

Abstract
Virus-like particles are powerful platforms for the development of functional hybrid materials. Here, we have grown a cross-linked polymer (cross-linked aminoethyl methacrylate) within the confines of the bacteriophage P22 capsid (P22-xAEMA) and functionalized the polymer with various loadings of paramagnetic manganese(III) protoporphyrin IX (MnPP) complexes for evaluation as a macromolecular magnetic resonance imaging contrast agent. The resulting construct (P22-xAEMA-MnPP) has r1,particle = 7,098 mM−1 s−1 at 298 K and 2.1 T (90 MHz) for a loading of 3,646 MnPP molecules per capsid. The Solomon-Bloembergen-Morgan theory for paramagnetic relaxivity predicts conjugating MnPP to P22, a supramolecular structure, would result in an enhancement in ionic relaxivity; however, all loadings experienced low ionic relaxivities, r1,ionic, ranging from 1.45 to 3.66 mM−1 s−1, similar to the ionic relaxivity of free MnPP. We hypothesize that intermolecular interactions between neighboring MnPP molecules block access of water to the metal site, resulting in low r1,ionic relaxivities. We investigated the effect of MnPP interactions on relaxivity further by either blocking or exposing water binding sites on MnPP. On the basis of these results, future design strategies for enhanced r1,ionic relaxivity are suggested. The measured r2,ionic relaxivities demonstrated an inverse relationship between loading and relaxivity. This results in a loading-dependent r2/r1 behavior of these materials indicating synthetic control over the relaxivity properties, making them interesting alternatives to current magnetic resonance imaging contrast agents.
Keywords: Bacteriophage P22 virus-like particle, Magnetic resonance imaging contrast agent, T1-enhanced contrast agent, Manganese(III) protoporphyrin IX, Atom-transfer radical polymerization
Introduction
Paramagnetic metal ions, such as gadolinium, manganese, and iron, have been extensively studied as magnetic resonance imaging (MRI) contrast agents owing to their ability to efficiently relax water protons. Gadolinium-based small molecules are predominantly used in medical imaging protocols as T1-enhanced MRI contrast agents [1, 2], but free gadolinium(III) ions are highly toxic and can cause nephrogenic systemic fibrosis in patients with renal failure [3, 4]. Thus, there are considerable efforts to develop effective alternative contrast agents, and manganese is a promising candidate [5], with limited toxicity at low concentrations. High metal-ligand complex stability is an important factor in contrast agent development to avoid metal-ion exchange or release and can be provided by macrocycles such as porphyrins [6].
Over the years, a variety of porphyrins chelated with manganese(III) ions have been investigated for their potential use as MRI contrast agents [7-11]. Recently, Winter et al. successfully engineered a heme protein, heme nitric oxide/oxygen-binding protein, to replace its native heme with manganese(III) protoporphyrin IX (MnPP) complexes [11]. The engineered heme nitric oxide/oxygen-binding protein exhibited enhanced relaxivity (r1,ionic = 12.0 mM−1 s −1) at 60 MHz compared with the protein containing the native heme (r1,ionic = 5.9 mM−1 s−1) and commercially available manganese- and gadolinium-based contrast agents (r1,ionic = 3.0 mM−1 s−1 for both). Although engineering of a heme protein as a scaffold of MnPP is an elegant approach, the number of MnPP molecules per protein is limited to the number of heme binding sites, i.e., one per protein in their work. To progress toward clinical application, we aim to establish a well-designed molecular system that has the capability to encapsulate such agents with much higher loading density, protect them from the external environment, and deliver them to a targeted tissue.
One approach has been to use covalent attachment of small-molecule contrast agents to macromolecular platforms [12, 13]. Various groups have explored dendrimers [14], liposomes [15, 16], protein cages [17, 18], and gold nanoparticles [19] as potential platforms for conjugation of small-molecule contrast agents. Virus-like particles (VLPs) derived from virus particles but devoid of their nucleic acid have an advantage over other systems because of their large macromolecular size and control over assembly, which allows high loading density of desired cargo molecules. We, and others, have explored these systems, such as Cowpea chlorotic mottle virus [20], Tobacco mosaic virus [21], Cowpea mosaic virus [22], bacteriophages Qb [23], MS2 [24, 25], and P22 [26-28] as macromolecular platforms for contrast-agent development. Although this research has resulted in the successful development of VLP-based MRI contrast agents, most of them utilized only the capsid surface to anchor imaging probes, not taking full advantage of the large interior cavity of the capsids.
Recently, we have successfully exploited the interior cavity of the P22 viral capsid and developed gadoliniumbased MRI contrast agents with significantly high ionic (per Gd ion) and particle (per P22 capsid) relaxivities. In those studies, organic polymers were synthesized in the confined environment of the interior cavity of P22 VLPs either by a series of azide-alkyne “click” reactions [27] or by atom-transfer radical polymerization [28], and were used as a scaffold to conjugate imaging molecules via a reactive functional group on the polymer. This resulted in a significant increase in loading capacity of the cargo by taking full advantage of the capsid volume. Here, we explore the use of MnPP complexed with P22 VLP-crosslinked aminoethyl methacrylate (xAMEA) polymer hybrids (P22-xAMEA) as potential MRI contrast agents. MnPP was conjugated to P22-xAEMA via a coupling reaction between carboxyl groups of the porphyrin and pendant amine groups of P22-xAEMA (Fig. 1), resulting in a high per-particle relaxivity for efficient T1-enhanced MRI contrast-agent development. Furthermore, as the ionic relaxivities of these constructs were lower than expected, we investigated MnPP aggregation, which we hypothesize is due to intermolecular interactions between neighboring MnPP molecules that can potentially block access of water to the metal site, and discuss potential solutions.
