

Abstract
Objective:
To determine whether droxidopa, an oral norepinephrine precursor, improves symptomatic neurogenic orthostatic hypotension (nOH).
Methods:
Patients with symptomatic nOH due to Parkinson disease, multiple system atrophy, pure autonomic failure, or nondiabetic autonomic neuropathy underwent open-label droxidopa dose optimization (100–600 mg 3 times daily), followed, in responders, by 7-day washout and then a 7-day double-blind trial of droxidopa vs placebo. Outcome measures included patient self-ratings on the Orthostatic Hypotension Questionnaire (OHQ), a validated, nOH-specific tool that assesses symptom severity and symptom impact on daily activities.
Results:
From randomization to endpoint (n = 162), improvement in mean OHQ composite score favored droxidopa over placebo by 0.90 units (p = 0.003). Improvement in OHQ symptom subscore favored droxidopa by 0.73 units (p = 0.010), with maximum change in “dizziness/lightheadedness.” Improvement in symptom-impact subscore favored droxidopa by 1.06 units (p = 0.003), with maximum change for “standing a long time.” Mean standing systolic blood pressure (BP) increased by 11.2 vs 3.9 mm Hg (p < 0.001), and mean supine systolic BP by 7.6 vs 0.8 mm Hg (p < 0.001). At endpoint, supine systolic BP >180 mm Hg was observed in 4.9% of droxidopa and 2.5% of placebo recipients. Adverse events reported in ≥3% of double-blind droxidopa recipients were headache (7.4%) and dizziness (3.7%). No patients discontinued double-blind treatment because of adverse events.
Conclusions:
In patients with symptomatic nOH, droxidopa improved symptoms and symptom impact on daily activities, with an associated increase in standing systolic BP, and was generally well tolerated.
Classification of evidence:
This study provides Class I evidence that in patients with symptomatic nOH who respond to open-label droxidopa, droxidopa improves subjective and objective manifestation of nOH at 7 days.
Neurogenic orthostatic hypotension (nOH) is a disabling disorder defined as a sustained blood pressure (BP) reduction when the patient stands, caused by impairment of compensatory autonomic reflexes.1–3 Additional hemodynamic abnormalities4–6 frequently include supine hypertension.7 The disorder results from deficient noradrenergic activation of vascular adrenoceptors due to degenerative loss of postganglionic sympathetic neurons, as in Parkinson disease (PD) and pure autonomic failure (PAF), or loss of central pathways that regulate sympathetic drive, as in multiple system atrophy (MSA).3,8–11 Symptoms represent hypoperfusion of the brain and other organs and tissues,8,12 and may include lightheadedness/dizziness (or actual syncope), visual disturbances, generalized weakness or fatigue, and diminished tolerance for standing or walking.2,9,13,14 In 1996, midodrine (ProAmatine; Shire, Newport, KY), an orally administered α1-adrenoceptor agonist prodrug,15–18 was granted US approval for the treatment of symptomatic OH.
Droxidopa (l-threo-3,4-dihydroxyphenylserine) (NORTHERA; Chelsea Therapeutics, Charlotte, NC) is an orally administered artificial amino acid converted both peripherally and centrally into norepinephrine.19–22 Because the enzyme responsible for this conversion, aromatic amino acid decarboxylase, is widely expressed, administration of droxidopa increases norepinephrine even if postganglionic sympathetic neurons are not intact.22–24 In February 2014, droxidopa received accelerated Food and Drug Administration (FDA) approval for the treatment of symptomatic nOH. Herein, we report the results of a large phase 3 clinical trial of droxidopa in symptomatic nOH, in which efficacy was assessed by measures including patient self-ratings on a validated nOH-specific scale encompassing symptoms and symptom impact on daily activities.
METHODS
Patients.
All patients were aged 18 years or older and were required to have a clinical diagnosis of symptomatic nOH due to PD, PAF, MSA, nondiabetic autonomic neuropathy (NDAN), or dopamine-β-hydroxylase deficiency, with a documented decrease ≥20 mm Hg in systolic or ≥10 mm Hg in diastolic BP upon standing ≤3 minutes.1 Key exclusion criteria were use of vasoconstrictor agents within 2 days before baseline; use of long-acting antihypertensives or norepinephrine reuptake inhibitors; sustained, severe supine hypertension; and significant systemic, hepatic, cardiac, or renal disease.
Study design.
This was a randomized, placebo-controlled, parallel-group trial conducted between August 2008 and July 2010 at 94 US, Canadian, and European centers. Open-label dose optimization lasting up to 14 days (but preferably ≤6) was followed by 7-day washout and then 7-day double-blind treatment.
During dose-optimization, open-label droxidopa initiated at 100 mg 3 times daily was titrated in 100-mg increments until each patient: (1) had a self-rating of 0 on a 0-to-10 Likert scale for “dizziness, lightheadedness, feeling faint, or feeling like you might black out” (item 1 of the Orthostatic Hypotension Questionnaire [OHQ],25 described below), plus an increase in standing systolic BP ≥10 mm Hg, compared with the baseline value, as measured 3 minutes poststanding (and 3 hours after a droxidopa dose); (2) reached the maximum permitted dosage, 600 mg 3 times daily; (3) had a sustained BP >180 mm Hg systolic or >110 mm Hg diastolic while standing, sitting, or supine; or (4) experienced intolerable side effects considered to be related to study drug.
