

Significance
DEAD-box proteins are ubiquitous in RNA metabolism and can act as RNA chaperones by promoting folding and rearrangements of structured RNAs. However, the mechanistic basis for these chaperone activities is not well understood. Here we show that the DEAD-box protein CYT-19 unfolds a large, structured RNA molecule in a stability-dependent fashion and is more active toward less compact conformations of the RNA. Our results indicate that CYT-19 functions predominantly by disrupting accessible RNA secondary structure and depends on spontaneous openings in tightly packed RNAs to gain access to RNA helices. Unwinding of exposed secondary structure promotes global RNA unfolding and facilitates formation of new structures. Analogous mechanisms are observed in other molecular chaperones and are likely widespread among DEAD-box helicases.
Keywords: RNA folding, RNA misfolding, RNA tertiary structure, RNA unwinding, superfamily 2 helicase
Abstract
DEAD-box proteins are nonprocessive RNA helicases and can function as RNA chaperones, but the mechanisms of their chaperone activity remain incompletely understood. The Neurospora crassa DEAD-box protein CYT-19 is a mitochondrial RNA chaperone that promotes group I intron splicing and has been shown to resolve misfolded group I intron structures, allowing them to refold. Building on previous results, here we use a series of tertiary contact mutants of the Tetrahymena group I intron ribozyme to demonstrate that the efficiency of CYT-19–mediated unfolding of the ribozyme is tightly linked to global RNA tertiary stability. Efficient unfolding of destabilized ribozyme variants is accompanied by increased ATPase activity of CYT-19, suggesting that destabilized ribozymes provide more productive interaction opportunities. The strongest ATPase stimulation occurs with a ribozyme that lacks all five tertiary contacts and does not form a compact structure, and small-angle X-ray scattering indicates that ATPase activity tracks with ribozyme compactness. Further, deletion of three helices that are prominently exposed in the folded structure decreases the ATPase stimulation by the folded ribozyme. Together, these results lead to a model in which CYT-19, and likely related DEAD-box proteins, rearranges complex RNA structures by preferentially interacting with and unwinding exposed RNA secondary structure. Importantly, this mechanism could bias DEAD-box proteins to act on misfolded RNAs and ribonucleoproteins, which are likely to be less compact and more dynamic than their native counterparts.
DEAD-box proteins constitute the largest family of RNA helicases and function in all stages of RNA metabolism (1, 2). In vivo, many DEAD-box proteins have been implicated in assembly and conformational rearrangements of large structured RNAs and ribonucleoproteins (RNPs), including the ribosome, spliceosome, and self-splicing introns (3). Thus, it is important to establish how these proteins use their basic mechanisms of RNA binding and helix unwinding to interact with and remodel higher-order RNA structures.
Structural and mechanistic studies have elucidated the basic steps of the ATPase cycle of DEAD-box proteins and have provided an understanding of the coupling between ATPase and duplex unwinding activities (4–11). The conserved helicase core consists of two flexibly linked RecA-like domains that contain at least 12 conserved motifs, including the D-E-A-D sequence in the ATP-binding motif II (3, 12). Binding of ATP and double-stranded RNA to domains 1 and 2, respectively, induces domain closure, which completes the formation of an ATPase active site at the domain interface and introduces steric clashes in the RNA binding site, leading to the displacement of one of the RNA strands (6, 7). ATP hydrolysis and inorganic phosphate release are then thought to regenerate the open enzyme conformation (4, 8, 13). Unlike conventional helicases, DEAD-box proteins have not been found to translocate, limiting the unwinding activity to short helices that can be disrupted in a single cycle of ATP binding and hydrolysis (4, 8, 9, 14–16). This mechanism is compatible with the physiological roles of DEAD-box proteins, because cellular RNAs rarely contain continuous base-paired regions that are longer than one or two helical turns.
The interactions of DEAD-box proteins with structured RNAs have been extensively studied using two homologous proteins that function as general RNA chaperones: CYT-19 from Neurospora crassa and Mss116 from Saccharomyces cerevisiae. In vivo, CYT-19 is required for efficient splicing of several mitochondrial group I introns and can promote splicing of group I and group II introns in yeast mutants that lack functional Mss116 (17, 18). Both proteins have been shown to act as general RNA chaperones during group I and group II intron folding in vitro and are thought to act primarily by reversing misfolding of the intron RNAs, although additional mechanisms may be used for some substrates (17–23). Importantly, the chaperone activities of these and other DEAD-box proteins correlate with their ATP-dependent helix unwinding activities, suggesting that DEAD-box proteins function by lowering the energy barriers for transitions between alternative structures that involve disruption of base pairs (24, 25).
In vitro studies using the group I intron ribozyme from Tetrahymena thermophila have been instrumental in probing the chaperone mechanism of CYT-19 (17, 26–28). This ∼400-nt RNA folds into a compact, globular structure composed of a conserved core and a series of peripheral elements that encircle the core by forming long-range tertiary contacts (Fig. 1) (29–31). Upon addition of Mg2+ ions, the majority of the ribozyme population becomes trapped in a long-lived misfolded conformation, which then slowly refolds to the native state (32). The misfolded intermediate is remarkably similar to the native ribozyme, forming a complete native network of secondary and tertiary interactions and a globally compact fold (33, 34). Despite these similarities, refolding to the native state requires extensive unfolding, including disruption of all five peripheral tertiary contacts and the core helix P3 (33, 35). To explain these results, a topological error has been proposed, wherein two single-stranded joining elements are crossed incorrectly in the core of the misfolded ribozyme, and transient disruption of the surrounding native structure is required for refolding (33, 35).
Fig. 1.
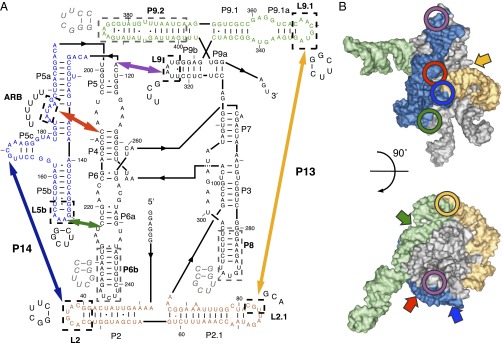
The Tetrahymena group I intron ribozyme. (A) Secondary structure and mutations. Peripheral elements are colored and thick arrows mark the long-range peripheral tertiary contacts. Paired regions (P) and loops that were mutated in this study (L) are labeled based on group I intron nomenclature in ref. 31. The mutated regions are enclosed in dashed boxes and labeled in bold, with sequence substitutions indicated nearby. Sequences that were deleted to construct the helix truncation mutants (Fig. 6) are enclosed in gray dashed boxes and the replacement nucleotides are shown in gray italic font. (B) Tertiary structure model of the ribozyme (31). Peripheral elements (colored surface) and the locations of the long-range peripheral tertiary contacts (circles) are highlighted using the same color scheme as in A. The ribozyme core is shown in silver. The block arrows indicate the approximate positions of tertiary contacts not visible in each respective view of the ribozyme. The figures were prepared using PyMOL.
Given the structural similarity between the native and misfolded ribozyme, it is interesting that CYT-19 can accelerate refolding of the misfolded intermediate by at least an order of magnitude without detectably unfolding the native ribozyme (26). Insights into this apparent preference for the misfolded ribozyme came from studies of two ribozyme mutants in which the tertiary structure was destabilized, making the stability of the native ribozyme comparable to that of the misfolded intermediate (28). CYT-19 unfolded the native and misfolded conformers of these mutants with comparable efficiencies, suggesting that the efficiency of chaperone-mediated unfolding depended on the stability of ribozyme tertiary structure. However, the mutations studied were concentrated in one region of the ribozyme, leaving open the possibility that CYT-19 recognizes local disruptions rather than global stability.
