

Abstract
The idiopathic inflammatory myopathies (IIM) are a heterogenous group of rare disorders that share many similarities. In addition to sporadic inclusion body myositis (IBM), these include dematomyositis (DM), polymyositis (PM), and autoimmune necrotizing myopathy (NM). For discussion of later three disorders, the reader is referred to the IIM review in this issue. IBM is the most common IIM after age 50. It typically presents with chronic insidious proximal leg and/or distal arm asymmetric muscle weakness leading to recurrent falls and loss of dexterity. Creatine kinase (CK) is up to 15 times elevated in IBM and needle electromyograhy (EMG) mostly shows a chronic irritative myopathy. Muscle histopathology demonstrates endomysial inflammatory exudates surrounding and invading non-necrotic muscle fibers often times accompanied by rimmed vacuoles and protein deposits. Despite inflammatory muscle pathology suggesting similarity with PM, it likely that IBM is has a prominent degenerative component as supported by refractoriness to immunosuppressive therapy. We review the evolution of our knowledge in IBM with emphasis on recent developments in the field and discuss ongoing clinical trials.
Keywords: Inclusion body myositis, idiopathic inflammatory myopathies, polymyositis, clinical presentation, diagnosis, pathology, pathophysiology, treatment, prognosis, rehabilitation, research
Epidemiology
IBM is a rare sporadic disorder with a male-to-female ratio of 2:1 to 3:1. Data on prevalence and incidence of IBM vary depending on methodology, countries and regions. In Western Australia, the overall prevalence was 9.3 per million while the age-adjusted prevalence of IBM in people over the age of 50 is 3.5/100,000, making it the most common IIM in this age group [1]. A recent study from South Australia yielded a higher overall prevalence rate of 51 per million, with an incidence of 2.9 per million.[2]The latter is comparable to the incidence observed in Sweden (2.2 per million) [3] but lower than the 7.9 per million recorded in Olmstead County, Minnesota adjusted for sex and age to the 2000 US Census population [4]. The highest reported prevalence is 71 per million inhabitants of Olmsted County, US, while it is lowest in the Netherlands at 4.9 per million and when age-adjusted to those older than 50, it is 16 per million Dutch inhabitants [3]. IBM is rare in African-Americans and in non-Caucasians. IBM should be considered in patients with appropriate symptoms who are older than 30. Symptom onset before age 60 occurs in 18% to 20% of patients [5,6].
Clinical Presentation
In most cases, IBM classically presents with insidious weakness in the proximal leg and/or distal arm [7,8]. There is typically five to eight year delay in presentation and diagnosis [3,5,7,9,10,11]. In the University of Kansas Medical Center (KUMC) IBM series fulfilling pathological criteria for probable IBM (Table 1), delay to diagnosis in 51 cases ranged from 1 to 15 years (mean 5.1 years) [12]. IBM typically manifests as slowly progressive quadriceps muscle more than hip flexor weakness leading to falls or difficulty standing and next common is finger flexor weakness leading to loss of dexterity [11] [Figure 1]. Falls were reported by 98% of questionnaire respondents, with 60% (37/62) falling frequently [13]. In most cases weakness is markedly asymmetric by at least one MRC grade and preferentially affects in the hand non-dominant distal phalangeal flexors (Table 1). Sparing of thenar, hypothenar and finger extensor muscles sets IBM apart from a myotomal disease such as amyotrophic lateral sclerosis or focal peripheral nerve disorders such as multifocal motor neuropathy. In general, one anticipates that wrist and finger flexors are weaker than the corresponding extensors, and wrist and finger flexors are more affected than shoulder abductors.
Table 1. Retrospective chart review of IBM from 2000 to 2010 at KUMC [12].
| Male:female ratio | 1.7:1 |
| Ethnicity (n=51) | 49 Caucasian; 2 Hispanics |
| Mean age at onset (yrs) | 61 (45-80) |
| Symptom onset before age 50 yrs: | 12% |
| Mean time to diagnosis (yrs) | 5.1 (1-15) |
| Mean follow up period (yrs) | 2.5 (0.5-8) |
| CK (IU/L) | 609 (59-3000) |
| Nerve conductions with axon loss neuropathy | 32% |
| Electromyography | 60% irritative myopathy 12% non-irritative myopathy 28% mixed neuropathic/myopathic pattern |
| Asymmetry | 90% |
| Non-dominant side weaker | 85% |
| Typical phenotype: Weak Finger Flexor (FF) and quadriceps (quads) |
39/51 (76%): 13 - Classic phenotype (FF and quads weakest) 11 - Classic FF, no preferential quads weakness 6 - Classic quads, no preferential FF weakness 9 - FF and quads weak but not weakest |
| Atypical phenotype | 12/51 (24%): 5/12: classic FF with leg weakness sparing quads 4/12: limb-girdle weakness 3/12: other atypical phenotypes (FF arm only, hip flexion/ankle dorsiflexion, facioscapulohumeral) |
| Muscle pathology | 43: inflammation and rimmed vacuoles 8: phenotypic IBM with inflammation |
| Mobility Outcome | 75%: recurrent falls 56%: assistive device use at mean 7.5 years 20%: wheelchair or scooter |
| Bulbar dysfunction | 51%: dysphagia 55%: facial weakness |
Adapted from Estephan B, Barohn RJ, Dimachkie MM et al. Sporadic IBM: a Case Cohort. Journal of Clinical Neuromuscular Disease. 2011; 12(3):18-19
Figure 1.
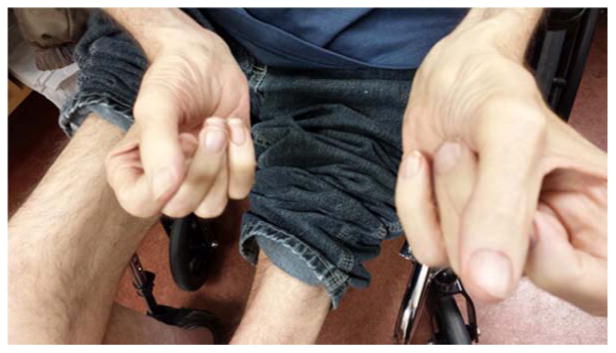
Finger flexor weakness and severe thigh muscle atrophy in IBM.
In our case series, 82% presented with limb weakness symptoms, most commonly restricted to the legs (34/51) [12]. Arm presentation was less common affecting 6/51 cases. Though it is reportedly a less frequent initial symptom, eight cases (16%) presented with dysphagia. Less typical initial complaints include foot drop as in two of our cases. Other rare presentations include sparing of the quadriceps muscles with prominent forearm muscle weakness, camptocormia [14] or even asymptomatic hyperCKemia. [15] There has been a reported case of primary respiratory failure in IBM. [16] On examination, all KUMC case series patients (39) with typical phenotype and most cases (10/12) with atypical phenotype had evidence of arm and leg weakness [12] (Table 1). However, 71% (36/51) demonstrated typical IBM weakness pattern in the arm or leg muscles. In nine cases, finger flexors and knee extensors were not weaker than finger extensors and hip flexors and four cases displayed limb-girdle pattern of weakness.
Dysphagia is a significant problem as it affects 50 to 70% of patients [5,17]. In our case series (Table 1), eight had dysphagia as the initial symptoms and in 7/8 cases dysphagia was the only presenting symptom of IBM for up to 10 years [12]. Ultimately, 51% of our cases experienced dysphagia. Mild to moderate facial weakness is frequently demonstrated (55%) and was the earliest IBM symptom for 20 years in 1/51 cases. Others have recently reported facial diplegia as a presenting manifestation of IBM. [18] The tibialis anterior muscle was involved in 70% of our IBM patients but in 12% ankle dorsiflexors were weaker than knee extensors. Scapular winging affected 8% of KUMC cases. Although mostly asymptomatic, a third of patients harbor a distal sensory loss gradient and/or electrophysiological evidence of a mild distal sensory axon loss peripheral polyneuropathy.