Responders were defined as having any improvement from baseline (i.e., ≥1 unit) on OHQ item 1, plus a ≥10 mm Hg increase from baseline in standing systolic BP. After 7-day washout, responders were assigned by a centralized, computerized randomization schedule to double-blind droxidopa or matching placebo (1:1) for 1 week at each patient's optimized dosage (100–600 mg 3 times daily). Patients in categories 3 or 4 received study drug at the highest tolerated dosage to which they were responders. All patients were counseled to take their last daily dose ≥4 hours before bedtime.
Efficacy measures.
The OHQ25 was administered at baseline, randomization, and end of study. Of its 10 items, 6 address nOH symptoms, namely, dizziness/lightheadedness, vision disturbance, weakness, fatigue, trouble concentrating, and head/neck discomfort. The other 4 ask the respondent to judge the nOH impact on daily activities requiring “standing a short time,” “standing a long time,” “walking a short time,” and “walking a long time.” Each item is scored on a Likert scale from 0 (not bothered/no interference) to 10 (worst possible/complete interference), describing the preceding week. The responses yield a composite symptom score and a composite symptom-impact score (each is the average of the item scores not rated 0 at baseline), and also an overall composite score (the average of the symptom and symptom-impact composite scores). The study's prespecified primary efficacy endpoint was the change in overall composite score from randomization to end of study. Secondary endpoints included changes in the symptom and symptom-impact composite scores, and in the individual OHQ items.
BP and heart rate were measured at screening, at baseline, on each day of dose optimization, at randomization, and at end of study (3 hours postdose during treatment periods). Brachial-arterial BP values were obtained by mercury, aneroid, or automated sphygmomanometry 3 times during 10 minutes while the patient was supine (with head and torso elevated approximately 30° from horizontal) and once 3 minutes poststanding. Change in standing systolic BP from randomization to end of study was an efficacy endpoint.
Safety was assessed by adverse events (AEs), clinical laboratory values, vital signs, and ECG.
Statistical analysis.
For OHQ data, mean change from randomization to end of study in the droxidopa and placebo groups was compared using analysis of covariance, with value at randomization as covariate and treatment group as main effect. Missing data were imputed by last observation carried forward, and statistical significance was set at the 2-sided, 5% level (at which n = 75 per group was predicted to have >80% power to detect a 1.2-unit group difference). BP changes from randomization to end of study were assessed by analysis of covariance, with missing data excluded. Post hoc analyses of change in OHQ composite score and standing systolic BP were performed in patient subsets defined by primary diagnosis and by concurrent dopa decarboxylase inhibitor (DDI) usage (carbidopa or benserazide). Relationships between changes in OHQ composite score and standing systolic BP were tested by Spearman correlation coefficients.
Classification of evidence.
This trial provides Class I evidence that droxidopa (100–600 mg 3 times daily) was significantly superior to placebo for treating nOH, both subjectively, including a mean OHQ composite-score improvement of 1.83 vs 0.93 units, a difference of 0.90 units (95% confidence interval [CI] 0.30–1.48; p = 0.003), and objectively, including a mean standing systolic BP increase of 11.2 vs 3.9 mm Hg, a difference of 7.3 mm Hg (95% CI 1.1–13.5; p < 0.001).
Standard protocol approvals, registrations, and patient consents.
The study protocol was approved by the institutional review board at each study site, and all patients provided written informed consent. The study was registered with ClinicalTrials.gov (NCT00782340).
RESULTS
Patients and study-drug dosage.
Of 263 patients participating in open-label droxidopa dose optimization, 162 responders (61.6%) were treated in the double-blind study phase, 80 with placebo and 82 with droxidopa (figure 1). The 101 nonrandomized patients had nOH characteristics and primary diagnoses resembling those of the 162 responders (table 1), but they differed in being older (mean age, 64.6 vs 56.6 years) and had slightly higher likelihoods of being male and being US participants. Among responders, 40.7% had PD, 33.3% PAF, 16.0% MSA, and 4.9% NDAN. None had dopamine-β-hydroxylase deficiency. One patient assigned to droxidopa but erroneously administered placebo is included in the droxidopa group for efficacy analysis and in the placebo group for safety reporting.
Figure 1. Patient disposition.

AE = adverse event; DB = double-blind; OL = open-label.
Table 1.
Demographic and baseline nOH characteristics (full-analysis set and unrandomized subjects)
Among randomized patients, the mean (SD) final study-drug dose was 430 (163) mg for droxidopa and 381 (144) for placebo, with 37.0% of droxidopa and 18.5% of placebo recipients taking the maximum 600 mg 3 times daily. By returned-capsule counts, mean dosage compliance was 99.2% for droxidopa and 100.7% for placebo. Among 65 patients using DDIs, the mean final study-drug dose was 438 (166) mg for droxidopa and 394 (154) for placebo.
OHQ outcomes.
