

Abstract
Substantial evidence suggests that the progressive loss of cardiomyocytes caused by apoptosis significantly contributes to the development of heart failure. β-Adrenergic receptor activation and subsequent persistent phosphodiesterase 3A (PDE3A) downregulation and concomitant inducible cAMP early repressor (ICER) upregulation (PDE3A/ICER feedback loop) has been proposed to play a key role in the pathogenesis of cardiomyocyte apoptosis. In contrast, insulin-like growth factor-1 can activate cell survival pathways, providing protection against cell death and restoring muscle function. In this study, we found that insulin-like growth factor-1 activates extracellular signal-regulated kinase 5 (ERK5) and inhibits PDE3A/ICER feedback loop. Insulin-like growth factor-1 normalized isoproterenol-mediated PDE3A downregulation and ICER upregulation via ERK5/MEF2 activation, and also inhibited isoproterenol-induced myocyte apoptosis. To determine the physiological relevance of ERK5 activation in regulating PDE3A/ICER feedback loop, we investigated the PDE3A/ICER expression and cardiomyocyte apoptosis in transgenic mice with cardiac specific expression of a constitutively active form of mitogen-activated protein (MAP)/extracellular signal-regulated protein kinase (ERK) kinase 5α (MEK5α) (CA-MEK5α-Tg). In wild-type mice, pressure overload– or doxorubicin-induced significant reduction of PDE3A expression and subsequent ICER induction. Cardiac specific expression of CA-MEK5α rescued pressure overload– or doxorubicin-mediated PDE3A downregulation and ICER upregulation and inhibited myocyte apoptosis as well as subsequent cardiac dysfunction in vivo. These data suggest that preventing the feedback loop of PDE3A/ICER by ERK5 activation could inhibit progression of myocyte apoptosis as well as cardiac dysfunction. These data suggest a new therapeutic paradigm for end stage of heart failure by inhibiting the PDE3A/ICER feedback loop via activating ERK5.
Keywords: ERK5, phosphodiesterase 3, inducible cAMP early repressor, heart failure, insulin-like growth factor-1
Cardiac remodeling, including dysregulated myocyte apoptosis, contributes to the development and progression of pathological remodeling after cardiac ischemia/reperfusion and the transition from cardiac hypertrophy to chronic heart failure.1–3 A number of observations suggest that cardiac myocyte loss by apoptosis contributes to the transition from cardiac hypertrophy to heart failure.4 Previously, we reported a key role of phosphodiesterase 3A (PDE3A) and inducible cAMP early repressor (ICER) in cardiomyocyte apoptosis.5 PDEs exist as a superfamily with multiple isoforms that differ in tissue distribution, biochemical properties, and sensitivity to chemical inhibitors.6 At least 21 genes encoding more than 50 different PDE isoforms have been identified and grouped into 11 broad families (PDE1 to PDE11).6 Each tissue/cell type expresses a distinct set of PDEs. In cardiomyocytes, PDE3, together with PDE4, accounts for >90% of the basal cAMP-hydrolyzing activity, although their relative contributions may differ among species.7 We have recently found that in both neonatal and adult cardiomyocytes, inhibition of PDE3 function but not PDE4 significantly increased cardiomyocyte apoptosis despite the fact that PDE4 inhibition elicited a more profound elevation of cAMP compared with PDE3 inhibition.5,7,8 These observations indicate that cardiomyocyte apoptosis is regulated by a subset of cAMP molecules that are preferentially coupled to PDE3, which are independent of the overall cellular cAMP levels in the cell.
ICERs are members of the cAMP-responsive element (CRE) modulator family. The expression of ICERs is transcriptionally induced by CRE-binding protein (CREB) via a CREB sequence in the ICER promoter.9 ICER proteins contain DNA-binding and leucine zipper domains but not the N-terminal transactivation domain, which renders them endogenous inhibitors of gene transcription driven by its cognates such as CREB and activating transcription factor 1 (ATF-1).9 ICER has been shown to function as an important inducer or mediator of apoptosis in various cell types including neuronal cells10 and cardiomyocytes.5,11 We have recently shown that angiotensin II (Ang II) and β-adrenergic receptor activation induced a sustained downregulation of PDE3A1 expression via a “positive-feedback loop,” where ICER represses PDE3A gene expression and PDE3A reduction then feeds back, leading to increased ICER expression resulting from protein kinase A (PKA) activation.8 This feedback regulatory mechanism is essential for sustained ICER induction and subsequent cardiomyocyte apoptosis. Our previous findings demonstrated a key role of PDE3A in the regulation of cardiomyocyte survival and suggested that strategies to block the PDE3A/ICER feedback loop could reduce cardiomyocyte apoptosis and benefit heart failure.