Associated Conditions
Though IBM is felt to be a neurodegenerative disorder, there is some association with autoimmune disorders in up to 15% of cases. Systemic lupus erythematosis, Sjogren's syndrome, thrompocytopenia, and sarcoidosis have been reported with IBM. The HLA-DRB1*03:01/*01:01 genotype confers the highest disease risk in inclusion body myositis [19,20] There is no increased risk of myocarditis, interstitial lung disease or malignancy in IBM [21].
Laboratory and Electrophysiologic Testing
Serum CK level may be normal or elevated, up to 12 to 15 times the upper normal limit. On occasion, it may be as high as 20 times the normal limit. ANA is positive in 20% of IBM patients and in some SSA may be elevated.
Needle electromyography typically shows an irritative myopathy that is associated with fibrillation pootentials. In our series, 60% showed an irritative myopathy pattern and 12% had a non-irritative myopathy [12]. In 28% of our IBM cases, the motor unit action potentials were mixed myopathic and neuropathic, with the latter being due to reinnervation of denervated and split muscle fibers. In some cases, the neurogenic motor unit action potentials in IBM may be sufficiently dense to overshadow the myopathic changes, leading to a misdiagnosis of motor neuron disease. However the clinical pattern of selective finger flexor weakness is helpful clinically in making that distinction. In our series, nerve conduction studies revealed a mild sensory axonal peripheral polyneuropathy in nearly a third of patients (Table 1).
Muscle Imaging
The role of MRI in neuromuscular diseases is expanding.[22] Specific patterns of muscular involvement can be identified on qualitative MRI of the lower limbs in patients with IBM. [23, 24, 25] Beyond diagnosis, quantitative MRI provides potentially very sensitive outcome measures, and has been used in clinical trials in other neuromuscular diseases. [26] In IBM targets for quantification include markers of both chronic and acute muscle pathology. Chronic muscle damage results in the infiltration of muscle tissue with fat and reduction in muscle size. Fatty infiltration is quantifiable using a number of MRI methods, notably the Dixon fat-water separation technique [27, 28] whereas atrophy can be quantified with any anatomical MRI sequence. Acute muscle pathology in IBM is qualitatively identifiable as hyperintensity on T2-weighted sequences with fat suppression. This can be quantified by measurement of the prolongation of the T2-relaxation time, as has been applied to juvenile dermatomyositis. [29] Degardin at al. performed magnetic resonance imaging studies on four IBM cases, two of whom had predominantly distal muscle involvement and two had asymmetric fat deposition [23]. Muscle involvement was typically found in the quadriceps, medial head of the gastrocnemius, and often in the soleus and tibialis anterior muscles. T2 hyperintensities was identified on short tau inversion recovery images and was associated with adjacent muscle fatty infiltration. In 32 IBM patients evaluated in 68 muscles of upper and lower extremities for muscle atrophy, fatty infiltration, and inflammation, fatty infiltration was far more common than inflammation. Fatty replacement most frequently affected the long finger flexors, anterior thigh muscles (relatively sparing the rectus femoris), and all muscles of the lower leg, preferentially affecting the medial gastrocnemius muscle [24]. Inflammation was present in 78% of the patients with a median of two inflamed muscles per patient. However, the amount of fatty infiltration correlated significantly with disease severity, disease duration and CK levels.
In a study of whole body positron emission tomography using Pittsburgh Compound B (PIB), an in vivo marker of amyloid-β in the brains of patients with Alzheimer's disease, six of seven IBM patients showed increased PIB levels in at least one gastrocnemius muscle [30]. The median gastrocnemius muscle PIB was significantly higher in IBM patients than in six non-IBM subjects. In two IBM patients with radiographically increased PIB uptake, gastrocmenius muscle biopsy showed several fibers with dense amyloid-β and PIB positive inclusions. However, another IBM patient with normal deltoid muscle PIB uptake was pathologically positive for amyloid-β without any detectable PIB positive inclusions.
Muscle Histopathology
IBM pathology demonstrates combined evidence of an inflammatory process with marked degenerative changes. Besides endomysial inflammation invading non-necrotic fibers (Figure 2a), the presence of small groups of atrophic fibers, eosinophilic cytoplasmic inclusions and importantly multiple myofibers with one or more rimmed vacuoles lined with granular material is highly supportive for the diagnosis of IBM (Figure 2b). Twenty percent of IBM patients are mislabeled as PM when no vacuoles are found even though they have classic IBM clinical phenotype [31]. Repeat muscle biopsies may be necessary to detect vacuoles in treatment-refractory patients with the phenotype of IBM and histopathology of PM [5,7]. To add to the complexity, patients who have steroid-responsive PM may have a few rimmed vacuoles [32]. Although better visualized on immunostaining of phosphorylated tau (with SMI-31), eosinophilic cytoplasmic inclusions are rarely seen in IBM.
Figure 2.
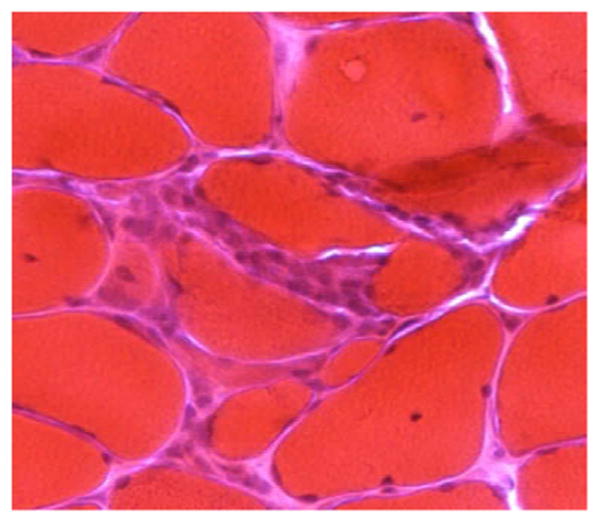
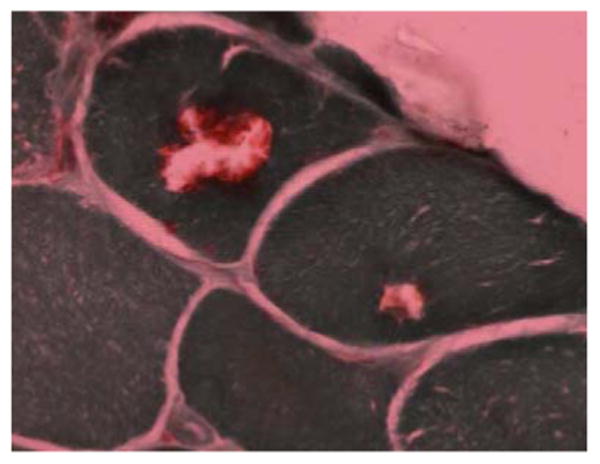
PM: Inflammatory infiltrates invading non-necrotic fibers - Hematoxylin and eosin; IBM muscle: Muscle fibers with rimmed vacuoles - Modified Gomori trichrome
Congo red method occasionally demonstrates positive material in vacuolated fibers that is likely to represent amyloid deposits. Ubiquitin-positive multiprotein-aggregates contain misfolded proteins in the β-pleated sheet conformation of amyloid especially composed of proteolytic Aβ42 within and next to the vacuoles. Fluorescent methods for detecting amyloid material are more sensitive than the Congo red. There is evidence for mitochondrial stress as demonstrated by abnormally increased number of ragged red fibers or of COX negative / SDH positive fibers. Some nuclei containing eosinophilic inclusions appear to be enlarged within or at the edge of the vacuoles. There is an increased likelihood of finding 15-18 nanometer (nm) tubulofilamentous cytoplasmic and intranuclear inclusions on electron microscopy (EM) when at least three vacuolated fibers are examined. The eosinophilic cytoplasmic inclusions correspond to the tubulofilamentous inclusions. Identifying more than one rimmed vacuole, more than one group of atrophic fibers per high-power field, and of endomysial inflammation is 95% predictive of finding the filamentous inclusions on EM [5].