Fig. 1.

a Manganese(III) protoporphyrin IX (MnPP) (1) was reacted with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)/N-hydroxysuccinimide (NHS) for 15 min in dimethyl sulfoxide (DMSO) to create an activated species (2). b The resulting product (2) was reacted with P22 virus-like particle-cross-linked aminoethyl methacrylate (xAEMA) polymer hybrids (P22-xAMEA) for 1 h in carbonate pH 9 buffer to couple MnPP to reactive amine groups on the P22-xAEMA polymer
Materials and methods
Materials
All materials were analytical grade and were purchased from either Sigma-Aldrich or Fisher Scientific, and were used as received unless otherwise noted. All water was deionized by use of a NANOpure water purification system. All protein samples that had been chemically modified were analyzed by UV-vis spectroscopy (model 8453 UVvis spectrophotometer, Agilent Technologies, Santa Clara, CA, USA) and dynamic light-scattering (90Plus particle size analyzer, Brookhaven Instruments, Holtsville, NY, USA). The synthesis of 2-bromoisobutyryl aminoethyl maleimide was reported in [28]. Manganese(III) protoporphyrin IX chloride was purchased from Frontier Scientific.
Mutagenesis, protein purification, and P22-xAEMA formation
The P22 (S39C) point mutation was made with established polymerase chain reaction protocols (Agilent Technologies) with pET-11a-based plasmids encoding genes for scaffolding and coat protein. The amplified DNA was transformed into Escherichia coli strain BL21 (DE3) and selected for ampicillin resistance [28]. P22 (S39C) procapsid, made up of 420 subunits, was produced by a heterologous expression system in E. coli. and purified by a previously described procedure [27]. The resulting virus pellet, after purification, was resuspended in 100 mM sodium phosphate, 50 mM NaCl, pH 7.0, and spun at 17,000 rpm for 20 min to remove particulates and lipid. The procapsid was heat-treated for 20 min at 65 °C to transform the protein into its expanded form as previously described and analyzed [29]. The samples of the expanded form were purified by pelleting via centrifugation at 45,000 rpm for 50 min in an ultracentrifuge (Sorvall), followed by resuspension in 100 mM sodium phosphate, 50 mM NaCl, pH 7.6 in preparation for protein labeling. The expanded form was labeled with an atom-transfer radical polymerization initiator molecule and xAEMA was synthesized inside the P22 VLP cavity, as previously described [28]. P22 procapsid, expanded, and polymerized (P22-xAEMA) samples were characterized by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transmission electron microscopy (Leo 912 AB), and dynamic light scattering (90Plus particle size analyzer, Brookhaven Instruments, Holtsville, NY, USA) [28]. The protein concentration was determined from the absorbance at 280 nm, where an extinction coefficient of 65,920 M−1 cm−1 (P22 subunit) was used. As described above, the expanded form of P22 was exclusively used for P22-xAEMA formation, and hereafter, we will refer to the unpolymerized expanded form of P22 as P22 and polymerized P22 as P22-xAEMA. All further labeling experiments with P22 and P22-xAEMA and sample analyses were conducted in sodium bicarbonate buffer (100 mM sodium bicarbonate, 50 mM NaCl, pH 9.0) because MnPP is more soluble in this buffer than in phosphate buffer at neutral pH.
MnPP labeling conditions: without EDC/NHS
P22 and P22-xAEMA were labeled with MnPP without the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS). First, a 30 mM stock solution of MnPP was made up in dimethyl sulfoxide and vortexed until it the MnPP had completely dissolved. From the 30 mM MnPP stock solution, 72 lL (1.40 mg, 2.14 lmol) MnPP was diluted with 144 lL water. Next, to 284 lL P22 or P22-xAEMA (0.0214 lmol subunit, 3.5 mg/mL, 100 mM sodium carbonate buffer, 50 mM NaCl, pH 9.0), 216 lL of diluted MnPP was added dropwise (100 MnPP molecules per P22 subunit, i.e. 42,000 MnPP molecules per P22 capsid) while the protein solution was being vortexed. The mixture was allowed to react for 1 h at room temperature, followed by purification away from excess MnPP/EDC/NHS with a Micro Bio-Spin column (Bio-Rad, Hercules, CA, USA) twice, resulting in 90 MnPP molecules per capsid (67.7 % yield) and 184 MnPP molecules per capsid (71.8 % yield), respectively.
MnPP labeling conditions: with EDC/NHS
Various loadings of MnPP molecules per P22-xAEMA (121-3,646 MnPP molecules per capsid) were achieved by using a molar excess ranging from one to 100 MnPP molecules per P22 subunit. The preparation of 100 molar excess of MnPP per P22 subunit was conducted as follows. To prepare P22-MnPP and P22-xAEMA-MnPP with EDC/NHS, a 1:1:1 molar ratio of MnPP, EDC, and NHS was created by adding 72 lL (30 mM in dimethyl sulfoxide, 1.40 mg, 2.14 lmol) MnPP to a solution of 72 lL EDC (30 mM in water, 0.25 mg, 2.14 lmol) and 72 lL NHS (30 mM in water, 0.41 mg, 2.14 lmol), and the mixture was stirred for 15 min at room temperature to activate carboxyl groups on MnPP. To 284 lL P22 or P22-xAEMA (0.0214 lmol subunit, 3.5 mg/mL, 100 mM sodium carbonate buffer, 50 mM NaCl, pH 9.0), 216 lL of the MnPP/EDC/NHS mixture was added dropwise while the protein solution was being vortexed. The mixture was allowed to react for 1 h at room temperature, followed by purification away from excess MnPP/EDC/NHS by use of a spin column twice, resulting in 780 MnPP molecules per capsid (82.0 % yield) and 1,200 MnPP molecules per capsid (92.3 % yield), respectively. The amount of labeling varies when the experiment is repeated at same molar excess (i.e., for molar excess of 100 MnPP molecules per P22 subunit, the labeling ranges from 1,200 to 3,646 MnPP molecules per capsid). All NMR data are based on the output, which is the labeling amount of MnPP molecules per capsid.
