic axons and cortical neurons in cortical arealization. Unexpectedly, forebrain wiring is normal in mice defective in Vangl1 and Vangl2, showing that, contrary to epithelial PCP, axon guidance can be Vangl independent in some contexts. Our results suggest that Celsr2-3 and Fzd3 regulate axonal navigation in the forebrain by using mechanisms different from classical epithelial PCP, and require interacting partners other than Vangl1-2 that remain to be identified.
From a functional perspective, the appropriate wiring of connections is arguably the most important aspect of brain development. The initial step in wiring is the directed extension of axons through a complex environment. Axonal growth cones are guided by a wide variety of attractive and repulsive cues. Some are secreted, diffusible, and act at a distance, whereas others are anchored locally to the extracellular matrix or to the surface of so-called guidepost cells or intermediate targets. Regulators of axon guidance include ligand/receptor partners such as Eph/ephrins, Slit/Robo, Semaphorins/Plexins-Neuropilins, Netrins/Unc5-Dcc (1), and the membrane proteins Celsr3 and Frizzled3, whose role was identified more recently (2–4). The seven-pass transmembrane proteins Celsr1-3 (Cadherin EGF LAG seven-pass G-type receptors 1–3) and Frizzled (Fzd) 3 and 6 belong to a set of proteins that regulate planar cell polarity (PCP) in epithelial sheets, where they work with Van Gogh-like (Vangl) 1 and 2, the cytoplasmic adaptors Dishevelled (Dvl) 1–3, Prickle (Pk) 1–4, and others whose role is less clearly defined (5–10). In epithelial PCP, expression of Frizzed-Celsr on one side of cells, and Vangl-Celsr on the other side, imparts a vectorial organization to the epithelium responsible, among other, for the coordinated growth of body hairs and cilia.
In this study, we analyzed genetically the contribution of Celsr2, Celsr3, Fzd3, and Vangl1, Vangl2 to axonal development in the forebrain, by focusing on the anterior commissure (AC), the corticospinal tract (CST), and the internal capsule (IC). The AC contains commissural axons from the anterior olfactory nuclei and from the temporal cortex, which cross the midline at embryonic day 13.5 (E13.5) to E14.5 (11–14). The IC contains three main axonal components. Thalamocortical axons (TCA) emerge from the thalamus—formerly called “dorsal” thalamus (15)—at E12.5. They run through the prethalamus (former “ventral” thalamus), turn and cross the diencephalon–telencephalon junction, progress through a corridor in the ventral telencephalon, and cross the pallial–subpallial boundary to reach the cortical anlage from E14.5 (16–19). Corticothalamic axons (CTA) emerge initially from neurons in the subplate and future cortical layer 6, around E13.5. They cross the pallial–subpallial boundary and progress in the ventral telencephalon, in opposite direction to TCA. They cross the diencephalon–telencephalon junction and begin to enter the thalamus at E14.5 (17, 19, 20). Subcerebral projections such as the CST begin to leave the cortical plate at E14.5–E15.5 to enter the IC, and diverge from CTA to form the cerebral peduncle, en route to their subcortical targets such as the spinal cord (21).
Previous work showed that Celsr3 is required for the development of the AC, IC, and CST (2), and Cre-mediated regional inactivation indicated that axon navigation requires Celsr3 expression in neurons of origin and/or in guidepost cells along the pathway (22). The Celsr3 mutant axonal phenotype is quite similar to that in Fzd3 mutant mice (3, 23), hinting that a PCP-like mechanism may regulate axon progression via interactions between growth cones and guidepost cells. To understand further the role of membrane-associated core PCP proteins in axon guidance, we used a panel of mutant mice and show genetically that Celsr2 and 3 regulate the formation of forebrain axon bundles in a redundant manner, and are required in the same cell populations as Fzd3. Unlike epithelial PCP, however, the action of Celsr2-3 and Fzd3 on forebrain axonal fascicles is Vangl1,2 independent. Inactivation of Celsr2-3 or Fzd3 in thalamus generates no evident phenotype, showing that the derailed TCA phenotype in constitutive mutants is non-cell autonomous. Furthermore, joint inactivation of Celsr2-3 or Fzd3 in thalamus and cortex perturbs the development of cortical maps.
Results
Celsr2 and Celsr3 Function Redundantly in Axon Guidance.
Inasmuch as Celsr2 and Celsr3 have redundant activity during ependymal ciliogenesis (24) and facial branchiomotor neuron migration (25), and both genes are coexpressed in postmitotic neurons, we wondered whether they also work together in axon guidance. Axonal bundles in Celsr2gt/gt mutant mice were similar to controls (Fig. 1 A and E). In contrast, examination of brains at postnatal day 0 (P0) showed that, in addition to the anomalies described previously in Celsr3−/− mutants (Fig. 1 B and F) (2), additional defects were prominent in double-mutant Celsr2gt/gt;Celsr3−/− or Celsr2−/−;Celsr3−/− samples, particularly the absence of stria medullaris and mammillothalamic tract, and a very diminutive fasciculus retroflexus (Fig. 1 C and G). Those features were also seen in Fzd3−/− mutants (Fig. 1 D and H), confirming a study with diffusion tensor imaging (26), and indicating that the wiring anomalies in Fzd3 mutants are very similar to those in Celsr2+3 double-mutant mice. This observation suggests strongly that Celsr3 and Celsr2, on one hand, and Fzd3, on the other hand, act similarly during axonal development, as they do during the migration of facial branchiomotor neurons (25) and in the steering of peripheral motor axons (27).
Fig. 1.

Redundancy between Celsr2 and Celsr3 in constitutive mutants at P0. Whereas no anomaly of axonal bundles is seen in Celsr2gt/gt brains (A and E) (n = 4), profuse defects are present in Celsr3−/− (B and F) as described (2). Celsr2 and 3 double mutants (C and G) (n = 6) display additional defects that mimic those in Fzd3−/− mice (D and H) (n = 3). (A–D) H&E staining; (E–H) neurofilament immunohistochemistry. fr, fasciculus retroflexus; ml, medial lemniscus; mt, mammillothalamic tract; sm, stria medullaris. (Scale bar: 500 µm.)
We next investigated the redundancy between Celsr2 and Celsr3 using Emx1-Cre mice, which express Cre in cortical projection neurons and their progenitors (28) as well as in limb ectoderm (22) (Fig. S1D), and Nex-Cre mice in which Cre is expressed in cortical projection neurons but not progenitors (29). We showed previously that TCA and CTA axonal components of the IC are preserved, and that subcerebral projections such as the CST are absent in Emx1-Cre;Celsr3f/− mice (22), indicating that Celsr3-deficient corticofugal axons are unable to project to subcerebral targets, yet can reach the thalamus. We examined Celsr2gt/gt;Emx1-Cre;Celsr3f/− mice, in which Celsr2 is mutated in the whole embryo and Celsr3 in cortical progenitors and projection neurons, as well as Emx1-Cre;Celsr2f/−;Celsr3f/− mice, in which both genes are inactivated by Emx1-Cre. Brains were studied at P0, a stage when hydrocephalus—which develops postnatally in those mutants (24)—does not perturb analysis. All components of the IC were well defined and of normal size in Celsr2gt/gt and Celsr2−/− animals. In contrast, in both Celsr2gt/gt;Emx1-Cre;Celsr3f/− and Emx1-Cre;Celsr2f/−;Celsr3f/− mutants, the IC was diminutive and abnormal looping fibers were seen in the external tier of the basal telencephalon, in the vicinity of the external capsule (Fig. 2 A–C). These looping axons were consistently labeled upon 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) placement in the diencephalon (Fig. 2 G–I), but not upon dye injection in cortex (Fig. 2 D–F), indicating that they contained mostly derailed TCA, which failed to progress to cortex, but few CTA fibers. Thus, Celsr3 and Celsr2 act redundantly in cortical neurons during development of the IC. Compared with the full defect in constitutive mutants, the partial IC phenotype observed upon cortical inactivation shows that the action of Celsr2 and 3 in cortical neurons, like that of Fzd3 described below (see Fig. 4), are partly non-cell autonomous.
Fig. 2.