There are several histopathologic similarities between PM and IBM [33]. In both, intact myofibers are surrounded and invaded by endomysial inflammatory cells that consist of macrophages and cytotoxic CD8+ T cells with MHC-1 expression on the surface of necrotic and non-necrotic myofibers. In addition, myeloid dendritic cells surround non-necrotic fibers and present antigen to CD8+ lymphocytes. However, mononuclear cells invade non-necrotic muscle fibers more frequently in IBM than in PM [31].
Patients who have typical IBM clinical features but few inflammatory cells or few rimmed vacuoles can be difficult to diagnose [7]. Both of the 1995 Griggs IBM diagnostic categories (definite and possible IBM) require inflammation with invasion of non-necrotic muscle fibers by mononuclear cells (see section below titled Diagnostic and Research Criteria) [34]. In addition to an endomysial inflammatory exudate, definite IBM histopathology includes the identification of vacuolated muscle fibers and either intracellular amyloid deposits or 15-18 nm tubulofilaments on electron microscopy. According to the 2010 IBM diagnostic criteria [35], the pathologic features of clinically defined IBM and of possible IBM require at least one of the following: invasion of non-necrotic fibers by mononuclear cells, rimmed vacuoles, or increased MHC-1 expression on the surface of intact muscle fibers. Since then, it was suggested that sarcoplasmic redistribution of Tar DNA binding protein 43 (TDP-43) is highly sensitive and specific in IBM [36]. Based on conventional stains in 36 patients with characteristic clinical features of IBM, 17 had Griggs definite IBM and 19 had possible IBM. Immunohistochemically, TDP-43 and p62 were the most sensitive markers, accumulating in all definite IBM and in 31% and 37%, respectively, of possible IBM cases.[37] Hence, the ENMC 2011 criteria introduce 3 additional alternative pathologic findings for the diagnosis of clinically defined IBM and probable IBM (Table 2) including 15 to 18 nm filaments, accumulation of amyloid (as demonstrated on Congo-red, crystal violet, or thioflavine T/S), or increase in other proteins (as evident on immunostains with antibodies to p62, SMI-31, or TDP-43).
Table 2. Proposed diagnostic criteria following ENMC Workshop 2011 [50].
| Clinical & laboratory features | Classification | Pathological features |
|---|---|---|
| Duration >12 months Age at onset > 45 yrs Quads weakness ≥ hip flex and/or FF weakness > should abd sCK no greater than 15xULN |
Clinicopathologically defined IBM | Endomysial inflammation & Rimmed vacuoles & Protein accumulation (amyloid or other proteins)* or 15-18nm filaments |
| Duration >12 months Age at onset > 45 yrs Quads weakness ≥ hip flex and FF weakness > should abd sCK no greater than 15xULN |
Clinically defined IBM |
One or more of: Endomysial inflammation or ↑ MHC1 or Rimmed vacuoles or Protein accumulation (amyloid or other proteins)* or 15-18nm filaments |
| Duration >12 months Age at onset > 45 yrs Quads weakness ≥ hip flex or FF weakness > should abd sCK no greater than 15xULN |
Probable IBM |
One or more of: Endomysial exudate or ↑ MHC1 or Rimmed vacuoles or Protein accumulation (amyloid or other proteins)* or 15-18nm filaments |
Amyloid or other protein accumulation by established methods: for amyloid Congo red, crystal violet, thioflavine T/S, and for other proteins p62, SMI-31, TDP-43.
Adapted from Machado P, Brady S, Hanna MG. Update in inclusion body myositis. Curr Opin Rheumatol. 2013 Nov;25(6):763-71.
Pathogenesis
Based on endomysial inflammation, IBM was originally believed to be a primary inflammatory myopathy. However, there is a significant body of evidence in support of a neurodegenerative etiology including the above deposition of p62 and redistribution of TDP-43. The exact contribution of these two pathways to the pathogenesis of IBM remains unknown.
Autoimmune modes of injury in IBM is supported by the identification of cytotoxic T cells, myeloid dendritic cells (mDCs), B cells, and the recently discovered IBM autoantibody [38, 39]. Like in PM, clonally restricted cytotoxic T-cells invade non-necrotic muscle fibers and destroy them through perforin, granzyme A, and granulysin pathways. The frequency of intact muscle fiber invasion in IBM is higher than that observed for vacuolated fibers or fibers with amyloid deposits. In addition, myeloid dendritic cells serve as antigen-presenting cells [33]. These mDCs help the maturation of naïve CD8+ T cells into cytotoxic autoaggressive T cells that surround and invade non-necrotic muscle fibers. Microarray studies showed an abundance of immunoglobulin transcripts in IBM muscle [40] and led to the recognition of antigen directed and clonally expanded plasma cells in IBM muscle [41]. Like in PM, type 1 interferon (IFN1) genes are modestly upregulated in IBM muscle but unlike PM, blood derived from IBM cases does not show this increase. Recently, Salajegheh et al. reported on plasma autoantibodies from 65 people, including 25 with IBM [39]. Immunoblots against normal human muscle demonstrate that thirteen of 25 (52%) IBM patient samples recognized a 43 kDa muscle protein. None of the other disease (N = 25) or healthy volunteer (N = 15) samples recognized this protein. Since then 2 separate groups confirmed the identity of the antigen to be cytosolic 5′-nucleotidase 1A (cN1A) [42, 43] Moderate reactivity of anti-cN1A autoantibodies was 70% sensitive and 92% specific, and high reactivity was 34% sensitive and 98% specific for the diagnosis of IBM.[42] cN1A reactivity by immunohistochemistry accumulated in perinuclear regions and rimmed vacuoles in IBM muscle, localizing to areas of myonuclear degeneration. In the Dutch study, high concentrations of cN1A autoantibodies were confirmed in 33% of IBM patient sera, whereas their prevalence in dermatomyositis, polymyositis, and other neuromuscular disorders appeared to be rare (4.2%, 4.5%, and 3.2%, respectively).[43]
Support for a degenerative pathophysiology originated from the lack of IBM response to immunomodulatory therapies. Immunohistochemical evidence backing the degenerative pathogenesis model of IBM stems from the identification in vacuolated muscle fibers of protein aggregates often associated with other neurodegenerative diseases. These aggregates include amyloid-β, hyperphosphorylated tau, ubiquitin, neurofilament heavy chain, presenilin, and parkin and are postulated to occur due to aberrant protein misfolding and accumulation [44]. Mechanisms contributing to this defect include the inhibition of the 26 S proteasome system, overexpression various heat shock proteins such as alpha B-crystallin (induced by the β amyloid precursor protein) [45], and impairment of autophagy [46]. Until recently, proponents of the autoimmune theory of IBM have countered about the lack of critically supported data demonstrating the presence of β amyloid proteins deposits on muscle Western Blot. In addition, β amyloid precursor protein which is secreted by inflammatory cells has also been demonstrated in PM tissues. Besides its presence in 10 IBM samples, tau-immunoreactivity was demonstrated in myonuclei of 10 normal subjects and 10 PM/dermatomyositis cases suggesting lack of specificity to tau of standard “anti-tau” antibodies including those directed at SMI-31 [47].
Subsequently, Askanas' group reported in 2010 that IBM muscle samples had accumulation of toxic low-molecular weight amyloid-β oligomers on dot-immunoblots with a variety of molecular weights and intensity but none of the control muscle biopsies had amyloid-β oligomers [48]. Nonfibrillar cytotoxic “Aβ-Derived Diffusible Ligands” originally derived from Aβ42 are prominently increased on dot-immunoblots, being consistent with the concept that intracellular toxicity of Aβ42 oligomers is likely an important aspect of IBM pathogenesis. Finally, they demonstrated in cultured human muscle fibers that inhibition of autophagy is a novel cause of Aβ oligomerization [48]. Of interest, a recent positron emission tomography study using PIB, a marker of amyloid-β, confirmed increased PIB uptake in the gastrocnemius muscle of IBM patients [30].