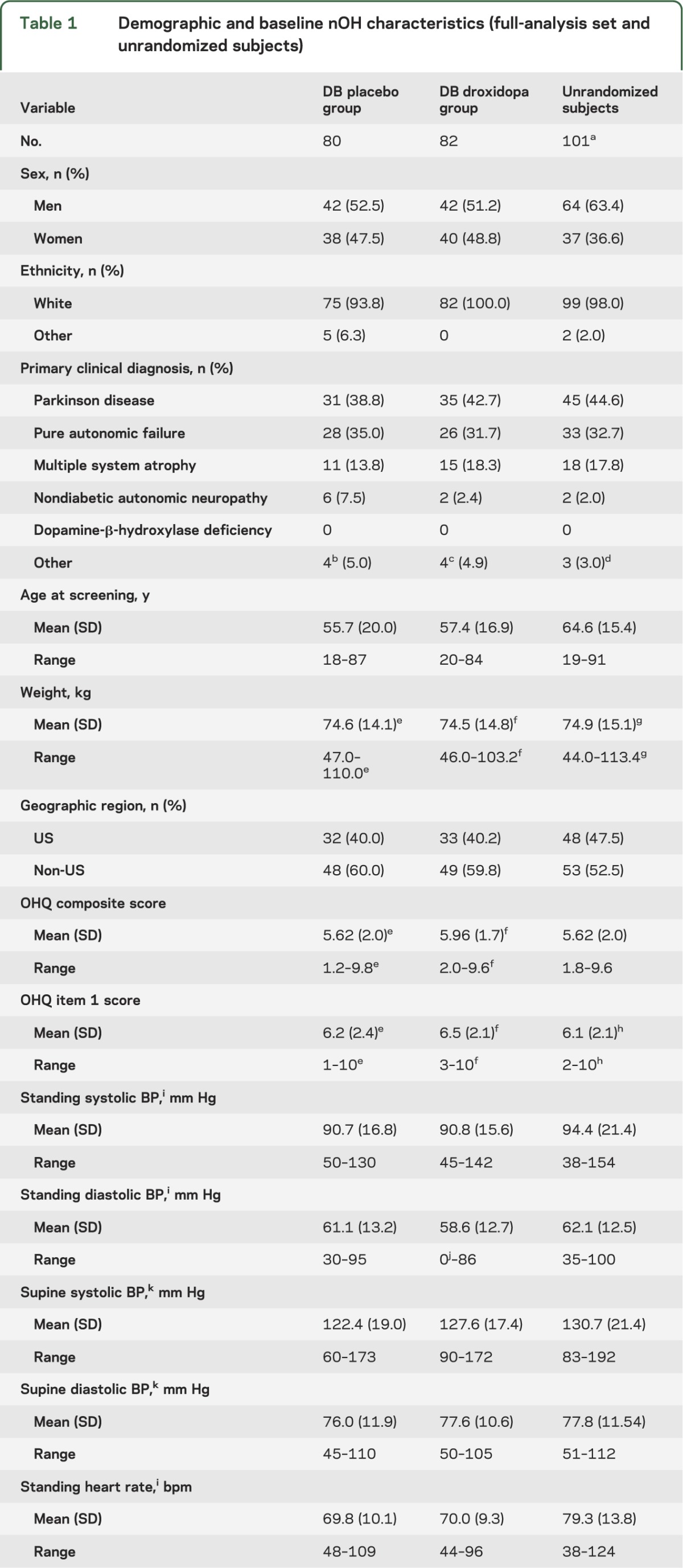
From randomization to end of study, droxidopa recipients had a mean (SD) change of −1.83 (2.07) units in OHQ composite score, vs −0.93 (1.69) among placebo recipients, favoring droxidopa by 0.90 units (95% CI 0.30–1.48; p = 0.003). The improvement was ≥3 units in 27.2% of droxidopa recipients vs 11.4% of placebo recipients (p = 0.016), and ≥4 units in 17.3% vs 2.5% (p = 0.003). For symptom composite score, mean change was −1.68 (2.13) vs −0.95 (1.90) units, favoring droxidopa by 0.73 units (95% CI 0.10–1.36; p = 0.010), with statistically significant differences from placebo on 4 of 6 symptom items (dizziness/lightheadedness, vision disturbance, weakness, and fatigue). For symptom-impact composite score, mean (SD) change was −1.98 (2.31) vs −0.92 (1.82) units, favoring droxidopa by 1.06 units (95% CI 0.41–1.71; p = 0.003), with statistically significant differences from placebo on all symptom-impact items (figure 2). Mean values at randomization and end of study for all OHQ and hemodynamic variables are shown in table e-1 on the Neurology® Web site at Neurology.org.
Figure 2. Treatment-group differences in OHQ score change, randomization to EOS, with 95% CIs (all treated patients; LOCF).
aAverage of nonzero item scores. bAverage of symptom and symptom-impact composite scores. *p < 0.05; **p < 0.01; ***p < 0.001; droxidopa vs placebo, analysis of covariance adjusted for treatment group and value at randomization. CI = confidence interval; EOS = end of study; LOCF = last observation carried forward; OHQ = Orthostatic Hypotension Questionnaire.
Hemodynamic outcomes.