Extracellular signal-regulated kinase 5/big mitogen-activated protein kinase 1 (ERK5/BMK1) is a member of the mitogen-activated protein kinase family, which is activated by oxidative and hyperosmotic stress, growth factors, and pathways involving certain G protein–coupled receptors.12 The upstream kinase that phosphorylates ERK5 has been identified as mitogen-activated protein kinase/ERK kinase (MEK5).13,14 Like many mitogen-activated protein kinase family members, ERK5 plays a significant role in cell growth and differentiation via activation of its downstream substrate myocyte enhancer factor 2 (MEF2), although emerging evidence suggests unique functional characteristics. Oxidative stress–mediated activation of ERK5 is documented to have an antiapoptotic effect15 and ERK5 knockout mice have impaired cardiac and vascular development.16 Recently we, along with Kasler et al, reported that ERK5 is not only a kinase but also possesses transcriptional activity.17,18 We also reported that ERK5 activation was significantly decreased in human heart failure.19 Although activation of ERK5 by the constitutively active form of MEK5α (CA-MEK5α) has been demonstrated to have a cardioprotective effect against acute ischemia/reperfusion in an isolated heart model,20 the exact role of ERK5 activation and its possible cardioprotective mechanism in chronic heart failure remains unclear.
An important cardiomyocyte survival factor is insulin-like growth factor-1 (IGF-1), but its effect on the PDE3A/ICER feedback loop remains largely unknown. IGF-1 is synthesized under the control of growth hormone by various cell types, including cardiac muscle.21 IGF-1 stimulates protein synthesis and cell growth and exerts antiapoptotic effects in many organs including cardiac muscle.22–24 Mehrhof et al have reported that IGF-1 activates Akt and ERK1/2 and inhibits apoptosis.25 Interestingly, although the importance of IGF-1/phosphatidylinositol 3-kinase activation in cell survival is well established,26–28 the specific role of Akt in heart failure has recently become more complicated.29–31
In this study, we found that ERK5 activation induced by IGF-1 significantly inhibited isoproterenol (ISO)-mediated apoptosis via regulating PDE3A/ICER feedback loop. In addition, activation of ERK5 in CA-MEK5α-Tg mice prevents pressure overload– and doxorubicin (Dox)-mediated induction of PDE3A/ICER feedback loop, cardiac apoptosis, as well as subsequent cardiac dysfunction in vivo. These data suggest the critical role of ERK5 activation on cardiac apoptosis and subsequent dysfunction via preventing the development of PDE3A/ICER feedback loop.
Materials and Methods
An expanded Materials and Methods section is available in the online data supplement at http://circres.ahajournals.org.
Details on reagents and adenovirus vectors, cultured rat neonatal cardiomyocytes, Western blot analysis, analysis of apoptosis, mouse model of thoracic aorta constriction (TAC), in vivo hemodynamic measurements with cardiac catheterization, protein extract from heart tissue, echocardiographic analysis, histology, ERK5 transcriptional activity (mammalian one-hybrid analysis), animal models, and statistical analysis are also provided in the online data supplement.
Results
ERK5 Activation by IGF-1 Inhibits the ISO-Induced Sustained Reduction of PDE3A and Concomitant Induction of ICER
Previously, we demonstrated that activation of ERK5 inhibits apoptosis in endothelial cells.32 There is increasing evidence that IGF-1 can be protective in cardiac injury, but the exact mechanism remains unclear. Therefore, we first investigated whether IGF-1 can increase ERK5 activation in cardiomyocytes. As shown in Figure 1A, we found that IGF-1 could significantly increase ERK5 kinase activation until at least 48 hours after stimulation. Because ERK5 is not just a “kinase” but also possesses strong transcriptional activity and increases MEF2 activity,17,18 we investigated whether IGF-1 can stimulate ERK5 transcriptional activity using a Gal4-ERK5 construct. As shown in Figure 1B, we found that IGF-1 significantly increased ERK5 transcriptional activity.
Figure 1.
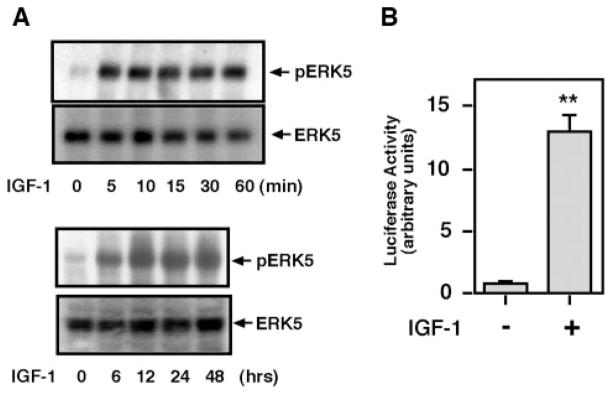
IGF-1 stimulated both ERK5 kinase and transcriptional activity. A, Cardiomyocytes were stimulated with IGF-1 (20 ng/mL), and phosphorylated (top) and total (bottom) ERK5 were measured by Western blot analysis at the indicated times. B, ERK5 transcriptional activity was measured by mammalian 1-hybrid assay with Gal4-ERK5 construct transfection, as we described previously.17
We have reported that ISO induced a sustained downregulation of PDE3A and upregulation of ICER, which are regulated by the PDE3A/ICER positive-feedback loop.8 The PDE3A/ICER feedback loop is essential for Ang II and ISO-induced apoptosis.5 Because IGF-1 stimulates ERK5 activation and inhibits cardiac apoptosis,21 we determined whether IGF-1–mediated ERK5 activation can inhibit both sustained reduction of PDE3A expression and induction of ICER following ISO treatment of cardiac myocytes. As shown in Figure 2A and 2B, ISO inhibited PDE3A expression and induced ICER expression after 24 hours of stimulation, and pretreatment with IGF-1 before 24 hours of ISO stimulation abolished ISO-mediated feedback loop of PDE3A reduction and ICER induction. Interestingly, transduction of dominant negative ERK5 (Ad-DN-ERK5) completely abolished the inhibitory effect of IGF-1 on ISO-mediated PDE3A reduction and ICER induction (Figure 2B). To confirm the role of endogenous ERK5 activation, ERK5 expression was reduced by ERK5 small interfering RNA (siRNA) (Figure 2C). We found that the reduction of ERK5 by ERK5 siRNA but not control siRNA abolished the inhibitory effect of IGF-1 on ISO-mediated reduction of PDE3A, indicating the importance of endogenous ERK5 in regulation of the PDE3A/ICER feedback loop.
