

Significance
Resistance of aggressive metastatic tumors to cisplatin therapy is partly related to extensive repair of cisplatin-modified DNA in the nucleus by the nucleotide excision repair machinery. Delivering cisplatin to the mitochondria to attack the mitochondrial genome lacking such repair machinery can lead to a rationally designed therapy for metastatic, chemoresistant cancers and might overcome the problems associated with conventional cisplatin treatment.
Keywords: click chemistry, brain cancer, OXPHOS, pharmacokinetics
Abstract
Chemoresistance of cisplatin therapy is related to extensive repair of cisplatin-modified DNA in the nucleus by the nucleotide excision repair (NER). Delivering cisplatin to the mitochondria to attack mitochondrial genome lacking NER machinery can lead to a rationally designed therapy for metastatic, chemoresistant cancers and might overcome the problems associated with conventional cisplatin treatment. An engineered hydrophobic mitochondria-targeted cisplatin prodrug, Platin-M, was constructed using a strain-promoted alkyne–azide cycloaddition chemistry. Efficient delivery of Platin-M using a biocompatible polymeric nanoparticle (NP) based on biodegradable poly(lactic-co-glycolic acid)-block-polyethyleneglycol functionalized with a terminal triphenylphosphonium cation, which has remarkable activity to target mitochondria of cells, resulted in controlled release of cisplatin from Platin-M locally inside the mitochondrial matrix to attack mtDNA and exhibited otherwise-resistant advanced cancer sensitive to cisplatin-based chemotherapy. Identification of an optimized targeted-NP formulation with brain-penetrating properties allowed for delivery of Platin-M inside the mitochondria of neuroblastoma cells resulting in ∼17 times more activity than cisplatin. The remarkable activity of Platin-M and its targeted-NP in cisplatin-resistant cells was correlated with the hyperpolarization of mitochondria in these cells and mitochondrial bioenergetics studies in the resistance cells further supported this hypothesis. This unique dual-targeting approach to controlled mitochondrial delivery of cisplatin in the form of a prodrug to attack the mitochondrial genome lacking NER machinery and in vivo distribution of the delivery vehicle in the brain suggested previously undescribed routes for cisplatin-based therapy.
The cellular powerhouse, mitochondria, are implicated in the process of carcinogenesis because of their vital roles in energy production and apoptosis. Mitochondria are the key players in generating the cellular energy through oxidative phosphorylation (OXPHOS) that produces reactive oxygen species (ROS) as by-products. Mitochondrial DNA (mtDNA) plays significant roles in cell death and metastatic competence. The close proximity of mtDNA to the ROS production site makes this genome vulnerable to oxidative damage. Mitochondrial dysfunction and associated mtDNA depletion possibly reversibly control epigenetic changes in the nucleus that contributes to cancer development (1). Thus, targeting mtDNA could lead to novel and effective therapies for aggressive cancers. Cisplatin, a Food and Drug Administration-approved chemotherapeutic agent, is most extensively characterized as a DNA-damaging agent and the cytotoxicity of cisplatin is attributed to the ability to form interstrand and intrastrand nuclear DNA (nDNA) cross-links (2). The nucleotide excision repair (NER) pathway plays major roles in repairing cisplatin-nDNA adducts (3). Resistance to cisplatin can result from several mechanisms, including decreased uptake and accelerated DNA repair by NER machinery. Limited studies have examined cisplatin activity on mtDNA of cancer cells (4). The lack of NER in the mitochondria and enhanced mtDNA mutation in aggressive cancer gives a strong rationale in directing cisplatin inside the mitochondrial matrix to provide an effective therapeutic option. However, the most important step to attack mtDNA by cisplatin requires a mitochondria-targeted drug and, even better, an optimal drug delivery system that is able to reach the innermost mitochondrial space, the mitochondrial matrix, where the mtDNA is located. We recently developed a biocompatible polymeric nanoparticle (NP) based on biodegradable poly(lactic-co-glycolic acid) (PLGA)-block (b)-polyethyleneglycol (PEG) functionalized with a terminal triphenylphosphonium (TPP) cation, which has remarkable activity to target mitochondria of cells due to its high lipophilic properties, presence of delocalized positive charge, and appropriate size range (5, 6). Here, we describe construction of a hydrophobic mitochondria-targeted cisplatin prodrug, Platin-M, using strain-promoted alkyne–azide cycloaddition (SPAAC) chemistry and its delivery using PLGA-b-PEG-TPP NPs to release cisplatin locally inside the mitochondrial matrix to attack mtDNA for overcoming resistance associated with conventional cisplatin-based chemotherapy (Fig. 1A).
Fig. 1.

(A) Schematic diagram for mitochondrial delivery of cisplatin prodrug using a targeted NP and the mechanism of action. (B) Synthesis of mitochondria-targeted Pt(IV) prodrug Platin-M.
Results and Discussion
Construction of Mitochondria-Targeted Cisplatin Prodrug.