Here we investigate the roles of RNA stability in CYT-19-mediated unfolding of the Tetrahymena ribozyme by using a series of ribozyme mutants with disruptions of each of the five peripheral tertiary contacts. We observe a strong correlation between CYT-19 activity and global stability of ribozyme tertiary structure. Further, we find that the RNA-dependent ATPase activity of CYT-19 depends on the accessibility of secondary structure in the ribozyme. Our results lead to a general model for recognition and remodeling of unstable or incorrectly folded RNAs by a DEAD-box protein.
Results
Efficiency of CYT-19–Mediated Unfolding Increases with Lower Ribozyme Stability.
To systematically address the roles of RNA stability in chaperone-mediated unfolding, we used a set of previously characterized Tetrahymena ribozyme variants in which the five long-range peripheral tertiary contacts are individually mutated (Fig. 1) (33, 36, 37). The mutations include substitutions of the A-rich bulge (ARB) of the metal ion core/metal ion core receptor interaction; the tetraloops L9 and L5b, which form two tetraloop/tetraloop receptor interactions; the L2 loop of the “kissing loop” interaction P14; and the L2.1 and L9.1 loops of the P13 kissing loop (Fig. 1). The ARB mutant was one of the ribozymes used in the previous study of CYT-19-mediated ribozyme unfolding (28). Footprinting approaches have shown increased Mg2+ requirements for tertiary folding by each of these and related tertiary contact mutants, indicating decreased global stability (37–39). Here we focused on the native, catalytically active state of each ribozyme, allowing us to accurately quantify the stability and chaperone-mediated unfolding by measuring oligonucleotide cleavage activity (28, 40). Although rearrangements of native group I intron structure are presumably not a physiological function of CYT-19, the principles of CYT-19-mediated unfolding are likely to be largely the same for resolving misfolded intermediates, which can strongly resemble their native counterparts.
To determine the stabilities of the mutant ribozymes in the native state, we measured the Mg2+-concentration dependences for equilibrium folding. We used a version of the previously described two-stage catalytic activity assay to determine the fraction of native ribozyme at each Mg2+ concentration (40, 41). After folding at 0–10 mM Mg2+, the ribozymes were transferred to 50 mM Mg2+, which blocks folding transitions from the native state and traps most of the nonnative ribozyme in the long-lived misfolded conformation, while permitting robust oligonucleotide cleavage activity by the native ribozyme. The oligonucleotide substrate, added subsequently in trace amount, is bound both by the native and the misfolded ribozymes but can only be cleaved by the native ribozyme. Thus, the fraction of substrate that is cleaved in a single turnover reflects the fraction of native ribozyme (40).
All of the mutants displayed greater Mg2+ requirements than the wild-type ribozyme, consistent with previous determinations by hydroxyl radical footprinting and oligonucleotide hybridization (37, 39, 42, 43) (Fig. 2A and Fig. S1). We calculated the native ribozyme stabilities based on the equilibrium values at 2 mM Mg2+, where all of the mutants populated quantifiable ratios of native to nonnative ribozyme, avoiding extrapolation-based approaches (Materials and Methods) (44). The native ribozyme stabilities at 2 mM Mg2+ ranged across ∼3 kcal/mol from the L9 mutant (most stable) to the ARB mutant (least stable). Note that the native wild-type ribozyme is too stable under these conditions for direct determination of the folding equilibrium.
Fig. 2.

CYT-19–mediated ribozyme unfolding. (A) Mg2+-dependent folding of WT (black) and mutant ribozymes. The mutants are designated by the tertiary contact component that is mutated: L9 (purple), L9.1 (orange circles), L2.1 (orange diamonds), L5b (green), L2 (blue), and ARB (red). For clarity, only representative isotherms are shown, with cumulative data shown in Fig. S1. The dashed line at 2 mM Mg2+ marks the condition used for unfolding experiments with CYT-19. The maximal measured fractions of native ribozyme were <1 and varied between ribozyme preparations, presumably because of a small and variable amount of damaged RNA (32). (B) Unfolding of the native ribozyme by CYT-19 (2 mM ATP⋅Mg2+). Representative time courses in the presence of CYT-19 buffer only (○) or 100 nM (△), 200 nM (◇), 300 nM (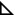 ), 400 nM (□), or 500 nM (▽) CYT-19 are shown. (C) Correlation of CYT-19–mediated unfolding rate constants with ribozyme stability in the presence of 100 µM and 2 mM ATP⋅Mg2+. Horizontal error bars indicate SEs from two or more determinations of ribozyme stability, and vertical error bars represent SEs from linear fits through cumulative data of CYT-19-dependent unfolding rates (Figs. S2B and S3; see Materials and Methods). In cases where the concentration dependences displayed upward curvature, the error bars correspond to the upper and lower limits of the unfolding efficiencies. For the L5b mutant, the reported efficiencies are derived from the fast phase of unfolding. The mutants are slightly less stable in 2 mM ATP⋅Mg2+ than in 100 µM ATP⋅Mg2+, most likely reflecting sequestration of Mg2+ ions by residual free ATP. The L5b mutant is the most sensitive to this change owing to its steep Mg2+ concentration dependence. The arrow indicates that the ∆G value for the L9 mutant is an upper limit because the amplitude of unfolding was too small under these conditions to measure the equilibrium accurately.
), 400 nM (□), or 500 nM (▽) CYT-19 are shown. (C) Correlation of CYT-19–mediated unfolding rate constants with ribozyme stability in the presence of 100 µM and 2 mM ATP⋅Mg2+. Horizontal error bars indicate SEs from two or more determinations of ribozyme stability, and vertical error bars represent SEs from linear fits through cumulative data of CYT-19-dependent unfolding rates (Figs. S2B and S3; see Materials and Methods). In cases where the concentration dependences displayed upward curvature, the error bars correspond to the upper and lower limits of the unfolding efficiencies. For the L5b mutant, the reported efficiencies are derived from the fast phase of unfolding. The mutants are slightly less stable in 2 mM ATP⋅Mg2+ than in 100 µM ATP⋅Mg2+, most likely reflecting sequestration of Mg2+ ions by residual free ATP. The L5b mutant is the most sensitive to this change owing to its steep Mg2+ concentration dependence. The arrow indicates that the ∆G value for the L9 mutant is an upper limit because the amplitude of unfolding was too small under these conditions to measure the equilibrium accurately.
To measure how stability affects CYT-19-mediated unfolding, we formed native ribozymes at 10 mM Mg2+ and initiated unfolding by diluting Mg2+ to 2 mM with simultaneous addition of CYT-19 and ATP. The fraction of native ribozyme at different time points was then determined by using the ribozyme activity assay, with proteinase K added to the folding quench to degrade CYT-19 before substrate cleavage (24, 28). We found that CYT-19 accelerated unfolding of all mutant ribozymes, with rates that increased with CYT-19 and ATP concentrations and with little or no acceleration in the absence of ATP (Fig. 2B and Figs. S2–S4) (28). Importantly, the apparent efficiencies of CYT-19-mediated unfolding, derived from CYT-19 concentration dependences of the unfolding rates, tracked with ribozyme stability (Fig. 2C and Figs. S2B and S3). The differences between the unfolding efficiencies of the most stable (L9) and the least stable (ARB) mutant were ∼70-fold and ∼20-fold at subsaturating (100 µM) and near-saturating (2 mM) ATP concentrations. Interestingly, the L5b tetraloop mutant showed one of the lowest CYT-19–mediated unfolding efficiencies despite an intermediate stability. This mutant also had one of the lowest spontaneous unfolding rates, suggesting that the intrinsic ribozyme propensity to unfold may play a role in CYT-19–mediated disruption (Fig. 2C and Fig. S5). Overall, our results indicate that CYT-19 efficiency is linked to the global stability of the ribozyme and suggest a role for spontaneous unfolding events in promoting CYT-19–mediated ribozyme unfolding.
ATPase Activity Increases with Lower Tertiary Stability.