Redundancy between Celsr2 and Celsr3 in cortical neurons. (A–C) Neurofilament immunohistochemistry (NF) at P0. Compared with control (A), mutant mice with regional inactivation of Celsr3 on Celsr2 mutant background (B) (n = 3) and with regional inactivation of Celsr2 and Celsr3 under Emx1-Cre (C) (n = 6) have significant atrophy of the IC (arrows), and looping fibers around the external capsule (arrowheads). (D–I) DiI injection at P0. Tracer administration in cortex (Ctx) (D–F) labels thalamic (Th) neurons retrogradely in control (n = 3) and mutants (n = 4 and 5). DiI in diencephalon (Dien) (G–I) labels TCA and cortical neurons in control brains (G) (n = 3); in mutants (H and I) (n = 4 and 5), it reveals abnormal looping axons (arrowheads). (Scale bars: 500 µm.)
Fig. 4.

Regional inactivation of Fzd3 phenocopies that of Celsr2+3. Compared with control brains (A, D, and G), inactivation of Fzd3 in cortex (B, E, and H) results in absence of anterior commissure (AC), diminutive IC (arrow in E) with external looping axons (arrowhead in E), and in absence of CST in the cervical spinal cord (white arrow in G, absent in H). Inactivation in basal forebrain does not affect AC formation but impairs development of all components of the IC with derailed thalamocortical axons (arrow in F), balling of cortical efferent fibers (white asterisk in F, black asterisk in C) and absence of CST (I). (A–C) H&E staining; (D–F) neurofilament immunohistochemistry; (G–I) Prkcg (protein kinase Cγ) immunohistochemistry. n = 3 for each mutant genotype. [Scale bars: 500 µm (A–F); 200 µm (G–I).]
Celsr2 and Celsr3 Act Independently of Vangl1 and Vangl2 in Forebrain Wiring.
Van Gogh is a key partner of Frizzled and Flamingo in Drosophila epithelial PCP (7). Similarly, in vertebrate epithelia such as the neuroepithelium, inner ear, epidermis, or ependyma, Vangl2 collaborates with Celsr1 or 2, and Fzd3 or 6 (24, 30–32). Furthermore, guidance defects of mesencephalic monoaminergic and spinal commissural fibers are present in Looptail Vangl2 mutant mice (33, 34).To assess whether Van Gogh-like proteins are implicated in forebrain axonal guidance, we inactivated both Vangl1 and Vangl2, using a constitutive Vangl1 genetrap allele known to be a null (35), and a conditional “floxed” Vangl2 allele (36). That Cre inactivation of the floxed Vangl2 allele proceeds as predicted was demonstrated by immunofluorescence (Fig. S1 A and B) and Western blot analysis at E13.5 (Fig. S1C), showing that no Vangl2 protein is detected in the forebrain of Foxg1-Cre;Vangl2f/− mice, and by analysis of Vangl1−/−;Emx1-Cre;Vangl2f/f mice in which hair whorls—a typical PCP phenotype (37)—are present in the hindfeet (Fig. S1E). We examined brains of Vangl1−/−;Foxg1-Cre;Vangl2f/f mice, in which Vangl1 is constitutively mutated and Vangl2 is inactivated in the whole forebrain except dorsal thalamus (38), at P0, a stage when all major axonal bundles are easily visualized. In those mutant animals, the AC was similar to that in controls (Fig. 3A), the IC, with CTA and TCA bundles, had a normal size and shape (Fig. 3B), and the CST, visualized in the hindbrain (Fig. 3C) and spinal cord, was indistinguishable from that in control mice. The observation that Vangl1 and Vangl2 are not required for the formation of forebrain axonal bundles was quite unexpected and stands in sharp contrast to the profuse anomalies in mice with forebrain-specific inactivation of Celsr3 (22) and Fzd3 mice described below (Fig. 4 and Fig. S2).
Fig. 3.
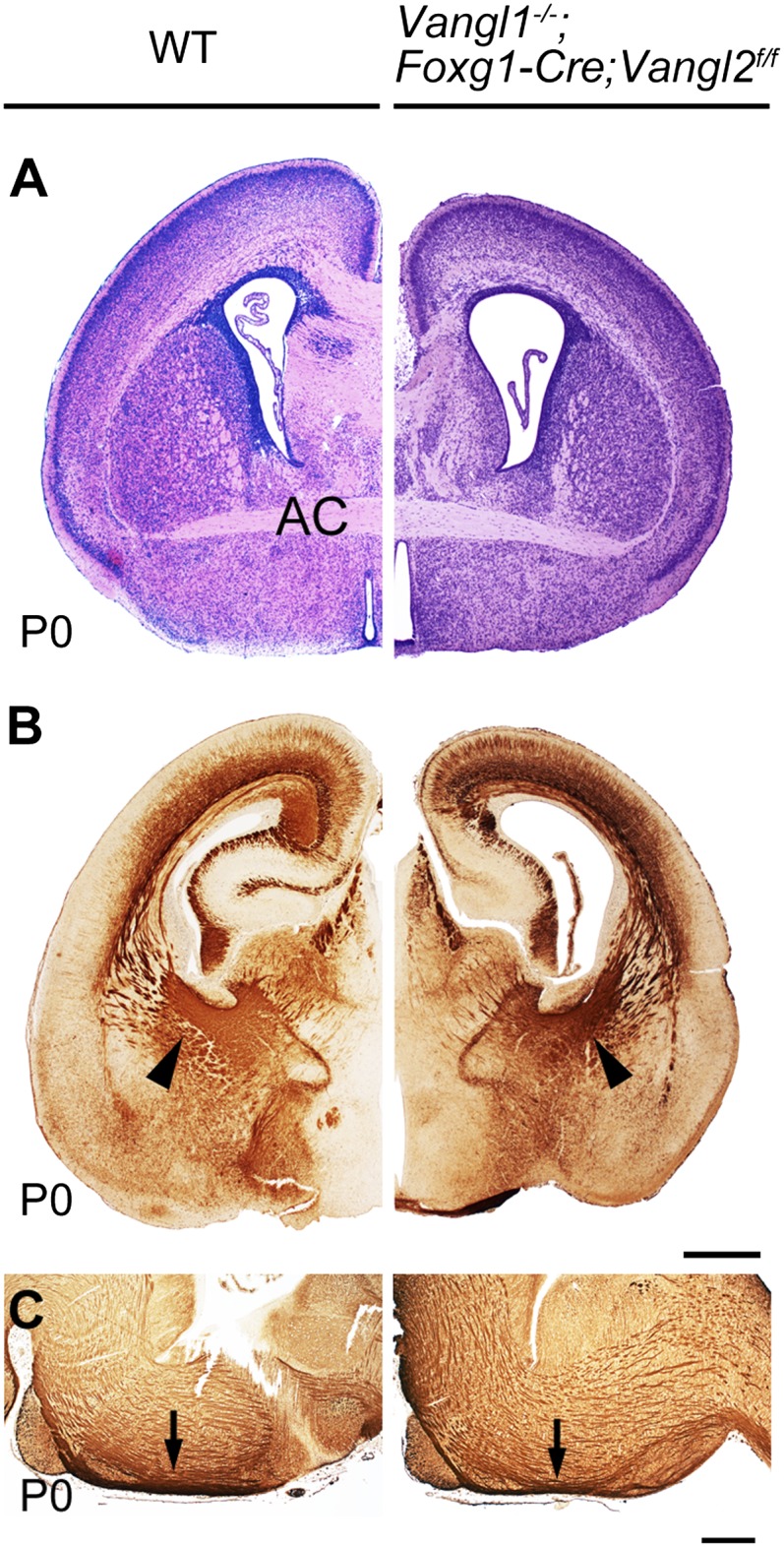
Forebrain axon bundles develop independently of Vangl1 and Vangl2. Comparison between wild type (WT) (Left) and Vangl−/−;Foxg1-Cre;Vangl2f/f mutant brains at P0 show normal development of the anterior commissure (AC in A; H&E staining), normal IC (arrowheads in B; neurofilament immunohistochemistry), and normal CST (arrows in C; neurofilament immunohistochemistry). n = 3. [Scale bars: 500 µm (A and B); 200 µm (C).]
Regional Inactivation of Fzd3 and Celsr3 Generates Similar Axonal Phenotypes.