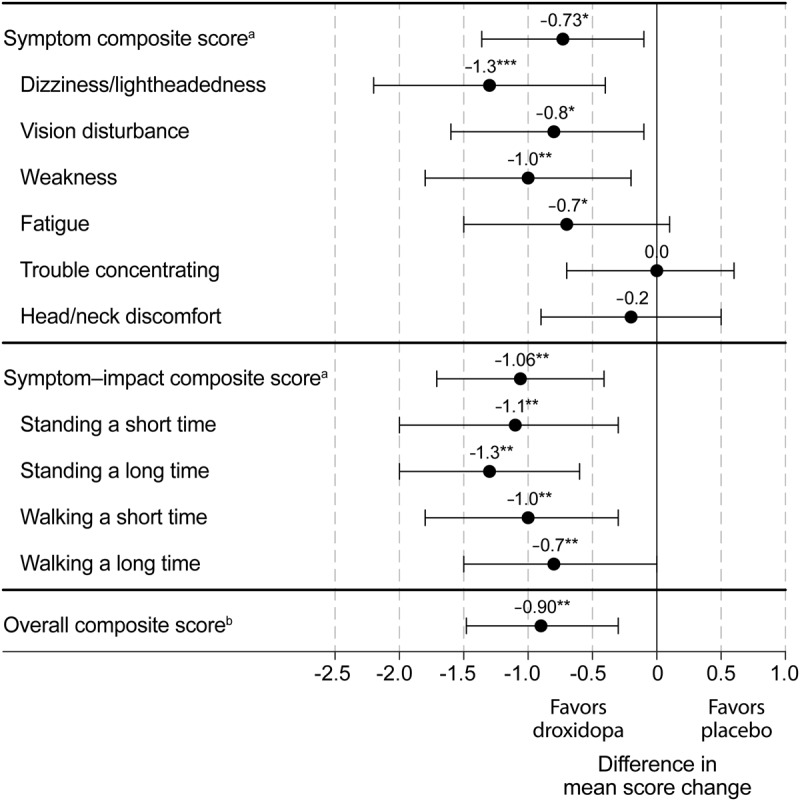
From randomization to end of study, standing systolic BP increased by a mean (SD) of 11.2 (22.9) mm Hg in droxidopa recipients vs 3.9 (16.3) in placebo recipients, a difference of 7.3 mm Hg (95% CI 1.1–13.5; p < 0.001). At endpoint, the mean (SD) standing systolic BP values were 107.4 (20.4) and 101.8 (22.3) mm Hg (figure 3).
Figure 3. Mean (SE) standing systolic BP during the study (all treated patients; LOCF).
*p < 0.001, droxidopa vs placebo from randomization to end of study; analysis of covariance adjusted for treatment group and value at randomization. BP = blood pressure; LOCF = last observation carried forward; SE = standard error.
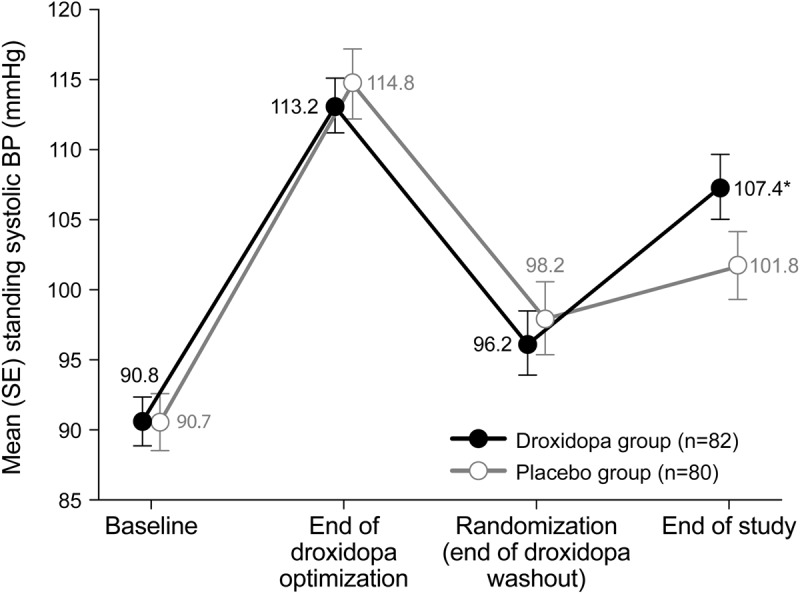
For droxidopa and for placebo, increase in standing systolic BP correlated with decrease in OHQ composite score (figure 4), with a Spearman correlation coefficient of −0.368 (p < 0.001) for droxidopa, −0.381 (p < 0.001) for placebo, and −0.426 (p < 0.001) overall.
Figure 4. Change in OHQ composite score vs standing systolic BP, randomization to EOS (missing values excluded).
The BP scale (but not the statistical analysis) omits one patient, a placebo recipient at (−125, −0.4). BP = blood pressure; EOS = end of study; OHQ = Orthostatic Hypotension Questionnaire.