There is also myonuclear degeneration early on in IBM since the majority of rimmed vacuoles are lined with nuclear membrane proteins. IBM myonuclei are often abnormally filled with neurofilaments and this may be the earliest detectable pathological change in IBM [21]. TDP-43, which acts as a scaffold for nuclear bodies through an interaction with survival motor neuron protein, is redistributed from nuclei to sarcoplasm in a large percentage of IBM myofibers reflecting a defect in nucleocytoplasmic shuttling. [36]. The extranuclear accumulation of TDP-43 is toxic to cells through RNA binding. p62, also known as sequestosome 1, is a shuttle protein transporting polyubiquinated proteins for their degradation by the proteasome and lysosome and is a component of the inclusions in several neurodegenerative disorders. TDP-43 and p62 accumulate in all definite IBM and in a third of possible IBM cases. [37] Thus, IBM muscle accumulates multiple toxic protein aggregates suggesting a disorder of protein homeostasis.
Diagnostic and Research Criteria
A multitude of IBM criteria have been proposed based a variety of clinical and histopathological features. Though these have been advanced for research use, they have permeated into the clinical realm. We will limit discussion to 4 most prominent criteria sets, namely those by Griggs, the ENMC 2000, MRC 2010 and ENMC 2011. The 1995 Griggs IBM criteria represent the first major effort to define diagnostic criteria for IBM and are heavily weighted towards muscle pathology since cases classified as definite IBM only need to demonstrate histopathological findings of inflammatory myopathy with mononuclear cell invasion of nonnecrotic muscle fibers, vacuolated muscle fibers and either intracellular amyloid deposits, or 15 to 18 nm tubulofilaments by electron microscopy (EM). The original 1995 criteria also included the category of possible IBM in patients with only inflammation on muscle biopsy but with some specific clinical features, age at onset exceeding 30 years, illness duration greater than 6 months, CK less than 12 times the upper normal limit and EMG must be consistent with inflammatory myopathy (long-duration potentials are acceptable) [34]. In possible IBM, muscle weakness must be in proximal and distal arm and leg muscles and must endorse at least one of the following patterns: finger flexor weakness, wrist flexor greater than wrist extensor weakness and quadriceps muscle weakness (MRC ≤ 4). In 2002, Griggs and Tawil added the category of probable IBM in patients fulfilling possible IBM criteria but additionally histopathological evidence of rimmed vacuoles. [49] Issues with the Griggs criteria include the low sensitivity of definite IBM, being heavily weighted towards inflammation and difficulty in admitting possible IBM in clinical trials.
In 2000 and to improve sensitivity, Badrising described the ENMC criteria for definite and probable IBM in cases of slowly progressive sporadic muscle weakness with mononuclear inflammatory infiltrates and invasion of non-necrotic muscle fibers [9]. Probable IBM requires in addition either weakness pattern (finger flexors or wrist flexors more than wrist extensors) or rimmed vacuoles. ENMC 2000 definite IBM requires both features or in the absence of typical weakness pattern, presence of both rimmed vacuoles on modified Gomori trichrome and tubulofilaments on EM. In this Dutch series, the sensitivity of Griggs definite IBM was 16% while that of ENMC 2000 definite IBM was 70% and this increase in sensitivity was due to lumping of Griggs probable IBM under ENMC definite IBM. An issue with the ENMC 2000 set of criteria is that definite IBM required presence of rimmed vacuoles. The absence of rimmed vacuoles despite inflammation and typical weakness pattern was seen in 20% (16/80) of the Mayo Clinic IBM cases [31]. This led to the next set of criteria in order to facilitate the diagnosis of IBM patients who fulfill clinical criteria for IBM but do not have strict pathologic features. As a result of the 2008 MRC Center for Neuromuscular Diseases IBM workshop, the MRC 2010 IBM diagnostic criteria were published [35]. Besides pathologically defined IBM (identical with Griggs definite IBM) and for suspected patients presenting with weakness onset after 35 years of age and lasting at least for 12 months, the two other categories are clinically defined IBM and possible IBM (essentially the same as defined by the Griggs definite criteria). In clinically defined IBM, weakness involves finger flexion more than shoulder abduction as well as knee extension more than hip flexion. Possible IBM is when weakness follows either one of the preceding two patterns. The pathologic criteria which are identical for possible and clinically defined IBM and require one of the following: invasion of non-necrotic fibers by mononuclear cells or rimmed vacuoles or increased MHC-1 expression on intact muscle fibers.
Finally, in 2011, twenty-four representatives from Europe, Australia and the USA including neurologists, rheumatologists, physiotherapists, industry representatives & patient representatives convened in Naarden. The MRC IBM criteria were further discussed and refined and this led to the newly proposed ENMC 2011criteria which are to be validated. [50] No longer is definite IBM diagnosis possible based on only pathologic features as the three new categories are clinico-pathologically defined IBM, clinically-defined IBM and probable IBM (Table 2). All three diagnostic categories require weakness duration >12 months, age at onset more than 45 yrs, and CK elevated up to 15 times. While weakness in clinico-pathologically defined IBM and probable IBM is in the quadriceps muscles more than hip flexors or in finger flexors more than shoulder abductors, both weakness patterns must coexist in clinically-defined IBM. Required pathologic features for clinically-defined IBM and probable IBM have been relaxed to include at least one of the following: endomysial inflammation, rimmed vacuoles, increased MHC-1, 15 to 18 nm filaments, or accumulation of amyloid or other proteins (Table 2). Hence, MRC 2010 possible IBM becomes ENMC 2011 probable IBM. Clinico-pathologically defined IBM is based on pathological demonstration of endomysial inflammation, rimmed vacuoles and one of the following: 15 to 18 nm filaments on EM, accumulation of amyloid (clarified to be on Congo-red, crystal violet, or thioflavine T/S) or build-up of other proteins (specified on p62, SMI-31, or TDP-43). The relative sensitivity and specificity of all of these methods in IBM remains unproven and require prospective validation.
Therapy
IBM is refractory to all treatments known to be effective in the idiopathic inflammatory myopathies including prednisone [5, 17]. On occasion, there may be a transient and mild improvement in response to corticosteroids (CS) early on in the course of the disease [3] or the initial response to CS may be more dramatic in some cases, but is unfortunately followed by progressive resistance to therapy over three to six years [51]. Furthermore, in a long-term observational study of 136 patients, those who received immunosuppressive treatments (52%) were more severely affected on disability scales and on the sporadic inclusion body myositis weakness composite index when compared to those that did not [52]. Progression towards walking handicap was more rapid among patients receiving immunosuppressive treatments. Since, immunosuppressive treatments do not ameliorate the natural course of IBM, it has become more controversial whether to offer CS early on in the course of IBM [9]. Despite an earlier encouraging report [5], randomized controlled trials of IVIG without CS [25,53] and with CS [54] did not show any benefit. IVIG and prednisone reduce some inflammatory and degenerative molecules in muscle of patients with IBM and in vitro, but do not sufficiently suppress myotoxic and cell stress mediators such as inducible nitric oxide synthase. [55] This may in part explain resistance of sporadic IBM to immunotherapy.
