

Significance
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas)-mediated genome engineering promises to revolutionize genetic studies in a variety of systems. Here we describe an optimized set of tools for CRISPR/Cas experiments in the model organism Drosophila melanogaster. These tools can be used for remarkably efficient germline transmission of (i) loss-of-function insertion and deletion mutations in essential genes or (ii) precise changes in the genome sequence introduced by homology-directed repair. These tools also permit efficient biallelic targeting of genes in somatic cells, thereby demonstrating a novel application of CRISPR/Cas in rapidly revealing mutant phenotypes within the organism. Our work also paves the way for high-throughput genetic screens in Drosophila with CRISPR/Cas.
Abstract
The type II clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) system has emerged recently as a powerful method to manipulate the genomes of various organisms. Here, we report a toolbox for high-efficiency genome engineering of Drosophila melanogaster consisting of transgenic Cas9 lines and versatile guide RNA (gRNA) expression plasmids. Systematic evaluation reveals Cas9 lines with ubiquitous or germ-line–restricted patterns of activity. We also demonstrate differential activity of the same gRNA expressed from different U6 snRNA promoters, with the previously untested U6:3 promoter giving the most potent effect. An appropriate combination of Cas9 and gRNA allows targeting of essential and nonessential genes with transmission rates ranging from 25–100%. We also demonstrate that our optimized CRISPR/Cas tools can be used for offset nicking-based mutagenesis. Furthermore, in combination with oligonucleotide or long double-stranded donor templates, our reagents allow precise genome editing by homology-directed repair with rates that make selection markers unnecessary. Last, we demonstrate a novel application of CRISPR/Cas-mediated technology in revealing loss-of-function phenotypes in somatic cells following efficient biallelic targeting by Cas9 expressed in a ubiquitous or tissue-restricted manner. Our CRISPR/Cas tools will facilitate the rapid evaluation of mutant phenotypes of specific genes and the precise modification of the genome with single-nucleotide precision. Our results also pave the way for high-throughput genetic screening with CRISPR/Cas.
Experimentally induced mutations in the genomes of model organisms have been the basis of much of our current understanding of biological mechanisms. However, traditional mutagenesis tools have significant drawbacks. Forward genetic approaches such as chemical mutagenesis lack specificity, leading to unwanted mutations at many sites in the genome. Traditional reverse genetic approaches, such as gene targeting by conventional homologous recombination, suffer from low efficiency and therefore are labor intensive. In recent years novel methods have been developed that aim to modify genomes with high precision and high efficiency by introducing double-stand breaks (DSBs) at defined loci (1). DSBs can be repaired by either nonhomologous end joining (NHEJ) or homology-directed repair (HDR). NHEJ is an error-prone process that frequently leads to the generation of small, mutagenic insertions and deletions (indels). HDR repairs DSBs by precisely copying sequence from a donor template, allowing specific changes to be introduced into the genome (2).
The type II clustered regular interspersed short palindromic repeat (CRISPR)/CRISPR-associated (Cas) system has emerged recently as an extraordinarily powerful method for inducing site-specific DSBs in the genomes of a variety of organisms. The method exploits the RNA-guided endonuclease Cas9, which plays a key role in bacterial adaptive immune systems. Target specificity of Cas9 is encoded by a 20-nt spacer sequence in the crisprRNA, which pairs with the transactivating RNA to direct the endonuclease to the complementary target site in the DNA (3). For genome engineering, crisprRNA and transactivating RNA can be combined in a single chimeric guide RNA (gRNA), resulting in a simple two-component system for the creation of DSBs at defined sites (3). Binding of the Cas9/gRNA complex at a genomic target site is constrained only by the requirement for an adjacent short protospacer-adjacent motif (PAM), which for the commonly used Streptococcus pyogenes Cas9 is NGG (4).
Several groups recently demonstrated CRISPR/Cas-mediated editing of the genome of Drosophila melanogaster (5–12), a key model organism for biological research. However, the rate of mutagenesis has varied widely both within and among different studies. Differences in the methods used to introduce Cas9 and gRNAs into the fly likely contribute significantly to different experimental outcomes. Kondo and Ueda (8) expressed both Cas9 and gRNA from transgenes stably integrated into the genome, but all other studies have used microinjection of expression plasmids or of in vitro-transcribed RNA into embryos to deliver one or both CRISPR/Cas components (5–7, 9–11). Much of the currently available evidence suggests that transgenic provision of Cas9 increases rates of germ-line transmission substantially (8, 10, 11). However, the influence of different regulatory sequences within cas9 transgenes on the rate of mutagenesis and on the location where mutations are generated within the organism has not been evaluated. The effect of different promoter sequences on the activities of gRNAs also has not been explored systematically. Therefore it is possible that suboptimal tools are being used currently for many CRISPR/Cas experiments in Drosophila.
Previous studies in Drosophila have focused on the use of CRISPR/Cas to create heritable mutations in the germ line. In principle, efficient biallelic targeting within somatic cells of Drosophila would represent a powerful system to dissect the functions of genes within an organismal context. However, the feasibility of such an approach has not been explored so far.
Here, we present a versatile CRISPR/Cas toolbox for Drosophila genome engineering consisting of a set of systematically evaluated transgenic Cas9 lines and gRNA-expression plasmids. We describe combinations of Cas9 and gRNA sources that can be used to induce, with high efficiency, loss-of-function mutations in nonessential or essential genes and integration of designer sequences by HDR. Finally, we show that our optimized transgenic tools permit efficient biallelic targeting in a variety of somatic tissues of the fly, allowing the characterization of mutant phenotypes directly in Cas9/gRNA-expressing animals.
Results
Generation and Evaluation of Cas9 Transgenes.
We generated a series of Cas9-expressing transgenes (Table S1) to compare their expression patterns and endonuclease activities. Expression plasmids were produced encoding S. pyogenes Cas9 [codon optimized for expression in human (13, 14) or Drosophila cells] fused to nuclear localization signals under the control of different regulatory sequences. We generated two constructs in which Cas9 was under the control of the nos promoter, which is active in the male and female germ line (15), and the nos 3′ UTR, which targets protein synthesis to the germ cells (15). These constructs are similar to those used by two previous studies to provide Cas9 transgenically (8, 10). We also generated two novel constructs: (i) act-cas9, in which Cas9 is fused to the ubiquitously expressed actin5C (act) promoter and the SV40 3′ UTR, and (ii) UAS-cas9, which has yeast upstream-activating sequences (UAS), allowing expression under control of the Gal4 transcriptional activator (16), and the p10 3′ UTR, which promotes efficient translation (17). These plasmids were integrated into the Drosophila genome at defined positions using the attB/attP/Phi31C system (18, 19). We also obtained two published stocks that express, from different genomic locations, a cas9 transgene under control of the vasa promoter and 3′ UTR that is purported to restrict expression of the endonuclease to the germ line (11).
To test the different Cas9 lines functionally, we designed a gRNA targeting the ebony (e) gene on chromosome 3 (referred to as “gRNA-e”) (Fig. 1A). Mutation of both wild-type e alleles leads to very dark coloration of the adult cuticle. gRNA-e targets Cas9 to the 5′ end of the e coding sequence, 25 bp after the translation initiation codon. A construct expressing gRNA-e from the promoter of the U6:3 spliceosomal snRNA gene (see below) was integrated stably at the attP2 site on chromosome 3 (Table S2). Transgenic supply of the gRNA was designed to eliminate the variability between experiments that is associated with direct injection of a plasmid or in vitro-transcribed RNA into embryos, thereby facilitating meaningful comparison of the activities of the different Cas9 lines.