To understand the mechanistic origins of the relationship between CYT-19–mediated unfolding and RNA tertiary stability, we tested two simple models. In the first model, less-stable ribozyme mutants would provide more opportunities for productive CYT-19 interactions, for example because their structures are more dynamic and on average less compact at 2 mM Mg2+. In the second model, CYT-19 would perform the same initial rearrangements in all mutants, with less-stable ribozymes being more likely to unfold following these rearrangements. We tested these two possibilities by measuring the RNA-dependent ATPase activity of CYT-19 under the conditions of the unfolding assays, with the prediction that the initial ATPase rates should increase with lower ribozyme stability (model 1) or remain constant regardless of stability (model 2).
The ATPase stimulation by each ribozyme was measured immediately following transfer of the prefolded native ribozyme to 2 mM Mg2+ under conditions identical to those of unfolding assays. We also measured ATPase stimulation by the native wild-type ribozyme, which is stably folded under these conditions (Fig. 2A), and a quintuple mutant, which contains mutations of all five peripheral tertiary contacts and has been shown by small-angle X-ray scattering (SAXS) to lack a stable compact structure (45). The ATPase rates showed a clear increase with decreasing native ribozyme stability, with larger differences observed in the presence of subsaturating ATP (Fig. 3 and Fig. S6). The strongest ATPase stimulation was measured with the quintuple mutant, indicating that the lack of compact tertiary structure stimulated CYT-19 activity. Thus, our results support the first model, in which CYT-19 has more opportunities to interact with the less-stable ribozymes.
Fig. 3.

Relationship between ATP hydrolysis rates and ribozyme stability (100 µM ATP, 500 nM CYT-19, 200 nM ribozyme, and 2 mM Mg2+). The color scheme for ribozyme mutants is the same as in Fig. 1. Dashed lines indicate the ATPase stimulation by the wild-type ribozyme (black, 1.37 ± 0.06 µM min−1) and the quintuple mutant (gray, 5.6 ± 0.4 µM min−1), as well as the RNA-independent ATPase rate (cyan, 0.05 ± 0.02 µM min−1). Vertical error bars indicate SE from at least four determinations.
CYT-19 Activity Tracks with Mg2+-Dependent Ribozyme Folding.
As an orthogonal approach to modulating ribozyme stability by mutations, we tested the effects of Mg2+ ion concentration on the ATPase stimulation. We hypothesized that the ATPase rates should mirror the Mg2+-dependent folding isotherms of each individual ribozyme, with the ATPase rate decreasing at increasing Mg2+ concentrations as more native ribozyme is present at equilibrium.
A subset of ribozyme variants (L2, L5b, and ARB mutants) were folded for ∼24 h at various Mg2+ concentrations and ATPase measurements were initiated by adding CYT-19 and 100 µM ATP. As expected, the ATPase rates decreased with increasing Mg2+ concentration around the respective folding midpoints of each mutant (Fig. S7). Although the ATPase rates increased with Mg2+ concentration at <2 mM Mg2+, this increase was independent of tertiary structure formation, as it was also observed with the quintuple mutant and oligonucleotide helices (Fig. S7) (8). The weak ATPase activity at low Mg2+ concentrations may result from slow release of RNA, limiting nucleotide turnover (46), or from Mg2+-dependent secondary structure stabilization (8). To isolate the effects of tertiary folding, we normalized the ATPase rates for each mutant by the corresponding rate for the quintuple mutant ribozyme at the same Mg2+ concentration (Fig. 4). The normalization uncovered a clear correspondence between the Mg2+-dependent fraction of native ribozyme and the ATPase stimulation across the whole tested range of Mg2+ concentrations. The small decreases in ATPase stimulation at high Mg2+ concentrations, beyond those required to support folding, could be caused by decreasing dynamics of the native ribozyme structures. Overall, the results support the role of global tertiary structure stability in modulating CYT-19 activity. Further, the observation that the largest changes in ATPase stimulation are centered on the folding midpoints, where the ratios of native to unfolded ribozyme are changing the most strongly, suggest that CYT-19 acts preferentially upon unfolded, noncompact ribozyme molecules within a population.
Fig. 4.
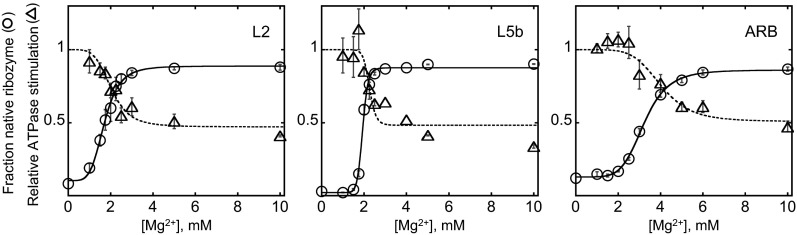
Correspondence between the ATPase stimulation (△) and the fraction of native ribozyme at equilibrium (○) for the L2, L5b, and ARB mutants. The ATPase rates were normalized by the rates measured at the same Mg2+ concentration with the quintuple mutant to account for tertiary folding-independent effects of Mg2+ on the ATPase activity (Fig. S7A). The Mg2+ concentration dependences of ATPase activity are reasonably well fit by the Hill equation (dashed curves), with the Hill coefficient fixed to match that of the folding isotherm for each mutant (solid curves) and the starting value fixed to 1, with minor deviation present for the L5b mutant.
To directly address the role of ribozyme compactness in the stability dependence of CYT-19 activity, we compared the ribozyme mutants by SAXS at 2 mM and 10 mM Mg2+. As expected, at 2 mM Mg2+ the radius of gyration (Rg) values of equilibrated ribozymes increased with lower native ribozyme stability, reflecting increased populations of noncompact forms (Fig. 5A and Fig. S8). Differences in compactness were also reflected in the shapes of the Kratky plots, where a pronounced parabolic peak indicates compact, globular particles, e.g., for the native wild-type ribozyme, whereas flat or ascending profiles indicate noncompact structures, e.g., for the quintuple mutant (47) (Fig. 5B). The peaks of the single tertiary contact mutants fall between those of the wild-type and the quintuple mutant ribozymes in an order corresponding to their stabilities (Fig. 5B). Further, at 10 mM Mg2+, which gives predominantly native folding in all ribozymes except the quintuple mutant, both the Rg values and the ATPase rates were clustered around the corresponding values for the wild-type ribozyme (Fig. 5 C and D), providing additional support for a link between compactness and ATPase stimulation. As expected, the quintuple mutant both maintained an expanded structure and continued to give strong stimulation of ATPase activity, and small enhancements in the Rg and ATPase stimulation were also observed for the ARB mutant ribozyme (Fig. 5 C and D). Together, the SAXS and ATPase measurements indicate that CYT-19 activity scales with ribozyme compactness, with noncompact ribozymes providing more productive interaction opportunities.
Fig. 5.

ATPase stimulation tracks with ribozyme compactness. (A) Relationship between the Rg value of the equilibrium ribozyme population and the ATPase stimulation by ribozymes equilibrated at 2 mM Mg2+. The Rg values were determined by P(R) analysis using Gnom (65). The error bars indicate SEs from two to three independent measurements. (B) Kratky plots of ribozymes equilibrated at 2 mM Mg2+. Scattering profiles are shown for the wild-type ribozyme (black), the quintuple mutant (gray), and the single tertiary contact mutants (color scheme as in Fig. 1, with the profiles of the P13 mutants L9.1 and L2.1 shown as solid and dotted lines, respectively). Representative Kratky plots acquired during the same beam time are shown. (C) Clustering of the Rg values and the ATPase rates at 10 mM Mg2+. The Rg values represent averages from measurements performed during a single beam time using 1 µM and 2 µM ribozyme. (D) Kratky plots of ribozymes equilibrated at 10 mM Mg2+. I, intensity (in arbitrary units); q, scattering vector (q = 2sinθ/λ, where 2θ is the scattering angle and λ is the X-ray wavelength).