A mitochondria-targeted Pt(IV)-prodrug of cisplatin, Platin-M, was designed by introducing two mitochondria-targeting delocalized lipophilic TPP cations in the axial positions (Fig. 1A). Recently, a Pt(II) compound, mtPt, with a mitochondria-targeting peptide for attacking mtDNA, was reported (7). Pt(IV) prodrugs are advantageous over the Pt(II) counterparts because of their greater stability and local activation, which allow a greater proportion of the active drug at the target site(s) (8). Mitochondrial functions including respiration are greatly reduced in cancer cells, and tumor microenvironments differ greatly from that of normal tissues. Mitochondrial membrane potential (ΔΨm) in most cancer cells is greater compared with that of normal cells (9). Therefore, TPP cation containing Platin-M will take advantage of the substantial negative ΔΨm across the inner mitochondrial membrane (IMM) to efficiently accumulate inside the matrix (Fig. 1A). Conventional coupling reaction with TPP ligands was found to be problematic due to reduction of Pt(IV). We recently developed a copper(I) free cycloaddition approach to introduce new functionalities on to Pt(IV) with high efficiency (10). A TPP moiety was introduced on azadibenzocyclooctyne (DBCO) derivative to result in DBCO-TPP (Fig. 1B and SI Appendix, Figs. S1–S4). A SPAAC reaction between an azide-Pt(IV) precursor Platin-Az (10) and DBCO-TPP resulted in Platin-M with high efficiency (Fig. 1B and SI Appendix, Figs. S5–S10). Feasible cellular reduction of Pt(IV) prodrug is required to release active cisplatin. Electrochemical studies performed at two different biologically relevant pH values of 7.4 and 6.4 demonstrated that Platin-M will release active cisplatin in the cellular environment (SI Appendix, Fig. S11).
Development of Mitochondria-Targeted NP for Platin-M.
Small molecules, in particular Pt-based therapeutics, show poor biodistribution (bioD) and pharmacokinetic (PK) properties, rapid clearance, and inactivation by biological nucleophiles before reaching the cellular targets. We anticipated that Platin-M would face similar challenges if administered in its pristine form. Self-assembled polymeric NPs composed of biodegradable PLGA-b-PEG block copolymer hold promise as carriers for small molecules (11). However, the NP system that can deliver Platin-M successfully to the mitochondria of cells can be challenging. We recently developed an engineered NP from a triblock copolymer PLGA-b-PEG-TPP and identified an optimized targeted formulation for mitochondrial delivery of small molecules (5, 6). We anticipated that engineering of NPs from PLGA-b-PEG-TPP polymers of different molecular weight would allow us to have a control over NP distribution in different mitochondrial compartments, outer mitochondrial membrane (OMM), IMM, the intermembrane space (IMS), and matrix and control the release kinetics of Platin-M from these NPs. Thus, we constructed two triblock copolymers, PLGALMW-b-PEG-TPP and PLGAHMW-b-PEG-TPP, based on a low–molecular-weight and a high–molecular-weight PLGA-COOH, respectively (SI Appendix, Figs. S12–S16). To understand the distribution properties of the NPs in different mitochondrial compartments, we fractionated mitochondria isolated from prostate cancer (PCa) PC3 cells treated with NPs from PLGAHMW-b-PEG-TPP and PLGALMW-b-PEG-TPP into OMM, IMM, IMS, and mitochondrial matrix. We incorporated 10% of PLGA-b-PEG-quantum dot (QD) (5) in both NPs; the NPs from PLGALMW-b-PEG-TPP had hydrodynamic diameter of 51.3 ± 0.8 nm and ζ potential of 44.0 ± 1.2 mV, and the NPs from PLGAHMW-b-PEG-TPP were 143.2 ± 3.2 nm in size with ζ potential of 28.1 ± 0.7 mV. Cadmium (Cd) quantification by inductively coupled plasma mass spectrometry (ICP-MS) and fluorescence imaging using an in vivo imaging system (IVIS) of cytosolic, IMM, OMM, IMS, and mitochondrial matrix indicated that the NPs from PLGALMW-b-PEG-TPP distributed most efficiently in the mitochondrial matrix and the NPs from PLGAHMW-b-PEG-TPP accumulated mainly in the OMM and cytosol (Fig. 2A). We used the NPs based on PLGALMW-b-PEG-TPP for efficient delivery of Platin-M inside the mitochondria for further studies.
Fig. 2.
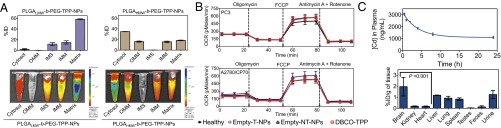
(A) Distribution of PLGAHMW-b-PEG-TPP and PLGALMW-b-PEG-TPP-NPs in mitochondrial compartments of PC3 cells by ICP-MS (Upper) and IVIS analysis (Lower). (B) Mitochondrial toxicity of DBCO-TPP, Empty-T-NPs, and Empty-NT-NPs in PC3 and A2780/CP70 cells by Mito Stress assay. (C) Variation of Cd concentration in plasma with time, tissue distribution, and cumulative excretion profiles following the administration of T-QD-NPs by i.v. to male rats.
Mitochondrial Toxicity of Mitochondria-Targeted NPs (T-NPs).