From randomization to end of study, systolic BP while supine increased by 7.6 (19.2) mm Hg in droxidopa recipients vs 0.8 (14.5) in placebo recipients, a difference of 6.8 mm Hg (95% CI 1.53–12.07; p < 0.001), to mean (SD) values of 133.7 (23.3) and 125.9 (22.2) mm Hg, respectively. Heart rate was not affected by treatment (table e-1).
Post hoc subgroup analyses.
From randomization to end of study, patients with PAF showed mean improvements in OHQ composite score and standing systolic BP significantly greater for droxidopa than for placebo (table e-2), by 1.67 units (95% CI 0.60–2.74; p = 0.001) and 6.7 mm Hg (95% CI −1.99 to +15.39; p = 0.009). However, each primary-diagnosis subgroup (PAF, PD, MSA, and NDAN) had 35 or fewer members. From randomization to end of study, DDI nonusers showed mean improvements in OHQ composite score and standing systolic BP significantly greater for droxidopa than for placebo (see table e-2), by 1.35 units (95% CI 0.56–2.14; p < 0.001) and 8.6 mm Hg (95% CI 0.54–16.66; p < 0.001). However, each DDI-usage subgroup had 50 or fewer members, and 57% of DDI nonusers had PAF while all DDI users had PD or MSA.
Safety.
During open-label dose escalation, 100 patients (38.0% of 263) reported AEs (table e-3), most frequently headache (9.9%), dizziness (6.5%), or nausea (4.6%). Cardiac AEs were reported in 3.0% of patients, most frequently palpitations (1.9%). Two patients reported serious AEs (SAEs): one experienced nausea and vomiting; the other experienced urinary tract infection and urinary obstruction. Thirteen patients (4.9%) had AEs leading to discontinuation, most frequently nausea (1.5%) or hypertension (0.8%).
During double-blind treatment, 15 droxidopa (18.5% of 81) and 12 placebo (14.8% of 81) recipients reported AEs (see table e-3), most frequently headache (in 7.4% of droxidopa and no placebo recipients), dizziness (in 3.7% vs 1.2%), fatigue (2.5% vs 2.5%), syncope (2.5% vs 1.2%), and falls (0.0% vs 3.7%). No cardiac AEs occurred, no SAEs were reported, and there were no discontinuations because of AEs.
At randomization, no patients assigned to droxidopa and 2 assigned to placebo (2.5%) had supine hypertension, defined as a systolic BP >180 mm Hg. At end of study, 4 droxidopa recipients (4.9%) and 2 placebo recipients (2.5%) had supine hypertension. Overall, a supine systolic value exceeded 200 mm Hg in 2 patients assigned to placebo and none assigned to droxidopa. Neither treatment group showed clinically significant trends in laboratory and ECG parameters.
DISCUSSION
In patients with symptomatic nOH, double-blind, placebo-controlled droxidopa yielded statistically significant improvement, as assessed by patient-reported outcome scores and by an increase in standing systolic BP. Among patient-reported outcomes, significant benefit was documented for 4 of 6 nOH symptoms on a validated nOH-specific severity scale,25 with greatest improvement for “dizziness, lightheadedness, feeling faint, or feeling like you might black out,” the cardinal nOH symptoms, and for the impact of symptoms on daily activities requiring walking or standing, with greatest improvement in tolerance for “standing a long time.”
In nOH studies, patient-reported outcomes deserve emphasis but pose interpretive challenges. The severity of nOH symptoms often varies day to day and throughout the day.13,26 Symptoms can also be affected by ambient temperature, physical exertion, and food and fluid intake.13,27,28 Most importantly, symptoms depend on posture. Study subjects feeling unwell from their primary neurologic illness may become reluctant to stand, easing or preventing nOH symptoms. Conversely, subjects feeling well may increase their standing and walking periods, provoking nOH symptoms.
Objective nOH outcome measures are also imperfect. BP values obtained when the OHQ is administered may not represent BP values during the time span to which the questionnaire refers (the previous week). Furthermore, hypotension is not the proximate cause of nOH symptoms; the direct relationship is with tissue hypoperfusion. Hence, between BP and nOH symptoms, the crucial intermediate factor is that the brain autoregulates its blood supply.29,30 Symptoms of nOH occur when BP levels decrease below the individual patient's critical cerebral perfusion pressure. Accordingly, a large treatment-related increase in standing BP is not necessarily more beneficial than a small increase, provided the small increase places BP within the patient's autoregulatory range.
Despite these potential confounders, the observed treatment effects of droxidopa were statistically robust and appear to be clinically meaningful. In psychometric analyses of data from a phase 4 midodrine study, including analyses anchored to Clinical Global Impression ratings of subjects' nOH, an improvement of 0.8 to 1.0 units was judged to constitute a minimal important change in OHQ composite score, symptom composite score, or symptom-impact composite score.25 In the present study, the mean changes from randomization to end of study on all these scores were 1.7 to 2.0 units for droxidopa (and 0.9–1.0 for placebo), and from baseline (i.e., predroxidopa inception) to end of study, the mean changes were 2.5 to 2.8.