Two Muscle Study Group randomized controlled studies of interferon β-1a at standard [56] or high doses did not reveal any efficacy in IBM [57]. A 48-week randomized controlled trial of methotrexate (MTX) in 44 IBM cases was also negative despite decrease in serum CK in the MTX group [58]. A 12- month small pilot trial comparing the effect of MTX combined with anti–thymocyte globulin (n=6) to that of MTX in five patients had suggested a mild benefit on muscle myometry in the group taking anti–thymocyte globulin [59]. A small randomized crossover pilot trial of placebo versus oxandrolone (an androgen receptor agonist) for 12 weeks did not reveal a statistically significant difference in the primary outcome measure of whole body maximal voluntary isometric contraction (MVICT). However, a significant benefit in the upper extremities MVICT was identified [60]. In a small pilot trial, there was no clinically meaningful improvement in handgrip after 12 months of etanercept administration [61]. A small open-label proof-of-principle study of alemtuzumab in IBM showed a reduction in muscle CD3+ lymphocytes but no significant improvement in strength or function [62]. An open-label pilot trial of 12 months oral simvastatin 40 mg daily confirmed its safety but none of the ten IBM patients had a significant clinical improvement [63]. IL-1β is upregulated in sIBM myofibers, co-localizes with APP and promotes the production of APP and amyloid deposits. In a small pilot study to examine whether anakinra, an IL1 receptor antagonist could benefit sIBM patients, four patients with biopsy-proven sIBM received this drug for a mean period of 7.7months. No improvement in muscle strength or stabilization was noted in any of the patients based on grip strength and MRC measurements. This treatment failure was felt to be due to insufficiency of anakinra to suppress the intramuscular IL1, short study duration, or irrelevance of IL1 in the disease process. [64]
Balloon dilation is often performed in IBM and dysphagia with variable response and often transient benefit.[65] Caution is suggested in recommending cricopharyngeal myotomy as there is a report of worsening after that procedure in a patient who also had a hiatal hernia. [66]
Ongoing Research
We developed the IBM functional rating scale (IBMFRS) which is 10-point functional rating scale for patients with IBM (Table 3). Based on analysis of 6-month data obtained in the high-dose beta interferon-1a trial [57, 67], the IBMFRS showed statistically significant correlations (P < 0.001) with maximal voluntary isometric contraction, manual muscle testing, and handgrip dynamometry. Compared to these other outcome measures, the IBMFRS was also the most sensitive measure of change.
Table 3. Inclusion Body Myositis Functional Rating Scale (IBMFRS) [67].
|
1. Swallowing – 4 Normal – 3 Early eating problems—occasional choking – 2 Dietary consistency changes – 1 Frequent choking – 0 Needs tube feeding |
6. Hygiene (bathing and toileting) – 4 Normal – 3 Independent but with increased effort or decreased activity – 2 Independent but requires use of assistive devices (shower chair, raised toilet seat, etc) – 1 Requires occasional assistance from caregiver – 0 Completely dependent |
|
2. Handwriting (with dominant hand prior to IBM onset) – 4 Normal – 3 Slow or sloppy; all words are legible – 2 Not all words are legible – 1 Able to grip pen but unable to write – 0 unable to grip pen |
7. Turning in bed and adjusting covers – 4 Normal – 3 Somewhat slow and clumsy but no help needed – 2 Can turn alone or adjust sheets, but with great difficulty – 1 Can initiate, but not turn or adjust sheets alone – 0 Unable or requires total assistance |
|
3. Cutting food and handling utensils – 4 Normal – 3 Somewhat slow and clumsy, but no help needed – 2 Can cut most foods, although clumsy and slow; some help needed – 1 Food must be cut by someone, but can still feed slowly – 0 Needs to be fed |
8. Sit to stand – 4 Independent (without use of arms) – 3 Performs with substitute motions (leaning forward, rocking) but without use of arms – 2 Requires use of arms – 1 requires assistance from a device or person – 0 Unable to stand |
|
4. Fine motor tasks (opening doors, using keys, picking up small objects) – 4 Independent – 3 Slow or clumsy in completing task – 2 Independent but requires modified techniques or assistive devices – 1 Frequently requires assistance from caregiver – 0 Unable |
9. Walking – 4 Normal – 3 Slow or mild unsteadiness – 2 Intermittent use of an assistive device (ankle–foot orthosis, cane, walker) – 1 Dependent on assistive device – 0 Wheelchair dependent |
|
5. Dressing – 4 Normal – 3 Independent but with increased effort or decreased efficiency – 2 Independent but requires assistive devices or modified techniques (Velcro snaps, shirts without buttons, etc) – 1 Requires assistance from caregiver for some clothing items – 0 total dependence |
10. Climbing stairs – 4 Normal – 3 Slow with hesitation or increased effort; uses hand rail intermittently – 2 Dependent on hand rail – 1 Dependent on hand rail and additional support (cane or person) – 0 Cannot climb stairs |
A 12-month trial of lithium chloride was completed aiming to decrease the activity of the glycogen synthase kinase (GSK), an enzyme that has a key role in the development of phosphorylated tau [68]. In addition, lithium in low doses is a well-known autophagy inducer that clears misfolded proteins and altered mitochondria from motor neurons [69]. Fifteen subjects were enrolled, four withdrew due to side effects and nine completed the 12-month study. Lithium treatment for one year produced no benefit. Despite a non-significant trend on quantitative muscle testing, the average MRC and IBMFRS scores did not improve significantly. Muscle GSK levels did not significantly change. Several experimental agents are being evaluated in ongoing clinical trials including arimoclomol, BYM338 and follistatin gene transfer therapy as listed on clinicaltrials.gov. Given the putative role of heat shock protein abnormalities in the pathogenesis of IBM, we conducted a two-center trial of arimoclomol, a heat shock protein 70 inducer that may therefore prevent protein misfolding. We completed this randomized controlled pilot study in 24 IBM subjects, 18 of whom received arimoclomol for four months and 8 were on placebo. Preliminary data analysis indicates that arimoclomol is well tolerated and safe in IBM [70]. We identified encouraging trends in some of the secondary outcome measures including the IBMFRS [71]. These encouraging preliminary signals support moving forward with a large multicenter study of arimoclomol in IBM.
The other two ongoing studies aim to increase muscle size strength and function using different approaches and for more details the reader is referred to clinicaltrials.gov. In the follistatin gene transfer therapy, follistatin gene carried by adeno-associated virus is injected into the thigh muscle of IBM and Becker muscular dystrophy patients. Using a different approach, Novartis is investigating the efficacy, safety and tolerability of BYM338 in patients with IBM as listed on clinicaltrials.gov.
Prognosis
IBM usually progresses to disability without affecting life expectancy. An earlier study had shown that at five years, 10/14 cases required a cane or support, and at ten years most cases (3/5) were wheelchair confined [72]. After mean disease duration of 7 years [12], 56% of our cases required an assistive device, with 20% requiring a wheelchair or motorized scooter (Table 1). More recently, two long term observational studies have shed more light on the rate of disease progression of IBM to disability [52,73]. After a mean disease duration of 20 years, the mean yearly decline in strength of 15 surviving IBM patients ranged from 3.5 to 5.4% as assessed by manual muscle testing and quantitative muscle testing, respectively [74]. This resulted in progressive impairment in activities of daily living and all 15 IBM patients were found after a mean disease duration of 20 years to be using a wheelchair, seven of them (47%) being completely wheelchair-bound. In another study of 136 cases followed in Paris and Oxford clinics between 1990 and 2008, 75% of patients had significant walking difficulties [52]. Thirty-seven percent used a wheelchair after a median duration from onset of 14 years, 95% confidence interval being 13 to 18 years. Disease progression towards walking handicap was more rapid among males, patients older at first symptoms and as noted above patients receiving immunosuppressive treatments [52]. At one-year follow-up of 23 IBM cases, manual muscle testing, quadriceps quantitative muscle testing (QMT) and IBMFRS significantly declined by 5.2%, 27.9%, and 13.8%, respectively. [74] QMT of the quadriceps muscle and IBMFRS were the most sensitive measures of disease progression. After a median time of seven years of disease duration, 63% of patients had lost independent ambulation. Disease onset after 55 years of age, but not sex or treatment, was found to be predictive of a shorter time to requirement of a cane.
Exercise
There certainly is a role of for physical therapy, orthotic devices, occupational therapy, a healthy well-balanced diet and exercise in IBM. Despite falls being a common occurrence for people with IBM, falls guidelines are not being followed, and referral rates to physiotherapy need to improve.[13] A tailored 12-week home exercise program, five days a week for 12 weeks in combination with stationary biking or walks, was found to be safe in seven patients [75]. There was no strength deterioration, no change in serum CK, and no increase in muscle inflammation on biopsy. However, the study was not able to show improved muscle strength or function.