Preclinical mitochondrial toxicology tests can have high predictive values. Thus, we studied whether empty PLGALMW-b-PEG-TPP-NPs (Empty-T-NPs) containing TPP molecules exhibit deleterious effects after entering mitochondria. TPP-based small molecules were described to disrupt ΔΨm, uncouple OXPHOS, and inhibit mitochondrial respiration (12). We examined Empty-T-NP, empty nontargeted PLGALMW-b-PEG-OH-NPs (Empty-NT-NPs), and DBCO-TPP–induced changes in mitochondrial respiration in PC3 and cisplatin-resistant ovarian cancer A2780/CP70 cells as a measure of mitochondrial toxicity (Fig. 2B and SI Appendix, Fig. S17). Oxygen consumption rate (OCR) of cells is an important indicator of normal mitochondrial functions. Mitochondrial bioenergetics in PC3 and A2780/CP70 cells treated with Empty-T-NPs (0.5 mg/mL), Empty-NT-NPs (0.5 mg/mL), and DBCO-TPP (10 μM) for 12 h were assessed using XF24 extracellular flux analyzer (SI Appendix). The effects of metabolic modulators oligomycin, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), antimycin A, and rotenone allowed determination of multiple parameters of mitochondrial functions (Fig. 2B and SI Appendix, Fig. S17). Empty-T-NPs, Empty-NT-NPs, and DBCO-TPP did not show any changes in the basal OCR and the OCR linked to ATP production in the treated cells. Several parameters linked to mitochondrial respiration; spare respiratory capacity, coupling efficiency, basal respiration, electron transport chain (ETC) accelerator response, and ATP coupler response were calculated from the OCR (picomoles per minute) vs. time (minutes) traces (Fig. 2B and SI Appendix, Fig. S17). The ATP synthase inhibitor oligomycin was injected to evaluate mitochondrial coupling upon accumulation of T-NPs inside the mitochondria. When proton flux through the ATP synthase is inhibited, phosphorylating respiration stops and residual oxygen consumption is primarily due to proton leak across the IMM. Oligomycin decreased OCR to the same level in healthy and Empty-T-NPs, Empty-NT-NPs, and DBCO-TPP–treated cells (Fig. 2B and SI Appendix, Fig. S17), indicating that the mitochondria remain coupled. Next, we injected FCCP, a H+ ionophore and uncoupler of OXPHOS, to examine maximal respiratory capacity. FCCP dissipates the proton gradient across the IMM and uncouples electron transport from OXPHOS; thus, in the presence of FCCP, OCR increases to the maximum extent supported by the ETC and substrate supply. Stimulation of respiration by FCCP in healthy cells and Empty-T-NPs, Empty-NT-NPs, and DBCO-TPP–treated cells were similar, indicating that bioenergetic functions are well preserved in the presence of T-NPs. Inhibition of mitochondrial flux by addition of rotenone, a complex I inhibitor and antimycin A, a complex III inhibitor in healthy, Empty-T-NP, Empty-NT-NP, and DBCO-TPP–treated cells indicated similar levels of mitochondrial and nonmitochondrial respiration. Collectively, these data suggested that several TPP containing T-NPs enter mitochondrial matrix efficiently; however, these NPs do not cause any mitochondrial function inhibition or toxicity. Heart cells contain hyperpolarized mitochondria; we therefore studied toxicity of mitochondria accumulating Empty-T-NPs in myogenic H9C2 cardiomyocytes derived from embryonic rat heart ventricle (SI Appendix, Fig. S18). MitoStress assay using mitochondrial inhibitors as described before on H9C2 cells after treatment with 0.5 mg/mL Empty-T-NPs and Empty-NT-NPs for 12 h demonstrated no significant effects on basal OCR levels in these cells (SI Appendix, Fig. S18). The basal OCR levels in cardiomyocytes were low compared with other cells indicating significant mitochondrial hyperpolarization. These properties make the T-NPs suitable for delivery of therapeutics inside the mitochondria.
Long-Circulating T-NPs Distribute in the Brain.
For potential in vivo translation of T-NPs in delivering Platin-M; bioD, excretion, and PK properties are the most critical parameters. We injected T-QD-NPs into Sprague–Dawley rats by a single-dose i.v. injection. Blood samples at predetermined time points up to 24 h postinjection, organs after 24 h, and cumulative urine, feces over 24 h were collected and analyzed for Cd by ICP-MS. Calculation of PK parameters by a two-compartment i.v. input model revealed a plasma elimination half life (t1/2) from the central compartment of 2.4 h (Fig. 2C and SI Appendix, Table S1). The total body clearance (CL) of T-NPs was ∼4.7 mL/h⋅kg in the central compartment and 0.05 mL/h⋅kg in the terminal phase (SI Appendix, Table S1). The high t1/2 and a small CL values indicated long-circulating properties of T-NPs. The significantly higher area under curve (AUC) of 34,784 ± 2,117 h⋅ng/mL further supported the long-circulation property of T-NPs. A peak plasma concentration (Cmax) of 3,237 ± 128 ng/mL indicated efficient distribution of T-NPs into bloodstream (SI Appendix, Table S1). A large volume of distribution (Vd) in the central compartment suggested that initially T-NPs distribute extensively into body tissues; however, a reduced Vd in the terminal compartment supported that, due to their unique composition with sterically hindered surface covered with –TPP moieties, these NPs exhibited decreased protein binding. All these parameters together supported that T-NP will be an excellent candidate for mitochondrial delivery of Platin-M.