CYT-19 Interacts with Exposed Ribozyme Helices.
Interestingly, the range of ATPase rates with the ribozyme mutants was much narrower than that of the unfolding efficiencies, and substantial ATPase stimulation was observed even with the most stable native ribozymes (approximately fourfold range from the wild-type ribozyme to the quintuple mutant) (Fig. 3). Considering CYT-19 activation by noncompact structures, we hypothesized that CYT-19 may interact with helices that are constitutively exposed on the surface of the folded ribozyme. The most likely candidates are P6b, P8, and P9.2, which prominently extend away from the ribozyme body and are not constrained by tertiary interactions (Fig. 6A). Therefore, we generated a set of mutants with these helices truncated (Fig. 1A). The mutants resembled the wild-type ribozyme in their stabilities and substrate cleavage rates, indicating that the structures were not perturbed beyond the truncated helices (Fig. S9). ATPase measurements revealed small but reproducible decreases in all individual truncation mutants and an additive, ∼30%, decrease with a P9.2 and P6b double truncation mutant (Fig. 6B). Counterintuitively, a triple truncation mutant showed no detectable decrease in ATPase stimulation relative to the wild-type ribozyme at 2 mM Mg2+. However, at 10 mM Mg2+ the ATPase stimulation decreased to 20% below WT, suggesting that at 2 mM Mg2+, decreased local stability of the mutant may oppose the decrease in ATPase rate owing to truncations (Fig. 6C). Adding the differences from the individual truncation mutants yields an estimated twofold reduction in ATPase stimulation relative to the wild-type ribozyme, suggesting that interactions with these helices underlie at least half of the ATPase stimulation by the folded ribozyme. This estimate is a lower limit because some residual ATPase activity for the mutants may arise from CYT-19 interactions with the “stumps” of the truncated helices, and for both the wild-type and mutant ribozymes small amounts of damaged, incompletely folded ribozyme may contribute to ATPase activity.
Fig. 6.
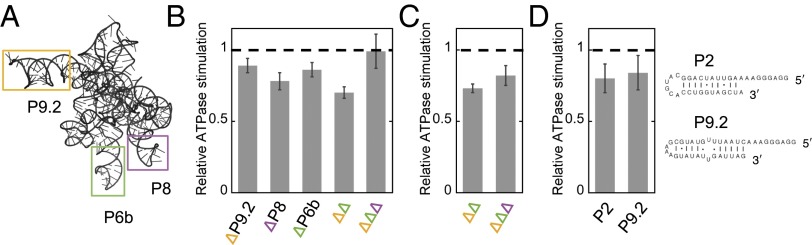
ATPase stimulation by exposed ribozyme helices. (A) Ribozyme model with helices P6b, P8, and P9.2 highlighted. In the truncation mutants, the helices were replaced with shorter stems and UUCG tetraloops (Fig. 1A). (B) ATPase stimulation by helix truncation mutants (100 µM ATP, 500 nM CYT-19, 200 nM native ribozyme, and 2 mM Mg2+). The ATPase rates were normalized by the rates measured with the wild-type ribozyme in the same experiment. The error bars indicate SEs from 5 to 6 measurements. ∆∆ and ∆∆∆ denote double (∆P9.2 + ∆P6b) and triple truncations, respectively. Two independent preparations of each ribozyme were measured to ensure that the small differences were not artifacts owing to differences between preparations. (C) ATPase stimulation by the double and triple truncation mutants at 10 mM Mg2+. ATPase rates are normalized by the value measured for the wild-type ribozyme in the same experiment. Error bars indicate SEs from four measurements using two ribozyme preparations. (D) ATPase stimulation by isolated RNA helices, corresponding to ribozyme helices P2 and P9.2. Shown are ATPase rates relative to the ATPase stimulation by the same molar concentration (200 nM) of wild-type ribozyme measured in the same experiment (100 µM ATP, 500 nM CYT-19, and 2 mM Mg2+).
To further address the roles of helix accessibility in stimulating CYT-19 activity, we compared ATPase stimulation by equimolar concentrations of the wild-type ribozyme and two isolated oligonucleotide helices corresponding to parts of the ribozyme (Fig. 6D). Interestingly, the oligonucleotides stimulated the ATPase activity to nearly the same level as the wild-type ribozyme (within 20%) (Fig. 6D). This result suggests that readily accessible secondary structure elements can interact productively with CYT-19 and that only a few favorable sites are available in the stably folded wild-type ribozyme. Together, the results in this section reveal three CYT-19 interaction sites within the fully folded Tetrahymena ribozyme and provide further support for a model in which CYT-19 interacts preferentially with exposed or protruding RNA helices on the surfaces of structured RNAs.
Discussion
Here, we investigated the ATP-dependent interactions of the DEAD-box protein CYT-19 with a group I intron ribozyme, a relative of the physiological group I intron substrates of CYT-19. We found that the efficiency of CYT-19–mediated unfolding tracks with the global tertiary structure stability of the ribozyme and that the ATPase activity of CYT-19 increases with decreasing compactness of the ribozyme structure. Based on these and previous results, we describe a model for general RNA chaperone activity in which CYT-19, and likely other DEAD-box proteins, remodels structured RNAs and RNPs primarily by binding and disrupting accessible secondary structure. Importantly, the preference for noncompact RNA structures can bias DEAD-box proteins to remodel misfolded RNAs and RNPs, in a manner conceptually similar to unrelated RNA and protein chaperones.
Model for DEAD-Box Protein-Mediated RNA Rearrangements.
The model that emerges for DEAD-box protein-mediated global rearrangements of group I intron structure is shown in Fig. 7. The basic features of this model are likely to apply generally to DEAD-box protein-mediated remodeling of misfolded RNAs as well as to native RNAs that undergo rearrangements during their functions. Based on our finding that compact tertiary packing of the ribozyme limits productive interactions with CYT-19, the first step is a spontaneous disruption of the compact RNA structure. This disruption, shown as a dynamic fluctuation at a peripheral tertiary contact, exposes one or more helices that can then be bound by the protein. A small number of helices that are exposed constitutively in the folded ribozyme can also interact and be unwound locally, as suggested by the ATPase activation by the folded ribozyme and the decreased ATPase activity upon truncation of extended helices. However, the strong dependence of unfolding efficiency on ribozyme stability suggests that unfolding of the Tetrahymena ribozyme is driven primarily by CYT-19 interactions with transiently exposed helices.
Fig. 7.

Model for CYT-19–mediated unfolding of structured RNA. The unfolding process is illustrated for the Tetrahymena ribozyme, shown schematically with black cylinders representing base-paired regions. The long-range peripheral tertiary contacts are shown as orange lines. Gray hairpins indicate the helices that are exposed and accessible for CYT-19 in the native structure but are not productive sites for unfolding. From left to right, unfolding is initiated as a spontaneous local disruption, which exposes helical segments that can be bound by CYT-19 (highlighted in yellow). ATP-dependent unwinding can induce further unfolding, exposing additional helices that can also be unwound by one or more CYT-19 monomers. The unfolding process is ultimately followed by refolding, giving formation of a new structure or a return to the initial structure.
The binding of an exposed helix and its subsequent unwinding by CYT-19 are both likely to contribute to larger-scale unfolding. By capturing a helix, CYT-19 can block reformation of tertiary contacts between the helix and the rest of the ribozyme, which may also destabilize structure that forms cooperatively, promoting further unfolding steps. ATP-dependent unwinding of the captured helix then furthers the overall unfolding, both by itself and because the distortions of local structure may propagate to adjacent or nearby helical segments. Both helix binding and unwinding are likely to result in the largest disruptions when involving transiently exposed helices because the tertiary interactions and cooperativity that hold these helices in place in the ground state are lost upon interaction with the DEAD-box protein.