Other investigators recently reported the benefits of a 16-week home exercise program performed twice per day in seven IBM patients, two of whom used a cane and another two a motorized scooter [76]. The exercises consisted of whole-body sit-to-stand exercises, biceps curl, shoulder press, heel lifts, isometric vastus medialis exercises, and ankle dorsiflexion. Surprisingly, patients improved in all muscle groups, including hip flexion, elbow extension, knee flexion and extension, and grip strength. Timed functional tests (to climb one flight of stairs and to walk 30 meters) were also improved. In another report, the same group of investigators described the effects of an aerobic exercise program using a stationary cycle ergometer at 80% of the initial maximal heart rate combined with the above mentioned resistance isometric and isotonic exercises of the upper and lower limbs in a group of seven IBM cases [77]. Besides demonstrating safety, they found this exercise routine to improve aerobic capacity and muscle strength in shoulder abduction, hip flexion, hip abduction, and knee flexion. However, no changes were noted in knee extension and grip strength and there were no significant changes in stair time or 30 meter walk test.
Given encouraging safety data, we recommend to our IBM patients mild-to-moderate intensity non-fatiguing exercises. There is a suggestion that exercise might lead to modestly improved or sustained muscle strength in some patients. However, there is conflicting data on the effect of exercise on the two muscle groups most severely affected in IBM (finger flexors and knee extensors) and on the potential for functional mobility benefit. Large multicenter controlled trials are needed to clarify any potential gains from exercise in people with IBM.
Conclusion
IBM is the most common inflammatory myopathy after age 50. Despite similarities with PM inflammatory pathology, IBM histopathology demonstrates marked degeneration and protein aggregation. The clinical phenotype of typical IBM is distinctive, manifesting as proximal leg or distal arm weakness though in our experience there are several phenotypic variants. IBM is refractory to all known immunosuppressive therapies. Low intensity exercise may slow down the rate of functional decline. IBM patients are very motivated and should be encouraged to participate in clinical trials.
-
-
IBM is the most common inflammatory myopathy after age 50.
-
-
Despite similarities with PM inflammatory pathology, IBM histopathology demonstrates marked degeneration and protein aggregation.
-
-
The clinical phenotype of typical IBM is distinctive, manifesting as proximal leg or distal arm weakness though in our experience there are several phenotypic variants.
-
-
IBM is refractory to all known immunosuppressive therapies.
-
-
Low intensity exercise may slow down the rate of functional decline.
-
-
IBM patients are very motivated and should be encouraged to participate in clinical trials.
Acknowledgments
This publication [or project] was supported by an Institutional Clinical and Translational Science Award, NIH/NCATS Grant Number UL1TR000001. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Mazen M. Dimachkie, Email: mdimachkie@kumc.edu, Department of Neurology, University of Kansas Medical Center, 3901 Rainbow Blvd, Mail Stop 2012, Kansas City, KS 66160, Phone: 913.588.6970, Fax: 913.588.0609.
Richard J. Barohn, Department of Neurology, University Distinguished Professor, University of Kansas Medical Center, 3901 Rainbow Blvd, Mail Stop 2012, Kansas City, KS 66160.
References
- 1.Phillips BA, Zilko PJ, Mastaglia FL. Prevalence of sporadic inclusion body myositis in Western Australia. Muscle Nerve. 2000;23(6):970–972. doi: 10.1002/(sici)1097-4598(200006)23:6<970::aid-mus20>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 2.Tan JA, Roberts-Thomson PJ, Blumbergs P, et al. Incidence and prevalence of idiopathic inflammatory myopathies in South Australia: a 30-year epidemiologic study of histology-proven cases. Int J Rheum Dis. 2013 Jun;16(3):331–8. doi: 10.1111/j.1756-185X.2011.01669.x. [DOI] [PubMed] [Google Scholar]
- 3.Lindberg C, Persson LI, Bjorkander J, et al. Inclusion body myositis: clinical, morphological, physiological and laboratory findings in 18 cases. Acta Neurol Scand. 1994;89:123–131. doi: 10.1111/j.1600-0404.1994.tb01647.x. [DOI] [PubMed] [Google Scholar]
- 4.Wilson FC, Ytterberg SR, Sauver JL, et al. Epidemiology of sporadic inclusion body myositis and polymyositis in Olmsted County, Minnesota. J Rheumatol. 2008;35(3):445–7. [PubMed] [Google Scholar]
- 5.Lotz BP, Engel AG, Nishino H, et al. Inclusion body myositis. Observations in 40 patients. Brain. 1989;112(Pt 3):727–747. doi: 10.1093/brain/112.3.727. [DOI] [PubMed] [Google Scholar]
- 6.Badrising UA, Maat-Schieman ML, van Houwelingen JC, et al. Inclusion body myositis. Clinical features and clinical course of the disease in 64 patients. Neurol. 2005;252(12):1448–1454. doi: 10.1007/s00415-005-0884-y. [DOI] [PubMed] [Google Scholar]
- 7.Amato AA, Gronseth GS, Jackson, et al. Inclusion body myositis: clinical and pathological boundaries. Ann Neurol. 1995;40:581–586. doi: 10.1002/ana.410400407. [DOI] [PubMed] [Google Scholar]
- 8.Barohn RJ, Amato AA. Inclusion body myositis. Curr Treat Options in Neurol. 2000;2:7–12. doi: 10.1007/s11940-000-0019-9. [DOI] [PubMed] [Google Scholar]
- 9.Badrising UA, Maat-Schieman M, van Duinen SG, et al. Epidemiology of inclusion body myositis in the Netherlands: a nationwide study. Neurol. 2000;55:1385–1387. doi: 10.1212/wnl.55.9.1385. [DOI] [PubMed] [Google Scholar]
- 10.Sayers ME, Chou SM, Calabrese LH. Inclusion body myositis: analysis of 32 cases. J Rheumatol. 1992;19:1385–1389. [PubMed] [Google Scholar]
- 11.Needham M, James I, Corbett A, et al. Sporadic inclusion body myositis: phenotypic variability and influence of HLA-DR3 in a cohort of 57 Australian cases. J Neurol Neurosurg Psychiatry. 2008;79(9):1056–1060. doi: 10.1136/jnnp.2007.138891. [DOI] [PubMed] [Google Scholar]
- 12.Estephan B, Barohn RJ, Dimachkie MM, et al. Sporadic IBM: a Case Cohort. Journal of Clinical Neuromuscular Disease. 2011;12(3):18–19. [Google Scholar]
- 13.Hiscock A, Dewar L, Parton M, et al. Frequency and circumstances of falls in people with inclusion body myositis: a questionnaire survey to explore falls management and physiotherapy provision. Physiotherapy. 2013 Aug 14; doi: 10.1016/j.physio.2013.06.002. pii: S0031-9406(13)00073-4. [DOI] [PubMed] [Google Scholar]
- 14.Goodman BP, Liewluck T, Crum BA, et al. Camptocormia due to inclusion body myositis. J Clin Neuromuscul Dis. 2012 Dec;14(2):78–81. doi: 10.1097/CND.0b013e3182650718. [DOI] [PubMed] [Google Scholar]
- 15.Finsterer J, Stöllberger C, Kovacs GG. Asymptomatic hyper-creatine-kinase-emia as sole manifestation of inclusion body myositis. Neurol Int. 2013 Jun 25;5(2):34–6. doi: 10.4081/ni.2013.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jethava A, Ali S, Dasanu CA. Primary respiratory failure due to inclusion body myositis: think outside the box. Conn Med. 2013 Mar;77(3):155–8. [PubMed] [Google Scholar]