Distribution of T-NPs in the major tissues, including spleen, liver, lungs, brain, heart, kidneys, and testes at 24 h postdose indicated maximum NP accumulation in the brain (Fig. 2C). The dichotomy between the brain capillary endothelium forming the blood–brain barrier (BBB) and endothelia in peripheral prevent passage of larger NPs with hydrophilic anionic surface. Pass from blood to brain of circulating NPs may only happen by transcellular mechanisms, which require a highly lipophilic NP with suitable size and charge. Brain endothelial cell surface and basement membrane components bearing highly anionic charges from sulfated proteoglycans are different from nonbrain endothelium and would allow the adsorptive-mediated transcytosis of cationic NPs (13). Thus, the small size and highly lipophilic surface of T-NPs helped their distribution in the brain. Furthermore, high mitochondrial density in cerebral endothelial cells than in peripheral endothelia further assisted these T-NPs to accumulate in the brain efficiently (14, 15). The distinctive properties of brain endothelium, high lipophilicity, and mitochondria-targeting properties of T-NPs provided selective targeting of these NPs to the brain. In many instances, positively charged NPs accumulate in the liver and the spleen by phagocytic cells present in the mononuclear phagocyte system located primarily in these organs. T-NPs, despite positively charged surface, showed a high brain-to-spleen ratio of ∼2.4 and a moderate brain-to-liver ratio of ∼1.6 (Fig. 2C). T-NPs demonstrated a high brain-to-kidney ratio of ∼10 and a brain-to-lung distribution ratio of ∼2.2. Retention of T-NPs was extremely low in the heart with a brain-to-heart ratio of 11, indicating that, although the heart cells have hyperpolarized mitochondria, the lipophilic properties of the T-NPs help preferential distribution in the brain. These highly positively charged NPs demonstrated hepatobiliary excretion.
Mitochondria-Targeted NP Formulation of Platin-M.
The rationale behind incorporation of Platin-M into a mitochondria-targeted delivery system such as T-NPs was to efficiently encapsulate hydrophobic Platin-M to increase its blood circulation; upon uptake by cancer cells with hyperpolarized ΔΨm, these NPs will deliver Platin-M with high accuracy and efficiency inside the mitochondrial matrix. However, any Platin-M released from the NPs before reaching mitochondria will take advantage of the TPP moieties present on Platin-M for mitochondrial uptake. Thus, this dual-targeted system expected to show effective mitochondrial accumulation. We constructed T-Platin-M-NPs by entrapping Platin-M inside PLGALMW-b-PEG-TPP polymer. As a single targeted control, we used Platin-M entrapped inside PLGALMW-b-PEG-OH to construct NT-Platin-M-NPs. We used a nanoprecipitation method (5) for entrapping Platin-M in these polymers and the NPs were characterized by dynamic light scattering to give the size, polydispersity index (PDI), and ζ potential of each preparation (Fig. 3A and SI Appendix, Figs. S19 and S20 and Tables S2 and S3). T-Platin-M-NPs and NT-Platin-M-NPs showed sizes in the range of 50–55 nm. T-Platin-M-NPs exhibited a highly positive ζ potential between 28 and 37 mV. NT-Platin-M-NPs showed a negative ζ potential between −22 and −34 mV. Morphology of T-Platin-M and NT-Platin-M-NPs was investigated using transmission electron microscopy (TEM) (Fig. 3A). Loading efficiencies of Platin-M at various added weight-percentage values of Pt(IV) to polymer indicated that Platin-M can be entrapped in these NPs with a very high loading and encapsulation efficiency (EE) (Fig. 3A). Percentage loading of Platin-M varied between 6 and 26% based on %feed with respect to feed of the polymer (Fig. 3A and SI Appendix, Tables S2 and S3). Given the amphiphilic character of Platin-M with two cationic TPP head groups and lipophilic DBCO moieties, we questioned whether high loading of Platin-M is due to the formation of self-micelles. Nanoprecipitation of an organic solution of Platin-M into water resulted in unstable macroparticles (size: 941.6 ± 589.5 nm; PDI: 0.799; ζ potential: −5.38 ± 1.66 mV) leaving out such a possibility. To the best of our knowledge, Platin-M showed very high loading efficiency among the known Pt(IV) complexes in a PLGA-PEG–based NP system (16, 17). Release kinetics of Platin-M from T and NT-NPs under physiological pH 7.4 in PBS at 37 °C demonstrated sustained release over a period of 72 h (Fig. 3B). A comparison of release kinetics of T and NT-NPs demonstrated that Platin-M released from NT-NPs at a slower rate (Fig. 3B). Positively charged Platin-M released from NT-NPs might get adsorb on the negatively charged NP surface and this noncovalent interaction might be responsible for slower release kinetics of Platin-M from NT-NPs. Platin-M was found to be released at a much faster rate at pH 6.0 compared with pH 7.4 (Fig. 3B). The pH-dependent release is possibly due to accelerated degradation of PLGA backbone and reduction of Platin-M at acidic pH.