Figure 2.

Role of ERK5 activation on IGF-1–mediated inhibition of PDE3A reduction and ICER induction (PDE3A/ICER feedback loop). A, Schematic diagram showing experimental protocol. Cardiomyocytes were transduced with Ad-LacZ, Ad-DN-ERK5, or Ad-DN-MEK1 at 50 multiplicities of infection for 24 hours (B) or ERK5 or control siRNA for 72 hours (C), followed by treatment with or without IGF-1 (20 ng/mL) for 24 hours and then stimulated with vehicle or ISO for 24 to 48 hours as indicated. Expression of PDE3A, ICER, and ERK5 activation were detected by Western blotting. B through D, Critical role of ERK5 activation on IGF-1–mediated inhibition on PDE3A/ICER feedback loop. B and C, Cardiomyocytes were transduced with Ad-LacZ or Ad-DN-ERK5, or ERK5 or control siRNA, followed by IGF-1 and ISO treatment, as described for A. D, Cardiomyocytes were transduced with Ad-LacZ or Ad-CA-MEK5α, followed by ISO treatment for 24 hours. E, Role of ERK5 activation on IGF-1–mediated inhibition on ISO-induced apoptosis. Cardiomyocytes were transduced with Ad-LacZ, Ad-CA-MEK5α, or Ad-DN-ERK5 at 50 multiplicities of infection for 24 hours, followed by treatment with or without IGF-1 (20 ng/mL) for 24 hours, and then stimulated with vehicle or ISO for 48 hours. IGF-1–induced ERK5 activation was critical for inhibiting ISO-mediated apoptosis. Data represent mean of 3 repeats (mean±SD). Similar results were obtained from at least 2 independent experiments. IB indicates immunoblot.
To detect whether activation of ERK5 can inhibit the ISO-mediated PDE3A/ICER feedback loop, we transduced cardiomyocytes with an adenovirus containing CA-MEK5α. As shown in Figure 2D, ERK5 activation by CA-MEK5α significantly inhibited ISO-mediated PDE3A reduction and ICER induction. These data indicate that ERK5 plays a critical role in the inhibitory effect of IGF-1 on the ISO-mediated PDE3A/ICER positive-feedback loop.
Role of ERK5 Activation in Mediating the IGF-1 Antiapoptotic Effect
Because persistent expression of ICER causes myocyte apoptosis and ERK5 activation inhibits ISO-mediated PDE3A reduction and ICER induction, we determined the role of IGF-1–mediated ERK5 activation in cardiomyocyte apoptosis. As shown in Figure 2E, ISO stimulation of 48 hours significantly increased cardiomyocyte apoptosis determined by TUNEL staining, which is consistent with observations reported previously.5 The role of ERK5 activation in ISO-induced apoptosis was determined by overexpressing CA-MEK5α in cardiomyocytes. As shown in Figure 2E, CA-MEK5α blocked ISO-induced apoptosis, suggesting the critical role of ERK5 activation in regulating cardiomyocyte apoptosis.
To determine the role of ERK5 in the IGF-1–mediated inhibitory effect on apoptosis, we transduced Ad-LacZ or Ad-DN-ERK5 into cardiomyocytes. Twenty four hours after transduction, we pretreated the cells with vehicle or IGF-1. Twenty-four hours later, the cells were stimulated by ISO for 48 hours and cardiomyocyte apoptosis was measured. As shown in Figure 2E, we found that pretreatment of cells by IGF-1 completely inhibited ISO-induced apoptosis. However, the antiapoptotic effect of IGF-1 was completely inhibited by Ad-DN-ERK5, indicating that ERK5 is critical in mediating the IGF-1 antiapoptotic effect in cardiomyocytes.