Sample analysis
P22 and P22-xAEMA samples and those with MnPP were analyzed by a variety of methods aimed at interrogating the purity, morphology, and composition of the samples. P22, P22-xAEMA, and P22-xAEMA-MnPP were imaged by transmission electron microscopy (Leo 912 AB, 100 kV) by negatively staining the sample with 1 % uranyl acetate on Formvar carbon-coated grids. These samples were also analyzed by SDS-PAGE on 10-20 % gradient tris(hydroxymethyl)aminomethane-glycine gels (Lonza). Protein was detected by staining with Coomassie blue.
Protein and manganese concentration
The protein concentrations of P22 and P22-xAEMA samples were determined from the absorbance at 280 nm (extinction coefficient 65,490 M−1 cm−1, P22 subunit). The protein concentration of the P22-xAEMA-MnPP samples was determined from the absorbance at 280 nm after the contribution from MnPP (extinction coefficient 19,820 M−1 cm−1) had been subtracted. The amount of MnPP conjugated with P22 VLPs was determined using an extinction coefficient of 25 mM−1 cm−1 at 462 nm [30].
Relaxivity measurements
T1 and T2 measurements were conducted with a broadband 19-MHz (0.447 T) vertical NMR spectroscopy system, Anasazi Fourter transform NMR 90-MHz (2.1 T) and Bruker DPX 300-MHz (7.0 T) spectrometers. Each P22-xAEMA-MnPP sample was diluted with 67 % D2O in H2O to yield a dilution series. The longitudinal relaxation rate constant (T1) was measured at each dilution by use of an inversion recovery pulse sequence and the transverse relaxation rate constant (T2) was measured by use of a Carr-Purcell-Meiboom-Gill sequence at 298 K, where the relaxation delay was set to six times the estimated T1 or T2. For 19 MHz (0.447 T), recovery times of 30 s were used for T1 relaxation times. Figure S1 shows a plot of 1/T1 versus manganese concentration (mM) for the three dilutions which was used to determine the relaxivity values (r1,ionic), where the ionic relaxivity (relaxivity per manganese ion) is equal to the slope of the line, and the particle relaxivity (relaxivity per P22) was calculated by multiplying the slope by the number of manganese ions per capsid. Similarly, a plot of 1/T2 versus manganese concentration (mM) was used to determine r2,ionic relaxivity values.
Imidazole titration experiments to block access of water to MnPP
To assess the influence of water accessibility on r1,ionic relaxivity of MnPP, we titrated free MnPP with imidazole. Imidazole was dissolved in carbonate buffer (100 mM sodium carbonate, 50 mM NaCl, pH 9.0) to create a stock solution of 10 M, which was added to 640 lM free MnPP in molar equivalents of 0, 10, 100, 500, or 1,000 excess to give a final manganese concentration of 390 lM. Samples were further characterized by UV-vis spectroscopy with 409 dilution. T1 relaxivity of the samples was measured as described in “Relaxivity measurements.”
Addition of acetone to disrupt intermolecular interactions of free MnPP
To evaluate the potential effect of MnPP aggregation within the P22 capsid on r1,ionic relaxivity, we compared free MnPP in solution and MnPP conjugated to P22-xA-EMA at the same concentration. The concentration of free MnPP was based on the local concentration of MnPP in the P22-xAEMA capsid, i.e., the volume of the spherical 64 nm P22 and number of manganese ions encapsulated as the P22-xAEMA-MnPP construct. Free MnPP was prepared in different concentrations, from 1.46 to 39.17 mM MnPP for P22-xAEMA-MnPP at loadings ranging from 121 to 3,646 MnPP molecules per capsid (100 mM sodium carbonate, 50 mM NaCl, pH 9.0). In addition, to evaluate the effect of disrupting interparticle interactions between MnPP molecules, we treated free MnPP molecules with two different concentrations of acetone, and measured r1,ionic relaxivity. At all concentrations of MnPP, deuterated acetone was added to achieve a final matrix of 60 % acetone-D6, 33.33 % H2O, and 6.67 % D2O or 20 % acetone-D6, 33.33 % H2O, and 46.67 % D2O. For 60 % acetone-D6, 252 lL acetone-D6 (99.5 % deuterated) was added to 140 lL MnPP and 28 lL D2O. For 20 % acetone-D6, 84 lL acetone-D6 (99.5 % deuterated) was added to 140 lL MnPP and 196 lL D2O. This solution was further diluted with the corresponding matrix to yield a dilution series of 1:3, 1:9, and 1:27. T1 relaxivity was measured as described in “Relaxivity measurements.”
Results and discussion
Synthesis
MnPP was attached to P22-xAEMA with various loadings by means of a simple coupling reaction [31]. The carboxylate groups of MnPP were activated with EDC/NHS and reacted with the amine functional groups of P22-xAEMA, encapsulated on the interior of the P22 capsid (Fig. 1). MnPP has two carboxyl groups and both could be coupled with the amine groups of P22-xAEMA. It is difficult to differentiate between the number of singly labeled carboxyl groups and the number of doubly labeled carboxyl group, and so, to minimize activation of both carboxyl groups on MnPP, a 1:1 molar ratio of EDC and NHS was added per MnPP molecule. To investigate conjugation of MnPP to the surface of the P22 capsid, the reaction was also conducted on P22. After removal of excess MnPP and the coupling agents, the concentration of protein and attached MnPP of the resulting materials (P22-xAEMA-MnPP or P22-MnPP) was determined by UV-vis spectroscopy using known molar absorbtivities.