Fig. 1.

Systematic evaluation of transgenic Cas9 strains. (A) Schematic of sequence and position of gRNA-e and gRNA-y target sites in the e and y loci. UTRs are shown in gray, and coding sequences are shown as colored blocks. Cas9 cut sites are indicated by arrowheads. (B and C) Representative examples of female flies expressing one copy of the different cas9 transgenes and one copy of the U6:3-gRNA-e (B) or U6:3-gRNA-y (C) transgene compared with wild-type controls (at least 200 adults of each genotype were examined). Body coloration darker (B) or lighter (C) than the control indicates CRISPR/Cas-mediated mutagenesis of the target gene in epidermal cells. The number of wild-type alleles present in each animal that must be mutated to give rise to the mutant phenotype is indicated below each image. Somatic targeting is widespread in animals expressing act-cas9 or vasa-cas9, is sporadic in animals expressing nos-cas9:GFP and nosG4VP16 UAS-cas9 (arrowheads), and is undetectable in animals expressing nos-cas9. (D and E) Assessment of germ-line transmission of nonfunctional e (D) and y (E) alleles induced by different cas9 transgenes, performed by crossing animals expressing cas9 and the appropriate gRNA to e or y mutant animals. Data represent mean and SEM from at least eight (D) or four (E) independent crosses for each cas9 line. Efficient germ-line transmission of CRISPR/Cas-induced mutations was observed for all cas9 transgenes, although statistically significant differences in mean values were apparent (*P < 0.05; **P < 0.01; ***P < 0.001; unpaired t test compared with act-cas9 with the same gRNA). Displayed values for U6:3-gRNA-y experiments are normalized to account for the 25% of y mutant offspring that would be expected in some crosses in the absence of CRISPR/Cas-mediated mutagenesis (Table S3). (F) Examples of sequences of CRISPR/Cas-induced germ-line mutations in e and y (found in the offspring of act-cas9 gRNA flies). Some mutations were observed in several different flies. The in-frame mutation in e (bottom sequence) was obtained from a fly with wild-type pigmentation. The start codon in this and subsequent figures is shown in bold text.
Flies expressing the U6:3-gRNA-e transgene were crossed to the transgenic Cas9 lines (see Fig. S1 for the general crossing scheme). Remarkably, all adult offspring expressing gRNA-e and act-cas9 had mosaic pigmentation in which the majority of cuticle exhibited very dark coloration (Fig. 1B). This result demonstrates highly efficient biallelic targeting of e in somatic cells. Surprisingly, the two independent genomic insertions of vasa-cas9 in combination with gRNA-e also resulted in e mutant tissue throughout most of the cuticle (Fig. 1B). Thus, Cas9 activity is not restricted to the germ line by either vasa-cas9 transgene. All nos-cas9:GFP U6:3-gRNA-e flies had patches of dark cuticle, but these patches were relatively small and infrequent (Fig. 1B). A similar phenotype was observed in ∼40% of flies in which U6:3-gRNA-e was combined with UAS-cas9 driven by the strong transcriptional activator Gal4VP16 under the control of nos regulatory sequences (nosG4VP16) (Fig. 1B). In contrast, all nos-cas9 U6:3-gRNA-e flies had wild-type coloration of the entire cuticle (Fig. 1B). The differential activity of nos-cas9:GFP and nos-cas9—both of which use the same nos promoter and 3′ UTR sequences and are inserted at the same genomic location—presumably reflects an influence of the GFP moiety, number of nuclear localization signals, or differences in the codon use for the Cas9 ORF in each construct (Table S1).
We next compared the somatic activity of the cas9 transgenes using a second gRNA transgene, which targets the X-linked yellow (y) gene 94 bp after the start codon (referred to as “gRNA-y”) (Fig. 1A). This gRNA also was expressed from the U6:3 promoter and was stably integrated at the attP2 site. In the absence of a wild-type y allele, the adult cuticle has lighter, yellow pigmentation. The combination of U6:3-gRNA-y with act-cas9 or vasa-cas9 led to a predominantly yellow cuticle in all animals (Fig. 1C). This was even the case in a vasa-cas9 U6:3-gRNA-y genotype in which four wild-type y alleles had to be mutated to give the yellow phenotype (Fig. 1C); note that different crosses had different numbers of y+ alleles because of differences in the genotypes of the Cas9 parental strains (Fig. 1C, Table S3, and Materials and Methods). In contrast, nos-cas9:GFP U6:3-gRNA-y animals had cuticle that contained only small clones of y mutant tissue (Fig. 1C), whereas the combination of U6:3-gRNA-y with nosG4VP16 > UAS-cas9 or nos-cas9 resulted in phenotypically wild-type cuticle throughout the animal (Fig. 1C). nos-cas9 U6:3-gRNA-y males, in which only a single wild-type y allele had to be mutated to produce yellow cuticle, also had wild-type pigmentation throughout the animal (Fig. S2). Together, the experiments with gRNA-e and gRNA-y demonstrate that act-cas9 and vasa-cas9 have substantial activity in cells that give rise to the cuticle, but activity in these cells is relatively low in nos-cas9:GFP and nosG4VP16 > UAS-cas9 flies and is not detectable in nos-cas9 flies.
We next assessed the germ-line transmission of CRISPR/Cas-induced mutations in e and y (Fig. 1 D and E and Table S4). Individual flies containing each cas9 transgene and U6:3-gRNA-e were crossed to a classical e mutant strain. All cas9 transgenes resulted in 42–54% of transmitted e alleles being nonfunctional (mean values from at least eight crosses per transgene) with the exception of nos-cas9, for which the mean value was 26 ± 3% (Fig. 1D and Table S4). Sequencing of nonfunctional e alleles from one of these crosses revealed a range of small indels in the vicinity of the predicted Cas9 cleavage site (Fig. 1F); this finding is consistent with molecular analysis of CRISPR/Cas-induced alleles in other studies (5–8, 10).
We assessed the transmission of CRISPR/Cas-induced y mutant alleles by crossing flies containing U6:3-gRNA-y and the different cas9 transgenes to a strain with a classical y mutant allele. In all cases, an average of >70% of the progeny expected to have wild-type pigmentation in the absence of mutagenesis had phenotypically yellow cuticle (Fig. 1E and Table S4). In the case of the act-cas9 and one of the vasa-cas9 transgenes, an average of 99–100% of such progeny were yellow (Fig. 1E and Table S4), demonstrating CRISPR/Cas-induced mutagenesis of wild-type y alleles in almost every germ cell. As expected, sequencing analysis of the progeny revealed indels at the predicted location of the y locus (Fig. 1F). Collectively our experiments indicate that act-cas9 and vasa-cas9 transgenes can induce mutations in somatic and germ-line cells with high frequencies and that nos-cas9 allows efficient germ-line transmission of mutations without widespread targeting in somatic cells.