Fig. 3.
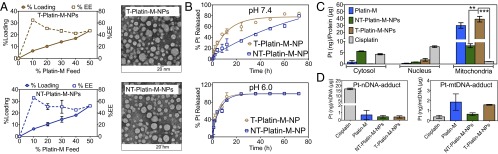
(A) Platin-M percentage loading, encapsulation efficiency (EE) in T- and NT-NPs, and TEM images of T-Platin-M-NPs and NT-Platin-M-NPs. (B) pH-dependent release kinetics of Platin-M from T- and NT-NPs in PBS at 37 °C. (C) Distribution of cisplatin, Platin-M, NT-Platin-M-NPs, and T-Platin-M-NPs in cytosolic, nuclear, and mitochondrial fractions of PC3 cells. (D) Comparison of Pt-nDNA and Pt-mtDNA adducts for cisplatin, Platin-M, NT-Platin-M-NP, and T-Platin-M-NPs in PC3 cells. ***P < 0.001; **P = 0.001–0.01.
Mitochondrial Accumulation of Platin-M and NPs.
Analysis of mitochondrial, cytosolic, and nuclear fractions isolated from PC3 cells treated with cisplatin, Platin-M, NT-Platin-M-NPs, and T-Platin-M-NPs showed that Pt concentration in the mitochondrial protein was 30 times higher than in the nuclear protein fractions for Platin-M or its T-NP formulation compared with cisplatin (Fig. 3C). Overall uptake of cisplatin was much lower than Platin-M or its NP formulations. The nontargeted system showed only approximately eight times greater accumulation of Platin-M in the mitochondrial fraction compared with cisplatin, a significant portion of NT-Platin-M-NPs was found in the cytosolic fractions, and the overall uptake was much lower than Platin-M or its T-NPs. (Fig. 3C). This further supported our hypothesis that dual targeting will enhance mitochondrial delivery efficiency of Platin-M. Further quantification and comparison of Pt bound to nDNA and mtDNA from the treated PC3 cells indicated that cisplatin released from Platin-M and T-Platin-M-NPs exhibited binding with mtDNA (Fig. 3D). Cisplatin adduct level in the nDNA was much higher than cisplatin-mtDNA adducts. Platin-M showed marginally higher mtDNA adduct compared with T-Platin-M-NP system. This might be due to the fact that, for T-NP system, Platin-M needs to be released from the NPs before reduction to cisplatin and subsequent mtDNA interaction and all Platin-M might not have released from the NPs during the course of this experiment; mtDNA isolation was carried out after 12 h, and we expect only ∼50% of Platin-M will be released from T-NPs during this time period (Fig. 3B). NT-Platin-M-NPs showed reduced levels of mtDNA and nDNA adduct formation. This may be due to the slow release of Platin-M from the NT-NPs; only ∼26% of Platin-M is expected to be released in a 12-h period (Fig. 3B).
Efficacy of Platin-M and NPs in Neuroblastoma and Cisplatin-Resistant Cells.
In vitro cytotoxicity of Platin-M, T-Platin-M-NPs, and NT-Platin-M-NPs was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide or MTT assay on PC3, human neuroblastoma SH-SY5Y, and cisplatin-resistant A2780/CP70 ovarian cancer cell lines (SI Appendix, Table S4 and Fig. S21). The rationale behind the use of androgen-independent PC3 cell line was the inherent resistance of these cells to cisplatin therapy. Furthermore, androgen-independent PCa with reduced normal mtDNA acquire a more progressive phenotype by inducing mutations to nDNA. Thus, targeting mtDNA could lead to effective therapies for aggressive androgen-independent PCa. The human neuroblastoma SH-SY5Y cell line displays several characteristics of neurons, and neuronal cells typically require an increased number of mitochondria because most neuronal ATP is generated through OXPHOS. Distribution of the T-NPs in the brain and increased number of mitochondria present in the brain cells prompted us to use SH-SY5Y cells in our studies. To understand the ability of Platin-M and its T-NPs to overcome resistance, we used cisplatin-resistant A2780/CP70 cells with hyperpolarized mitochondria and efficient cisplatin-nDNA repair machinery. In all the cell lines, Platin-M and its NPs showed significantly enhanced cytotoxicity compared to cisplatin. In the resistant cells, Platin-M activity was ∼16 times better than cisplatin. Incorporation of Platin-M in T-NPs further enhanced this activity; the potency of T-Platin-M-NPs was ∼85 times better than cisplatin in the resistance cells. Incorporation of Platin-M in a nontargeted NP system showed an increase in efficacy of approximately six times compared with cisplatin in the resistance