As additional helices are exposed by initial unfolding events they are also expected to become productive sites for DEAD-box protein binding and unwinding. Multiple protein monomers may participate in structure disruption, and the upward curvature in the CYT-19 concentration dependence of both the unfolding and ATPase activities for some ribozymes is consistent with this hypothesis (Figs. S2B, S3, and S6B) (25). It is not clear whether multiple monomers function independently or cooperatively, or whether they can act antagonistically in some RNA folding transitions. As unfolding progresses, spontaneous formation of new contacts and reformation of disrupted structure compete with the unfolding steps, and the ribozyme can ultimately fold to the native state or the long-lived misfolded conformation.
A key feature of the model described here is that CYT-19 does not directly disrupt tertiary contacts, but instead relies on spontaneous losses of tertiary structure. Although it remains possible that CYT-19 can actively disrupt some types of tertiary interactions, the dependence of ATPase activity on overall compaction of the ribozyme indicates that the dominant activity of CYT-19 is to interact with accessible secondary structure and promote ATP-dependent unwinding. Further, the dependence of unfolding efficiency on ribozyme stability indicates that binding and unwinding of these helical segments leads to global unfolding of the RNA. Thus, it seems that even in the context of RNAs with extensive tertiary structure the ATP-dependent activity of CYT-19 is primarily as a helicase, with ATP being used to unwind helical segments during RNA structural rearrangements.
Implications for Misfolded RNAs and RNPs.
Analogous to the tertiary contact mutants studied here, misfolded RNAs will typically have less-compact structures than the intact native RNA, either dynamically, owing to lower stability and thus increased frequency of fluctuations, or constitutively, owing to incorrect packing of helices that may leave some helices unprotected and accessible to DEAD-box proteins. The increased accessibility of the helical segments is expected to lead to preferential disruption of misfolded structures by DEAD-box proteins, providing the RNA with new opportunities to refold to the native state. Ultimately, the native conformer can accumulate if it is more stable and therefore protects its helical segments more effectively against fortuitous interactions with DEAD-box proteins.
In addition to signaling incorrect tertiary folding, exposed helices could also indicate failure to bind proteins during RNP assembly, analogous to recognition of exposed single-stranded termini by Ro and La chaperones (48–50). The preferential disruption of exposed secondary structure by DEAD-box proteins could play a role, for example, in proofreading mechanisms and structural transitions of the spliceosome or during ribosome maturation, which require multiple helicases (51, 52).
Comparisons with Other Chaperones.
Our results are reminiscent of previous observations for other general RNA chaperones, which often act by recognizing structural elements that are protected by stable folding and RNP formation but can be exposed in defective RNAs. The retroviral nucleocapsid peptide and the unwinding domain of the human hnRNP A1 protein bind single-stranded RNA and contain motifs for exposed guanosine bases (53, 54). These proteins are proposed to chaperone rearrangements during dimerization of retroviral RNA by interacting with accessible guanosines in the monomer structure (55). The ATP-independent bacterial protein StpA shows increased refolding activity with destabilized mutants of a viral group I intron and may rely on enhanced exposure of potential interaction sites within the structurally unstable introns (56). A key difference is the use of ATP by DEAD-box proteins, which allows them to efficiently disrupt structure while also being able to switch via ATP hydrolysis and Pi release to a state that dissociates rapidly from the single-stranded RNA after unwinding (4, 8).
Despite important differences between structural properties of RNA and proteins, the preference of CYT-19 for exposed RNA helices also resembles substrate recognition modes by several protein chaperones. For instance, Hsp70 chaperones bind to hydrophobic regions, which are buried in native proteins and, when exposed, are common markers for protein misfolding (57). Hsp90 chaperones preferentially bind unstable, flexible protein structures, and the heat-shock cognate protein 70 initiates clathrin coat disassembly by capturing conformational fluctuations at the C termini of clathrin units (58–60). Hence, stimulation by open, flexible structures appears to be a general theme in recognition of folding errors by molecular chaperones.
Materials and Methods
Materials.
Ribozyme mutants were prepared using the QuikChange protocol (Agilent Technologies) with primers from Integrated DNA Technologies. All mutations were confirmed by sequencing. The ribozymes were transcribed by T7 RNA polymerase and purified using Qiagen RNeasy columns as described (61). Concentrations were determined by absorbance at 260 nm using an extinction coefficient of 3.9 × 106 M−1 cm−1 for the wild-type ribozyme and tertiary contact mutants (61). Extinction coefficients for the truncation mutants were calculated based on sequence changes relative to the wild-type ribozyme and were 2.5–12.4% lower.
The RNA oligonucleotides (Dharmacon) were deprotected following the manufacturer’s instructions and purified by ethanol precipitation. Oligonucleotide concentrations were determined using extinction coefficients supplied by the manufacturer (P2: 371,400 M−1 cm−1; P9.2: 421,500 M−1 cm−1). The ribozyme substrate rSA5 (CCCUCUA5) was 5′-end-labeled with [γ-32P]ATP (Perkin-Elmer) using T4 polynucleotide kinase (62), purified by nondenaturing PAGE [20%; 7 M urea, 1× TBE buffer (89 mM Tris-borate, 2 mM EDTA, pH 8.3)], and stored at −20 °C in TE buffer (10 mM Tris⋅Cl, pH 8.0, and 1 mM EDTA).
CYT-19 was expressed and purified as described (28) and stored in single-use aliquots at −80 °C in storage buffer (CYT-19 buffer) containing 20 mM Tris⋅Cl (pH 7.5), 500 mM KCl, 1 mM EDTA, 0.2 mM DTT, and 50% (vol/vol) glycerol. The protein concentration was determined via Bradford assay using BSA as a standard.
Stability Determination of Tetrahymena Ribozyme Mutants.
The Mg2+ dependence for native ribozyme folding was measured using catalytic activity essentially as described (40). Briefly, the ribozymes (200 nM) were incubated with 0–10 mM Mg(OAc)2, 50 mM Na-Mops (pH 7.0), 10% CYT-19 buffer [final 2 mM Tris⋅Cl (pH 7.5), 50 mM KCl, 0.1 mM EDTA, 0.02 mM DTT, and 5% glycerol (vol/vol)], and 2 mM ATP⋅Mg2+ at 25 °C. ATP and CYT-19 buffer were included in the equilibrium incubations to match the conditions used to probe CYT-19–dependent unfolding. Here and below, ATP was added in an equimolar mix with Mg(OAc)2. After ∼24 h, aliquots of the folding reactions were transferred to a twofold excess of folding quench, containing 70–75 mM MgCl2 (to give a final concentration of 50 mM Mg2+), 750 µM guanosine (500 µM final), and 50 mM Na-Mops (pH 7.0). After ≥2 min at 25 °C to ensure that the RNA was trapped in the stable native or misfolded conformations, a trace amount of 5′-[32P]-labeled substrate was added (one-seventh total volume) and the cleavage reaction was stopped after 1 min by adding two volumes of 72% formamide (vol/vol), 100 mM EDTA, and 0.4 mg/mL each of xylene cyanol and bromophenol blue. The fraction of product generated in a burst in a single-turnover reaction was previously shown to provide a good measure of the fraction of native ribozyme, because the native and misfolded conformers bind the substrate oligonucleotide with comparable rates but only the native ribozyme cleaves the substrate (32). Control reactions showed that a 1-min time point was representative of the burst amplitude for all mutants. The 5′ cleavage product was separated from the uncleaved substrate by 20% denaturing PAGE (7 M urea and 1× TBE) and quantified using a phosphorimager (GE Healthcare). Data were quantified using ImageQuant 5.2 (GE Healthcare) and analyzed with Kaleidagraph (Synergy Software).