It was intriguing that for each cas9 transgene the frequency of transmission of nonfunctional y alleles was higher than that observed for e alleles (Fig. 1 D and E). To determine whether the ability of e to tolerate in-frame mutations at the gRNA-e target site contributes to this difference, the target site of eight phenotypically wild-type progeny from act-cas9 U6:3-gRNA-e parents was sequenced. Seven flies had small indels that retain the overall reading frame (e.g., Fig. 1F); only a single fly inherited an unmodified e locus. Thus, the frequency of nucleotide changes in gRNA-e experiments is substantially higher than suggested by phenotypic screening.
Optimized Single and Double gRNA Expression Vectors.
To date, gRNAs targeting the Drosophila genome have been transcribed from one of the RNA polymerase III-dependent promoters of the U6 snRNA genes (5, 9–11). There are three U6 genes in the fly: U6:1, U6:2, and U6:3. The great majority of published CRISPR/Cas experiments have used gRNAs produced by the U6:2 promoter. The U6:1 promoter has been assessed in a single experiment using direct plasmid injection to deliver the gRNA construct (10), and the U6:3 promoter has not been used previously. To compare the activity of the three U6 promoters in CRISPR/Cas experiments, plasmids were generated harboring each U6 promoter followed by the gRNA core sequence and a BbsI restriction-site cassette (Fig. 2A and Fig. S3). The plasmids also contained an attB site to allow integration at defined positions in the Drosophila genome and an eye pigmentation marker to permit screening for insertion events. We used these plasmids to generate constructs expressing gRNA-y under the control of each U6 promoter. To avoid positional effects on gRNA expression, each construct was stably integrated at the attP40 site on chromosome 2.
Fig. 2.

Versatile gRNA-expression vectors. (A) Schematic of the U6 locus. U6 genes are shown as orange blocks; intervening sequences and neighboring genes are shown in gray. Sequences 5′ to each U6 gene were cloned in front of a core gRNA sequence to generate plasmids pCFD1–3. The asterisk indicates positions of the BbsI cloning cassette for insertion of target sequence. (B) Differential activity of U6-gRNA constructs in epidermal cells. Female flies are shown that are heterozygous for act-cas9 and a gRNA-y transgene under the control of the U6:1, U6:2, or U6:3 promoter. Lighter body coloration indicates CRISPR/Cas-mediated mutagenesis of the two wild-type y alleles present in these flies. Arrowheads show examples of wild-type pigmentation with the U6:1 promoter and mutant pigmentation with the U6:2 promoter. (C) Mean germ-line transmission rates using different gRNA transgenes. nos-cas9 U6-gRNA-y females were crossed to y mutant males, and y mutant offspring were counted (three crosses per genotype). Displayed values are adjusted to account for the 25% of y mutant offspring expected in the absence of mutagenesis. Error bars represent SEM. (D) Targeting of two genes in the same animal with a double gRNA vector, pCFD4. Representative images are shown of flies that express act-cas9 in the absence or presence of a single transgene expressing gRNAs to e and cu. The arrowhead indicates the curled wing of a fly with extensive ebony pigmentation. (E) pCFD4 allows mutagenesis by offset-nicking in combination with act-cas9D10A. (Upper Left) Target sites of gRNAs (arrowheads indicate location of nicks). (Right) Summary of results from injecting pCFD4 encoding the two y gRNAs into transgenic act-cas9D10A embryos and crossing to y flies to assess germ-line transmission of loss-of-function y alleles. In these and other figures, “founder” refers to an animal that transmitted mutant alleles to the next generation. In the four crosses, 22 of 22 files, 11 of 56 flies, 27 of 31 files, and none of 35 flies inherited a nonfunctional y allele. (Lower Left) Sequence of six different indels found in 20 analyzed yellow progeny.
Flies expressing each gRNA-y transgene were crossed to flies expressing the act-cas9 transgene. All progeny expressing act-cas9 and U6:3-gRNA-y developed a completely yellow cuticle (Fig. 2B). This finding indicates extremely efficient targeting of the two wild-type y alleles in this cross, consistent with results of combining this cas9 transgene with U6:3-gRNA-y integrated at another genomic site (Fig. 1C). All act-cas9 U6:1-gRNA-y animals developed cuticle that was mostly yellow but had small spots of wild-type tissue (Fig. 2B). In contrast, all act-cas9 U6:2-gRNA-y flies retained large amounts of wild-type cuticle with many small yellow patches (Fig. 2B). We also produced flies expressing nos-cas9 and each of the U6-gRNA-y constructs and assessed the germ-line transmission of nonfunctional y alleles to the next generation as described above. Of the progeny expected to have wild-type pigmentation in the absence of CRISPR/Cas-mediated targeting, 69 ± 2%, 41 ± 3%, and 99 ± 1% were phenotypically yellow when the U6:1, U6:2 and U6:3 promoters were used, respectively (Fig. 2C and Table S4). Collectively, these results demonstrate that the different U6 promoters differ substantially in their ability of drive mutagenesis in both somatic cells and the germ line, with U6:3 having the strongest, U6:2 having the weakest, and U6:1 having intermediate activity.
Simultaneous expression of two gRNAs can be used to create defined deletions (5, 8), to target two genes simultaneously (20), or to mutagenize a single gene by offset nicking (21, 22). The last method increases CRISPR/Cas specificity by combining a Cas9 “nickase” mutant [Cas9D10A (3)], which cuts only one DNA strand, with two gRNAs to the target gene. DSBs are induced by this protein only when two molecules are guided to opposite strands of the DNA in close proximity, an event that is extremely unlikely at an off-target site. This method has not been used previously in Drosophila. To enable simultaneous expression of two gRNAs, we produced a plasmid containing both the U6:1 and U6:3 promoters adjacent to gRNA core sequences (Fig. 2D). Different promoters were chosen to avoid the risk associated with recombination between identical sequences during cloning exercises. The plasmid was designed to allow one-step cloning of the two gRNAs by introducing target sequences in PCR primers (Fig. S3) and therefore allows more convenient and rapid workflow than a previously reported strategy for the same task, which used sequential cloning of gRNAs adjacent to two U6:2 promoters (8).
We used our dual gRNA plasmid to make a transgenic line simultaneously expressing gRNAs targeting e or curled (cu), which causes a curled wing phenotype when mutated (23), and crossing that line to act-cas9 flies. All 120 of the analyzed act-cas9 gRNA-e gRNA-cu flies had large patches of dark cuticle, and 52% had one or two curled wings (Fig. 2D). Thus, the dual gRNA plasmid can be used to target two genes in the same animal efficiently.
We also integrated two sequences targeting opposite strands of the y locus into the dual gRNA vector and injected the plasmid directly into transgenic embryos expressing the Cas9D10A nickase under the control of the act promoter (Table S1). Forty-two percent of the offspring from injected animals received a nonfunctional y allele, which we confirmed was associated with indels at the expected position (Fig. 2E). All transgenic animals expressing both act-cas9D10A and the strong single-target U6:3-gRNA-y transgene had wild-type coloration of the cuticle, indicating that single DNA nicks have low mutagenic potential in flies. We conclude that the dual gRNA vector can be used in combination with act-cas9D10A flies to create mutagenic DSBs through offset nicking.
Efficient Germ-Line Transmission of Mutations in Essential Genes.