In Drosophila, Frizzled and Flamingo (Celsr) are thought to work in cis and can interact physically upon transfection in S2 cells (39). To test genetically whether an analogous action may account for the role of Fzd3 and Celsr2-3 in axon guidance, we examined the AC and IC in mice with regional inactivation of Celsr2+3 and Fzd3 in the whole forebrain using Foxg1-Cre, in the cortex using Emx1-Cre or Nex-Cre, and in the ventral forebrain using Dlx5/6-Cre (40). In comparison with control mice (Fig. 4 A, D, and G), inactivation of Fzd3 upon Foxg1-Cre expression resulted in complete absence of AC and IC (Fig. S2 A and B), and inactivation upon Emx1-Cre or Nex-Cre expression resulted in the following: (i) absence of the AC (Fig. 4B); (ii) strongly reduced size but normal trajectory of CTA and TCA fiber bundles in the IC, except for the presence of aberrant looping fibers in basal forebrain (Fig. 4E and Fig. S2D); and (iii) complete absence of CST in the spinal cord (Fig. 4H). This phenotype is similar to that described above in Emx1-Cre;Celsr2f/−;Celsr3f/− and Nex-Cre;Celsr2f/−;Celsr3f/− mutant mice (Fig. 2). When Fzd3 was inactivated in the basal forebrain using Dlx5/6-Cre, the AC developed normally (Fig. 4C) but all components of the IC were absent. Prominent neurofilament-rich CTA bundles were derailed in the basal telencephalon. TCA failed to turn at the diencephalon–telencephalon junction and were also misrouted to the ventral aspect of the basal telencephalon; some crossed to the other side in the preoptic and hypothalamic region (Fig. 4F), and no CST was present in the spinal cord (Fig. 4I). The Dlx5/6-Cre;Fzd3f/− axonal phenotype appears somewhat more marked than that described previously in Dlx5/6-Cre;Celsr3f/− samples. It also differs in some aspects such as a less prominent whorl of neurofilament-rich axons in the external tier of the basal forebrain. Despite those differences, the overall similarity of axonal bundle malformations in Dlx5/6-Cre;Fzd3f/− and Dlx5/6-Cre;Celsr3f/− mutant mice is remarkable. Together with an independent study using conditional Fzd3 inactivation (23), our observations that the regional inactivation of Celsr2+3, or Fzd3, using different Cre strains, generates similar phenotypes suggests strongly that Celsr2, 3, and Fzd3 proteins are both required in the same cells.
Inactivation of Celsr2 and 3, or Fzd3 in Thalamus Does Not Affect Forebrain Wiring.
To further evaluate the role of Celsr2, Celsr3, and Fzd3 in thalamocortical wiring, we searched for mouse strains that express Cre in thalamus, and ideally not in prethalamus and more rostral areas. We first used Wnt3a-Cre and Nkx6.2-Cre, which are expressed early in thalamus in a complementary manner (41, 42). However, Nkx6.2 is also expressed focally in the ganglionic eminences, around the sulcus that separates the lateral and medial ganglionic eminences, and Wnt3a in the hippocampal hem. Brains from Celsr2gt/gt;Wnt3a-Cre;Nkx6-Cre;Celsr3f/− mice, with constitutive inactivation of Celsr2 and inactivation of Celsr3 in thalamus upon Wnt3a and Nkx6.2-Cre, were similar to control. To palliate difficulty associated with Wnt3a and Nkx6.2 expression outside thalamus, we obtained more specific Cre expression using Foxb1-Cre, which is active in the dorsal thalamus from an early developmental stage (E9.5–E10.5), but not expressed in prethalamus or telencephalon (43) (Fig. S3). Again, all forebrain axonal bundles and the CST developed normally in Foxb1-Cre;Celsr2f/−;Celsr3f/− (Fig. 5 A, E, and I, and Table 1) and Foxb1-Cre;Fzd3f/− mutants (Fig. 5 C, G, and K, and Table 1). Together with previous observations using Rora-Cre mice (22), and given the restrictions related to the timing and efficiency of Cre-induced recombination, the present data provide strong evidence that, although TCA are prominently derailed in constitutive Celsr3 and Fzd3 mutants, and upon conditional inactivation in ventral telencephalon (Dlx5/6-Cre), deletion of Celsr2+3 or Fzd3 in their thalamic neurons of origin has no impact on their trajectory.
Fig. 5.

Inactivation in thalamus does not affect wiring, and joint inactivation in cortex and thalamus affects it mildly. At P0, hematoxylin–eosin (H&E) (A–D), neurofilament immunostaining (NF) (E–H), and protein kinase Cγ immunofluorescence (Prkcg) (I–L) show that, upon inactivation in thalamus using Foxb1-Cre (A, C, E, G, I, and K) (n = 5 and 3, respectively), the anterior commissure (AC), the IC (arrows in E and G), and the CST (arrows in I and K) are present. Joint inactivation in thalamus and cortex (n = 7 and 3, respectively) results in absence of AC crossing (B and D), atrophy of the IC (arrows in F and H) with some looping of axons close to the external capsule (arrowhead in F), and absence of CST in the spinal cord (J and L). These anomalies are similar to those obtained upon cortex-specific inactivation. [Scale bars: 500 µm (A–H); 200 µm (I–L).]
Table 1.
Summary of cortical and thalamic projection phenotypes
| Genotype | Wiring phenotype |
| Celsr2gt/gt | No phenotype (Fig. 1 A and E) |
| Celsr2gt/gt;Celsr3−/− | Full defect of IC (Fig. 1 C, D, G, and H) |
| Celsr2−/−;Celsr3−/− | |
| Fzd3−/− | |
| Celsr2gt/gt;Emx1-Cre;Celsr3f/− | Decreased IC with partially blocked/delayed TCA (Figs. 2 and 4 B and E) |
| Emx1-Cre;Celsr2f/−;Celsr3f/− | Looping of TCA in external basal forebrain (Fig. 2 and Fig. S2D) |
| Nex-Cre;Celsr2f/−;Celsr3f/− | Absence of CST (Fig. 4H) |
| Emx1-Cre;Fzd3f/− | Layer 5 axons routed to thalamus (Fig. 7H) |
| Nex-Cre;Fzd3f/− | Normal barrel field (Fig. S5 A, B, E, and F) |
| Foxb1-Cre;Celsr2f/−;Celsr3f/− | No phenotype (Fig. 5 A, C, E, G, I, and K and Fig. S5 C, D, G, and H) |
| Foxb1-Cre;Fzd3f/− | |
| Nex-Cre;Foxb1-Cre;Celsr2f/−;Celsr3f/− | Decreased IC with partially blocked/delayed TCA (Fig. 5 B, D, F, and H) |
| Nex-Cre;Foxb1-Cre;Fzd3f/− | Looping of TCA and CTA in external basal forebrain (Figs. 5 and 6) |
| Absence of CST (Fig. 5 J and L) | |
| Layer 5 axons routed to thalamus (Fig. 7I) | |
| Abnormal barrel field (Fig. 8) |
Joint Inactivation in Cortex and Thalamus Enhance Wiring Phenotypes Marginally.
As described above, the action of Celsr2-3 and Fzd3 is nonautonomous in thalamic neurons and TCA, and partially nonautonomous in cortical neurons of origin of CTA. How can we explain a fully cell-autonomous action in neurons of origin of the AC and CST, and non–cell-autonomous effect on CTA and TCA? First, a trivial explanation that the Cre drivers used are not fully active in the context of early cortical and thalamic neurons cannot be formally excluded but appears rather unlikely, as discussed later. Second, unlike AC and CST neurons, expression of Celsr2-3 and Fzd3 may not be required in cortical and thalamic neurons, and CTA and TCA projections could rely more than others on expression in guidepost cells. A third possibility could be that a reciprocal interaction between CTA and TCA, mediated by other molecules, palliate unilateral Celsr2-3 or Fzd3 deficiency. The third model, reminiscent of the “handshake” or “rendezvous” (44, 45), predicts that joint inactivation in neurons of origins of CTA and TCA should yield a more severe phenotype than the addition of phenotypes observed upon single inactivation in cortex or thalamus.