cells. These differences in activities between Platin-M and its mitochondria-targeted and nontargeted NP formulations can be correlated to the differences in ΔΨm values of A2780/CP70, PC3, and SH-SY5Y cells. A simple OCR study on these cells using Mito Stress assay supported this hypothesis (SI Appendix, Fig. S22). Cisplatin-resistant A2780/CP70 cells showed low basal OCR due to the hyperpolarized ΔΨm compared with the other two cell lines. This hyperpolarization assisted Platin-M to accumulate into the matrix efficiently. Incorporation of Platin-M inside a mitochondria-targeted NP system with multiple –TPP molecules on the NP surface increased the accumulation, further demonstrating an enhanced activity in the resistant cells. However, NT-Platin-M-NP was not able to accumulate in the hyperpolarized resistant cells and high glutathione levels in the resistant cells (18) facilitated reduction of released Platin-M to generate cisplatin in the cytosol, and hence NT-Platin-M-NPs showed less activity compared with Platin-M and T-Platin-M-NPs in resistant cells. These observations further supported our rationale of using dual-targeting approach for effective delivery of cisplatin inside the mitochondrial matrix. Neuroblastoma accounts for ∼15% of all childhood cancer deaths (19). Cisplatin is widely used to treat patients with high-risk neuroblastoma. Thus, the distribution of T-NPs in the brain prompted us to explore the use of Platin-M and its NPs for possible use in neuroblastoma. Platin-M activity in neoroblastoma SH-SY5Y cell line was 5.5 times higher compared with cisplatin. T-Platin-M-NPs demonstrated a response that was ∼17 times greater than the effects shown by cisplatin (SI Appendix, Table S4 and Fig. S21). NT-Platin-M-NPs also showed enhanced activity over cisplatin. The increased efficiency of Platin-M and its T-NPs over cisplatin might be due to the increased number of mitochondria present in the SH-SY5Y cells. This was further supported by our data that these neuroblastoma cells showed higher OCR levels due to the presence of increased number of mitochondria (SI Appendix, Fig. S22). Toxicity of Platin-M and its NPs in human mesenchymal stem cells (hMSCs) was investigated to understand the effect of these formulations in noncancerous cells. Toxicity of Platin-M in hMSCs was found to be approximately three times less than its activity in the resistance cells. Incorporation of Platin-M inside the T-NPs reduced the toxicity in stem cells further; T-Platin-M-NPs showed ∼5.5 times less toxicity in stem cells compared with the activity in the resistant cells (SI Appendix, Table S4 and Fig. S21). This remarkable ability of Platin-M and its T-NP formulation to overcome cisplatin resistance will play significant roles in the success of this technology. The cell death phenotype analysis in SH-SY5Y cells upon treatment with Platin-M and its NPs was carried out by Alexa Fluor 488–Annexin-V–propidium iodide (PI) staining (SI Appendix, Fig. S23). Twelve hours posttreatment with cisplatin, Platin-M, NT-Platin-M-NPs, and T-Platin-M-NPs, cells were stained with Annexin-V and PI, and analyzed by flow cytometry. Early apoptotic cells characterized by a high Annexin-V signal in the absence of PI staining were observed only in Platin-M– and T-Platin-M-NP–treated cells under the experimental conditions. The pivotal role of mitochondria in initiating apoptotic cell decay might be responsible for more apoptotic population with Platin-M and T-Platin-M-NPs because both of these formulations accumulate inside the mitochondria in significant amounts.
Given the substantial Pt adduct formation with mtDNA, cell death, and cellular apoptosis after treatment with Platin-M or its T-NPs, we performed citrate synthase activity to assess whether these activities translate into functional impairment of the ETC (SI Appendix, Fig. S24). Citrate synthase activity as a mitochondrial marker in SH-SY5Y cells treated with 1 μM cisplatin, 1 μM Platin-M, 1 μM NT-Platin-M-NPs, and 1 μM T-Platin-M-NPs for 12 h at 37 °C was studied. A diminished citrate synthase activity was observed with Platin-M– and T-Platin-M-NP–treated cells, confirming disruption of ETC.
Mitochondrial Bioenergetics.