To calculate the folding equilibria, we normalized the equilibrium fraction of native ribozyme at 2 mM Mg2+ (fN,obs) by the maximal value (fN,max; formed at 10 mM Mg2+), which ranged from 0.8 to 0.9 and varied between ribozyme preparations, presumably owing to a small fraction of damaged RNA. The calculations also took into account the small fraction of native ribozyme that forms in the 50 mM Mg2+ folding quench (∼10% of the unfolded ribozyme evades the long-lived misfolded conformation and folds to the native state) (36). This “partitioning” value (fpart) was derived for each mutant from the average fraction of native ribozyme measured in the bottom plateau of the Mg2+-dependence curve, where essentially all of the ribozyme is unfolded. Overall, the fraction of native ribozyme present at equilibrium at 2 mM Mg2+ (fN) can be derived from the observed fraction as follows:
| [1] |
| [2] |
The final equation used to calculate native ribozyme stabilities at 2 mM Mg2+ was
| [3] |
where
| [4] |
The correction for partitioning in Eqs. 1 and 2 assumes that the nonnative ribozyme species that accumulate have the same partitioning value as the fully unfolded ribozyme, as suggested by the similarity of the K1/2 values measured here with values measured previously by chemical footprinting under similar conditions (37, 41). After establishing that 2 mM Mg2+ was appropriate for determining the folding equilibria of all mutants, we obtained the equilibrium values from the endpoints of ribozyme unfolding time courses in the absence of CYT-19 (see next section), measured in parallel with CYT-19–dependent unfolding. Stabilities were calculated using the same equations as above (Eqs. 1–4).
We found that varying the ATP⋅Mg2+ concentration in the folding reactions had a small but reproducible effect on the folding equilibrium, with greater ATP concentrations leading to lower stability of the native ribozyme (Fig. 2C). This effect is most likely caused by residual free ATP in the added equimolar mixtures of ATP and Mg2+. We therefore report the ribozyme stabilities separately for experiments performed in the presence of 100 µM and 2 mM ATP.
Monitoring Native Ribozyme Unfolding by Cleavage Activity.
Unfolding time courses were measured essentially as described (28). The ribozymes (2 µM) were prefolded to the native state by incubating them in submerged tubes at 50 °C for 45 min–1 h with 10 mM Mg(OAc)2 and 50 mM Na-Mops (pH 7.0) and stored on ice until use. Unfolding was initiated by diluting the ribozyme 10-fold into the reaction mix, bringing the concentrations to 200 nM ribozyme, 100–500 nM CYT-19, 2 mM Mg(OAc)2, 50 mM Na-Mops (pH 7.0), 0–2 mM ATP⋅Mg2+, and 10% (vol/vol) CYT-19 buffer. CYT-19 was kept on ice at 10× final concentration and added to the reaction mix immediately before ribozyme addition. Unfolding was monitored at 25 °C and the fraction of native ribozyme over time was determined as above (Stability Determination of Tetrahymena Ribozyme Mutants), except that the quench also included 1 mg/mL Proteinase K. The zero time points for the unfolding reactions were obtained by adding prefolded native ribozyme directly to the quench solution.
The unfolding data were fit by a single-exponential equation, with the exception of the L5b mutant ribozyme, which unfolded in two phases, presumably owing to loss of CYT-19 activity during the slow unfolding of this mutant. The slow phase had the same rate constant as spontaneous unfolding of the L5b ribozyme (Figs. S2B and S3). Loss of CYT-19 activity was also observed with other mutants when folding was monitored over longer times (>20 min), as indicated by a slow return to the intrinsic equilibrium value. To account for the variable endpoints in the unfolding reactions, the rate constants obtained from single exponential fits (or each phase of the double exponential fit in the case of L5b unfolding) were normalized by the ratio of the maximal possible unfolding amplitude (Amax = fN,prefold – fN,part) and the observed amplitude (Aobs = fN,prefold – fN,end), where fN,prefold represents the initial fraction of native ribozyme, formed by folding at 10 mM Mg2+, 50 °C; fN,part represents the fraction of native ribozyme formed in the folding quench, which limits the observed amplitude of unfolding (Stability Determination of Tetrahymena Ribozyme Mutants); and fN,end is the observed endpoint. This approach is analogous to obtaining initial rates from linear fits of the earliest data points and normalizing by the observable range (fN,prefold – fN,part); however, exponential fits were more accurate because they were constrained by a greater number of available time points.
The rate constants for spontaneous unfolding were calculated from the observed rate constants as follows. The observed rate constant, kobs, is defined as the sum of the unfolding rate constant, kunf, and the rate constant for folding, kfldg:
The endpoint of the unfolding reaction yields the equilibrium Keq = kfldg/kunf, allowing substitution of kfldg with Keq⋅kunf. Thus, the unfolding rate constant was calculated as
The unfolding efficiencies were derived from linear fits of the CYT-19 concentration dependences of unfolding rates (Figs. S2B and S3). Upper and lower limits of the slopes were determined where systematic deviations from linearity occurred.
ATPase Assays.
ATP hydrolysis was monitored under conditions identical to those of the folding reactions but in the presence of trace [γ-32P]ATP (Perkin-Elmer) in addition to unlabeled ATP. Reactions were initiated by adding prefolded native ribozyme and time points were collected during the initial linear phase, not exceeding ∼20% of the total ATP concentration, with a quench of 100 mM (final) EDTA. RNA-independent ATPase rates were measured by substituting TE buffer for the RNA. Reactions were analyzed by TLC, using a mobile phase of 1 M formic acid and 0.5 M LiCl, and quantified using a phosphorimager and Kaleidagraph software (8).
To measure ATPase stimulation by ribozymes pre-equilibrated at various Mg(OAc)2 concentrations, the RNAs (250 nM) were folded for ∼24 h at 25 °C in the presence of 0–10 mM Mg(OAc)2, 50 mM Na-Mops (pH 7.0), and 10% CYT-19 buffer (vol/vol). ATPase reactions were initiated by adding CYT-19 and ATP (with trace [γ-32P]ATP) in the desired Mg2+ concentration, bringing final concentrations to 200 nM RNA, 500 nM CYT-19, 100 µM ATP⋅Mg2+, and 10% CYT-19 buffer (vol/vol).
The oligonucleotide helices (2 µM) were incubated for 5 min at 25 °C with 10 mM Mg(OAc)2 and 50 mM Na-Mops (pH 7.0) and kept on ice until use. The helices were diluted 10-fold into the ATPase reaction mix containing final 1–10 mM Mg(OAc)2, 100 µM ATP⋅Mg2+, 50 mM Na-Mops (pH 7.0), with 500 nM CYT-19 (final) added immediately before the RNA.
SAXS Experiments.
Data were collected at the Advanced Photon Source (APS) beamline 12-ID-C, using a sample–detector distance of 2 m. To probe the compactness of ribozyme variants at 2 mM Mg2+, the ribozymes (600 nM) were equilibrated at 25 °C in the presence of 2 mM Mg(OAc)2, 50 mM Na-Mops (pH 7.0), and 10% CYT-19 buffer (vol/vol). The samples were concentrated using Amicon Ultra filters (EMD Millipore; 30 kDa or 50 kDa molecular weight cutoff), followed by multiple cycles of washing with the folding buffer to ensure that the free Mg2+ concentration remained at 2 mM even with the micromolar ribozyme concentrations required for SAXS measurements (63, 64). The samples were flash-frozen in liquid nitrogen and shipped to the APS on dry ice. To measure scattering from ribozymes at 10 mM Mg2+, the RNAs were folded on site by incubation at 45–50 °C for 1 h [2 µM ribozyme, 50 mM Na-Mops (pH 7.0), and 10 mM Mg(OAc)2] and cooled on ice. All SAXS measurements were performed at 25 °C. Scattering was measured using 1 µM and 2 µM ribozyme, and no systematic increases were observed in the Rg values with increasing ribozyme concentration. Twenty exposures of 1 s (12 keV) were collected and averaged for each sample. Blanks consisted of either the concentrator flow-through (2 mM Mg2+) or were prepared from the ingredients of the ribozyme folding solution, by substituting TE buffer for the ribozyme (10 mM Mg2+). The data were circularly averaged at the beamline and further analyzed using IgorPro (WaveMetrics) and Gnom (65).