We tested this latter hypothesis by inactivating Celsr3 upon Emx1, Wnt3a, and Nkx6.2-Cre expression, on a Celsr2 mutant background (Celsr2gt/gt;Triple Cre;Celsr3f/−) (Fig. 6). The axonal phenotype in those mice was nearly similar to that observed in mice with Emx1-Cre–induced inactivation alone (Fig. 2), except that derailed looping fibers in the laterobasal telencephalon could be labeled not only by DiI administration in diencephalon, but also in cortex (Fig. 6 D and F). We then used Foxb1-Cre in conjunction with Nex-Cre to inactivate Celsr2+3 or Fzd3 in both cortex and thalamus. Brains of Foxb1-Cre;Nex-Cre;Celsr2f/−;Celsr3f/− and Foxb1-Cre;Nex-Cre;Fzd3f/− mice were characterized by anomalies related to cortical-specific inactivation, particularly absence of AC and CST and atrophy of IC (Fig. 5 B, F, and J, and D, H, and L, and Table 1). To study this further, we examined E14.5 embryos using DiI injection in cortex or diencephalon. In control embryos, both TCA and CTA reached their target area as expected (Fig. 7 A and D). In Nex-Cre;Celsr2f/−;Celsr3f/− embryos, DiI in cortex labeled cortical efferent fibers that did not pass the level of the lateral ganglionic eminence, and TCA labeled upon diencephalic injection did not extend beyond the pallial–subpallial boundary (Fig. 7 B and E). Inactivation in thalamus in addition to cortex (Foxb1-Cre;Nex-Cre;Celsr2f/−;Celsr3f/−) resulted in a slightly more pronounced IC phenotype, with no CTA passing the pallial–subpallial boundary and no TCA leaving the basal telencephalon (Fig. 7 C and F). This modest synergistic effect of inactivation upon both Nex-Cre and Foxb1-Cre was confirmed in P30 animals labeled with the Thy1-YFP transgene (46). In control brains, most layer 5 and some layer 6 neurons were labeled, cortical efferent fibers from layer 5 descended in the cerebral peduncle as subcerebral projections but never went to thalamus, and a small number of labeled fibers from layer 6 ran to the thalamus (Fig. 7G). In Thy1;Nex-Cre;Celsr2f/−;Celsr3f/− mice, layer 5 was diminutive, presumably due to defective subcerebral projections and resulting neuronal death, yet a significant number of labeled axons from layer 5 ran abnormally to the thalamus (Fig. 7H). The phenotype was somewhat accentuated in Thy1;Nex-Cre;Foxb1-Cre;Celsr2f/−;Celsr3f/− mice, in which the atrophy of layer 5 was more pronounced, and derailed fibers to thalamus less abundant that in Thy1;Nex-Cre;Celsr2f/−;Celsr3f/− animals (Fig. 7I).
Fig. 6.

Inactivation in cortex and thalamus using triple Cre confirm addition of phenotypes. (A and B) At P0, neurofilament immunohistochemistry (NF) shows that, compared with Celsr2 gt/gt brains (n = 4)—which are normal—there is clear atrophy of the IC with looping fibers in triple Cre mutants (arrowhead in B) (n = 4). (C–F) DiI injections at P0 show that looping axons are labeled in triple Cre mutants following tracer injection both in cortex (Ctx) (arrow in D) (n = 4) and in diencephalon (Dien) (arrowhead in F) (n = 4); compare with Fig. 2 D–I. (Scale bars: 500 µm.)
Fig. 7.

Connection patterns upon inactivation of Celsr2+3 in both cortex and thalamus. In control E14.5 embryos (A and D), DiI in cortex labels CTA and retrogradely fills neurons in thalamus (Th), whereas DiI in diencephalon (Dien) prominently labels TCA. Upon cortical inactivation (B and E), DiI in cortex labels CTA that cross the pallial–subpial boundary (PSPB) (dotted line) but do not extend much beyond it, whereas DiI in Dien labels TCA in basal forebrain that do not reach cortex. The blunted growth of both CTA and TCA is more severe upon joint inactivation in cortex and thalamus (C and F). n = 3 for each DiI experiment. Using the Thy1-YFP transgene in P30 animals, control brains (G) (n = 7) show prominent labeling of cortical layer 5 (Ctx) and subcerebral projections (arrows), none of which reaches the thalamus (Th). In cortical mutants (H) (n = 10), layer 5 is atrophic and some of its axons are derailed to the thalamus (arrowheads). The phenotype is more severe upon joint inactivation in cortex and thalamus (I) (n = 7). [Scale bars: 200 µm (A–F); 500 µm (G–I).]
Altogether, these observations show that the wiring defects generated upon cortical inactivation of Celsr2+3 and Fzd3 are only marginally increased when target genes are also inactivated in thalamus, arguing against a rescue via CTA–TCA interactions through Celsr2-3/Fzd3 and for the importance of expression in guidepost cells.
Celsr2-3 and Fzd3 Are Required in TCA and Cortical Neurons for Fine Cortical Arealization.
We then considered whether the expression of Celsr2-3 and Fzd3 in thalamus might influence the formation of cortical areas, which are known to depend on thalamocortical input (47). To assess this, we studied the barrel field, an area in primary somatosensory cortex corresponding to whisker-related input, where thalamic afferents project to barrel centers and synapse with dendrites of excitatory spiny stellate and pyramidal neurons in cortical layer 4 (48). We compared barrel fields in P8 mice of the following genotypes: control, Nex-Cre;Celsr2f/−;Celsr3f/−, Foxb1-Cre;Celsr2f/−;Celsr3f/−, Nex-Cre;Foxb1-Cre;Celsr2f/−;Celsr3f/−, Nex-Cre;Fzd3f/−, Foxb1-Cre;Fzd3f/−, and Nex-Cre;Foxb1-Cre;Fzd3f/−. All mice had a similar organization of whiskers on the snout (Fig. S4). We found that barrel fields were similar to control in all brains with inactivation of Celsr2-3 or Fzd3 upon single Nex or Foxb1-Cre expression (Fig. 8 A and B, and Fig. S5). In contrast, although the barrel field was present in its expected position, it was diminutive in Nex-Cre;Foxb1-Cre;Celsr2f/−;Celsr3f/− and Nex-Cre;Foxb1-Cre;Fzd3f/− mice (Fig. 8 C–F). In tangential and coronal sections, a pattern of rows and columns was present in the primary sensory field, but the fine organization was diffuse, fuzzy, and less sharply defined than in control and single Cre brains. Intriguingly, this architectonic disorganization looked more pronounced in the anterolateral than the posteromedial barrel subfields (Fig. 8 C and E). These data indicate that Celsr2-3 and Fzd3 mediate interactions between thalamocortical afferents and their cortical targets, which are required for the fine mapping of TCA after they reach appropriate cortical areas.
Fig. 8.

Abnormal organization of the barrel field upon combined inactivation in cortex and thalamus. (A and B) In control mice, the somatosensory map, studied using vGlut2 immunostaining (“presynaptic” component, green) and DAPI (“postsynaptic” component, blue) at P8, appears normal, with five rows of barrels (A–E) in the posteromedial subfield corresponding to whiskers (A, tangential section) and vGlut2-positive TCA terminal clusters in layer IV (B, coronal section). In contrast, the barrel fields are diminutive and architectonically disorganized when Celsr2+3 (C and D) (n = 11) or Fzd3 (E and F) (n = 3) are jointly inactivated in both cortex (Nex-Cre) and thalamus (Foxb1-Cre). The vGlut2-positive TCA terminals do not form clear clusters in layer IV in both mutants (D and F). Asterisks: anterolateral fields; II/III, IV, V: cortical layers. [Scale bars: 500 µm (A, C, and E); 200 µm (B, D, and F).]
Discussion
Celsr2 and Celsr3 Act in Redundant Manner in Axon Guidance.