ERK5/MEF2 Activation Is Critical for the IGF-1–Mediated Inhibition of PDE3A/ICER Feedback Loop
It is well established that MEF2 is an important downstream substrate for ERK5 kinase.33 Therefore, we investigated the role of MEF2 on the IGF-1–mediated inhibitory effect on PDE3A/ICER feedback loop. First, we transduced cardiomyocytes with an adenovirus containing a dominant negative form of MEF2C (R3T mutant, a mutation in the MADS box of MEF2C that eliminates DNA binding without affecting dimerization) (Ad-DN-MEF2) or Ad-LacZ as a control. The dominant negative form of R3T MEF2C mutant can dimerize with endogenous MEF2 proteins including MEF2A and MEF2C and inhibit their activities.34 After 24 hours of transduction, cardiomyocytes were stimulated with IGF-1 or vehicle followed by ISO or vehicle stimulation as shown in Figure 2A. As shown in Figure 3A, IGF-1 inhibited ISO-mediated PDE3A reduction and ICER induction, and we found that Ad-DN-MEF2 transduction abolished this inhibitory effect of IGF-1 on PDE3A reduction and ICER induction. To further confirm the importance of MEF2 activation, we cotransduced Ad-CA-MEK5α and Ad-DN-MEF2, and, after 24 hours of transduction, we stimulated the cells with ISO. As shown in Figure 3B, activation of ERK5 inhibited ISO-mediated PDE3A reduction and ICER induction. Transduction of Ad-DN-MEF2 did not inhibit Ad-CA-MEK5α–mediated ERK5 activation as expected (Figure 3B, third panel from the bottom). Transduction with Ad-DN-MEF2 abolished the inhibitory effect of IGF-1 on ISO-mediated PDE3A downregulation and ICER upregulation, suggesting a critical role for MEF2 activation in IGF-1/ERK5-mediated inhibition on PDE3A/ICER feedback loop.
Figure 3.
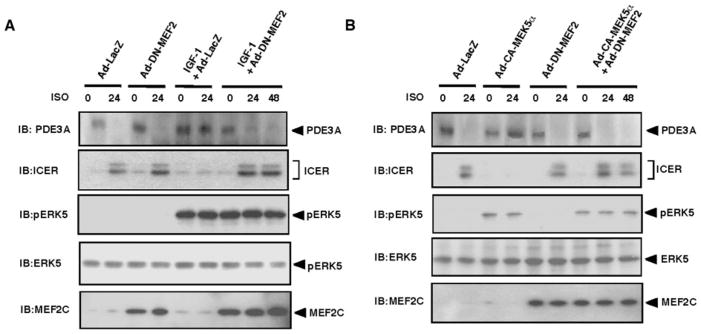
Critical role of MEF2 activation on IGF-1–mediated inhibition on PDE3A/ICER feedback loop. Cardiomyocytes were transduced with Ad-LacZ, Ad-DN-MEF2 (A, B), or Ad-CA-MEK5α (B), followed by treatment with or without IGF-1 (20 ng/mL) for 24 hours (A), as described in Figure 2A, and then stimulated with vehicle or ISO for 24 to 48 hours as indicated. Expression of PDE3A, ICER, and ERK5 activation were detected by Western blotting. IB indicates immunoblot.
Role of ERK5 in the Regulation of ICER Protein Stability, Which Inhibits ERK5-Mediated Antiapoptotic Effect and PDE3A Expression
In this study, we found that activation of ERK5 can induce PDE3A expression and inhibit ICER induction. Previously, we have reported that overexpression of ICER inhibits PDE3A expression and forms a PDE3A/ICER feedback loop.8 The expression level of ICER is regulated by CREB-dependent ICER gene transcription as well as proteasome-dependent ICER protein ubiquitination and degradation.35 We have found the critical role of PKA activation in stabilizing the ICER protein.8 However, the role of ERK5 activation on ICER protein stability remains unknown. To determine the role of ERK5 activation on ICER stability, we examined the effect of ERK5 activation on forskolin-mediated ICER protein stability. As shown in Figure 4A, cardiomyocytes were transduced with Ad-LacZ or Ad-CA-MEK5α for 24 hours and then transduced with Ad-ICER for 12 hours (this time allowing a sufficient expression of ICER; data not shown). After Ad-ICER transduction, cells were treated with cycloheximide for 12 hours and then stimulated with forskolin for 16 hours. As shown in Figure 4B and 4C, in the presence of lacZ, forskolin increased ICER expression, although cells were pretreated with cycloheximide. ICER was induced exogenously by Ad-ICER transduction and forskolin-mediated ICER induction was investigated under the treatment with the protein synthesis inhibitor cycloheximide. Therefore, the increase of ICER by forskolin was solely attributable to ICER protein stabilization but not attributable to the regulation of transcriptional machinery or RNA stability, as we described previously.8 CA-MEK5α significantly decreased forskolin-mediated ICER stabilization in both cardiomyocytes and CHO cells (Figure 4B and 4C), indicating that ERK5 plays an important role in destabilizing the ICER protein, which is opposite of the effect of PKA activation on ICER stability. The exact mechanism of ICER stabilization induced by PKA remains unclear. It is possible that ERK5 activation inhibits the process of PKA-mediated ICER stabilization. However, further investigation is required to address this question.
Figure 4.

Role of ERK5 activation on ICER stability and PDE3A/ICER feedback loop regulation. A, Schematic diagram showing experimental protocol. Cardiomyocytes were transduced with Ad-LacZ or Ad-CA-MEK5α for 24 hours and then transduced with Ad-ICER. Twelve hours after Ad-ICER transduction, cells were pretreated with cycloheximide, then stimulated with forskolin (10 μmol/L) or vehicle for 16 hours. B, Western blotting showing ICER and cAMP-responsive element modulator (CREM) protein expression in cardiomyocytes treated with the above protocol. C, The intensity of the band representing ICER was normalized to the intensity of Ad-LacZ and vehicle-treated control samples, which was designated as 1. Data represent mean±SD of 3 samples. *P<0.05. D, Effect of ERK5 activation on ICER-mediated apoptosis. Cardiomyocytes were transduced with Ad-LacZ, Ad-CA-MEK5α, or Ad-ICER at 50 multiplicities of infection for 24 hours. ERK5 activation could not reverse ICER-mediated apoptosis. Data represent mean of 3 repeats (mean±SD). Similar results were obtained from at least 2 independent experiments. E, Cardiomyocytes were transduced with Ad-LacZ, Ad-ICER, or Ad-CA-MEK5α. After 24 hours of transduction, expression of PDE3A, ICER, and ERK5 activation were detected by Western blotting. IB indicates immunoblot.