To determine the extent of noncovalent interactions between MnPP and the polymer and/or P22, both P22 and P22-xAEMA were incubated with MnPP (100 MnPP molecules per P22 subunit) in the absence of EDC and NHS. Successful attachment of MnPP via EDC/NHS coupling was clear from the visible characteristics of the samples (Fig. 2a), and was confirmed spectroscopically by comparison with the noncovalent interaction controls (Fig. 2b, c). After purification by means of a spin column, the P22-xAEMA-MnPP sample exhibited a UV-vis spectrum (Fig. 2c) in which the expected peaks for MnPP at 365, 472, and 562 nm [11] were clearly visible, confirming the formation of P22-xAEMA-MnPP.
Fig. 2.

a Photographs of each sample: 1 P22-MnPP without EDC, 2 P22-MnPP with EDC, 3 P22-xAEMA-MnPP without EDC, and 4 P22-xAEMA-MnPP with EDC (1,200 MnPP molecules per capsid). All four samples were incubated with 100 MnPP molecules per P22 subunit. Purification was performed using spin columns (twice) to remove unreacted MnPP from P22 and P22-xAEMA treated without (no color) and with (reddish color) EDC/NHS. UV-vis spectra of b P22-MnPP with (red line) and without (blue line) EDC and c P22-xAEMA-MnPP with (red line) and without (blue line) EDC. The peaks expected for MnPP (green line) appear at 365, 462, and 562 nm for both P22-MnPP and P22-xAEMA-MnPP with EDC/NHS (red), whereas the peaks are less prevalent for controls without EDC/NHS (blue), and are not present for P22 (black line)
Characterization
The MnPP loading per P22 capsid was determined for each of the constructs from their UV-vis spectra. As shown in Table S1, the MnPP loading per capsid in P22-xAEMA-MnPP ranged from 121 MnPP molecules per capsid to 3,646 MnPP molecules per capsid and was tunable on the basis of the molar excess of MnPP added per capsid during the reaction. P22-MnPP resulted in 778 MnPP molecules per capsid, the result of labeling endogenous lysines in the P22 capsid. There was some evidence of noncovalent interactions between MnPP and P22, with loadings of 155 MnPP molecules per capsid and 90 MnPP molecules per capsid for P22-xAEMA-MnPP without EDC and P22-MnPP without EDC, respectively. All of the following characterization was done on a loading of 1,200 MnPP molecules per capsid as a representative sample of P22-xAEMA-MnPP.
To confirm that the reaction with MnPP/EDC/NHS did not alter the morphology of P22 via interparticle cross-linking after EDC activation or aggregation, the size of P22-xA-EMA-MnPP was characterized. The hydrodynamic diameter, as measured by dynamic light scattering, remained essentially unchanged for the covalently linked MnPP samples (Fig. 3a-c). Particle diameters of 68 ± 6 nm (P22-xAEMA) and 64 ± 7 nm (P22-xAEMA-MnPP) were observed as compared with 68 ± 4 nm for P22, suggesting there was no interparticle cross-linking mediated by the EDC reaction. When the particles were visualized by transmission electron microscopy, the morphology of P22 appeared unchanged before and after the MnPP conjugation reaction (Fig. 3d-f), further confirming that the cage-like structure of the P22 VLPs was unaffected by the coupling reaction. Analysis by SDS-PAGE (Fig. 4) showed no appearance of new bands before (Fig. 4, lane 3) and after (Fig. 4, lane 4) modification of P22-xAEMA with MnPP. The streaking and appearance of higher molecular weight bands for P22-xAEMA (Fig. 4, lane 3) compared with P22 (Fig. 4, lane 2) is expected and indicative of the formation of intraparticle subunit-subunit coupling due topolymer cross-linking onthe interiorof the capsid. Together these data suggest that the overall P22 morphology was unaffected after conjugation with MnPP.
Fig. 3.

Average diameters determined by dynamic light-scattering for P22 (a), P22-xAEMA (b), and P22-xAEMA-MnPP (1,200 MnPP molecules per capsid) (c). The P22-xAEMA-MnPP construct retains its size, indicative of a monodisperse population after conjugation of MnPP. The transmission electron microscopy images of P22 (d), P22-xAEMA (e), and P22-xAEMA-MnPP (1,200 MnPP molecules per capsid) (f) show that P22 maintains its overall morphology after conjugation to MnPP
Fig. 4.