- 17.Barohn RJ, Amato AA, Sahenk Z, et al. Inclusion body myositis: explanation for poor response to immunosuppressive therapy. Neurol. 1995;45(7):1302–1304. doi: 10.1212/wnl.45.7.1302. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh PS, Laughlin RS, Engel AE. Inclusion body myositis presenting with facial diplegia. Muscle Nerve. 2013 Aug 27; doi: 10.1002/mus.24060. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 19.Garlepp MJ, Laing B, Zilko PJ, et al. HLA associations with inclusion body myositis. Clin Exp Immunol. 1994t;98:40–5. doi: 10.1111/j.1365-2249.1994.tb06604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rothwell S, Cooper RG, Lamb JA, et al. Entering a new phase of immunogenetics in the idiopathic inflammatory myopathies. Curr Opin Rheumatol. 2013 Nov;25(6):735–41. doi: 10.1097/01.bor.0000434676.70268.66. [DOI] [PubMed] [Google Scholar]
- 21.Amato AA, Barohn RJ. Inclusion body myositis: old and new concepts. J Neurol Neurosurg Psychiatry. 2009 Nov;80(11):1186–93. doi: 10.1136/jnnp.2009.173823. [DOI] [PubMed] [Google Scholar]
- 22.Mercuri E, Pichiecchio A, Allsop J, et al. Muscle MRI in inherited neuromuscular disorders: past, present, and future. J Magn Reson Imaging. 2007 Feb;25(2):433–40. doi: 10.1002/jmri.20804. [DOI] [PubMed] [Google Scholar]
- 23.Degardin A, Morillon D, Lacour A, et al. Morphologic imaging in muscular dystrophies and inflammatory myopathies. Skeletal Radiol. 2010;39(12):1219–27. doi: 10.1007/s00256-010-0930-4. [DOI] [PubMed] [Google Scholar]
- 24.Cox FM, Reijnierse M, van Rijswijk CS, et al. Magnetic resonance imaging of skeletal muscles in sporadic inclusion body myositis. Rheumatol. 2011;50(6):1153–61. doi: 10.1093/rheumatology/ker001. [DOI] [PubMed] [Google Scholar]
- 25.Amato AA, Barohn RJ, Jackson CE, et al. Inclusion body myositis: treatment with intravenous immunoglobulin. Neurol. 1994;44(8):1516–1518. doi: 10.1212/wnl.44.8.1516. [DOI] [PubMed] [Google Scholar]
- 26.Wagner KR, Fleckenstein JL, Amato AA, et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008 May;63(5):561–71. doi: 10.1002/ana.21338. [DOI] [PubMed] [Google Scholar]
- 27.Glover GH, Schneider E. Three-point Dixon technique for true water/fat decomposition with B0 inhomogeneity correction. Magn Reson Med. 1991 Apr;18(2):371–83. doi: 10.1002/mrm.1910180211. [DOI] [PubMed] [Google Scholar]
- 28.Hiba B, Richard N, Hébert LJ, et al. Quantitative assessment of skeletal muscle degeneration in patients with myotonic dystrophy type 1 using MRI. J Magn Reson Imaging. 2012 Mar;35(3):678–85. doi: 10.1002/jmri.22849. [DOI] [PubMed] [Google Scholar]
- 29.Maillard SM, Jones R, Owens C, et al. Quantitative assessment of MRI T2 relaxation time of thigh muscles in juvenile dermatomyositis. Rheumatology (Oxford) 2004 May;43(5):603–8. doi: 10.1093/rheumatology/keh130. [DOI] [PubMed] [Google Scholar]
- 30.Maetzler W, Reimold M, Schittenhelm J, et al. Increased [11C]PIB-PET levels in inclusion body myositis are indicative of amyloid beta deposition. J Neurol Neurosurg Psychiatry. 2011;82(9):1060–2. doi: 10.1136/jnnp.2009.197640. [DOI] [PubMed] [Google Scholar]
- 31.Chahin N, Engel AG. Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurol. 2008;70(6):418–424. doi: 10.1212/01.wnl.0000277527.69388.fe. [DOI] [PubMed] [Google Scholar]
- 32.Van der Meulen MF, Hoogendijk JE, Moons KG, et al. Rimmed vacuoles and the added value of SMI-31 staining in diagnosing sporadic inclusion body myositis. Neuromuscul Disord. 2001;11:447–451. doi: 10.1016/s0960-8966(00)00219-4. [DOI] [PubMed] [Google Scholar]
- 33.Greenberg SA, Pinkus GS, Amato AA, et al. Myeloid dendritic cells in inclusion-body myositis and polymyositis. Muscle Nerve. 2007;35(1):17–23. doi: 10.1002/mus.20649. [DOI] [PubMed] [Google Scholar]
- 34.Griggs RC, Askanas V, DiMauro S, et al. Inclusion body myositis and myopathies. Ann Neurol. 1995 Nov;38(5):705–13. doi: 10.1002/ana.410380504. [DOI] [PubMed] [Google Scholar]
- 35.Hilton-Jones D, Miller A, Parton M, et al. Inclusion body myositis: MRC Centre for Neuromuscular Diseases, IBM workshop, London, 13 June 2008. Neuromuscul Disord. 2010;20(2):142–7. doi: 10.1016/j.nmd.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 36.Salajegheh M, Pinkus JL, Taylor JP, et al. Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve. 2009;40:19–31. doi: 10.1002/mus.21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dubourg O, Wanschitz J, Maisonobe T, et al. Diagnostic value of markers of muscle degeneration in sporadic inclusion body myositis. Acta Myol. 2011 Oct;30(2):103–8. [PMC free article] [PubMed] [Google Scholar]
- 38.Greenberg SA. Inclusion body myositis. Curr Opin Rheumatol. 2011;23(6):574–8. doi: 10.1097/BOR.0b013e32834b53cc. [DOI] [PubMed] [Google Scholar]
- 39.Salajegheh M, Lam T, Greenberg SA. Autoantibodies against a 43 kDa muscle protein in inclusion body myositis. PLoS One. 2011;6:e20266. doi: 10.1371/journal.pone.0020266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greenberg SA, Sanoudou D, Haslett JN, et al. Molecular profiles of inflammatory myopathies. Neurol. 2002;59(8):1170–82. doi: 10.1212/wnl.59.8.1170. [DOI] [PubMed] [Google Scholar]
- 41.Greenberg SA, Bradshaw EM, Pinkus JL, et al. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurol. 2005;65(11):1782–1787. doi: 10.1212/01.wnl.0000187124.92826.20. [DOI] [PubMed] [Google Scholar]
- 42.Larman HB, Salajegheh M, Nazareno R, et al. Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol. 2013 Mar;73(3):408–18. doi: 10.1002/ana.23840. [DOI] [PubMed] [Google Scholar]
- 43.Pluk H, van Hoeve BJ, van Dooren SH, et al. Autoantibodies to cytosolic 5′-nucleotidase 1A in inclusion body myositis. Ann Neurol. 2013 Mar;73(3):397–407. doi: 10.1002/ana.23822. [DOI] [PubMed] [Google Scholar]
- 44.Askanas V, Engel WK. Inclusion-body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition. Neurology. 2006;66(2 Suppl 1):S39–48. doi: 10.1212/01.wnl.0000192128.13875.1e. [DOI] [PubMed] [Google Scholar]
- 45.Wojcik S, Engel WK, McFerrin J, et al. Overexpression and proteasome inhibition increase alpha B-crystallin in cultured human muscle: relevance to inclusion-body myositis. Neuromuscul Disord. 2006 Dec;16(12):839–44. doi: 10.1016/j.nmd.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Askanas V, Engel WK. Sporadic inclusion-body myositis: conformational multifactorial ageing-relateddegenerative muscle disease associated with proteasomal and lysosomal inhibition, endoplasmic reticulum stress, and accumulation of amyloid-β42 oligomers and phosphorylated tau. Presse Med. 2011;40(4 Pt 2):e219–35. doi: 10.1016/j.lpm.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 47.Salajegheh M, Pinkus JL, Nazareno R, et al. Nature of “Tau” immunoreactivity in normal myonuclei and inclusion body myositis. Muscle Nerve. 2009;40(4):520–8. doi: 10.1002/mus.21471. [DOI] [PubMed] [Google Scholar]