Although ERK5 can regulate ICER destabilization, the involvement of ICER as a downstream effector of ERK5 activation during the ERK5-mediated antiapoptotic effect and PDE3A expression remains unclear. As shown in Figure 2D, we found that activation of ERK5 could inhibit ISO-mediated ICER induction. To determine the contribution of ERK5-mediated ICER reduction to the antiapoptotic effect, we cotransduced cardiomyocytes with Ad-LacZ or Ad-CA-MEK5α with Ad-ICER and assessed apoptosis. In agreement with our previous reports,5 we found that apoptosis was evoked by ICER overexpression (Figure 4D). However, Ad-CA-MEK5α transduction could not inhibit apoptosis induced by ICER overexpression (Figure 4D). Furthermore, we found that activation of ERK5 by CA-MEK5α could not restore PDE3A reduction by ICER overexpression (Figure 4E), suggesting that the reduction of ICER and induction of PDE3A is a critical downstream event mediated by ERK5 activation, which is the mechanism for the observed ERK5 antiapoptotic effect in cardiomyocytes.
Pressure Overload and Doxorubicin-Mediated Reduction of PDE3A and ICER Induction Were Inhibited in the CA-MEK5α-Tg Heart
Previously, we found that PDE3A reduction and subsequent ICER induction is critical for ISO-mediated apoptosis and that pressure overload by TAC decreased PDE3A expression and increased ICER expression.5 Therefore, we next examined whether ERK5 activation by CA-MEK5α prevents pressure overload–induced PDE3A reduction and subsequent ICER induction. As shown in Figure 5A (functional data in Table I of the online data supplement; Figure 7), pressure overload inhibited PDE3A expression and significantly increased ICER induction in nontransgenic littermate control (NLC) mice. In contrast, PDE3A expression was preserved in CA-MEK5α-Tg mice, and we did not find any significant increase of ICER expression induced by pressure overload in CA-MEK5α-Tg mice. There were no significant differences in Akt and ERK1/2 activation among NLC and CA-MEK5α-Tg mice. To determine the general role of ERK5 activation on PDE3A/ICER feedback loop in heart failure, we also examined PDE3A and ICER expression in a Dox-mediated mouse heart failure model. As shown in Figure 5B, we also found a significant reduction of PDE3A and ICER induction after 5 days of Dox treatment, which were completely abolished in CA-MEK5α-Tg mice. No significant differences of Akt and ERK1/2 activation among NLC and CA-MEK5α-Tg mice were observed in this model. These data support the critical role of ERK5 activation in regulating the PDE3A/ICER feedback loop in a heart failure model.
Figure 5.
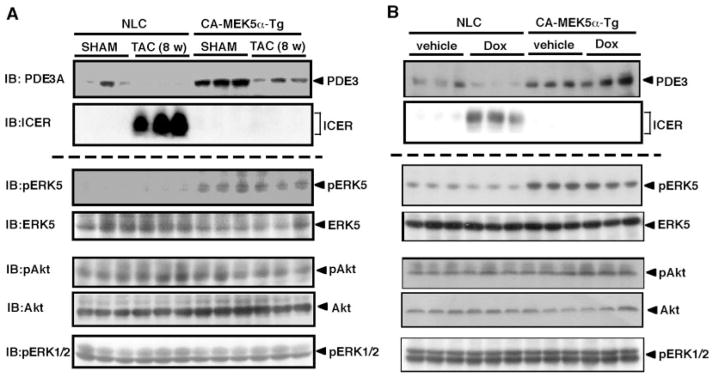
Pressure overload and Dox-mediated PDE3A reduction and ICER induction in NLC and CA-MEK5α-Tg mice. A, Western blot showing PDE3A and ICER expression and ERK5, Akt, and ERK1/2 kinase activity in sham and 8-week TAC mouse hearts in NLC and CA-MEK5α-Tg mice. B, Western blot showing PDE3A and ICER expression and ERK5, Akt, and ERK1/2 kinase activity in vehicle and Dox-treated (5 days) hearts in NLC and CA-MEK5α-Tg mice. IB indicates immunoblot.
Figure 7.

Cardioprotective role of ERK5 activation in Dox- and TAC-mediated heart failure model. A, Representative M-mode echocardiogram of NLC and CA-MEK5α-Tg mice treated with vehicle or Dox. Dd indicates diameter of diastole; Ds, diameter of systolic. B, Percentage of fractional shortening (FS) in NLC and CA-MEK5α-Tg mice after vehicle or Dox treatment. C and D, Hemodynamic measurements in NLC and CA-MEK5a-Tg mice after Dox treatment (C) or TAC (D). All data are expressed as mean±SD. **P<0.01.