Change in OHQ composite score correlated with change in standing systolic BP both in droxidopa and in placebo recipients. This reassuringly implies that the subjective and objective findings are related—i.e., that nOH symptoms change in parallel (but opposite) direction to standing BP—strongly supporting the validity of OHQ composite score as an nOH severity measure. Nevertheless, there was an impressive placebo effect, both in symptoms, as measured by OHQ, and in BP. In patients with nOH, classic placebo response presumably would have a limited capacity to affect BP consistently. More plausibly, the placebo recipients may have improved just by their study participation, e.g., via increased physical activity, adherence to sleeping in a head-up tilt position, or increased water and salt intake (for which the study did not control). It is apparent that whenever an intervention alleviates or worsens nOH, it acts through BP changes.
Although post hoc subgroup analyses were hampered by small sample sizes and lack of appropriate matching, the results are suggestive. Across all primary diagnoses, symptomatic improvement was numerically greater for droxidopa than for placebo, but statistically significant only in PAF (see table e-2). For its part, improvement in standing systolic BP was numerically greater both in PAF and in PD than in MSA (see table e-2). These patterns are not surprising: patients with PAF and PD have significant postganglionic noradrenergic denervation, whereas patients with MSA have mostly preganglionic neuronal loss.11 Accordingly, a more pronounced droxidopa pressor effect might be expected in PAF or PD than in MSA, attributable to adrenoceptor supersensitivity to circulating norepinephrine.22,24 This hypothesis should be explored in clinical studies with adequate power.
Because DDIs block peripheral decarboxylation of droxidopa to norepinephrine, there has been concern that DDI usage might blunt the pressor effect of droxidopa.22 The literature is inconclusive: in one study, high carbidopa doses (200 mg) abolished the pressor effect,22 but for the lower DDI doses routinely used for parkinsonism, another study implies no reduction of pressor effect.21 Although the present study suggests the possibility of lessened droxidopa benefit among DDI users, the subgroups of patients taking and not taking DDIs were not matched for diagnosis. In particular, most of the DDI nonusers had PAF, while none of the DDI users had it, a difference that may have contributed to the droxidopa benefit observed in the nonusers. The issue of DDI comedication should be addressed in studies designed for that purpose.
Potential limitations of the present study include the absence of continuous BP monitoring (e.g., overnight, or beyond 3 minutes of standing), the absence of diary-based AE reporting, and the possibility of treatment unmasking (because of open-label pretreatment). Another limitation was the 1-week duration of double-blind treatment. The efficacy, safety, and tolerability of long-term droxidopa therapy will require separate investigation. In consonance with current standards for clinical research, in which pharmacotherapies are increasingly tested for benefit in patient subsets, the study utilized an enrichment design.31 The criteria for prerandomization discontinuation included subjective response to droxidopa without an objective BP improvement (to exclude “placebo” response, plus patients with nOH-like symptoms not due to hemodynamic causes). Patients were also excluded for safety reasons. Nonrandomized subjects resembled the randomized subjects at least in their primary diagnoses and baseline nOH characteristics. During optimization, however, their mean OHQ item 1 score improved less, by 2.8 (n = 100) vs 5.0 (n = 162) units, and their mean standing systolic BP increased less, by 6.0 (n = 93) vs 23.2 (n = 161) mm Hg. The results emphasize the need to individualize nOH therapy.
Droxidopa was generally well tolerated, including a low incidence or no occurrence of SAEs and of discontinuations because of AEs. The incidence of supine hypertension was also low, with no values >200 mm Hg. In a dose-response study of midodrine in nOH, 17% of patients exhibited such values after a 10-mg dose (and 41% did so after 20 mg).18
Overall, this short-term, multicenter trial showed that droxidopa treatment led to significant amelioration of multiple symptoms of nOH (dizziness, vision disturbance, weakness, and fatigue) and of nOH impact on activities requiring standing or walking, with an associated increase in standing systolic BP. These phase 3 findings expand the evidence supporting droxidopa for treatment of nOH.
Supplementary Material
ACKNOWLEDGMENT
Editorial assistance was provided by M.F., Senior Medical Writer, The Curry Rockefeller Group, LLC, Tarrytown, NY, and was funded by Chelsea Therapeutics, Inc. Statistical analysis was provided by J.M., Director, Biostatistics and Statistical Programming, Americas, Chiltern, Wilmington, NC. H.K. drafted the first version of the manuscript. All authors contributed substantially to the study design, the analysis and interpretation of data, and/or the drafting or critical revision of the manuscript, and all authors gave final approval for submission.