The phenotype in double Celsr2 and Celsr3 mutants is more pronounced than the single-mutant phenotypes, indicating that both genes act in a partly redundant manner in the PCP-related pathway that regulates axon guidance. Further arguments are provided by observations that Celsr2+3 and Fzd3 inactivation also generates similar anomalies during migration of facial branchiomotor neurons (25) and in the patterning of peripheral motor nerves (27). At first sight, this is in contradiction with previously published data attributing opposite roles to Celsr2 and Celsr3 in the control of neurite growth (49, 50). Using RNA interference in brain slices, Celsr2 down-regulation in neurons resulted in shorter and less profuse basal dendrites, whereas down-regulating Celsr3 had opposite effects. Reciprocal results were observed when normal neurons were cultured in contact with Celsr2- or Celsr3-expressing cells. Experiments carried out by swapping of Celsr2 and 3 ectodomains showed that the transmembrane domains and C terminus determine the dendrite enhancing or suppressing action (49). How can Celsr2 and Celsr3 affect dendrite development in opposite manner, while being redundant in the control of axon guidance? Presumably, downstream intracellular pathways are context and/or time dependent, and may differ in dendritic and axonal compartments of a same neuron. Also, signals that promote and inhibit neurite growth in vitro may end up having similar global effects on axon guidance in vivo, where growing axons encounter a more complex environment. Studies of the downstream signaling events controlled by Celsr/Fzd complexes are needed to understand that issue further.
Forebrain Axon Guidance Is Independent of Vangl1 and Vangl2.
Our results show that forebrain axonal bundles require Celsr2, Celsr3, and Fzd3, but not Vangl1 and Vangl2. During epidermal and eye development in flies, Fmi, Fz, Dsh, Van Gogh, and Pk interact closely (51–53). Mutations in Van Gogh and Fz also affect branching of axons of mushroom body neurons, albeit with independent effects (54). Similarly, there is ample evidence that Celsr1, Fzd6, and Vangl1,2 act together to regulate skin development (30, 32, 37) and that Celsr1, Fzd6, Dvl, and Vangl2 collaborate in the regulation of neural tube closure (31, 55, 56), and in the planar organization of cilia in the mouse ependyma (24, 57), inner ear (31, 58), and Xenopus skin (59). Furthermore, Looptail (Lp) mice, which have a G1391A missense mutation in Vangl2, display abnormal growth of mesencephalic monoaminergic axons in homozygous mutant fetuses (33), and of spinal commissural fibers in homozygous and to a lesser extent in heterozygous embryos (34). Our finding that axon guidance in the anterior commissure, thalamocortical, corticothalamic, and corticospinal tracts proceeds normally in the absence of both Vangl1 and Vangl2 was thus unexpected. A first note of caution is that, although the validation data (Fig. S1) make it rather unlikely, the Emx1-Cre and Foxg1-Cre drivers may not be fully active in the Vangl2 floxed genomic context. Second, given its complex regulation, axon guidance may require Vangl proteins in some anatomic systems but not, or less, in others. However, we want to emphasize that the Lp allele is known to have strong dominant-negative activity (36, 60). An abnormally folded Vangl2 protein may have not only dominant-negative but also some gain-of-function activity, for example by perturbing folding and/or hampering membrane targeting of Celsr and/or Fzd proteins. Furthermore, homozygous Lp embryos have a fully open neural tube, and some axonal abnormalities in the spinal cord and midbrain could be secondary to the dysraphic malformation itself, rather than reflect the direct action of Vangl2 in axon guidance. In Drosophila, Van Gogh cooperates closely with the Prickle adaptor (61). The observation that Fzd3 and Celsr2, 3 guide forebrain axons in a Vangl1,2-independent manner leads to the testable prediction that this role is also independent of Prickle-like proteins. Van Gogh and Fz affect neurite growth independently in Drosophila (54), and Van Gogh, Prickle, and Dishevelled mutant neurons in Caenorhabditis elegans display supernumerary neurites (62). An additional role of Vangl2 in synapse formation was recently demonstrated (63, 64). These observations suggest that Vangl1,2 are likely to affect brain development by acting in different pathways, in addition to epithelial PCP.
Celsr2, 3, and Fzd3 Act in the Same Cell Populations.
The fact that Celsr and Fzd regulate axon guidance independently of Vangl suggests that mechanisms are different from those that regulate PCP in epithelial sheets. Phenotypes generated upon conditional inactivation of Celsr2 and 3 show that axonal growth cones need to interact with guidepost cells and that this interaction requires expression of Celsr2 and/or Celsr3 in guidepost cells or in growth cones, depending on the pathway (22). Together with another study (23), our present work confirms the essential role of Fzd3 in axons, guidepost cells, or both (3). The fact that inactivation of Fzd3 with different Cre-expressing strains consistently mimics that of Celsr2+3 suggests that both proteins act in the same cell populations, indicating that Fzd3 may participate in a complex in the membrane, together with Celsr2/3. This would be in line with genetic and biochemical data in Drosophila, particularly the observation that Fz and Fmi coimmunoprecipitate when expressed in S2 cells (39). Like in Drosophila, the formation of a complex containing Celsr2, 3, and Fzd3 does not preclude expression of Celsr2, 3 in other cells and/or other sectors of the plasma membrane. However, the fact that Celsr and Fzd act probably in cis and independently of Vangl, raises the crucial question of interacting partners in trans.
Another question concerns the signal generated at the growth cone when it encounters guidepost cells, and how this signal steers the growth cone in the right direction. Both Celsr2 and Celsr3 are able to trigger calcium signals in transfected cells, with different kinetics leading to activation of different target genes (49). It would be interesting to assess whether analogous calcium signals are induced by Fzd3, and to test whether calcium waves triggered by Celsr2, 3–Fzd3 complexes can differ in dendritic and axonal compartments.
What Mechanisms Could Account for Non–Cell-Autonomous Action in Thalamus and Cortex?
Our results demonstrate that inactivation of Celsr2+3 or Fzd3 in dorsal thalamus has no effect on reciprocal thalamocortical projections, and that inactivation in cortex prevents the formation of subcerebral projections and leads to atrophy of the IC, yet does not generate the full defect that occurs in mice with constitutive mutations or conditional inactivation in basal forebrain (22). A rather similar situation was described in ubiquitin ligase Phr1 mutant mice, in which constitutive inactivation leads to complete absence of IC, whereas cortical inactivation with Emx1-Cre yields a partial thalamocortical phenotype (65, 66). As in all studies with the Cre-loxP system, an intrinsic limitation and concern is that Cre expression may not occur early enough with sufficient activity to generate a full phenotype. As the activity of the Cre drivers used was validated in several studies in addition to our data, this is rather unlikely. Non-cell autonomy upon inactivation in cortex or thalamus could be explained by postulating that Celsr2, 3, and Fzd3 have an crucial role in cortical and thalamic neurons, like in neurons of origin of the AC and CST, but that CTA and TCA interact reciprocally and rescue each other in case of unilateral Celsr2, 3, and Fzd3 inactivation. Upon inactivation in thalamus, nothing should prevent CTA from initiating their growth out of the cortex, in the ventral corridor (18) toward the thalamus. Reciprocally, cortical inactivation should not perturb the initial growth and turning of TCA. Thus, in “single-side” mutants, CTA and TCA could meet at some point along their trajectory and assist progression of the mutant partner (44, 45). The observation that the joint inactivation of Celsr2, 3, or Fzd3 in both cortex and thalamus adds little to single inactivation and fails even remotely to recapitulate the constitutive or conditional Dlx5/6-Cre phenotypes, argues against a crucial role of TCA–CTA fiber interactions. We therefore believe that interactions of axons with intermediate guidepost cells are particularly important and need to be investigated further, using more specific Cre driver strains and in vitro culture systems (17, 44, 67).
Whereas it does not impact much on axonal wiring, inactivation of Celsr2, 3, or Fzd3 in thalamus in addition to cortex results in a disorganization of the barrel field. This is not observed upon single inactivation in cortex or thalamus: expression of Celsr2, 3, or Fzd3 in one partner, either TCA or cortical neurons, is sufficient for fine cortical arealization. The phenotype observed is reminiscent of that generated by constitutive inactivation of Bhlhb5 (68), by inactivation of Ebf1 in basal forebrain (69), or by cortex-specific inactivation of Lmo4 (70). As far as we know, however, areal anomalies generated specifically upon joint gene inactivation in thalamus, the presynaptic component, and cortical neurons, the postsynaptic partner, have not been reported. Whether the tangential organization of other cortical areas is also affected was not tested but would appear likely. That a barrel field does form at the right location in double conditional mutants shows that TCA are still able to ascend to the cortex and find their way through deep cortical layers. The simplest explanation is therefore that a reciprocal interaction between thalamic afferents and target neurons in layer 4 is involved. The absence of phenotype upon unilateral inactivation suggests that, besides Celsr2, 3, and Fzd3, other unidentified molecules participate in that interaction. Interestingly, a top-down interaction between sensory cortical neurons and thalamus was recently demonstrated (71).