The experiments described above identified Cas9 and gRNA reagents that can target nonessential genes in somatic and/or germ-line tissues with different efficiencies. We next sought to identify Cas9/gRNA combinations that can give efficient germ-line transmission of mutations in genes that are essential for viability. Doing so necessitates finding conditions in which transmission is not compromised by deleterious effects of biallelic gene targeting in somatic tissues. We generated transgenic fly lines expressing gRNAs targeting the genes encoding either the secreted signaling molecule Wingless (Wg) or the Wg secretion factor Wntless (Wls). Both the wg and wls genes are essential in the soma for development of Drosophila to adult stages (24–26). Both gRNAs were under the control of the U6:3 promoter and integrated at the attP40 site. gRNA-wg directs Cas9 to cut just after the region of the target gene that encodes the signal sequence, and gRNA-wls directs Cas9 to cut within the signal sequence-encoding region of the target gene (Fig. 3A).
Fig. 3.

Efficient targeting of essential genes. (A) Schematic of sequence and position of target sites of gRNA-wg and gRNA-wls. (B) Summary of fate of animals that developed to pupal stage and that coexpressed U6:3-gRNA-wg or U6:3-gRNA-wls with different cas9 transgenes. (C) Representative examples of phenotypes observed in adult cas9 gRNA-wg and cas9 gRNA-wls flies. Defects consistent with perturbed Wg signaling include wing notches (Upper) and malformation of the legs and abdominal segments (Lower). Images at the left and center left show severe malformations; image on the top right shows mild malformations. The arrowhead indicates a wing notch, which was found in 5% of nos-cas9 U6:3-gRNA-wls flies. (D) Summary of results assessing transmission of wg loss-of-function alleles (wgLOF) by genetic complementation tests with a known wg null allele (in the four crosses, four of four flies, one of six flies, three of five flies, and four of four flies inherited a wgLOF allele). wgCRISPR alleles that retain function are not detected by this method. (E) Assessment of transmission of nonfunctional wls alleles. A fertile nos-cas9 gRNA-wls fly was crossed to a balancer stock, and the wls locus of 15 heterozygous progeny was sequenced. Ten sequencing chromatograms could be read unambiguously; all contained indels at the target site. The mutated sequence is shown below the wild-type sequence. The 27-bp deletion was recovered in two different flies.
No offspring survived past the pupal stage when U6:3-gRNA-wg or U6:3-gRNA-wls flies were crossed to the ubiquitous act-cas9 line (Fig. 3B), presumably because of effective biallelic targeting of wg and wls in the soma. Crossing vasa-cas9, nos-cas9:GFP, and nosG4VP16 > UAS-cas9 to each gRNA also resulted in substantial lethality, with the few animals that progressed to adulthood exhibiting a range of morphological defects consistent with defects in Wg signaling (Fig. 3C). Unsurprisingly, many of these adults died shortly after eclosion, and those that survived often had poor fertility. In contrast, no significant lethality was observed for nos-cas9 U6:3-gRNA-wg and nos-cas9 U6:3-gRNA-wls pupae (Fig. 3B). All the eclosed adults appeared morphologically normal, with the exception of 5% of nos-cas9 U6:3-gRNA-wls flies that had small notches in the wing (Fig. 3C). This phenotype indicates a local impairment of Wg signaling, suggesting that not all Cas9 activity is restricted to the germ line when nos-cas9 is combined with this gRNA. This series of experiments demonstrates that only the nos-cas9 line has sufficiently germ-line–restricted Cas9 activity to allow efficient development of flies to adulthood when either essential gene is targeted.
We next evaluated germ-line transmission of CRISPR/Cas-induced mutations in wg and wls using nos-cas9. Nineteen randomly selected offspring of nos-cas9 U6:3-gRNA-wg flies were tested for wg mutations by genetic complementation with a previously characterized wg null allele (27). The wg null allele was not complemented in 12 of the 19 crosses, demonstrating efficient creation of nonfunctional wg mutations by CRISPR/Cas (Fig. 3D).
The nos-cas9 U6:3-gRNA-wls females were poorly fertile, which is compatible with the requirement for maternal Wls during embryonic development (25, 26). The nos-cas9 U6:3-gRNA-wls males also had low fertility, raising the possibility of a previously undescribed role for Wls in the male germ line. Nonetheless, we were able to sequence wls alleles from PCR products of 15 randomly selected offspring of an outcrossed nos-cas9 U6:3-gRNA-wls fly that was fertile. The sequencing chromatograms had double peaks around the gRNA target site in every case. This pattern indicates the presence of a CRISPR/Cas-induced wls mutant allele in each fly, along with the wild-type allele inherited from the other parent. Peaks from the mutagenized and wild-type wls alleles could be called unambiguously in the sequencing traces from 10 of the 15 samples; each of these 10 traces revealed an indel mutation, nine of which were unique (Fig. 3E). Many of the indels created frameshifts or removed the ATG start codon, presumably resulting in loss of wls function. Together these results show that our optimized CRISPR/Cas system allows the generation of loss-of-function alleles in essential genes at frequencies that make screening with methods other than direct DNA sequencing unnecessary.
Precise Genome Modification by HDR.
The work described above identified tools for generating loss-of-function indel mutations in both the soma and germ line of Drosophila. In many cases, it is desirable to dissect gene function further by introducing specific sequence changes into the genome. In Drosophila it has been possible for several years to create specific genome modifications by HDR, although targeting typically occurs with very low efficiency (28). Recent studies have suggested that zinc finger, transcriptional activator-like effector (TALE), and Cas9 nucleases can increase the efficiency of HDR in flies, implying that the generation of site-specific DSBs is rate limiting (11, 12, 27, 29, 30). Therefore we tested whether the highly effective creation of DSBs by our transgenic CRISPR/Cas system could be exploited to facilitate efficient HDR.
We first attempted to introduce a missense mutation into the wls locus using an ssDNA oligonucleotide donor (ssODN). We designed an ssODN harboring a G-to-C transversion at the gRNA-wls target site, which would cause a Gly11Ala mutation in Wls and remove the PAM sequence (Fig. 4A). The ssODN was injected into nos-cas9 U6:3-gRNA-wls preblastoderm embryos, with six of the resulting flies outcrossed. Sequence analysis of the wls locus from the offspring of these crosses revealed that four of the six flies had transmitted the designed change, with 28% of the 46 tested progeny receiving the mutation (Fig. 4 B and C). Sixty-one percent of the total progeny instead inherited an indel mutation at the target site, and the other animals received an unmodified wls allele. We also introduced an 11-bp sequence containing a restriction site into the e locus by ssODN injection into act-cas9 U6:3-gRNA-e embryos; 11% of the 76 progeny analyzed contained the desired mutation (Fig. S4). These data demonstrate that our CRISPR/Cas tools can support HDR using oligonucleotide-derived sequences with efficiencies compatible with direct screening for targeting events by PCR and sequencing. We also injected a circular double-stranded plasmid containing the GFP-coding sequence flanked by 1.4- and 1.7-kb homology arms from the wg locus into nos-cas9 U6:3-gRNA-wg embryos (Fig. S5A). The donor template is designed to produce an in-frame insertion of GFP within the wg coding region, leading to a secreted Wg::GFP protein. Thirty-eight percent of the offspring of injected embryos expressed a GFP protein in the Wg expression pattern, which appeared to be secreted (Fig. S5 B–D). PCR-based analysis of the wg locus in five of these animals confirmed successful ends-out targeting in all cases (Fig. S5E). These data provide evidence that our CRISPR/Cas tools also can be used to introduce longer exogenous sequences into an essential gene with high efficiency.