Cardiac Expression of CA-MEK5α Reduces Apoptosis Induced by Pressure Overload or Dox
Apoptosis is believed to be important for the transition from hypertrophy to heart failure.4 It has been reported that although apoptotic cells are not detectable in normal mouse LV, they are widespread in the LV after 8-week TAC.36 Similarly, we found very few TUNEL-positive cells in control and 4-week TAC mouse hearts. In contrast, in hearts after 8-weeks of TAC, the number of in situ TUNEL-positive cells was increased to 0.33±0.06% (Figure 6A, left). We found significantly decreased TUNEL-positive cells in CA-MEK5α-Tg mice after 8 weeks of TAC (Figure 6A, left). In a Dox-mediated heart failure model, we also found that Dox-induced apoptosis was significantly inhibited in CA-MEK5α-Tg mice, supporting the general antiapoptotic role of ERK5 activation in heart failure (Figure 6A, right). To confirm these data, we further evaluated the process of apoptosis in vivo by examining cleaved caspase-3 expression. As shown in Figure 6B, cleaved caspase-3 expression was significantly increased in both TAC and Dox-mediated heart failure models, and activation of ERK5 by CA-MEK5α abolished cleaved caspase-3 expression in these heart failure models. Of note, these same samples did not have any significant differences in ERK1/2 and Akt expression or activation (Figure 5). These data further support the antiapoptotic role of ERK5 activation during heart failure.
Figure 6.

Pressure overload– and Dox-induced myocardial apoptosis in NLC and CA-MEK5-Tg mice. A, Pressure overload–mediated (left) or Dox-mediated (right) percentage of TUNEL-positive nuclei in NLC and CA-MEK5α-Tg mice. B, Pressure overload–mediated (left) or Dox-mediated (right) cleaved caspase-3 expression in NLC and CA-MEK5α-Tg mice. The intensity of the band representing cleaved caspase-3 was normalized to the intensity of 1 control samples that was designated as 1. Data represent mean±SD of 3 samples.** P<0.01 compared with NLC sham (left) or vehicle treatment (right).
CA-MEK5α–Mediated ERK5 Activation Prevents Dox or TAC-Mediated Cardiac Dysfunction
Because we found a critical inhibitory role for ERK5 activation on PDE3A and the ICER feedback loop and apoptosis, we next investigated whether ERK5 activation can inhibit the development of heart failure in vivo. Figure 7A shows a representative echocardiogram after vehicle or Dox administration in NLC and CA-MEK5α-Tg mice. As shown in supplemental Table I, left ventricular (LV) end-systolic diameter was markedly increased in NLC mice after Dox injection, but significantly smaller in Dox-injected CA-MEK5α-Tg mice compared with Dox-injected NLC mice. Fractional shortening was also better preserved in Dox-injected CA-MEK5α mice than Dox-NLC (supplemental Table I; Figure 7B). In agreement with our previous reports, there was no difference in basal cardiac function between NLC and CA-MEK5α-Tg mice.20 After Dox injection, we observed a significant decrease of developed pressure and dP/dtmax in NLC mice. In contrast, Dox-injected CA-MEK5α-Tg mice had normal developed pressure and dP/dtmax (Figure 7C, supplemental Table II).
To further determine the role of ERK5 activation in heart failure, we performed TAC in both NLC and CA-MEK5α-Tg mice. As shown in supplemental Table III, LV weight was increased by 37% in 4-week TAC mice and by 89% in 8-week TAC mice compared with age-matched controls. At these stages of hypertrophy, there were no differences between CA-MEK5α-Tg and NLC in body weight, LV weight, and LV/body weight ratio. In the 4-week TAC mice, absolute LV systolic pressure was elevated in both CA-MEK5α-Tg and NLC mice. Interestingly, in the 8-week TAC NLC mice, absolute LV systolic pressure was lower than in the 4-week TAC mice despite a greater magnitude of hypertrophy. However, we found that in CA-MEK5α-Tg mice, absolute LV systolic pressure did not decrease compared with 4-week TAC CA-MEK5α-Tg mice. Furthermore, in the 8-week TAC mice, LV systolic developed pressure per gram (LV weight) was significantly depressed in NLC mice compared with the sham-operation group. In contrast, LV systolic developed pressure per gram was significantly higher in the 8-week TAC CA-MEK5α-Tg mice. Furthermore, we found that the absolute values for developed pressure, dP/dtmax, and dP/dtmin in the 8-week TAC CA-MEK5α-Tg mice were greater than those in the 8-week TAC NLC mice (Figure 7D, and supplemental Table III). Heart rate was similar between the groups (supplemental Table III). These data suggest the presence of depressed systolic performance in the 8-week TAC NLC mice compared with the 4-week TAC NLC mice, and ERK5 activation induced by CA-MEK5α-Tg inhibits the development of cardiac dysfunction by pressure overload.
Discussion
We have previously reported that the PDE3A expression and activity was significantly decreased during the transition from compensated hypertrophy to decompensated heart failure.5 Because PDE3A expression and activity as well as subsequent inhibition of ICER expression are crucial for inhibiting apoptosis, the reduction of PDE3A may have a significant impact on pressure overload– and Dox-mediated apoptosis and subsequent cardiac dysfunction. In fact, we found that activation of ERK5 significantly inhibited PDE3A reduction as well as ICER induction and subsequent apoptosis and partially normalized cardiac function. These data suggest an important role for the PDE3A/ICER (Figure 8) feedback loop in the transition from compensated hypertrophy to decompensated heart failure.