We next investigated whether accumulation of Platin-M inside the mitochondrial matrix of PC3, A2780/CP70, and SH-SY5Y cells altered mitochondrial respiration by measuring OCRs as a way of assessing OXPHOS (Fig. 4 and SI Appendix, Fig. S25). PC3, A2780/CP70, and SH-SY5Y cells were treated with cisplatin, Platin-M, NT-Platin-M-NPs, and T-Platin-M-NPs, for 12 h. The basal OCR levels of Platin-M– or T-Platin-M-NP–treated cells were found to be significantly less than control cells, indicating a loss in total mitochondrial mass. To support that the reduced OCR upon treatment with T-Platin-M-NPs is due to total mitochondrial mass, we decided to use 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide or JC-1 red/green fluorescence measurements in SH-SY5Y cells, which would allow careful discrimination between a shift in fluorescence due to reduced potential in the total cell population and reduced JC-1 red/green fluorescence due to cell death (SI Appendix, Fig. S26). Our results from flow cytometry analyses confirmed the loss of ΔΨm and mitochondrial mass in T-Platin-M-NP–treated cells in a greater extent compared with Platin-M–, NT-Platin-M-NPs–, and cisplatin-treated cells (SI Appendix, Fig. S26). The ATP synthase inhibitor oligomycin was injected to evaluate mitochondrial coupling upon accumulation of Platin-M inside the mitochondria of these cells. Addition of oligomycin showed that the levels of ATP-linked respiration were attenuated in control cells or cells treated with cisplatin, NT-Platin-M-NPs; Platin-M–treated cells showed less reduction, and T-Platin-M-NPs did not show any significant changes. Stimulation of mitochondrial respiration with FCCP after oligomycin was comparable for cisplatin, NT-Platin-M-NPs, and control cells, but Platin-M and T-Platin-M-NPs showed no enhancement in OCR levels. Finally, injection of a combination of mitochondrial complex III inhibitor antimycin A and mitochondrial complex I inhibitor rotenone significantly inhibited the OCR due to the formation of mitochondrial ROS and nonmitochondrial O2 consumption. Overall, Platin-M and T-Platin-M-NPs showed inhibition of mitochondrial respiration in A2780/CP70 and SH-SY5Y cells to a greater extent compared with PC3 cells. This mitochondrial respiration programming by Platin-M and T-Platin-M-NPs indicated enormous potential of the current technology in the treatment of chemoresistance tumor with inherently hyperpolarized mitochondria and of neuroblastoma with increased number of mitochondria. Effects of cisplatin, Platin-M, NT-Platin-M-NPs, and T-Platin-M-NPs on hyperpolarized H9C2 cardiomyocytes under identical conditions showed similar trends as observed with other cell lines (SI Appendix, Fig. S27).
Fig. 4.

Mitochondrial bioenergetics in (A) SH-SY5Y and (B) A2780/CP70 cells in response to cisplatin, Platin-M, NT-Platin-M-NPs, and T-Platin-M-NPs. ***P < 0.001; **P = 0.001–0.01; *P = 0.01–0.05; nonsignificant (ns), P > 0.05.
Distribution of T-Platin-M-NPs in the Brain.
We next assessed whether the brain-penetrating T-NPs could deliver Platin-M to the brain by crossing the BBB in in vivo settings. We administered Platin-M, NT-Platin-M-NPs, and T-Platin-M-NPs to male rats by i.v. injection. The serum Pt concentrations from T-Platin-M-NPs and NT-Platin-M-NPs indicated long circulatory properties compared with Platin-M delivered without any delivery system (SI Appendix, Table S5, and Fig. 5). Pt remaining in systemic circulation 1 h postadministration from T-Platin-M-NPs was significantly higher than from Platin-M (SI Appendix, Table S5, and Fig. 5). The high t1/2, significantly higher AUC, and a large Vd of T-Platin-M-NPs supported that T-NP is an excellent candidate for mitochondrial delivery of Platin-M. Similar to Empty-T-NPs, Pt distribution was significantly higher in the brain compared with kidneys, heart, lungs, and spleen when T-Platin-M-NP was used (Fig. 5). Platin-M from NT-Platin-M-NPs was mostly distributed in the spleen, liver, and kidneys, and a significant amount of Pt from these samples was found in urine collected at 24 h directly from the animal bladders (SI Appendix, Fig. S28A). The significant differences in bioD patterns between Platin-M, NT-Platin-M-NPs, and T-Platin-M-NPs further supported the rationale for using a dual-targeting system for delivery of Platin-M. The bioD data indicated that both liver and kidney are involved in Pt elimination (SI Appendix, Fig. S28A) of these NPs. No significant alterations of clinical chemistry parameters in rats after treatment with Platin-M and its NPs was observed (SI Appendix, Fig. S28B). A comparison of T-QD-NPs (Fig. 2C) and T-Platin-M-NPs (Fig. 5) bioD profiles showed very similar distribution pattern, further confirming that Platin-M is associated with the NPs throughout the study. Plasma Pt levels coupled with unique distribution pattern, which involved significant Pt accumulation in the brain, demonstrated that T-Platin-M-NPs have the potential to demonstrate significant clinical impact.
Fig. 5.

Pt concentration variation in plasma and organs following the administration of Platin-M, NT-Platin-M-NPs, and T-Platin-M-NPs i.v. to male rats. ***P < 0.001; **P = 0.001–0.01; nonsignificant (ns), P > 0.05.
Here, we outlined a strategy for precise mitochondrial delivery of cisplatin for chemoresistance aggressive cancers—from construction of a mitochondria-targeted cisplatin prodrug to its formulation in a targeted delivery vehicle—that can be implemented in cisplatin resistance settings and in cancers of central nervous system. These studies represent an initial development of previously undescribed routes for cisplatin-based therapy. Because most late-stage cancers are resistant to cisplatin treatment because of the development of chemoresistance, therefore, delivery of cisplatin inside the mitochondrial matrix to attack mtDNA lacking NER using a mitochondria targeted Pt(IV) prodrug and a targeted NP, as outlined here, can be extremely beneficial in providing a previously unidentified therapeutic strategy to tackle otherwise-resistant advanced cancers.
Materials and Methods
Description of materials, synthesis of DBCO-TPP, Platin-M, polymers, and NP formulations, and other methods are described in SI Appendix.
Animals.