- 48.Nogalska A, D'Agostino C, Engel WK, et al. Novel demonstration of amyloid-β oligomers in sporadic inclusion-body myositis muscle fibers. Acta Neuropathol. 2010;120(5):661–6. doi: 10.1007/s00401-010-0737-3. [DOI] [PubMed] [Google Scholar]
- 49.Tawil R, Griggs RC. Inclusion body myositis. Curr Opin Rheumatol. 2002;14:653–7. doi: 10.1097/00002281-200211000-00004. [DOI] [PubMed] [Google Scholar]
- 50.Machado P, Brady S, Hanna MG. Update in inclusion body myositis. Curr Opin Rheumatol. 2013 Nov;25(6):763–71. doi: 10.1097/01.bor.0000434671.77891.9a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verma A, Bradley WG, Ringel SP. Treatment-responsive polymyositis transforming into inclusion body myositis. Neurol. 2008:P060–19. [Google Scholar]
- 52.Benveniste O, Guiguet M, Freebody J, et al. Long-term observational study of sporadic inclusion body myositis. Brain. 2011;134(Pt 11):3176–84. doi: 10.1093/brain/awr213. [DOI] [PubMed] [Google Scholar]
- 53.Dalakas MC, Sonies B, Dambrosia J, et al. Treatment of inclusion-body myositis with IVIg: a double-blind, placebo-controlled study. Neurol. 1997;48:712–716. doi: 10.1212/wnl.48.3.712. [DOI] [PubMed] [Google Scholar]
- 54.Dalakas MC, Koffman B, Fujii M, et al. A controlled study of intravenous immunoglobulin combined with prednisone in the treatment of IBM. Neurol. 2001;56:323–327. doi: 10.1212/wnl.56.3.323. [DOI] [PubMed] [Google Scholar]
- 55.Zschüntzsch J, Voss J, Creus K, et al. Provision of an explanation for the inefficacy of immunotherapy in sporadic inclusion body myositis: quantitative assessment of inflammation and β-amyloid in the muscle. Arthritis Rheum. 2012 Dec;64(12):4094–103. doi: 10.1002/art.37692. [DOI] [PubMed] [Google Scholar]
- 56.Muscle Study Group. Randomized pilot trial of betaINF1a (Avonex) in patients with inclusion body myositis. Neurol. 2001;57:1566–1570. doi: 10.1212/wnl.57.9.1566. [DOI] [PubMed] [Google Scholar]
- 57.Muscle Study Group. Randomized pilot trial of high-dose betaINF-1a in patients with inclusion body myositis. Neurol. 2004;63:718–720. doi: 10.1212/01.wnl.0000134675.98525.79. [DOI] [PubMed] [Google Scholar]
- 58.Badrising UA, Maat-Schieman ML, Ferrari, et al. Comparison of weakness progression in inclusion body myositis during treatment with methotrexate or placebo. Ann Neurol. 2002;51:369–372. doi: 10.1002/ana.10121. [DOI] [PubMed] [Google Scholar]
- 59.Lindberg C, Trysberg E, Tarkowski A, et al. Anti-T-lymphocyte globulin treatment in inclusion body myositis: a randomized pilot study. Neurol. 2003;61:260–262. doi: 10.1212/01.wnl.0000071852.27182.c7. [DOI] [PubMed] [Google Scholar]
- 60.Rutkove SB, Parker RA, Nardin RA, et al. A pilot randomized trial of oxandrolone in inclusion body myositis. Neurol. 2002;58(7):1081–1087. doi: 10.1212/wnl.58.7.1081. [DOI] [PubMed] [Google Scholar]
- 61.Barohn RJ, Herbelin L, Kissel JT, et al. Pilot trial of etanercept in the treatment of inclusion-body myositis. Neurol. 2006;66(2 Suppl 1):S123–4. doi: 10.1212/01.wnl.0000192258.32408.54. [DOI] [PubMed] [Google Scholar]
- 62.Dalakas MC, Rakocevic G, Schmidt J, et al. Effect of Alemtuzumab (CAMPATH 1-H) in patients with inclusion-body myositis. Brain. 2009;132(Pt 6):1536–1544. doi: 10.1093/brain/awp104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sancricca C, Mora M, Ricci E, et al. Pilot trial of simvastatin in the treatment of sporadic inclusion-body myositis. Neurol Sci. 2011;32(5):841–7. doi: 10.1007/s10072-011-0657-6. [DOI] [PubMed] [Google Scholar]
- 64.Kosmidis ML, Alexopoulos H, Tzioufas AG, et al. The effect of anakinra, an IL1 receptor antagonist, in patients with sporadic inclusion body myositis (sIBM): A small pilot study. J Neurol Sci. 2013 Aug 14; doi: 10.1016/j.jns.2013.08.007. pii: S0022-510X(13)02856-6. [DOI] [PubMed] [Google Scholar]
- 65.Murata KY, Kouda K, Tajima F, et al. Balloon dilation in sporadic inclusion body myositis patients with Dysphagia. Clin Med Insights Case Rep. 2013;6:1–7. doi: 10.4137/CCRep.S10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sanei-Moghaddam A, Kumar S, Jani P, et al. Cricopharyngeal myotomy for cricopharyngeus stricture in an inclusion body myositis patient with hiatus hernia: a learning experience. BMJ Case Rep. 2013 Jan 22;2013 doi: 10.1136/bcr-2012-008058. pii: bcr2012008058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jackson CE, Barohn RJ, Gronseth G, Muscle Study Group et al. Inclusion body myositis functional rating scale: a reliable and valid measure of disease severity. Muscle Nerve. 2008 Apr;37(4):473–6. doi: 10.1002/mus.20958. [DOI] [PubMed] [Google Scholar]
- 68.Saperstein DS, Levine T, Hank N, et al. Pilot Trial of Lithium Treatment in Inclusion Body Myositis. Neurology. 2011;76(Suppl 4):A106. [Google Scholar]
- 69.Pasquali L, Longone P, Isidoro C, et al. Autophagy, lithium, and amyotrophic lateral sclerosis. Muscle Nerve. 2009 Aug;40(2):173–94. doi: 10.1002/mus.21423. [DOI] [PubMed] [Google Scholar]
- 70.Wang Y, He J, McVey AL, et al. Twelve-month Change of IBMFRS in the Arimocolomol Inclusion Body Myositis Pilot Study. Poster 7.255 at the American Academy of Neurology annual Meeting; April 26 2012 at 5 pm; New Orleans. [Google Scholar]
- 71.Wang Y, He J, McVey AL, Pasnoor M, et al. Twelve-month Change of IBMFRS in the Arimocolomol Inclusion Body Myositis Pilot Study. Ann Neurol. 2013 Dec;74(Suppl 17):S95–96. [Google Scholar]
- 72.Sekul EA, Dalakas MC. Inclusion body myositis: new concepts. Semin Neurol. 1993;13(3):256–63. doi: 10.1055/s-2008-1041132. [DOI] [PubMed] [Google Scholar]
- 73.Cox FM, Titulaer MJ, Sont JK, et al. A 12-year follow-up in sporadic inclusion body myositis: an end stage with major disabilities. Brain. 2011;134(Pt 11):3167–75. doi: 10.1093/brain/awr217. [DOI] [PubMed] [Google Scholar]
- 74.Cortese A, Machado P, Morrow J, et al. Longitudinal observational study of sporadic inclusion body myositis: implications for clinical trials. Neuromuscul Disord. 2013 May;23(5):404–12. doi: 10.1016/j.nmd.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 75.Arnardottir S, Alexanderson H, Lundberg IE, et al. Sporadic inclusion body myositis: pilot study on the effects of a home exercise program on muscle function, histopathology and inflammatory reaction. J Rehabil Med. 2003;35(1):31–35. doi: 10.1080/16501970306110. [DOI] [PubMed] [Google Scholar]
- 76.Johnson GL, et al. The effectiveness of an individualized, home-based functional exercise program for patients with sporadic inclusion body myositis. J Clin Neuromusc Dis. 2007;8:187–194. [Google Scholar]
- 77.Johnson LG, Collier KE, Edwards DJ, et al. Improvement in aerobic capacity after an exercise program in sporadic inclusion body myositis. J Clin Neuromuscul Dis. 2009;10(4):178–184. doi: 10.1097/CND.0b013e3181a23c86. [DOI] [PubMed] [Google Scholar]