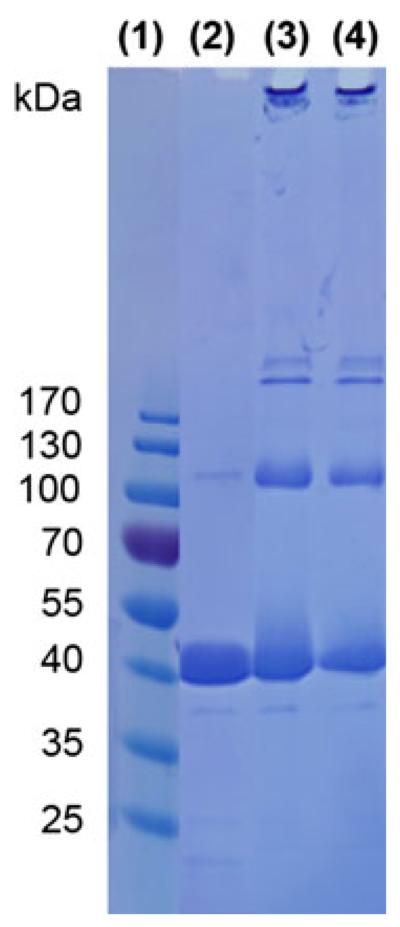
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel with marker (1), P22 (2), P22-xAEMA (3), and P22-xAEMA-MnPP (1,200 MnPP molecules per capsid) (4)
NMR and T1 and T2 relaxivity
We investigated T1 relaxivity as a function of MnPP loading on a per manganese ion basis (r1,ionic) and per P22 capsid basis (r1,particle), over a range of loadings from 90 to 3,646 MnPP molecules per capsid at 2.1 T (90 MHz). The loading factor appeared to have almost no impact on r1,ionic relaxivity, which remained below 5 mM−1 s−1 across the range of loadings (Fig. 5a). However, r1,particle increased with the loading factor, and the high loading capacity compensates for lower ionic relaxivities, with the highest observed r1,particle being 7,100 mM−1 s−1 for this system (Fig. 5b).
Fig. 5.

Ionic (a) and particle (b) relaxivities r1 (blue circles) and r2 (red triangles) of P22-xAEMA-MnPP at different loadings of MnPP, ranging from 121 to 3,646 MnPP molecules per capsid, with P22-xAEMA-MnPP without EDC (155 MnPP molecules per capsid) represented in pink, P22-MnPP with EDC (778 MnPP molecules per capsid) represented in orange, and P22-MnPP without EDC (90 MnPP molecules per capsid) represented in green. Loading has no impact on r1,ionic, which remains below 5 mM−1 s−1, whereas r2,ionic dramatically decreases as the loading increases
The r2,ionic relaxivity was also evaluated for this system and was observed to have an inverse relationship with the MnPP loading factor (Fig. 5a). The r2,ionic relaxivity decreased significantly with increasing MnPP loading factor at lower loading and gradually leveled off at higher loading. A similar phenomenon was reported in an investigation of r2,ionic relaxivity at the initial stages of iron uptake in ferritin [32]. In that article, it was proposed that r2,ionic relaxivity decreases with increasing iron loading factor owing to the formation of antiferromagnetically coupled clusters. Similarly, in our P22 system there may be more MnPP clusters at higher loading factors that can couple antiferromagnetically and which decrease the overall r2,ionic relaxivity of the sample. The negative impact of antiferromagnetic coupling could also affect r1,ionic relaxivity [33], but we did not see a corresponding decrease in r1,ionic relaxivity across the MnPP loading range we examined. It could be interesting to explore this phenomenon further in the future.
At lower loadings, the sample exhibits high r2/r1 (see Table S1), suggesting that this system could potentially be used as a T2-enhanced contrast agent. However, at higher loadings, r2/r1 approaches 1, thus shifting toward a T1-enhanced contrast agent. Therefore, P22-xAEMA loaded with MnPP can be tuned to be either a T1 or a T2 contrast agent by controlling the loading factor.
On the basis of the Solomon-Bloembergen-Morgan analytical model for understanding paramagnetic relaxivity [34-39], we expected that the r1,ionic relaxivity would be enhanced after conjugation of MnPP to the macromolecular P22 capsid [27, 28, 40]. But contrary to expectations, we did not observe an enhancement. The Solomon-Bloembergen-Morgan model predicts that the relaxivity is dominated by three important parameters: the number of metal-bound water molecules (q), the mean residence lifetime for metal-bound water (sM), and the rotational correlation time (sR). We, and others, have effectively used this model to predict and design new contrast agents that optimize these important parameters [17, 27, 41]. Both sR and sM of free MnPP (sR = 50-80 ps, sM = 10 ns) [42] are comparable to those of free gadolinium diethylene-triaminepentaacetic acid (sR = 70 ps, sM = 16 ns) [27]. We have shown previously that conjugating a gadolinium diethylenetriaminepentaacetic acid to a similar P22-polymer system does not alter sM of the small-molecule contrast agent [27] significantly, but does dramatically alter sR. Thus, we assumed the same sR for the P22-xAEMA-MnPP system and assumed that sM would be similar to that of free MnPP. The nuclear magnetic relaxation dispersion profile for MnPP conjugated to a large particle, such as the P22-xAEMA particle (sR around 10 ns) [17, 27], is predicted to have enhanced relaxivity at low field (20 MHz) [36]. Thus, the ionic relaxivity of P22-xAEMA-MnPP (see Table S1) was measured at 0.45, 2.1, and 7 T (19, 90, and 300 MHz) (Fig. 6) with the expectation that we would see a relaxivity enhancement, particularly at 0.45 T (19 MHz). For comparison, the relaxivity of free MnPP at 14.5 mM was also measured. The concentration of free MnPP was based on the local concentration of MnPP in the P22-xAEMA capsid, i.e., the volume of the spherical 64 nm P22 and number of manganese ions encapsulated as the P22-xA-EMA-MnPP construct (1,200 MnPP molecules per capsid). The results show that the relaxivity changes only slightly across field strengths, and not as significantly as expected on the basis of the nuclear magnetic relaxation dispersion profiles of other macromolecular manganese complexes [36, 43] and our previous work with macro-molecular gadolimium-based agents [27].
Fig. 6.