Figure 8.
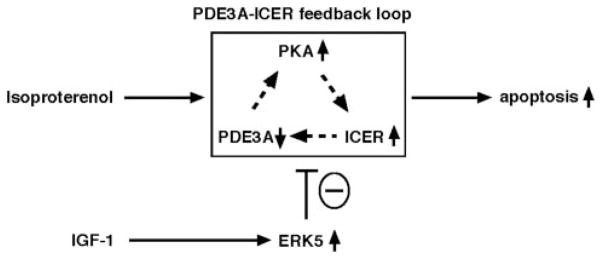
Scheme of ERK5/MEF2 activation and PDE3A/ICER feedback loop. ISO induced sustained downregulation of PDE3A expression and upregulation of ICER, which is regulated by PDE3A/ICER-positive feedback loop. IGF-1–induced ERK5 activation leads to PDE3A expression caused by MEF2-dependent PDE3A promoter activation and inhibits induction of PDE3A/ICER autoregulatory positive-feedback loop, which plays a key role in cardiomyocyte apoptosis.
To our knowledge this is the first report to show that IGF-1 activates ERK5 in cardiomyocytes. IGF-1/ERK5 inhibits the ISO-mediated PDE3A/ICER feedback loop and plays a critical role in regulating ISO-induced apoptosis. Based on both DN-ERK5 and ERK5 siRNA results (Figure 2), IGF-1–mediated ERK5 activation is definitely “necessary” to inhibit ISO-mediated reduction of PDE3A. Furthermore, we found that the antiapoptotic effect of ERK5 activation was lost by overexpression of ICER by Ad-ICER transduction, suggesting that ERK5 activation acts upstream of the PDE3A/ICER feedback loop, consistent with the IGF-1 results reported in Figures 2 and 3. The ability of IGF-1 to inhibit cell death by both apoptosis and necrosis has previously been shown in experimental ischemic cardiomyopathy in vivo.37 In addition, Welch et al have reported that cardiac specific IGF-1 expression prevented apoptosis and improved hemodynamics in tropomodulin-overexpressing transgenic mice.26 Because IGF-1 activates phosphatidylinositol 3-kinase, which in turn phosphorylates the downstream effector molecule Akt and inhibits apoptosis, Akt has been the main focus of the antiapoptotic effect of IGF-1.31 However, the role of Akt in heart failure becomes complicated, because overexpression of Akt in the heart may become maladaptive.29,30 Although future studies will be required for the dependency of phosphatidylinositol 3-kinase on ERK5 activation, our data suggest a critical role for ERK5 activation in relaying the IGF-1–mediated cardioprotective effect. Furthermore, because we found the possible involvement of other kinases in regulating PDE3A expression (C.Y., and J.-i.A., unpublished data, 2006), we assume that activation of ERK5 kinase alone may not be “sufficient” to increase PDE3 expression. Therefore, CA-MEK5α did not change the basal expression of PDE3A (Figure 2D) but only prevented its loss and induction of ICER after ISO treatment (Figure 2B and 2C). In contrast, we observed increased PDE3 expression in CA-MEK5α-Tg mice in vivo (Figure 5A), suggesting that ERK5 may be among the critical factors to regulate PDE3 expression in vivo. To better understand the role of ERK5 activation in IGF-1 signaling and PDE3A expression, further studies would be required using IGF-1 transgenic mice with knock out of ERK5.
An expanded Discussion section is available in the online data supplement
Supplementary Material
Acknowledgments
We thank Drs Thunder Jalili, Paul S. Brookes, and Yisang Yoon for critical reading of the manuscript.
Sources of Funding
This work was supported by grants from the America Heart Association (Grant-in-Aid 0455847T to C.Y.; Scientist Development Grant 0630155N to B.D.; Scientist Development Grant 0435437T to B.C.B.; Postdoctoral Fellowship 0325769T to S.I.; and Postdoctoral Fellowship 0625957T to C.-H.W.) and from the NIH (HL-66919, GM-071485, and HL-077789 to J.-i.A.; HL-077789 to C.Y.).
Footnotes
Disclosures
None.