Materials and Methods
Animals.
Experiments were carried out according to guidelines from the Animal Ethics Committees of the University of Louvain and Jinan University. In addition to the Celsr2 gene trap (Celsr2gt) allele used previously (24), we generated constitutive and conditional Celsr2 mutant mice. A Neo cassette flanked by LoxP and Frt sites was inserted into intron 15, and a LoxP site into intron 28 to generate allele Celsr2fn. This allele proved to be a null, and the Neo cassette was removed by crosses with ROSA-Flp mice (72), yielding a conditional floxed Celsr2f allele. The null allele Celsr2− was obtained by crosses with PGK-Cre germline deleter mice (73) (Fig. S6A). Due to the deletion and resulting frameshift, the predicted mutated protein should be truncated before the GPS domain and the seven-transmembrane segments. No Celsr2 protein was detected using a C-terminal antibody (Fig. S6B), and the migration of facial branchiomotor neurons was similarly affected in Celsr2−/− and in Celsr2gt/gt embryos (Fig. S6 D and G), showing that this allele is a null, although the expression of an N-terminal partial protein sequence cannot be excluded. That the Celsr2f allele behaves as predicted upon Cre-mediated recombination is also shown by the abnormal migration of FBM neurons in Isl1-Cre;Celsr2f/− mutant embryos (Fig. S6H). We also generated a “knockout first” Fzd3 allele using a targeting vector from the Eucomm Consortium (Project 78503), which proved a hypomorph. A conditional allele was derived by crosses with ROSA-Flp females, and a null allele was then obtained by crosses with PGK-Cre females. In the null allele, exon 3 (197 nt) is deleted (Fig. S2E), generating a frameshift and a reading frame that encodes 67 N-terminal amino acids of the Fzd3 protein sequence, or 43 residues after removal of the signal peptide.
Other mouse strains were described and validated in the following publications: Celsr3 (22), Dlx5/6-Cre (40), Emx1-Cre (28), Foxb1-Cre (43), Foxg1-Cre (38), Nex-Cre (29), Nkx6.2-Cre (74), Wnt3a-Cre (75), Vangl1 and Vangl2 (35, 36), and Thy1-YFP transgenics (46). Primers for genotyping are listed in Table S1.
To palliate issues related to potential perturbations due to Cre expression, Cre drivers were always heterozygotes and control mice with Cre expression were consistently used. For example, Emx1-Cre;Celsr3f/− mice were compared with controls that included Emx1-Cre;Celsr3f/+ mice. We also verified that Cre expression does not generate apoptosis, by immunostaining for activated Caspase3 (Fig. S7). For comparison of genotypes and DiI experiments, at least three animals were sectioned serially and examined, and phenotypes were fully penetrant.
Histological Procedures.
Brains were fixed by intracardial perfusion with 4% (vol/vol) paraformaldehyde (PFA) in PBS, followed by fixation in the same mixture, overnight for adult and P0 brains, and for a few hours for E14.5 embryos. Brains were embedded in paraffin and sectioned serially at 8 µm. Paraffin sections were stained with hematoxylin–eosin (H&E) or by immunohistochemistry, with detection using an ABC kit (DAKO). For cryostat sections, dissected brains were cryoprotected in 30% (wt/vol) sucrose and embedded in OCT. For study of barrels, just after PFA perfusion, hemispheres were dissected, flattened between two slides, and further fixed overnight at 4 °C. Seventy-micrometer-thick sections were prepared using a vibratome, stained by immunohistochemistry with antibody to Vglut2, and counterstained with DAPI, before photography under a fluorescence or a confocal microscope. For immunohistochemistry, we used the following primary antibodies: guinea pig polyclonal antibody to Vglut2 [1:1,000 (vol/vol); Millipore], mouse monoclonal antibody to neurofilament 2H3 (1:500; Developmental Studies Hybridoma Bank), rabbit monoclonal antibody to PKCγ/Prkcg (1:400; Abcam), goat polyclonal antibody N-13 to Vangl2 (1:50; Santa-Cruz; sc-46561), rabbit polyclonal antibody to cleaved (activated) caspase3 (1:200; Cell Signaling). Signal detection was carried out using goat anti-guinea pig-Fluorescein (1:500; Vector) or donkey anti-rabbit-Alexa Fluor 488 (1:1,000; Molecular Probes). DiI crystals (D282; Molecular Probes) were implanted in target areas of PFA-fixed P0 or E14.5 brains using tungsten needles. Brains were subsequently incubated at 37 °C for 4 wk for P0 brains or 3 wk for E14.5 brains, and sectioned in the coronal plane using a vibratome. Sections were mounted in medium with DAPI and examined with fluorescence microscopy.
Western Blot.
Whole brains of Celsr2+/+, Celsr2−/−, Celsr2gt/gt, or forebrain and hindbrain regions of Foxg1-Cre;Vangl2f/− embryos aged E13.5 were homogenized in NuPAGE LDS sample buffer (Thermo Fisher) and cleared by centrifugation at 16,000 × g for 15 min at 4 °C. Samples containing 30 µg of total protein were analyzed on 4–15% Mini-PROTEAN TGX Gel (Bio-Rad) and transferred to nitrocellulose membrane (BioScience) by electroblotting (Invitrogen). Membranes were blocked with 5% (wt/vol) skimmed milk and 0.1% Tween 20, in PBS, for 30 min, and incubated with a mouse antibody to Celsr2 (76) or goat polyclonal antibody N-13 to Vangl2 (1:500; Santa-Cruz; sc-46561) at 4 °C overnight. Signal was detected using an HRP-conjugated goat anti-mouse Ig (DAKO) followed by chemiluminescence using a SuperSignal West Pico kit (Pierce) and Hyperfilm ECL (Amersham Biosciences).
Supplementary Material
Acknowledgments
We thank Jean Hébert, Kevin Jones, Nicoletta Kessaris, and Klaus Amin Nave for Cre mice; Nicolas Parmentier for help with Western blots; and Esther Paitre, Isabelle Lambermont, Valérie Bonte, and Rachid El Kaddouri for technical assistance. This work was supported by grants from the National Natural Science Foundation of China [31200826 (to Y.Q.) and 31070955 (to L.Z.)], National Basic Research Program of China [973 Program, 2014CB542205 (to Y.Q.) and 2011CB504402 (to L.Z.)], Guangdong Natural Science Foundation [S2012040006744 (to Y.Q.)], Guangzhou Science and Technology Project [13200068 (to Y.Q.)], and Jinan University Research and Innovation Foundation [21612345 (to Y.Q.)], and by Belgian grants Actions de Recherches Concertées (ARC-10/15-026), Fonds de la Recherche Scientifique Medicale 3.4550.11, Fonds National de La Recherche Scientifique (FNRS) T0002.13, Interuniversity Poles of Attraction (Services Fédéraux des Affaires Scientifiques, Techniques, et Culturelles, PAI p6/20 and PAI7/20), Fondation Médicale Reine Elisabeth, Fondation JED-Belgique, and WELBIO-CR-2012A-07 from the Région Wallonne (to F.T. and A.M.G.). F.T. is a senior research associate of the Belgian FNRS.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1402105111/-/DCSupplemental.