Fig. 4.

Efficient incorporation of an exogenous sequence in wls by HDR following Cas9-induced DSBs. (A) Schematic of the donor DNA in relation to the gRNA target site at the wls locus. The donor was a single-stranded oligonucleotide with 60-nt homology to the wls locus at either side of the Cas9 cut site and a G-to-C transversion (red, lowercase) that disrupts the PAM sequence. Donor DNA was injected into embryos that were the progeny of nos-cas9 females and U6:3-gRNA-wls males. (B) Summary of results from screening flies for HDR events by PCR and sequencing. In the six crosses, none of eight flies, two of eight flies, three of eight flies, six of seven flies, none of seven flies, and two of eight flies inherited the wlsG32C allele. (C) Example of the verification of the precise integration of the donor DNA in the wls locus by direct sequencing of a PCR product amplified from a heterozygous fly. Double peaks in the chromatogram represent an overlay of the sequence from the mutant and wild-type wls alleles. The asterisk indicates the position of the mutation.
CRISPR/Cas as a Tool for Analyzing Loss-of-Function Mutant Phenotypes in Somatic Tissues.
An important result from the evaluation of our tools is that transgenic CRISPR/Cas with U6:3-gRNAs can result in highly effective biallelic mutagenesis in the soma. We noticed in our experiments targeting y and e that cuticles frequently contained large patches of cells with the same mutant phenotype (Fig. 1 B and C), presumably resulting from clonal inheritance of the same CRISPR/Cas-induced lesion. Analysis of classical Drosophila mutations in genetic mosaics has been invaluable for understanding functions of genes that act at multiple stages during development as well as cell autonomous and nonautonomous mechanisms (31). However, generating the stocks required to induce clones by traditional methods is time consuming (Discussion). Therefore we wondered if our CRISPR/Cas tools could be used for the rapid generation of clones in which both alleles of essential genes are mutated.
We first analyzed wing imaginal discs from act-cas9 U6:3-gRNA-wls third-instar larvae using an antibody that recognizes the C-terminal tail of the Wls protein (32). Endogenous Wls is expressed ubiquitously in the wing imaginal disc, where it is required for the secretion of the Wg signaling molecule from Wg-producing cells (25, 26, 32). All wing imaginal discs from control act-cas9 U6:3-gRNA-e animals had wild-type Wls protein levels and distribution (Fig. 5A). In contrast, all act-cas9 U6:3-gRNA-wls wing imaginal discs examined contained patches of cells with partially reduced Wls levels, with no Wls protein, or with wild-type Wls levels (Fig. 5B). The first two scenarios presumably reflect clonal tissue that has one or two wls mutant alleles, respectively, that fail to produce full-length Wls protein. Wg protein was retained in Wg-producing cells within clones that lacked detectable Wls protein (Fig. 5B), confirming biallelic disruption of wls.
Fig. 5.
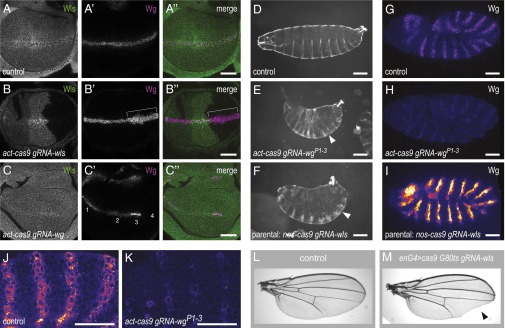
Revealing mutant phenotypes through efficient biallelic targeting with transgenic CRISPR/Cas. (A–C) Genetic mosaics induced in third-instar wing imaginal discs by somatic CRISPR/Cas. (A) Control act-cas9 U6:3-gRNA-e wing discs have ubiquitous expression of Wls and expression of Wg in a stripe along the dorsal–ventral boundary. (B) act-cas9 U6:3-gRNA-wls discs have patches of cells with wild-type Wls levels, partially reduced Wls expression, and no detectable Wls protein. Wg protein accumulates intracellularly in Wg-producing cells in the absence of Wls (bracket). (C) Clones with different distributions of endogenous Wg protein can be observed in act-cas9 U6:3-gRNA-wg discs: 1, wild type; 2, nuclear accumulation; 3, increased Wg levels; and 4, no Wg protein. A second example with abnormal nuclear Wg staining is shown at higher magnification in Fig. S6. Images are representative of >10 discs of each genotype. (D–K) Biallelic targeting by transgenic CRISPR/Cas can reveal mutant phenotypes during embryogenesis. (D–F) Cuticle preparations at the end of embryogenesis. (D) act-cas9 U6:3-gRNA-e control animal. (E and F) All act-cas9 gRNA-wgP1–3 embryos (E) and most embryos from nos-cas9 U6:3-gRNA-wls parents (F) have naked region of cuticle replaced by denticle bands (exemplified by arrowheads). (G–K) Analysis of Wg protein in stage 9/10 embryos by immunostaining (“fire” lookup table facilitates comparison of Wg signal intensity in different genotypes). Compared with control nos-cas9 U6:3-gRNA-e embryos (G), Wg signal is strongly reduced in act-cas9 gRNA-wgP1–3 embryos (H) and is strongly increased in embryos from nos-cas9 U6:3-gRNA-wls parents (I). (J and K) Higher-magnification views of Wg protein in embryos, showing variation in levels in act-cas9 gRNA-wgP1–3 embryos (K), presumably caused by independent CRISPR/Cas targeting events in subsets of cells. act-cas9 U6:3-gRNA-e is shown as a control (J). Images are representative of >50 embryos examined. (L and M) Cas9 activity can be focused on specific tissues by Gal4/UAS. Expression of gRNA-wls together with UAS-cas9 (line CFD5 in Table S1) under the control of enGal4 and Gal80ts gives rise to viable flies that have wing notches posteriorly in the adult wing (M; arrowhead). Control wing (L) is from an animal that did not inherit the gRNA-wls transgene. All genotypes in this figure refer to one copy of each transgene. (Scale bars: A–C, 40 μm; D–F, 100 μm; G–K, 30 μm.)
We next examined wing imaginal discs from act-cas9 U6:3-gRNA-wg animals using an anti-Wg antibody (33). The Wg expression domain normally includes a stripe of cells along the dorsal–ventral boundary of the wing pouch, a pattern always observed in act-cas9 U6:3-gRNA-e discs (Fig. 5A). In contrast, act-cas9 U6:3-gRNA-wg discs had disrupted patterns of Wg expression (Fig. 5C). Although clones that lost all detectable Wg protein were common, presumably reflecting biallelic loss-of-function mutations, we also observed clones with increased abundance or altered subcellular distribution of the protein (Fig. 5C and Fig. S6). These latter clones likely contain in-frame mutations at the gRNA target site that confer new properties to the Wg protein. This finding suggests that CRISPR/Cas also can shed light on the functional roles of specific regions of a protein.