GLOSSARY
- AE
adverse event
- BP
blood pressure
- CI
confidence interval
- DDI
dopa decarboxylase inhibitor
- FDA
Food and Drug Administration
- MSA
multiple system atrophy
- NDAN
nondiabetic autonomic neuropathy
- nOH
neurogenic orthostatic hypotension
- OHQ
Orthostatic Hypotension Questionnaire
- PAF
pure autonomic failure
- PD
Parkinson disease
- SAE
serious adverse event
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Kaufmann was involved in the study concept and design, the acquisition, analysis, and interpretation of data, and writing the first draft of the manuscript, as well as critical revision of the manuscript for important intellectual content, and was the study principal investigator. Dr. Freeman was involved in the study concept and design and in the acquisition, analysis, and interpretation of data, as well as critical revision of the manuscript for important intellectual content, and was a study investigator. Dr. Biaggioni was involved in the study concept and design and in the acquisition, analysis, and interpretation of data, as well as critical revision of the manuscript for important intellectual content. Dr. Low was involved in the study concept and design and in the acquisition, analysis, and interpretation of data, as well as critical revision of the manuscript for important intellectual content, and was a study investigator. Dr. Pedder was involved in the research project as part of his duties as an employee of Chelsea Therapeutics, Inc. He had substantial involvement in the study concept, design, oversight, and management, and in critical revision of the manuscript for important intellectual content. Dr. Hewitt was involved in the research project as part of his duties as an employee of Chelsea Therapeutics, Inc. He had substantial involvement in the study concept, design, oversight, and management, and in critical revision of the manuscript for important intellectual content. Mr. Mauney performed the study's statistical analyses. Mr. Feirtag assisted in drafting and revising the manuscript, and in responding to reviewer comments. Dr. Mathias was involved in the study concept and design and in the acquisition, analysis, and interpretation of data, as well as critical revision of the manuscript for important intellectual content.
STUDY FUNDING
Sponsored by Chelsea Therapeutics, Inc. The article processing charge was paid by Chelsea Therapeutics.
DISCLOSURE
H. Kaufmann serves on a scientific advisory board for Chelsea Therapeutics, Inc.; serves as editor-in-chief of Clinical Autonomic Research; receives research support from the NIH (U54NS065736), the FDA (FD-R-3731-01), and the Dysautonomia Foundation, Inc.; and has received compensation as a consultant/advisory board member for Eli Lilly, Pfizer, and AstraZeneca. R. Freeman has served on scientific advisory boards for Abbott, Alnylam, Bristol-Myers Squibb, Chelsea Therapeutics, Inc., Johnson & Johnson, PamLab, Pfizer, and Sanofi-Aventis; has received funding from National Institute of Neurological Disorders and Stroke and NHLBI; and has received personal compensation for his editorial activities (editor) at Autonomic Neuroscience: Basic and Clinical. I. Biaggioni is a consultant for Chelsea Therapeutics, Inc., and AstraZeneca, and receives research support from AstraZeneca and Forest Laboratories. P. Low served on a scientific advisory board for Chelsea Therapeutics, Inc., served as a clinical editor for Autonomic Neuroscience, has received royalties from publication of the book Clinical Autonomic Disorders, served as a consultant for WR Medical, and is funded by NIH grants P01NS 44233 and U54 NS065736. S. Pedder is an employee of Chelsea Therapeutics, Inc., receiving a salary, a variable annual bonus, and stock options in the company as part of his compensation package. L. Hewitt is an employee of Chelsea Therapeutics, Inc., receiving a salary, a variable annual bonus, and stock options in the company as part of his compensation package. J. Mauney is an employee of Chiltern, the Contract Research Organization that provided statistical and other services for the study, as contracted by Chelsea Therapeutics, Inc. M. Feirtag is an employee of The Curry Rockefeller Group, which provided support for preparing this manuscript, as contracted by Chelsea Therapeutics, Inc. C. Mathias serves on a scientific advisory board for and has received funding from Chelsea Therapeutics, Inc. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Kaufmann H. Consensus statement on the definition of orthostatic hypotension, pure autonomic failure and multiple system atrophy. Clin Auton Res 1996;6:125–126 [DOI] [PubMed] [Google Scholar]
- 2.Freeman R. Clinical practice: neurogenic orthostatic hypotension. N Engl J Med 2008;358:615–624 [DOI] [PubMed] [Google Scholar]
- 3.Freeman R, Wieling W, Axelrod FB, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin Auton Res 2011;21:69–72 [DOI] [PubMed] [Google Scholar]
- 4.Mann S, Altman DG, Raftery EB, Bannister R. Circadian variation of blood pressure in autonomic failure. Circulation 1983;68:477–483 [DOI] [PubMed] [Google Scholar]