Ionic relaxivities r1 (blue circles) and r2 (red triangles) of P22-xAEMA-MnPP at a loading of 1,200 MnPP molecules per capsid (closed symbols) and free MnPP (open symbols) at 14.5 mM (the same local concentration as its P22-xAEMA-MnPP counterpart, see Table S1) at three different field strengths of 0.45, 2.1, and 7 T. The trend shows relatively small changes in relaxivity across field strengths
To understand why no increase in r1,ionic relaxivity was observed for MnPP after conjugation to P22 VLPs as compared with free MnPP, we explored the interactions between MnPP molecules further. The presence of intermolecular MnPP interactions could structurally block access of water to the manganese ion, resulting in a lower effective q and thus a diminished r1,ionic relaxivity [42]. This clustering of MnPP molecules could account for the observed lack of r1,ionic relaxivity enhancement for P22-xAEMA-MnPP when compared with free MnPP. We conducted the two experiments described in the following sections to explore this possibility.
Imidazole titration experiments
To probe the effects of access of water to the manganese ion on the relaxivity, we titrated free MnPP with imidazole, which is expected to bind the axial sites in MnPP and compete with water access. This competitive binding experiment was monitored by both changes in the optical spectrum and changes in the ionic relaxivity. Figure 7a and Fig. S2 show the changes in the UV-vis spectrum of MnPP on titration with imidazole [44]. At 640 lM MnPP, imidazole was added in a range up to 1,000 molar excess with regard to MnPP. The UV-vis spectrum shows an emerging peak at 482 nm, the intensity of which increased with increasing imidazole concentration, clearly indicating interactions between the imidazole and the MnPP complex (Fig. S2). We further investigated the effects of imidazole binding to MnPP by measuring the 1H relaxivity at 2.1 T (90 MHz) at a range of added imidazole concentrations. We found an inverse relationship between the change in absorbance at 482 nm (imidazole binding to MnPP) and the measured relaxivity (Fig. 7b). This is consistent with imidazole binding to both axial sites on MnPP at high imidazole concentrations, where these sites are essentially saturated and the effective value of q approaches 0. Thus, imidazole appears to influence intermolecular MnPP interactions and blocks water access, which has a clear, dominant influence on the relaxivity.
Fig. 7.

UV-vis absorbance at 482 nm (a) and r1,ionic relaxivity at 2.1 T (90 MHz) (b). Measurements were taken at 0, 10, 100, 500, and 1,000 equiv of added imidazole to 640 lM MnPP. There is an inverse relationship between the increase in absorbance at 482 nm due to imidazole binding to MnPP and the decrease in relaxivity. Even at 10 equiv of imidazole, a significant difference in relaxivity was observed
Acetone-D6
In an attempt to disrupt the intermolecular interactions between neighboring MnPP molecules and potentially increase water access and therefore the number of bound and exchangeable water molecules (q), we investigated the effects of adding acetone on the r1,ionic relaxivity of free MnPP at concentrations based on the same local concentration of MnPP as in our P22-xAEMA-MnPP system. First, r1,ionic relaxivities of MnPP and P22-xAEMA-MnPP were investigated at 2.1 T (90 MHz). MnPP and P22-xAEMA-MnPP exhibited similar r1,ionic values at all concentrations, with free MnPP having slightly smaller r1,ionic values than P22-xAEMA-MnPP (Fig. 8). Second, r1,ionic relaxivity of MnPP in the presence of 20 or 60 % acetone-D6 was measured. These experiments were conducted only as a proof of concept, as acetone is neither a suitable solvent to achieve protein stability nor a suitable solvent for in vivo applications. The free MnPP was treated with acetone-D6 to disperse the MnPP molecules and observe the impact on relaxivity (Fig. 8). After free MnPP had been treated with 20 % acetone-D6, the ionic relaxivities did not show a significant change, with only a slight increase in relaxivity at the lower MnPP concentrations. However, with 60 % acetone-D6, a dramatic increase in ionic relaxivity was observed at all concentrations, with a trend toward the highest increase in relaxivity for the lowest concentration of MnPP. These data correlate well with published data [42] and indicate that the acetone disrupts intermolecular MnPP interactions, resulting in greater water exchange at the manganese centers and an increase in the effective value of q. Extrapolation from these data suggestsd that the high local concentrations of MnPP inside P22-xAEMA-MnPP probably results in strong intermolecular interactions between neighboring MnPP molecules, resulting in a lower observed ionic relaxivity compared with the ionic relaxivity when the interactions are disrupted, as in the case of free MnPP in the presence of acetone. This result implies that we could increase the ionic relaxivity of MnPP by disrupting the intermolecular interactions between MnPP molecules. For example, modification of the porphyrin moiety, in a manner analogous to the designed picket-fence porphyrins [45], could minimize MnPP interactions.
Fig. 8.