Animals were obtained from Harlan Laboratory and handled in accordance with Guide for the Care and Use of Laboratory Animals (20) of American Association for Accreditation of Laboratory Animal Care, Animal Welfare Act, and other applicable federal and state guidelines. All animal work presented here was approved by Institutional Animal Care and Use Committee of University of Georgia.
Statistics.
All data were expressed as mean ± SD. Statistical analysis was performed using GraphPad Prism software, version 5.00. Comparisons between two values were performed using an unpaired Student t test. A one-way ANOVA with a post hoc Tukey test was used to identify significant differences among the groups.
Supplementary Material
Acknowledgments
We thank Dr. Nagesh Kolishetti for helpful discussions. We thank Prof. Rick Tarleton for the Seahorse analyzer. We thank Walter J. Lunsmann for assistance with PK parameter calculations and Brittany Feldhaeusser for plasma clinical chemistry analyses. This work was supported in part by the Center for Metalloenzyme Studies center grant from the National Institutes of Health (P30GM092378) as a startup fund (to S.D.) and by the Office of the Vice President for Research (University of Georgia; S.D.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1405244111/-/DCSupplemental.
References
- 1.Smiraglia DJ, Kulawiec M, Bistulfi GL, Gupta SG, Singh KK. A novel role for mitochondria in regulating epigenetic modification in the nucleus. Cancer Biol Ther. 2008;7(8):1182–1190. doi: 10.4161/cbt.7.8.6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jamieson ER, Lippard SJ. Structure, recognition, and processing of cisplatin-DNA adducts. Chem Rev. 1999;99(9):2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 3.Damia G, Imperatori L, Stefanini M, D’Incalci M. Sensitivity of CHO mutant cell lines with specific defects in nucleotide excision repair to different anti-cancer agents. Int J Cancer. 1996;66(6):779–783. doi: 10.1002/(SICI)1097-0215(19960611)66:6<779::AID-IJC12>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 4.Yang Z, et al. Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: Possible role in apoptosis. Clin Cancer Res. 2006;12(19):5817–5825. doi: 10.1158/1078-0432.CCR-06-1037. [DOI] [PubMed] [Google Scholar]
- 5.Marrache S, Dhar S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc Natl Acad Sci USA. 2012;109(40):16288–16293. doi: 10.1073/pnas.1210096109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marrache S, Tundup S, Harn DA, Dhar S. Ex vivo programming of dendritic cells by mitochondria-targeted nanoparticles to produce interferon-gamma for cancer immunotherapy. ACS Nano. 2013;7(8):7392–7402. doi: 10.1021/nn403158n. [DOI] [PubMed] [Google Scholar]
- 7.Wisnovsky SP, et al. Targeting mitochondrial DNA with a platinum-based anticancer agent. Chem Biol. 2013;20(11):1323–1328. doi: 10.1016/j.chembiol.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pathak RK, Marrache S, Choi JH, Berding TB, Dhar S. The prodrug platin-A: Simultaneous release of cisplatin and aspirin. Angew Chem Int Ed Engl. 2014;53(7):1963–1967. doi: 10.1002/anie.201308899. [DOI] [PubMed] [Google Scholar]
- 9.Summerhayes IC, et al. Unusual retention of rhodamine 123 by mitochondria in muscle and carcinoma cells. Proc Natl Acad Sci USA. 1982;79(17):5292–5296. doi: 10.1073/pnas.79.17.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pathak RK, McNitt CD, Popik VV, Dhar S. Copper-free click-chemistry platform to functionalize Cisplatin prodrugs. Chem Eur J. 2014;20(23):6861–6865. doi: 10.1002/chem.201402573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peer D, et al. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 12.Reily C, et al. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol. 2013;1(1):86–93. doi: 10.1016/j.redox.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.dos Santos WL, Rahman J, Klein N, Male DK. Control of lymphocyte adhesion to brain and aortic endothelium: ICAM-1, VCAM-1 and negative charge. J Neuroimmunol. 1996;66(1-2):125–134. doi: 10.1016/0165-5728(96)00037-9. [DOI] [PubMed] [Google Scholar]
- 14.Oldendorf WH, Cornford ME, Brown WJ. The large apparent work capability of the blood-brain barrier: A study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol. 1977;1(5):409–417. doi: 10.1002/ana.410010502. [DOI] [PubMed] [Google Scholar]
- 15.Hervé F, Ghinea N, Scherrmann JM. CNS delivery via adsorptive transcytosis. AAPS J. 2008;10(3):455–472. doi: 10.1208/s12248-008-9055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci USA. 2008;105(45):17356–17361. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolishetti N, et al. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc Natl Acad Sci USA. 2010;107(42):17939–17944. doi: 10.1073/pnas.1011368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Godwin AK, et al. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc Natl Acad Sci USA. 1992;89(7):3070–3074. doi: 10.1073/pnas.89.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol. 1999;17(7):2264–2279. doi: 10.1200/JCO.1999.17.7.2264. [DOI] [PubMed] [Google Scholar]
- 20. Committee for the Update on the Guide for the Care and Use of Laboratory Animals (2011) Guide for the Care and Use of Laboratory Animals (The National Academies Press, Washington, DC), 8th Ed.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.