The Rg values were determined using Gnom and verified by the Guinier approximation (47, 65). All Gnom solutions had scores of 0.594 or higher, representing reasonable fits, as defined by the program. The Rg values at 2 mM Mg2+ are averages of results from two to three independently prepared samples measured during separate beam times. The data at 10 mM Mg2+ were obtained during a single beamtime and the reported Rg values are averages of samples measured at 1 and 2 µM ribozyme. For Kratky analysis, the scattering profiles were normalized by the forward scattering (I0) values obtained through Guinier analysis.
Supplementary Material
Acknowledgments
We thank Dan Herschlag for providing the plasmid for the quintuple mutant and members of the R.R. laboratory for helpful comments and discussions. This work was supported by National Institute of General Medical Sciences Grant GM070456 (to R.R.) and Welch Foundation Grant F-1563.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1404307111/-/DCSupplemental.
References
- 1.Cordin O, Banroques J, Tanner NK, Linder P. The DEAD-box protein family of RNA helicases. Gene. 2006;367:17–37. doi: 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 2.Jankowsky E, Fairman ME. RNA helicases—one fold for many functions. Curr Opin Struct Biol. 2007;17(3):316–324. doi: 10.1016/j.sbi.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 3.Linder P, Jankowsky E. From unwinding to clamping - the DEAD box RNA helicase family. Nat Rev Mol Cell Biol. 2011;12(8):505–516. doi: 10.1038/nrm3154. [DOI] [PubMed] [Google Scholar]
- 4.Liu F, Putnam A, Jankowsky E. ATP hydrolysis is required for DEAD-box protein recycling but not for duplex unwinding. Proc Natl Acad Sci USA. 2008;105(51):20209–20214. doi: 10.1073/pnas.0811115106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hilbert M, Karow AR, Klostermeier D. The mechanism of ATP-dependent RNA unwinding by DEAD box proteins. Biol Chem. 2009;390(12):1237–1250. doi: 10.1515/BC.2009.135. [DOI] [PubMed] [Google Scholar]
- 6.Mallam AL, Del Campo M, Gilman B, Sidote DJ, Lambowitz AM. Structural basis for RNA-duplex recognition and unwinding by the DEAD-box helicase Mss116p. Nature. 2012;490(7418):121–125. doi: 10.1038/nature11402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell. 2006;125(2):287–300. doi: 10.1016/j.cell.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, et al. DEAD-box proteins can completely separate an RNA duplex using a single ATP. Proc Natl Acad Sci USA. 2008;105(51):20203–20208. doi: 10.1073/pnas.0811075106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henn A, Cao W, Hackney DD, De La Cruz EM. The ATPase cycle mechanism of the DEAD-box rRNA helicase, DbpA. J Mol Biol. 2008;377(1):193–205. doi: 10.1016/j.jmb.2007.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henn A, Bradley MJ, De La Cruz EM. ATP utilization and RNA conformational rearrangement by DEAD-box proteins. Annu Rev Biophys. 2012;41:247–267. doi: 10.1146/annurev-biophys-050511-102243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jarmoskaite I, Russell R. DEAD-box proteins as RNA helicases and chaperones. Wiley Interdiscip Rev RNA. 2011;2(1):135–152. doi: 10.1002/wrna.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fairman-Williams ME, Guenther UP, Jankowsky E. SF1 and SF2 helicases: Family matters. Curr Opin Struct Biol. 2010;20(3):313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao W, et al. Mechanism of Mss116 ATPase reveals functional diversity of DEAD-Box proteins. J Mol Biol. 2011;409(3):399–414. doi: 10.1016/j.jmb.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogers GW, Jr, Richter NJ, Merrick WC. Biochemical and kinetic characterization of the RNA helicase activity of eukaryotic initiation factor 4A. J Biol Chem. 1999;274(18):12236–12244. doi: 10.1074/jbc.274.18.12236. [DOI] [PubMed] [Google Scholar]
- 15.Bizebard T, Ferlenghi I, Iost I, Dreyfus M. Studies on three E. coli DEAD-box helicases point to an unwinding mechanism different from that of model DNA helicases. Biochemistry. 2004;43(24):7857–7866. doi: 10.1021/bi049852s. [DOI] [PubMed] [Google Scholar]
- 16.Uhlmann-Schiffler H, Jalal C, Stahl H. Ddx42p—a human DEAD box protein with RNA chaperone activities. Nucleic Acids Res. 2006;34(1):10–22. doi: 10.1093/nar/gkj403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohr S, Stryker JM, Lambowitz AM. A DEAD-box protein functions as an ATP-dependent RNA chaperone in group I intron splicing. Cell. 2002;109(6):769–779. doi: 10.1016/s0092-8674(02)00771-7. [DOI] [PubMed] [Google Scholar]
- 18.Huang HR, et al. The splicing of yeast mitochondrial group I and group II introns requires a DEAD-box protein with RNA chaperone function. Proc Natl Acad Sci USA. 2005;102(1):163–168. doi: 10.1073/pnas.0407896101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halls C, et al. Involvement of DEAD-box proteins in group I and group II intron splicing. Biochemical characterization of Mss116p, ATP hydrolysis-dependent and -independent mechanisms, and general RNA chaperone activity. J Mol Biol. 2007;365(3):835–855. doi: 10.1016/j.jmb.2006.09.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Potratz JP, Del Campo M, Wolf RZ, Lambowitz AM, Russell R. ATP-dependent roles of the DEAD-box protein Mss116p in group II intron splicing in vitro and in vivo. J Mol Biol. 2011;411(3):661–679. doi: 10.1016/j.jmb.2011.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zingler N, Solem A, Pyle AM. Dual roles for the Mss116 cofactor during splicing of the ai5γ group II intron. Nucleic Acids Res. 2010;38(19):6602–6609. doi: 10.1093/nar/gkq530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karunatilaka KS, Solem A, Pyle AM, Rueda D. Single-molecule analysis of Mss116-mediated group II intron folding. Nature. 2010;467(7318):935–939. doi: 10.1038/nature09422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Russell R, Jarmoskaite I, Lambowitz AM. Toward a molecular understanding of RNA remodeling by DEAD-box proteins. RNA Biol. 2013;10(1):44–55. doi: 10.4161/rna.22210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Del Campo M, et al. Do DEAD-box proteins promote group II intron splicing without unwinding RNA? Mol Cell. 2007;28(1):159–166. doi: 10.1016/j.molcel.2007.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Del Campo M, et al. Unwinding by local strand separation is critical for the function of DEAD-box proteins as RNA chaperones. J Mol Biol. 2009;389(4):674–693. doi: 10.1016/j.jmb.2009.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tijerina P, Bhaskaran H, Russell R. Nonspecific binding to structured RNA and preferential unwinding of an exposed helix by the CYT-19 protein, a DEAD-box RNA chaperone. Proc Natl Acad Sci USA. 2006;103(45):16698–16703. doi: 10.1073/pnas.0603127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grohman JK, et al. Probing the mechanisms of DEAD-box proteins as general RNA chaperones: The C-terminal domain of CYT-19 mediates general recognition of RNA. Biochemistry. 2007;46(11):3013–3022. doi: 10.1021/bi0619472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhaskaran H, Russell R. Kinetic redistribution of native and misfolded RNAs by a DEAD-box chaperone. Nature. 2007;449(7165):1014–1018. doi: 10.1038/nature06235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cate JH, et al. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science. 1996;273(5282):1678–1685. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- 30.Golden BL, Gooding AR, Podell ER, Cech TR. A preorganized active site in the crystal structure of the Tetrahymena ribozyme. Science. 1998;282(5387):259–264. doi: 10.1126/science.282.5387.259. [DOI] [PubMed] [Google Scholar]