Ionic relaxivities at different concentrations of free MnPP compared with P22-xAEMA-MnPP in carbonate pH 9 buffer (see Table S1 for the corresponding loading factors). The free MnPP was further treated with either 20 or 60 % acetone-D6 to try to disperse the MnPP molecules and see the impact of acetone on the relaxivity. MnPP (green bars) at the same local concentrations of MnPP as for P22-xAEMA-MnPP (blue) exhibits similar r1,ionic values, with those for free MnPP being slightly smaller than those for P22-xAEMA-MnPP. After free MnPP had been treated with 20 % acetone-D6 (red bars), the ionic relaxivities did not change much, increasing only slightly for the lower concentrations. At 60 % acetone-D6 (orange bars), a dramatic increase in ionic relaxivity was observed at all concentrations, with a trend toward the highest increase in relaxivity for the lowest concentration of MnPP
Conclusion
In summary, we have covalently encapsulated a large number of MnPP molecules inside P22 VLP-polymer hybrids and demonstrated potential for this construct as a tunable MRI contrast agent. This system could be a good alternative to gadolinium-based contrast agents, which are known to be linked to nephrogenic systemic fibrosis. We can conjugate more MnPP molecules to P22-xAEMA versus P22 alone using EDC and NHS to chemically introduce MnPP to the P22 capsid up to 3,646 MnPP molecules per capsid. The improvement in labeling is important for the delivery of contrast agents on a per-particle basis, allowing high concentration delivery of the contrast agent. Although we do not see an improvement in r1,ionic relaxivity, the high per-particle relaxivity of P22-xAEMA-MnPP, resulting from the high loading density, could be beneficial when these particles are imparted with cell-targeting capabilities. Our results suggest that we could also increase the r1,ionic relaxivity of MnPP by disrupting the intermolecular interactions between MnPP molecules. In future design iterations of these hybrid materials, we might overcome the intermolecular interactions of MnPP molecules by altering the porphyrin moiety to minimize MnPP interactions [45]. Alternatively, incorporation of other manganese porphyrins which have larger r1,ionic relaxivity, such as manganese(III) tetrakis(4-sulfonatophenyl)porphine [42, 46, 47], may enable us to utilize more fully the advantages of the P22 VLP-polymer platform.
Supplementary Material
Acknowledgments
This research was supported in part by grants from the National Institutes of Health (R01-EB012027) and the American Heart Association (13SDG16970075).
Abbreviations
- EDC
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide
- MnPP
Manganese(III) protoporphyrin IX
- MRI
Magnetic resonance imaging
- NHS
N-Hydroxysuccinimide
- P22-xAMEA
P22 virus-like particle-cross-linked aminoethyl methacrylate polymer hybrid
- VLP
Virus-like particle
- SDS-PAGE
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- xAEMA
Cross-linked aminoethyl methacrylate polymer
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00775-013-1075-4) contains supplementary material, which is available to authorized users.
Contributor Information
Shefah Qazi, Chemistry and Biochemistry Department, Montana State University, Bozeman, MT 59717, USA.
Masaki Uchida, Chemistry and Biochemistry Department, Montana State University, Bozeman, MT 59717, USA.
Robert Usselman, National Institute of Standards and Technology, Boulder, CO 80305, USA.
Riley Shearer, Chemistry and Biochemistry Department, Montana State University, Bozeman, MT 59717, USA.
Ethan Edwards, Chemistry and Biochemistry Department, Montana State University, Bozeman, MT 59717, USA.
Trevor Douglas, Chemistry and Biochemistry Department, Montana State University, Bozeman, MT 59717, USA; Department of Chemistry, Indiana University, 800 East Kirkwood Ave., Bloomington, IN 47405, USA.
References
- 1.Caravan P, Ellison J, McMurry T, Lauffer R. Chem Rev. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- 2.Werner EJ, Datta A, Jocher CJ, Raymond KN. Angew Chem Int Ed. 2008;47:8568–8580. doi: 10.1002/anie.200800212. [DOI] [PubMed] [Google Scholar]
- 3.Broome DR. Eur J Radiol. 2008;66:230–234. doi: 10.1016/j.ejrad.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 4.Lin SP, Brown JJ. J Magn Reson Imaging. 2007;25:884–899. doi: 10.1002/jmri.20955. [DOI] [PubMed] [Google Scholar]
- 5.Pan D, Caruthers SD, Senpan A, Schmieder AH, Wickline SA, Lanza GM. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2010;3:162–173. [Google Scholar]
- 6.Falk JE. Porphyrins and metalloporphyrins. Elsevier; Amsterdam: 1975. pp. 15–18. [Google Scholar]
- 7.Chen CW, Cohen JS, Myers CE, Sohn M. FEBS Lett. 1984;168:70–74. doi: 10.1016/0014-5793(84)80208-2. [DOI] [PubMed] [Google Scholar]
- 8.Fiel RJ, Musser DA, Mark EH, Mazurchuk R, Alletto JJ. Magn Reson Imaging. 1990;8:255–259. doi: 10.1016/0730-725x(90)90097-l. [DOI] [PubMed] [Google Scholar]
- 9.Nasu H, Takehara Y, Isogai S, Kodaira N, Takeda H, Saga T, Nakajima S, Sakata I, Sakahara H. J Magn Reson Imaging. 2004;20:294–299. doi: 10.1002/jmri.20112. [DOI] [PubMed] [Google Scholar]