The experiments described above demonstrated that act-cas9 and U6:3-gRNA can be used in imaginal discs to induce biallelic targeting of essential genes in a stochastic fashion. To investigate whether our transgenic CRISPR/Cas tools can reveal somatic mutant phenotypes at earlier developmental stages, we attempted to disrupt wg function in the embryo. wg is expressed zygotically in the embryo and is required for segment polarity; classical wg null mutants fail to complete embryogenesis and have regions of naked cuticle replaced with an array of denticles (24). Our earlier finding that many act-cas9 U6:3-gRNA-wg embryos develop to pupal stages demonstrates that they retain significant wg function (Fig. 3B). We therefore generated a gRNA transgene that simultaneously expresses three gRNAs targeting the wg promoter (gRNA-wgP1–3). This construct, in which the gRNAs were under the control of the U6:1, U6:2, and U6:3 promoters, was integrated at the attP2 site. All act-cas9 gRNA-wgP1–3 animals arrested before the end of embryogenesis with a strong segment polarity phenotype (Fig. 5 D and E). This result presumably reflects biallelic disruption of wg expression, because a single functional copy of wg is sufficient for normal development. Indeed, immunostaining of act-cas9 gRNA-wgP1–3 embryos revealed strongly reduced expression of Wg protein compared with act-cas9 U6:3-gRNA-e controls (Fig. 5 G, H, J, and K), indicative of interference with transcription of both alleles. We conclude that somatic CRISPR/Cas can be used to induce biallelic mutations in genes that are expressed zygotically in embryos.
Additional observations indicated that transgenic CRISPR/Cas also can be used to study loss-of-function embryonic phenotypes of maternally contributed genes. As described above, nos-cas9 U6:3-gRNA-wls adults had low fertility. Closer inspection revealed that intercrosses of these flies resulted in many eggs being laid but with less than 5% developing to larval stages. Wls normally is contributed maternally to the embryo. Although zygotic wls null mutants from heterozygous mothers develop until pupal stages, offspring of mothers with a homozygous wls-mutant germ line die at embryonic stages with cuticle phenotypes typical of perturbed Wg signaling (25, 26). The great majority of embryos from nos-cas9 U6:3-gRNA-wls mothers arrested at the end of embryogenesis with segment polarity phenotypes reminiscent of wls germ-line mutant clones (Fig. 5F). This observation indicates biallelic disruption of wls in the germ line of nos-cas9 U6:3-gRNA-wls females. Consistent with this notion, immunostaining revealed a strong accumulation of Wg protein within producing cells of embryos derived from nos-cas9 U6:3-gRNA-wls mothers (Fig. 5I), presumably resulting from defective Wg protein secretion in the absence of Wls.
Finally, we explored whether biallelic gene targeting in specific somatic cell types was possible by coupling CRISPR/Cas to the Gal4/UAS system. UAS-cas9 was combined with engrailed-Gal4 (enGal4), which drives UAS expression specifically in posterior compartments of the animal, and gRNA-wls. We also included a temperature-sensitive Gal80 transgene (Gal80ts) to restrict Cas9 activity further by temporal control of Gal4. Following a temperature shift of second-instar larvae of this genotype to 29 °C, many adult flies had pronounced notches specifically in the posterior half of the wing (Fig. 5 L and M). This phenotype was not observed in the absence of gRNA-wls (Fig. 5L). These results show that biallelic targeting of essential genes can be focused on specific cell types by regulating expression of Cas9 with Gal4/UAS. However, the first generation of UAS-cas9 constructs requires optimization. Substantial lethality was observed when UAS-cas9 transgenic lines were crossed to strong, ubiquitous Gal4 drivers, and this lethality was not dependent on the presence of a gRNA. This toxicity also was not dependent on the endonuclease activity of Cas9, because it also was observed with a catalytically dead mutant protein. Furthermore, when UAS-cas9 was combined with gRNA-wls, we observed some wls− clones in wing discs that were independent of Gal4 activity. Nonetheless this effect, which presumably is caused by leaky expression from the UAS promoter, was not sufficient to produce wing notching in the adult. Future efforts will be aimed at producing UAS-cas9 variants that have reduced expression levels but still retain sufficient activity for biallelic targeting in response to Gal4.
Discussion
CRISPR/Cas promises to revolutionize genome engineering in a large variety of organisms. Although proof-of-principle studies have demonstrated CRISPR/Cas-mediated mutagenesis in Drosophila melanogaster (5–12), a systematic evaluation of different tools has not been reported. We have developed a collection of tools to harness further the potential of CRISPR/Cas for functional studies in Drosophila. We have characterized different Cas9 and gRNAs expression constructs and revealed combinations that allow mutagenesis of essential and nonessential genes with high rates, support efficient integration of exogenous sequences, and can reveal mutant phenotypes readily in somatic tissues.
To assess the activity of different constructs, we used a fully transgenic CRISPR/Cas system in which Cas9 and gRNA are expressed from plasmid sequences stably integrated at defined positions in the genome. The advantage of this approach for comparative analysis is the low variability between experiments. For example, we found that 100% of flies expressing transgenic Cas9 and gRNA constructs had efficient germ-line mutagenesis of the target site. In contrast, the number of animals that transmit mutations to the next generation using delivery of one or both CRISPR/Cas components by RNA or DNA microinjection is highly variable and often is less than 50% (5, 6, 9–11). However, injection-based CRISPR/Cas may be advantageous under certain circumstances, such as when one wishes to generate a mutation directly in a complex genetic background. Our gRNA-expression plasmids are also suitable for this purpose.
Our quantitative analysis using transgenic Cas9 and gRNA constructs reveals that, of the three U6 promoters, the previously uncharacterized U6:3 promoter leads to the strongest gRNA activity. This observation suggests that targeting rates of previous approaches have been limited by the selection of the U6:2 or U6:1 promoter. In combination with highly active Cas9 lines (e.g., act-cas9, vasa-cas9), transgenic U6:3-gRNA can be used to transmit mutations in nonessential genes through the germ line with remarkably high efficiency (i.e., with 50–100% of offspring receiving a nonfunctional CRISPR-generated allele). However, these Cas9 lines also are active outside the germ line, and biallelic targeting in the soma effectively prevents their use for transmission of mutations in essential genes in combination with U6:3-gRNA transgenes. The ubiquitous activity of vasa-cas9 presumably was masked in a previous study in which a U6:2-gRNA plasmid targeting the eye pigmentation gene rosy was injected at the posterior of the embryo, thereby restricting its activity in the vicinity of the future germ cells (11). It remains to be evaluated if activity of CRISPR/Cas is sufficiently restricted by this approach to allow efficient germ-line transmission of mutations in essential genes. We demonstrate that efficient targeting of essential genes is possible using U6:3-gRNA transgenes in combination with the transgenic nos-cas9 line in which endonuclease activity is largely germ-line restricted. This finding held true even for wls, which is required in the germ line, because fertility (although much reduced) was not abolished.