- 5.Okamoto LE, Gamboa A, Shibao C, et al. Nocturnal blood pressure dipping in the hypertension of autonomic failure. Hypertension 2009;53:363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Voichanski S, Grossman C, Leibowitz A, et al. Orthostatic hypotension is associated with nocturnal change in systolic blood pressure. Am J Hypertens 2012;25:159–164 [DOI] [PubMed] [Google Scholar]
- 7.Goldstein DS, Pechnik S, Holmes C, Eldadah B, Sharabi Y. Association between supine hypertension and orthostatic hypotension in autonomic failure. Hypertension 2003;42:136–142 [DOI] [PubMed] [Google Scholar]
- 8.Horowitz DR, Kaufmann H. Autoregulatory cerebral vasodilation occurs during orthostatic hypotension in patients with primary autonomic failure. Clin Auton Res 2001;11:363–367 [DOI] [PubMed] [Google Scholar]
- 9.Maule S, Papotti G, Naso D, Magnino C, Testa E, Veglio F. Orthostatic hypotension: evaluation and treatment. Cardiovasc Hematol Disord Drug Targets 2007;7:63–70 [DOI] [PubMed] [Google Scholar]
- 10.Ziegler MG, Lake CR, Kopin IJ. The sympathetic-nervous-system defect in primary orthostatic hypotension. N Engl J Med 1977;296:293–297 [DOI] [PubMed] [Google Scholar]
- 11.Goldstein DS, Sharabi Y. Neurogenic orthostatic hypotension: a pathophysiological approach. Circulation 2009;119:139–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novak V, Novak P, Spies JM, Low PA. Autoregulation of cerebral blood flow in orthostatic hypotension. Stroke 1998;29:104–111 [DOI] [PubMed] [Google Scholar]
- 13.Low PA, Singer W. Management of neurogenic orthostatic hypotension: an update. Lancet Neurol 2008;7:451–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathias CJ, Kimber JR. Postural hypotension: causes, clinical features, investigation, and management. Annu Rev Med 1999;50:317–336 [DOI] [PubMed] [Google Scholar]
- 15.Kaufmann H, Brannan T, Krakoff L, Yahr MD, Mandeli J. Treatment of orthostatic hypotension due to autonomic failure with a peripheral alpha-adrenergic agonist (midodrine). Neurology 1988;38:951–956 [DOI] [PubMed] [Google Scholar]
- 16.Jankovic J, Gilden JL, Hiner BC, et al. Neurogenic orthostatic hypotension: a double-blind, placebo-controlled study with midodrine. Am J Med 1993;95:38–48 [DOI] [PubMed] [Google Scholar]
- 17.Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension: a randomized, double-blind multicenter study. Midodrine Study Group. JAMA 1997;277:1046–1051 [PubMed] [Google Scholar]
- 18.Wright RA, Kaufmann HC, Perera R, et al. A double-blind, dose-response study of midodrine in neurogenic orthostatic hypotension. Neurology 1998;51:120–124 [DOI] [PubMed] [Google Scholar]
- 19.Biaggioni I, Robertson D. Endogenous restoration of noradrenaline by precursor therapy in dopamine-beta-hydroxylase deficiency. Lancet 1987;2:1170–1172 [DOI] [PubMed] [Google Scholar]
- 20.Freeman R, Landsberg L, Young J. The treatment of neurogenic orthostatic hypotension with 3,4-DL-threo-dihydroxyphenylserine: a randomized, placebo-controlled, crossover trial. Neurology 1999;53:2151–2157 [DOI] [PubMed] [Google Scholar]
- 21.Mathias CJ, Senard JM, Braune S, et al. L-threo-dihydroxyphenylserine (L-threo-DOPS; droxidopa) in the management of neurogenic orthostatic hypotension: a multi-national, multi-center, dose-ranging study in multiple system atrophy and pure autonomic failure. Clin Auton Res 2001;11:235–242 [DOI] [PubMed] [Google Scholar]
- 22.Kaufmann H, Saadia D, Voustianiouk A, et al. Norepinephrine precursor therapy in neurogenic orthostatic hypotension. Circulation 2003;108:724–728 [DOI] [PubMed] [Google Scholar]
- 23.Goldstein DS, Holmes C, Kaufmann H, Freeman R. Clinical pharmacokinetics of the norepinephrine precursor L-threo-DOPS in primary chronic autonomic failure. Clin Auton Res 2004;14:363–368 [DOI] [PubMed] [Google Scholar]
- 24.Esler M. An explanation of the unexpected efficacy of L-DOPS in pure autonomic failure. Clin Auton Res 2004;14:356–357 [DOI] [PubMed] [Google Scholar]
- 25.Kaufmann H, Malamut R, Norcliffe-Kaufmann L, Rosa K, Freeman R. The Orthostatic Hypotension Questionnaire (OHQ): validation of a novel symptom assessment scale. Clin Auton Res 2012;22:79–90 [DOI] [PubMed] [Google Scholar]
- 26.Granger JP, Alexander BT, Llinas M. Mechanisms of pressure natriuresis. Curr Hypertens Rep 2002;4:152–159 [DOI] [PubMed] [Google Scholar]
- 27.Lipsitz LA, Ryan SM, Parker JA, Freeman R, Wei JY, Goldberger AL. Hemodynamic and autonomic nervous system responses to mixed meal ingestion in healthy young and old subjects and dysautonomic patients with postprandial hypotension. Circulation 1993;87:391–400 [DOI] [PubMed] [Google Scholar]
- 28.Shannon JR, Diedrich A, Biaggioni I, et al. Water drinking as a treatment for orthostatic syndromes. Am J Med 2002;112:355–360 [DOI] [PubMed] [Google Scholar]
- 29.Berne RM, Winn HR, Rubio R. The local regulation of cerebral blood flow. Prog Cardiovasc Dis 1981;24:243–260 [DOI] [PubMed] [Google Scholar]
- 30.Aaslid R, Lindegaard KF, Sorteberg W, Nornes H. Cerebral autoregulation dynamics in humans. Stroke 1989;20:45–52 [DOI] [PubMed] [Google Scholar]
- 31.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research, Center for Devices and Radiological Health. Guidance for industry: enrichment strategies for clinical trials to support approval of human drugs and biological products. Draft guidance, December 2012. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM332181.pdf. Accessed December 6, 2013
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.