- 10.Nelson JA, Schmiedl U. Magn Reson Med. 1991;22:366–371. doi: 10.1002/mrm.1910220243. [DOI] [PubMed] [Google Scholar]
- 11.Winter MB, Klemm PJ, Phillips-Piro CM, Raymond KN, Marletta MA. Inorg Chem. 2013;52:2277–2279. doi: 10.1021/ic302685h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caravan P. Chem Soc Rev. 2006;35:512–523. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]
- 13.Cormode DP, Jarzyna PA, Mulder WJM, Fayad ZA. Adv Drug Deliv Rev. 2010;62:329–338. doi: 10.1016/j.addr.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bumb A, Brechbiel M, Choyke P. Acta Radiol. 2010;51:751–767. doi: 10.3109/02841851.2010.491091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamaly N, Miller A. Int J Mol Sci. 2010;11:1759–1776. doi: 10.3390/ijms11041759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mulder W, Strijkers G, van Tilborg G, Griffioen A, Nicolay K. NMR Biomed. 2006;19:142–164. doi: 10.1002/nbm.1011. [DOI] [PubMed] [Google Scholar]
- 17.Liepold L, Abedin M, Buckhouse E, Frank J, Young M, Douglas T. Nano Lett. 2009;9:4520–4526. doi: 10.1021/nl902884p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abedin M, Liepold L, Suci P, Young M, Douglas T. J Am Chem Soc. 2009;131:4346–4354. doi: 10.1021/ja8079862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferreira MF, Mousavi B, Ferreira PM, Martins CIO, Helm L, Martins JA, Geraldes CFGC. Dalton Trans. 2012;41:5472–5475. doi: 10.1039/c2dt30388d. [DOI] [PubMed] [Google Scholar]
- 20.Liepold L, Anderson S, Willits D, Oltrogge L, Frank J, Douglas T, Young M. Magn Reson Med. 2007;58:871–879. doi: 10.1002/mrm.21307. [DOI] [PubMed] [Google Scholar]
- 21.Bruckman MA, Hern S, Jiang K, Flask CA, Yu X, Steinmetz NF. J Mater Chem B. 2013;1:1482–1490. doi: 10.1039/C3TB00461A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prasuhn D, Yeh R, Obenaus A, Manchester M, Finn MG. Chem Commun. 2007:1269–1271. doi: 10.1039/b615084e. [DOI] [PubMed] [Google Scholar]
- 23.Pokorski J, Breitenkamp K, Liepold L, Qazi S, Finn MG. J Am Chem Soc. 2011;133:9242–9245. doi: 10.1021/ja203286n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson E, Isaacman S, Peabody D, Wang E, Canary J, Kirshenbaum K. Nano Lett. 2006;6:1160–1164. doi: 10.1021/nl060378g. [DOI] [PubMed] [Google Scholar]
- 25.Hooker J, Datta A, Botta M, Raymond K, Francis M. Nano Lett. 2007;7:2207–2210. doi: 10.1021/nl070512c. [DOI] [PubMed] [Google Scholar]
- 26.Min J, Jung H, Shin H-H, Cho G, Cho H, Kang S. Biomacromolecules. 2013;14:2332–2339. doi: 10.1021/bm400461j. [DOI] [PubMed] [Google Scholar]
- 27.Qazi S, Liepold LO, Abedin MJ, Johnson B, Prevelige P, Frank JA, Douglas T. Mol Pharm. 2013;10:11–17. doi: 10.1021/mp300208g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lucon J, Qazi S, Uchida M, Bedwell GJ, LaFrance B, Prevelige PE, Jr, Douglas T. Nat Chem. 2012;4:781–788. doi: 10.1038/nchem.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teschke CM, McGough A, Thuman-Commike PA. Biophys J. 2003;84:2585–2592. doi: 10.1016/S0006-3495(03)75063-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yonetani T, Asakura T. J Biol Chem. 1968;243:3996–3998. [PubMed] [Google Scholar]
- 31.Hermanson GT. Bioconjugate techniques. 2nd edn. Elsevier; Rockford: 2008. [Google Scholar]
- 32.Herynek V, Bulte JWM, Douglas T, Brooks RA. J Biol Inorg Chem. 2000;5:51–56. doi: 10.1007/s007750050007. [DOI] [PubMed] [Google Scholar]
- 33.Koenig SH, Brown RD, Gibson JF, Ward RJ, Peters TJ. Magn Reson Med. 1986;3:755–767. doi: 10.1002/mrm.1910030511. [DOI] [PubMed] [Google Scholar]
- 34.Schaefle N, Sharp R. J Phys Chem A. 2005;109:3267–3275. doi: 10.1021/jp046227o. [DOI] [PubMed] [Google Scholar]
- 35.Sharp R, Abernathy SM, Lohr LL. J Chem Phys. 1997;107:7620–7629. [Google Scholar]
- 36.Lauffer RB. Chem Rev. 1987;87:901–927. [Google Scholar]
- 37.Helm L, Merbach AE. Chem Rev. 2005;105:1923–1960. doi: 10.1021/cr030726o. [DOI] [PubMed] [Google Scholar]
- 38.Helm L. Prog NMR Spectrosc. 2006;49:45–64. [Google Scholar]
- 39.Helm L, Nicolle G, Merbach AE. Adv Inorg Chem. 2005;57:327–379. [Google Scholar]
- 40.Caravan P, Farrar CT, Frullano L, Uppal R. Contrast Media Mol Imaging. 2009;4:89–100. doi: 10.1002/cmmi.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Datta A, Hooker J, Botta M, Francis M, Aime S, Raymond K. J Am Chem Soc. 2008;130:2546–2552. doi: 10.1021/ja0765363. [DOI] [PubMed] [Google Scholar]
- 42.Kellar KE, Foster N. Inorg Chem. 1992;31:1353–1359. [Google Scholar]
- 43.Fanali G, Fesce R, Agrati C, Ascenzi P, Fasano M. FEBS J. 2005;272:4672–4683. doi: 10.1111/j.1742-4658.2005.04883.x. [DOI] [PubMed] [Google Scholar]
- 44.Neya S, Morishima I, Yonezawa T. Biochemistry. 1981;20:2610–2614. doi: 10.1021/bi00512a037. [DOI] [PubMed] [Google Scholar]
- 45.Collman JP, Gagne RR, Reed C, Halbert TR, Lang G, Robinson WT. J Am Chem Soc. 1975;97:1427–1439. doi: 10.1021/ja00839a026. [DOI] [PubMed] [Google Scholar]
- 46.McMillan JH, Cox GG, Kimler BF, Spicer JS, Batnitzky S. Magn Reson Imaging. 1991;9:553–558. doi: 10.1016/0730-725x(91)90042-k. [DOI] [PubMed] [Google Scholar]
- 47.Sur SK, Bryant RG. J Phys Chem. 1995;99:4900–4905. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.