Our optimized CRISPR/Cas system also allowed the integration of designer mutations with high efficiency. By injecting oligonucleotide donors and donor plasmids into embryos expressing cas9 and gRNA transgenes, we achieved precise modification of the target site in 11–38% of all offspring analyzed. A previous study in which an oligonucleotide was injected together with cas9 and gRNA plasmids found that 0.3% of all offspring integrated the exogenous sequence at the target site (5). Very recently, Gratz et al. (11) have reported integration rates for larger constructs that range from 0–11% by injecting the donor and gRNA plasmid into vasa-cas9 transgenic embryos. It is difficult to compare the integration efficiency of our method with that in previous studies directly because of the use of different gRNAs, donors, and target genes. Nonetheless, the increased reproducibility and efficiency of site-specific DSB generation with our optimized, fully transgenic CRISPR/Cas system is likely to facilitate integration of exogenous sequences by HDR as compared with methods that deliver Cas9 or gRNA by direct embryo injection. The frequency of integration is of paramount importance when the goal is to introduce subtle changes in the DNA sequence, such as individual point mutations, because a low rate of integration necessitates screening by cointegration of a marker gene, which can affect function of the targeted locus [note that exogenous sequences are still retained following removal of markers by site-specific recombinases (34)]. We demonstrate here that optimized transgenic CRISPR/Cas can facilitate the introduction of precise changes at endogenous loci with rates that make it practical to screen solely by PCR and Sanger sequencing. We envisage that one important application of our tools will be the introduction of disease mutations into the Drosophila genome to model human pathologies.
We also demonstrate a novel application of CRISPR/Cas in rapidly analyzing gene function in somatic cells. Our optimized tools permit efficient generation of genetic mosaics in which clones of cells have biallelic gene disruption. Mitotic recombination using the yeast site-specific recombinase Flp is typically used for this purpose in Drosophila (34). This method requires recombination of the mutation of interest onto a chromosome containing the FLP recombinase target (FRT) site; this process is time consuming, particularly for genes located close to the FRT site. Somatic CRISPR/Cas requires only one cross to give rise to mosaics. Furthermore, unlike the FRT/Flp system, there is no a priori reason for CRISPR/Cas to require cell division, and hence this method should be better suited for interrogating gene function in differentiated tissues such as the adult brain. One advantage of FRT/Flp in mosaic analysis is that clones can be marked readily by the presence or absence of a linked marker gene, such as gfp. Here we identified mutant cells with antibodies specific to the proteins studied, but for many proteins no antibody will be available. Under these circumstances we envisage mutant tissue being identified first by using CRISPR/Cas to generate a line in which a fluorescent protein or short epitope tag has been knocked into the endogenous locus. In any case, such a reagent would be valuable for characterizing the function of the protein. We anticipate that CRISPR/Cas and FRT-Flp will coexist as complementary methods for mosaic analysis of gene function in the future.
As is the case for somatic CRISPR/Cas, RNAi can reveal mutant phenotypes after a single cross. However, RNAi often reduces gene expression only partially, meaning that some mutant phenotypes can be incompletely penetrant or missed all together because of residual protein. Somatic CRISPR/Cas readily provides access to null-mutant phenotypes caused by biallelic out-of-frame mutations. Consequently, somatic targeting of all genes tested (e, y, cu, wg, or wls) revealed the classical loss-of-function mutant phenotype with high penetrance. Furthermore, RNAi is notorious for off-target effects (35), which can complicate phenotypic analysis significantly. Early studies in human cells did suggest that off-target mutagenesis also is prevalent for CRISPR/Cas (36, 37). However, experiments in Drosophila so far have failed to detect induced mutations at sites without perfect complementarity (6, 11). These findings suggest that CRISPR/Cas operates with high fidelity in flies, although addressing the issue of specificity definitively will require genome-wide searches for induced indels. Nonetheless, it is encouraging that nonspecific phenotypes were not detected in any of our experiments targeting e, y, cu, wg, or wls. A recent study showed that truncating the target sequence of gRNAs results in improved fidelity of CRISPR/Cas in human cells (38), suggesting a straightforward approach to minimize the risk of off-target effects in Drosophila. Our generation of tools for offset nicking-based mutagenesis in flies provides a further option for increasing specificity. Because of the ease with which CRISPR/Cas experiments can be performed, the specificity of mutant phenotypes generated with wild-type Cas9 also can be tested using multiple independent gRNAs.
High-throughput genetic screens are likely to be another important application of somatic gene targeting by CRISPR/Cas. The generation of gRNA-expression plasmids through cloning of annealed short oligonucleotides is an easy and economic one-step process that is scalable to generate a library of targeting vectors for thousands of genes. CRISPR/Cas-based screens will be particularly powerful if mutagenesis can be restricted in time and space. We have taken a step in this direction by demonstrating that expressing Cas9 under the control of the Gal4/UAS system can enrich biallelic targeting within a defined group of cells.
Materials and Methods
Addition materials and methods are described in SI Materials and Methods.
Plasmid Construction.
Unless otherwise noted, cloning was performed with the Gibson Assembly Master Mix (New England Biolabs). PCR products were produced with the Q5 High-Fidelity 2× Master Mix (New England Biolabs). All inserts were verified by sequencing. Details of plasmid construction are available in SI Materials and Methods. Primers used for plasmid construction are listed in Table S5.
Drosophila Genetics.
Details of transgenes and fly stocks are given in Tables S1–S3 and S6. Throughout this study, transgenic cas9 virgin females were crossed to U6-gRNA–expressing males.
Assessing relative activity of Cas9 and gRNA lines.
All crosses involving gRNA-e produced offspring with two wild-type e alleles that can be subjected to gene targeting by CRISPR/Cas. Because some Cas9 strains were generated in a y mutant background and some attP sites are marked by y+ transgenes, offspring from the different gRNA-y crosses inherit different numbers of y alleles from their parents (Fig. 1C and Table S3). As a result, either 0% or 25% of F2 offspring from the different crosses would be expected to be phenotypically yellow in the absence of CRISPR/Cas-mediated mutagenesis. To correct for this imbalance, we subtracted 25% of the phenotypically yellow animals when calculating germ-line transmission rates of nonfunctional alleles from data collected from the latter type of cross.
Genetic complementation of wg loss-of-function alleles.
nos-cas9/+; gRNA-wg/+ flies were crossed to a Sp/CyO balancer strain. Individual flies of the genotype wgtest/CyO then were crossed to wgTV-Cherry/CyO flies. wgTV-Cherry is a wg null allele in which the first exon of wg is replaced with a homologous recombination targeting cassette (27). The next generation of the crosses was screened for the presence or absence of flies without the CyO chromosome. Cultures in which all flies had the CyO chromosome indicated that the wgtest allele could not genetically complement wgTV-Cherry and hence did not encode functional Wg protein.
Molecular Characterization of Target Loci.
To identify the molecular nature of CRISPR/Cas-induced mutations, genomic DNA was extracted from individual flies or larvae by crushing them in 15 μL microLysis-plus (Microzone) and releasing the DNA in a thermocycler according to the supplier’s instructions. We used 0.5 μL of the supernatant in 25-μL PCR reactions (Q5 2× Master Mix kit; New England Biolabs) using primers binding 300–500 bp upstream and downstream of the target site. PCR products were gel purified and were either sent directly for Sanger sequencing or cloned into pBluescript-SK(+) by Gibson assembly. In the latter case, single colonies were selected as templates for PCR amplification, and PCR products were sequenced. Direct sequencing of PCR products from genomic DNA of heterozygous flies usually yields an overlay of the DNA sequence from both chromosomes. Indels can be observed as regions with double peaks in the trace. In the majority of such regions the sequence of both alleles can be called unambiguously (see Fig. 4C and Fig. S4D for examples).