References
- 1.Dickson BJ. Molecular mechanisms of axon guidance. Science. 2002;298(5600):1959–1964. doi: 10.1126/science.1072165. [DOI] [PubMed] [Google Scholar]
- 2.Tissir F, Bar I, Jossin Y, De Backer O, Goffinet AM. Protocadherin Celsr3 is crucial in axonal tract development. Nat Neurosci. 2005;8(4):451–457. doi: 10.1038/nn1428. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Thekdi N, Smallwood PM, Macke JP, Nathans J. Frizzled-3 is required for the development of major fiber tracts in the rostral CNS. J Neurosci. 2002;22(19):8563–8573. doi: 10.1523/JNEUROSCI.22-19-08563.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tissir F, Goffinet AM. Shaping the nervous system: Role of the core planar cell polarity genes. Nat Rev Neurosci. 2013;14(8):525–535. doi: 10.1038/nrn3525. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Nathans J. Tissue/planar cell polarity in vertebrates: New insights and new questions. Development. 2007;134(4):647–658. doi: 10.1242/dev.02772. [DOI] [PubMed] [Google Scholar]
- 6.Simons M, Mlodzik M. Planar cell polarity signaling: From fly development to human disease. Annu Rev Genet. 2008;42:517–540. doi: 10.1146/annurev.genet.42.110807.091432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodrich LV, Strutt D. Principles of planar polarity in animal development. Development. 2011;138(10):1877–1892. doi: 10.1242/dev.054080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goodrich LV. The plane facts of PCP in the CNS. Neuron. 2008;60(1):9–16. doi: 10.1016/j.neuron.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu X, et al. PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature. 2004;430(6995):93–98. doi: 10.1038/nature02677. [DOI] [PubMed] [Google Scholar]
- 10.Lee J, et al. PTK7 regulates myosin II activity to orient planar polarity in the mammalian auditory epithelium. Curr Biol. 2012;22(11):956–966. doi: 10.1016/j.cub.2012.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sturrock RR. Development of the mouse anterior commissure. Part II. A comparison of glial differentiation in the anterior and posterior limbs of the anterior commissure of the mouse brain during myelination using semithin light microscopic sections. Zentralbl Veterinarmed [C] 1976;5(2):113–121. doi: 10.1111/j.1439-0264.1976.tb00778.x. [DOI] [PubMed] [Google Scholar]
- 12.Sturrock RR. Development of the mouse anterior commissure. Part I. A comparison of myelination in the anterior and posterior limbs of the anterior commissure of the mouse brain. Zentralbl Veterinarmed [C] 1976;5(1):54–67. doi: 10.1111/j.1439-0264.1976.tb00656.x. [DOI] [PubMed] [Google Scholar]
- 13.Ito A, et al. Tsukushi is required for anterior commissure formation in mouse brain. Biochem Biophys Res Commun. 2010;402(4):813–818. doi: 10.1016/j.bbrc.2010.10.127. [DOI] [PubMed] [Google Scholar]
- 14.Falk J, et al. Dual functional activity of semaphorin 3B is required for positioning the anterior commissure. Neuron. 2005;48(1):63–75. doi: 10.1016/j.neuron.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 15.Scholpp S, Lumsden A. Building a bridal chamber: Development of the thalamus. Trends Neurosci. 2010;33(8):373–380. doi: 10.1016/j.tins.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robichaux MA, et al. EphB receptor forward signaling regulates area-specific reciprocal thalamic and cortical axon pathfinding. Proc Natl Acad Sci USA. 2014;111(6):2188–2193. doi: 10.1073/pnas.1324215111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molnár Z, Garel S, López-Bendito G, Maness P, Price DJ. Mechanisms controlling the guidance of thalamocortical axons through the embryonic forebrain. Eur J Neurosci. 2012;35(10):1573–1585. doi: 10.1111/j.1460-9568.2012.08119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.López-Bendito G, et al. Tangential neuronal migration controls axon guidance: A role for neuregulin-1 in thalamocortical axon navigation. Cell. 2006;125(1):127–142. doi: 10.1016/j.cell.2006.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Price DJ, Clegg J, Duocastella XO, Willshaw D, Pratt T. The importance of combinatorial gene expression in early mammalian thalamic patterning and thalamocortical axonal guidance. Front Neurosci. 2012;6:37. doi: 10.3389/fnins.2012.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, Macklis JD. Molecular logic of neocortical projection neuron specification, development and diversity. Nat Rev Neurosci. 2013;14(11):755–769. doi: 10.1038/nrn3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arlotta P, et al. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45(2):207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- 22.Zhou L, et al. Early forebrain wiring: Genetic dissection using conditional Celsr3 mutant mice. Science. 2008;320(5878):946–949. doi: 10.1126/science.1155244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hua ZL, Jeon S, Caterina MJ, Nathans J. Frizzled3 is required for the development of multiple axon tracts in the mouse central nervous system. Proc Natl Acad Sci USA. 2014;111:E3005–E3014. doi: 10.1073/pnas.1406399111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tissir F, et al. Lack of cadherins Celsr2 and Celsr3 impairs ependymal ciliogenesis, leading to fatal hydrocephalus. Nat Neurosci. 2010;13(6):700–707. doi: 10.1038/nn.2555. [DOI] [PubMed] [Google Scholar]
- 25.Qu Y, et al. Atypical cadherins Celsr1-3 differentially regulate migration of facial branchiomotor neurons in mice. J Neurosci. 2010;30(28):9392–9401. doi: 10.1523/JNEUROSCI.0124-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Zhang J, Mori S, Nathans J. Axonal growth and guidance defects in Frizzled3 knock-out mice: A comparison of diffusion tensor magnetic resonance imaging, neurofilament staining, and genetically directed cell labeling. J Neurosci. 2006;26(2):355–364. doi: 10.1523/JNEUROSCI.3221-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hua ZL, Smallwood PM, Nathans J. Frizzled3 controls axonal development in distinct populations of cranial and spinal motor neurons. eLife. 2013;2(0):e01482. doi: 10.7554/eLife.01482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorski JA, et al. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22(15):6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goebbels S, et al. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis. 2006;44(12):611–621. doi: 10.1002/dvg.20256. [DOI] [PubMed] [Google Scholar]
- 30.Devenport D, Fuchs E. Planar polarization in embryonic epidermis orchestrates global asymmetric morphogenesis of hair follicles. Nat Cell Biol. 2008;10(11):1257–1268. doi: 10.1038/ncb1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Guo N, Nathans J. The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J Neurosci. 2006;26(8):2147–2156. doi: 10.1523/JNEUROSCI.4698-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ravni A, Qu Y, Goffinet AM, Tissir F. Planar cell polarity cadherin Celsr1 regulates skin hair patterning in the mouse. J Invest Dermatol. 2009;129(10):2507–2509. doi: 10.1038/jid.2009.84. [DOI] [PubMed] [Google Scholar]
- 33.Fenstermaker AG, et al. Wnt/planar cell polarity signaling controls the anterior-posterior organization of monoaminergic axons in the brainstem. J Neurosci. 2010;30(47):16053–16064. doi: 10.1523/JNEUROSCI.4508-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shafer B, Onishi K, Lo C, Colakoglu G, Zou Y. Vangl2 promotes Wnt/planar cell polarity-like signaling by antagonizing Dvl1-mediated feedback inhibition in growth cone guidance. Dev Cell. 2011;20(2):177–191. doi: 10.1016/j.devcel.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torban E, et al. Genetic interaction between members of the Vangl family causes neural tube defects in mice. Proc Natl Acad Sci USA. 2008;105(9):3449–3454. doi: 10.1073/pnas.0712126105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song H, et al. Planar cell polarity breaks bilateral symmetry by controlling ciliary positioning. Nature. 2010;466(7304):378–382. doi: 10.1038/nature09129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo N, Hawkins C, Nathans J. Frizzled6 controls hair patterning in mice. Proc Natl Acad Sci USA. 2004;101(25):9277–9281. doi: 10.1073/pnas.0402802101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hébert JM, McConnell SK. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol. 2000;222(2):296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- 39.Chen WS, et al. Asymmetric homotypic interactions of the atypical cadherin flamingo mediate intercellular polarity signaling. Cell. 2008;133(6):1093–1105. doi: 10.1016/j.cell.2008.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stenman J, Toresson H, Campbell K. Identification of two distinct progenitor populations in the lateral ganglionic eminence: Implications for striatal and olfactory bulb neurogenesis. J Neurosci. 2003;23(1):167–174. doi: 10.1523/JNEUROSCI.23-01-00167.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Louvi A, Yoshida M, Grove EA. The derivatives of the Wnt3a lineage in the central nervous system. J Comp Neurol. 2007;504(5):550–569. doi: 10.1002/cne.21461. [DOI] [PubMed] [Google Scholar]