- 31.Lehnert V, Jaeger L, Michel F, Westhof E. New loop-loop tertiary interactions in self-splicing introns of subgroup IC and ID: A complete 3D model of the Tetrahymena thermophila ribozyme. Chem Biol. 1996;3(12):993–1009. doi: 10.1016/s1074-5521(96)90166-0. [DOI] [PubMed] [Google Scholar]
- 32.Russell R, Herschlag D. Probing the folding landscape of the Tetrahymena ribozyme: Commitment to form the native conformation is late in the folding pathway. J Mol Biol. 2001;308(5):839–851. doi: 10.1006/jmbi.2001.4751. [DOI] [PubMed] [Google Scholar]
- 33.Russell R, et al. The paradoxical behavior of a highly structured misfolded intermediate in RNA folding. J Mol Biol. 2006;363(2):531–544. doi: 10.1016/j.jmb.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 34.Russell R, Millett IS, Doniach S, Herschlag D. Small angle X-ray scattering reveals a compact intermediate in RNA folding. Nat Struct Biol. 2000;7(5):367–370. doi: 10.1038/75132. [DOI] [PubMed] [Google Scholar]
- 35.Mitchell D, 3rd, Jarmoskaite I, Seval N, Seifert S, Russell R. The long-range P3 helix of the Tetrahymena ribozyme is disrupted during folding between the native and misfolded conformations. J Mol Biol. 2013;425(15):2670–2686. doi: 10.1016/j.jmb.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wan Y, Suh H, Russell R, Herschlag D. Multiple unfolding events during native folding of the Tetrahymena group I ribozyme. J Mol Biol. 2010;400(5):1067–1077. doi: 10.1016/j.jmb.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benz-Moy TL, Herschlag D. Structure-function analysis from the outside in: Long-range tertiary contacts in RNA exhibit distinct catalytic roles. Biochemistry. 2011;50(40):8733–8755. doi: 10.1021/bi2008245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shcherbakova I, Brenowitz M. Perturbation of the hierarchical folding of a large RNA by the destabilization of its Scaffold’s tertiary structure. J Mol Biol. 2005;354(2):483–496. doi: 10.1016/j.jmb.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 39.Treiber DK, Williamson JR. Concerted kinetic folding of a multidomain ribozyme with a disrupted loop-receptor interaction. J Mol Biol. 2001;305(1):11–21. doi: 10.1006/jmbi.2000.4253. [DOI] [PubMed] [Google Scholar]
- 40.Wan Y, Mitchell D, 3rd, Russell R. Catalytic activity as a probe of native RNA folding. Methods Enzymol. 2009;468:195–218. doi: 10.1016/S0076-6879(09)68010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Russell R, Tijerina P, Chadee AB, Bhaskaran H. Deletion of the P5abc peripheral element accelerates early and late folding steps of the Tetrahymena group I ribozyme. Biochemistry. 2007;46(17):4951–4961. doi: 10.1021/bi0620149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pan J, Woodson SA. The effect of long-range loop-loop interactions on folding of the Tetrahymena self-splicing RNA. J Mol Biol. 1999;294(4):955–965. doi: 10.1006/jmbi.1999.3298. [DOI] [PubMed] [Google Scholar]
- 43.Celander DW, Cech TR. Visualizing the higher order folding of a catalytic RNA molecule. Science. 1991;251(4992):401–407. doi: 10.1126/science.1989074. [DOI] [PubMed] [Google Scholar]
- 44.Sattin BD, Zhao W, Travers K, Chu S, Herschlag D. Direct measurement of tertiary contact cooperativity in RNA folding. J Am Chem Soc. 2008;130(19):6085–6087. doi: 10.1021/ja800919q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Das R, et al. The fastest global events in RNA folding: electrostatic relaxation and tertiary collapse of the Tetrahymena ribozyme. J Mol Biol. 2003;332(2):311–319. doi: 10.1016/s0022-2836(03)00854-4. [DOI] [PubMed] [Google Scholar]
- 46.Liu F, Putnam AA, Jankowsky E. DEAD-box helicases form nucleotide-dependent long-lived complexes with RNA. Biochemistry. 2013;53(2):423–433. doi: 10.1021/bi401540q. [DOI] [PubMed] [Google Scholar]
- 47.Doniach S. Changes in biomolecular conformation seen by small angle X-ray scattering. Chem Rev. 2001;101(6):1763–1778. doi: 10.1021/cr990071k. [DOI] [PubMed] [Google Scholar]
- 48.Shi H, O’Brien CA, Van Horn DJ, Wolin SL. A misfolded form of 5S rRNA is complexed with the Ro and La autoantigens. RNA. 1996;2(8):769–784. [PMC free article] [PubMed] [Google Scholar]
- 49.Fuchs G, Stein AJ, Fu C, Reinisch KM, Wolin SL. Structural and biochemical basis for misfolded RNA recognition by the Ro autoantigen. Nat Struct Mol Biol. 2006;13(11):1002–1009. doi: 10.1038/nsmb1156. [DOI] [PubMed] [Google Scholar]
- 50.Reinisch KM, Wolin SL. Emerging themes in non-coding RNA quality control. Curr Opin Struct Biol. 2007;17(2):209–214. doi: 10.1016/j.sbi.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 51.Staley JP, Guthrie C. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell. 1998;92(3):315–326. doi: 10.1016/s0092-8674(00)80925-3. [DOI] [PubMed] [Google Scholar]
- 52.Strunk BS, Karbstein K. Powering through ribosome assembly. RNA. 2009;15(12):2083–2104. doi: 10.1261/rna.1792109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rajkowitsch L, et al. RNA chaperones, RNA annealers and RNA helicases. RNA Biol. 2007;4(3):118–130. doi: 10.4161/rna.4.3.5445. [DOI] [PubMed] [Google Scholar]
- 54.Russell R. RNA misfolding and the action of chaperones. Front Biosci. 2008;13:1–20. doi: 10.2741/2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grohman JK, et al. A guanosine-centric mechanism for RNA chaperone function. Science. 2013;340(6129):190–195. doi: 10.1126/science.1230715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grossberger R, et al. Influence of RNA structural stability on the RNA chaperone activity of the Escherichia coli protein StpA. Nucleic Acids Res. 2005;33(7):2280–2289. doi: 10.1093/nar/gki515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475(7356):324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 58.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11(7):515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 59.Böcking T, Aguet F, Harrison SC, Kirchhausen T. Single-molecule analysis of a molecular disassemblase reveals the mechanism of Hsc70-driven clathrin uncoating. Nat Struct Mol Biol. 2011;18(3):295–301. doi: 10.1038/nsmb.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taipale M, et al. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. 2012;150(5):987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Russell R, Herschlag D. New pathways in folding of the Tetrahymena group I RNA enzyme. J Mol Biol. 1999;291(5):1155–1167. doi: 10.1006/jmbi.1999.3026. [DOI] [PubMed] [Google Scholar]
- 62.Zaug AJ, Grosshans CA, Cech TR. Sequence-specific endoribonuclease activity of the Tetrahymena ribozyme: Enhanced cleavage of certain oligonucleotide substrates that form mismatched ribozyme-substrate complexes. Biochemistry. 1988;27(25):8924–8931. doi: 10.1021/bi00425a008. [DOI] [PubMed] [Google Scholar]
- 63.Bai Y, et al. Quantitative and comprehensive decomposition of the ion atmosphere around nucleic acids. J Am Chem Soc. 2007;129(48):14981–14988. doi: 10.1021/ja075020g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pabit SA, et al. Counting ions around DNA with anomalous small-angle X-ray scattering. J Am Chem Soc. 2010;132(46):16334–16336. doi: 10.1021/ja107259y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Cryst. 1992;25:495–503. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.