Embryo Injections.
Embryos were injected using standard procedures (further details are given in SI Materials and Methods). For the delivery of plasmid DNA for the production of transgenes with the Phi31C system, 150 ng/μL of DNA in sterile dH2O was injected. ssODNs designed to modify the wls locus were injected into the posterior region of nos-cas9/+; U6-3-gRNA-wls/+ preblastoderm embryos as a 750-ng/μL solution in dH2O.
Supplementary Material
Acknowledgments
We thank C. Alexandre, A. Baena-Lopez, K. Beumer, D. Carroll, M. Harrison, K. Koles, K. O’Connor-Giles, A. Rodal, J. P. Vincent, J. Wildonger, and others from the Drosophila genome engineering community for generously sharing unpublished results and reagents; S. Gratz for further details of published y HDR experiments; M. Reimao-Pinto and T. Samuels for help with cloning and genotyping; and S. Aldaz, K. Basler, J. Bischof, S. Collier, J. Crooker, N. Perrimon, K. Röper, D. Stern, and R. Yagi for additional reagents. This study was supported by a Marie-Curie IntraEuropean Fellowship (to F.P.), the Swiss National Science Foundation (to F.P.), Howard Hughes Medical Institute (H.-M.C. and T.L.), and by UK Medical Research Council Project U105178790 (S.L.B.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1405500111/-/DCSupplemental.
References
- 1.Carroll D. Genome engineering with targetable nucleases. Annu Rev Biochem. 2014;2(83):409–439. doi: 10.1146/annurev-biochem-060713-035418. [DOI] [PubMed] [Google Scholar]
- 2.Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annu Rev Genet. 2006;40:363–383. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
- 3.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507(7490):62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gratz SJ, et al. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 2013;194(4):1029–1035. doi: 10.1534/genetics.113.152710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bassett AR, Tibbit C, Ponting CP, Liu J-L. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Reports. 2013;4(1):220–228. doi: 10.1016/j.celrep.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu Z, et al. Highly efficient genome modifications mediated by CRISPR/Cas9 in Drosophila. Genetics. 2013;195(1):289–291. doi: 10.1534/genetics.113.153825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kondo S, Ueda R. Highly improved gene targeting by germline-specific Cas9 expression in Drosophila. Genetics. 2013;195(3):715–721. doi: 10.1534/genetics.113.156737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sebo ZL, Lee HB, Peng Y, Guo Y. A simplified and efficient germline-specific CRISPR/Cas9 system for Drosophila genomic engineering. Fly (Austin) 2014;8(1):52–57. doi: 10.4161/fly.26828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ren X, et al. Optimized gene editing technology for Drosophila melanogaster using germ line-specific Cas9. Proc Natl Acad Sci USA. 2013;110(47):19012–19017. doi: 10.1073/pnas.1318481110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gratz SJ, et al. Highly specific and efficient CRISPR/Cas9-catalyzed homology-directed repair in Drosophila. Genetics. 2014;196(4):961–971. doi: 10.1534/genetics.113.160713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu Z, et al. Various applications of TALEN- and CRISPR/Cas9-mediated homologous recombination to modify the Drosophila genome. Biol Open. 2014;3(4):271–280. doi: 10.1242/bio.20147682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jinek M, et al. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Doren M, Williamson AL, Lehmann R. Regulation of zygotic gene expression in Drosophila primordial germ cells. Curr Biol. 1998;8(4):243–246. doi: 10.1016/s0960-9822(98)70091-0. [DOI] [PubMed] [Google Scholar]
- 16.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118(2):401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 17.Pfeiffer BD, Truman JW, Rubin GM. Using translational enhancers to increase transgene expression in Drosophila. Proc Natl Acad Sci USA. 2012;109(17):6626–6631. doi: 10.1073/pnas.1204520109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Groth AC, Fish M, Nusse R, Calos MP. Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics. 2004;166(4):1775–1782. doi: 10.1534/genetics.166.4.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci USA. 2007;104(9):3312–3317. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153(4):910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mali P, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31(9):833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ran FA, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154(6):1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grönke S, Bickmeyer I, Wunderlich R, Jäckle H, Kühnlein RP. Curled encodes the Drosophila homolog of the vertebrate circadian deadenylase Nocturnin. Genetics. 2009;183(1):219–232. doi: 10.1534/genetics.109.105601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nüsslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287(5785):795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 25.Bänziger C, et al. Wntless, a conserved membrane protein dedicated to the secretion of Wnt proteins from signaling cells. Cell. 2006;125(3):509–522. doi: 10.1016/j.cell.2006.02.049. [DOI] [PubMed] [Google Scholar]
- 26.Bartscherer K, Pelte N, Ingelfinger D, Boutros M. Secretion of Wnt ligands requires Evi, a conserved transmembrane protein. Cell. 2006;125(3):523–533. doi: 10.1016/j.cell.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 27.Baena-Lopez LA, Alexandre C, Mitchell A, Pasakarnis L, Vincent J-P. Accelerated homologous recombination and subsequent genome modification in Drosophila. Development. 2013;140(23):4818–4825. doi: 10.1242/dev.100933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rong YS, Golic KG. Gene targeting by homologous recombination in Drosophila. Science. 2000;288(5473):2013–2018. doi: 10.1126/science.288.5473.2013. [DOI] [PubMed] [Google Scholar]
- 29.Beumer KJ, et al. Efficient gene targeting in Drosophila by direct embryo injection with zinc-finger nucleases. Proc Natl Acad Sci USA. 2008;105(50):19821–19826. doi: 10.1073/pnas.0810475105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beumer KJ, et al. Comparing zinc finger nucleases and transcription activator-like effector nucleases for gene targeting in Drosophila. G3 (Bethesda) 2013;3(10):1717–1725. doi: 10.1534/g3.113.007260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolfner MF, Goldberg ML. Harnessing the power of Drosophila genetics. Methods Cell Biol. 1994;44:33–80. doi: 10.1016/s0091-679x(08)60906-1. [DOI] [PubMed] [Google Scholar]
- 32.Port F, et al. Wingless secretion promotes and requires retromer-dependent cycling of Wntless. Nat Cell Biol. 2008;10(2):178–185. doi: 10.1038/ncb1687. [DOI] [PubMed] [Google Scholar]
- 33.Brook WJ, Cohen SM. Antagonistic interactions between wingless and decapentaplegic responsible for dorsal-ventral pattern in the Drosophila Leg. Science. 1996;273(5280):1373–1377. doi: 10.1126/science.273.5280.1373. [DOI] [PubMed] [Google Scholar]
- 34.Bischof J, Basler K. Recombinases and their use in gene activation, gene inactivation, and transgenesis. Methods Mol Biol. 2008;420:175–195. doi: 10.1007/978-1-59745-583-1_10. [DOI] [PubMed] [Google Scholar]
- 35.Ma Y, Creanga A, Lum L, Beachy PA. Prevalence of off-target effects in Drosophila RNA interference screens. Nature. 2006;443(7109):359–363. doi: 10.1038/nature05179. [DOI] [PubMed] [Google Scholar]
- 36.Fu Y, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31(9):822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pattanayak V, et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31(9):839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32(3):279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.