- 42.Fogarty M, et al. Spatial genetic patterning of the embryonic neuroepithelium generates GABAergic interneuron diversity in the adult cortex. J Neurosci. 2007;27(41):10935–10946. doi: 10.1523/JNEUROSCI.1629-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao T, Zhou X, Szabó N, Leitges M, Alvarez-Bolado G. Foxb1-driven Cre expression in somites and the neuroepithelium of diencephalon, brainstem, and spinal cord. Genesis. 2007;45(12):781–787. doi: 10.1002/dvg.20356. [DOI] [PubMed] [Google Scholar]
- 44.Deck M, et al. Pathfinding of corticothalamic axons relies on a rendezvous with thalamic projections. Neuron. 2013;77(3):472–484. doi: 10.1016/j.neuron.2012.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Molnár Z, Blakemore C. How do thalamic axons find their way to the cortex? Trends Neurosci. 1995;18(9):389–397. doi: 10.1016/0166-2236(95)93935-q. [DOI] [PubMed] [Google Scholar]
- 46.Feng G, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28(1):41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 47.Grove EA, Fukuchi-Shimogori T. Generating the cerebral cortical area map. Annu Rev Neurosci. 2003;26:355–380. doi: 10.1146/annurev.neuro.26.041002.131137. [DOI] [PubMed] [Google Scholar]
- 48.Petersen CC. The functional organization of the barrel cortex. Neuron. 2007;56(2):339–355. doi: 10.1016/j.neuron.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 49.Shima Y, et al. Opposing roles in neurite growth control by two seven-pass transmembrane cadherins. Nat Neurosci. 2007;10(8):963–969. doi: 10.1038/nn1933. [DOI] [PubMed] [Google Scholar]
- 50.Shima Y, Kengaku M, Hirano T, Takeichi M, Uemura T. Regulation of dendritic maintenance and growth by a mammalian 7-pass transmembrane cadherin. Dev Cell. 2004;7(2):205–216. doi: 10.1016/j.devcel.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 51.Das G, Reynolds-Kenneally J, Mlodzik M. The atypical cadherin Flamingo links Frizzled and Notch signaling in planar polarity establishment in the Drosophila eye. Dev Cell. 2002;2(5):655–666. doi: 10.1016/s1534-5807(02)00147-8. [DOI] [PubMed] [Google Scholar]
- 52.Strutt D, Johnson R, Cooper K, Bray S. Asymmetric localization of frizzled and the determination of notch-dependent cell fate in the Drosophila eye. Curr Biol. 2002;12(10):813–824. doi: 10.1016/s0960-9822(02)00841-2. [DOI] [PubMed] [Google Scholar]
- 53.Adler PN. Planar signaling and morphogenesis in Drosophila. Dev Cell. 2002;2(5):525–535. doi: 10.1016/s1534-5807(02)00176-4. [DOI] [PubMed] [Google Scholar]
- 54.Ng J. Wnt/PCP proteins regulate stereotyped axon branch extension in Drosophila. Development. 2012;139(1):165–177. doi: 10.1242/dev.068668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Curtin JA, et al. Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr Biol. 2003;13(13):1129–1133. doi: 10.1016/s0960-9822(03)00374-9. [DOI] [PubMed] [Google Scholar]
- 56.Wang J, et al. Dishevelled genes mediate a conserved mammalian PCP pathway to regulate convergent extension during neurulation. Development. 2006;133(9):1767–1778. doi: 10.1242/dev.02347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guirao B, et al. Coupling between hydrodynamic forces and planar cell polarity orients mammalian motile cilia. Nat Cell Biol. 2010;12(4):341–350. doi: 10.1038/ncb2040. [DOI] [PubMed] [Google Scholar]
- 58.Montcouquiol M, et al. Asymmetric localization of Vangl2 and Fz3 indicate novel mechanisms for planar cell polarity in mammals. J Neurosci. 2006;26(19):5265–5275. doi: 10.1523/JNEUROSCI.4680-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park TJ, Mitchell BJ, Abitua PB, Kintner C, Wallingford JB. Dishevelled controls apical docking and planar polarization of basal bodies in ciliated epithelial cells. Nat Genet. 2008;40(7):871–879. doi: 10.1038/ng.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yin H, Copley CO, Goodrich LV, Deans MR. Comparison of phenotypes between different vangl2 mutants demonstrates dominant effects of the Looptail mutation during hair cell development. PLoS One. 2012;7(2):e31988. doi: 10.1371/journal.pone.0031988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bastock R, Strutt H, Strutt D. Strabismus is asymmetrically localised and binds to Prickle and Dishevelled during Drosophila planar polarity patterning. Development. 2003;130(13):3007–3014. doi: 10.1242/dev.00526. [DOI] [PubMed] [Google Scholar]
- 62.Sanchez-Alvarez L, et al. VANG-1 and PRKL-1 cooperate to negatively regulate neurite formation in Caenorhabditis elegans. PLoS Genet. 2011;7(9):e1002257. doi: 10.1371/journal.pgen.1002257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nagaoka T, et al. The Wnt/planar cell polarity pathway component Vangl2 induces synapse formation through direct control of N-cadherin. Cell Reports. 2014;6(5):916–927. doi: 10.1016/j.celrep.2014.01.044. [DOI] [PubMed] [Google Scholar]
- 64.Yoshioka T, Hagiwara A, Hida Y, Ohtsuka T. Vangl2, the planar cell polarity protein, is complexed with postsynaptic density protein PSD-95 [corrected] FEBS Lett. 2013;587(10):1453–1459. doi: 10.1016/j.febslet.2013.03.030. [DOI] [PubMed] [Google Scholar]
- 65.Lewcock JW, Genoud N, Lettieri K, Pfaff SL. The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron. 2007;56(4):604–620. doi: 10.1016/j.neuron.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 66.Bloom AJ, Miller BR, Sanes JR, DiAntonio A. The requirement for Phr1 in CNS axon tract formation reveals the corticostriatal boundary as a choice point for cortical axons. Genes Dev. 2007;21(20):2593–2606. doi: 10.1101/gad.1592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garel S, Rubenstein JL. Intermediate targets in formation of topographic projections: Inputs from the thalamocortical system. Trends Neurosci. 2004;27(9):533–539. doi: 10.1016/j.tins.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 68.Joshi PS, et al. Bhlhb5 regulates the postmitotic acquisition of area identities in layers II-V of the developing neocortex. Neuron. 2008;60(2):258–272. doi: 10.1016/j.neuron.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lokmane L, et al. Sensory map transfer to the neocortex relies on pretarget ordering of thalamic axons. Curr Biol. 2013;23(9):810–816. doi: 10.1016/j.cub.2013.03.062. [DOI] [PubMed] [Google Scholar]
- 70.Kashani AH, et al. Calcium activation of the LMO4 transcription complex and its role in the patterning of thalamocortical connections. J Neurosci. 2006;26(32):8398–8408. doi: 10.1523/JNEUROSCI.0618-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zembrzycki A, Chou SJ, Ashery-Padan R, Stoykova A, O’Leary DD. Sensory cortex limits cortical maps and drives top-down plasticity in thalamocortical circuits. Nat Neurosci. 2013;16(8):1060–1067. doi: 10.1038/nn.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Farley FW, Soriano P, Steffen LS, Dymecki SM. Widespread recombinase expression using FLPeR (flipper) mice. Genesis. 2000;28(3-4):106–110. [PubMed] [Google Scholar]
- 73.Lallemand Y, Luria V, Haffner-Krausz R, Lonai P. Maternally expressed PGK-Cre transgene as a tool for early and uniform activation of the Cre site-specific recombinase. Transgenic Res. 1998;7(2):105–112. doi: 10.1023/a:1008868325009. [DOI] [PubMed] [Google Scholar]
- 74.Kessaris N, et al. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. 2006;9(2):173–179. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yoshida M, Assimacopoulos S, Jones KR, Grove EA. Massive loss of Cajal-Retzius cells does not disrupt neocortical layer order. Development. 2006;133(3):537–545. doi: 10.1242/dev.02209. [DOI] [PubMed] [Google Scholar]
- 76.Shima Y, et al. Differential expression of the seven-pass transmembrane cadherin genes Celsr1-3 and distribution of the Celsr2 protein during mouse development. Dev Dyn. 2002;223(3):321–332. doi: 10.1002/dvdy.10054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.