References
- 1.Kang PM, Izumo S. Apoptosis and heart failure: a critical review of the literature. Circ Res. 2000;86:1107–1113. doi: 10.1161/01.res.86.11.1107. [DOI] [PubMed] [Google Scholar]
- 2.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–1141. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 3.Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003;93:896–906. doi: 10.1161/01.RES.0000102042.83024.CA. [DOI] [PubMed] [Google Scholar]
- 4.Wollert KC, Drexler H. Regulation of cardiac remodeling by nitric oxide: focus on cardiac myocyte hypertrophy and apoptosis. Heart Fail Rev. 2002;7:317–325. doi: 10.1023/a:1020706316429. [DOI] [PubMed] [Google Scholar]
- 5.Ding B, Abe J, Wei H, Huang Q, Walsh RA, Molina CA, Zhao A, Sadoshima J, Blaxall BC, Berk BC, Yan C. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: implication in heart failure. Circulation. 2005;111:2469–2476. doi: 10.1161/01.CIR.0000165128.39715.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shakur Y, Holst LS, Landstrom TR, Movsesian M, Degerman E, Manganiello V. Regulation and function of the cyclic nucleotide phosphodiesterase (PDE3) gene family. Prog Nucleic Acid Res Mol Biol. 2001;66:241–277. doi: 10.1016/s0079-6603(00)66031-2. [DOI] [PubMed] [Google Scholar]
- 7.Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, Huston E, Hannawacker A, Lohse MJ, Pozzan T, Houslay MD, Zaccolo M. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res. 2004;95:67–75. doi: 10.1161/01.RES.0000134629.84732.11. [DOI] [PubMed] [Google Scholar]
- 8.Ding B, Abe J, Wei H, Xu H, Che W, Aizawa T, Liu W, Molina CA, Sadoshima J, Blaxall BC, Berk BC, Yan C. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc Natl Acad Sci U S A. 2005;102:14771–14776. doi: 10.1073/pnas.0506489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mioduszewska B, Jaworski J, Kaczmarek L. Inducible cAMP early repressor (ICER) in the nervous system—a transcriptional regulator of neuronal plasticity and programmed cell death. J Neurochem. 2003;87:1313–1320. doi: 10.1046/j.1471-4159.2003.02116.x. [DOI] [PubMed] [Google Scholar]
- 10.Jaworski J, Mioduszewska B, Sanchez-Capelo A, Figiel I, Habas A, Gozdz A, Proszynski T, Hetman M, Mallet J, Kaczmarek L. Inducible cAMP early repressor, an endogenous antagonist of cAMP responsive element-binding protein, evokes neuronal apoptosis in vitro. J Neurosci. 2003;23:4519–4526. doi: 10.1523/JNEUROSCI.23-11-04519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomita H, Nazmy M, Kajimoto K, Yehia G, Molina CA, Sadoshima J. Inducible cAMP early repressor (ICER) is a negative-feedback regulator of cardiac hypertrophy and an important mediator of cardiac myocyte apoptosis in response to beta-adrenergic receptor stimulation. Circ Res. 2003;93:12–22. doi: 10.1161/01.RES.0000079794.57578.F1. [DOI] [PubMed] [Google Scholar]
- 12.Gutkind JS. Regulation of mitogen-activated protein kinase signaling networks by G protein-coupled receptors. Sci STKE. 2000;2000:RE1. doi: 10.1126/stke.2000.40.re1. [DOI] [PubMed] [Google Scholar]
- 13.Zhou G, Bao ZQ, Dixon JE. Components of a new human protein kinase signal transduction pathway. J Biol Chem. 1995;270:12665–12669. doi: 10.1074/jbc.270.21.12665. [DOI] [PubMed] [Google Scholar]
- 14.Lee JD, Ulevitch RJ, Han J. Primary structure of BMK1: a new mammalian MAP kinase. Biochem Biophys Res Commun. 1995;213:715–724. doi: 10.1006/bbrc.1995.2189. [DOI] [PubMed] [Google Scholar]
- 15.Suzaki Y, Yoshizumi M, Kagami S, Koyama AH, Taketani Y, Houchi H, Tsuchiya K, Takeda E, Tamaki T. Hydrogen peroxide stimulates c-Src-mediated big mitogen-activated protein kinase 1 (BMK1) and the MEF2C signaling pathway in PC12 cells: potential role in cell survival following oxidative insults. J Biol Chem. 2002;277:9614–9621. doi: 10.1074/jbc.M111790200. [DOI] [PubMed] [Google Scholar]
- 16.Regan CP, Li W, Boucher DM, Spatz S, Su MS, Kuida K. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc Natl Acad Sci U S A. 2002;99:9248–9253. doi: 10.1073/pnas.142293999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akaike M, Che W, Marmarosh NL, Ohta S, Osawa M, Ding B, Berk BC, Yan C, Abe J. The hinge-helix 1 region of peroxisome proliferator-activated receptor gamma1 (PPARgamma1) mediates interaction with extracellular signal-regulated kinase 5 and PPARgamma1 transcriptional activation: involvement in flow-induced PPARgamma activation in endothelial cells. Mol Cell Biol. 2004;24:8691–8704. doi: 10.1128/MCB.24.19.8691-8704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kasler HG, Victoria J, Duramad O, Winoto A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol Cell Biol. 2000;20:8382–8389. doi: 10.1128/mcb.20.22.8382-8389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeishi Y, Huang Q, Abe J, Che W, Lee JD, Kawakatsu H, Hoit BD, Berk BC, Walsh RA. Activation of mitogen-activated protein kinases and p90 ribosomal S6 kinase in failing human hearts with dilated cardiomyopathy. Cardiovasc Res. 2002;53:131–137. doi: 10.1016/s0008-6363(01)00438-2. [DOI] [PubMed] [Google Scholar]
- 20.Cameron SJ, Itoh S, Baines CP, Zhang C, Ohta S, Che W, Glassman M, Lee JD, Yan C, Yang J, Abe J. Activation of big MAP kinase 1 (BMK1/ERK5) inhibits cardiac injury after myocardial ischemia and reperfusion. FEBS Lett. 2004;566:255–260. doi: 10.1016/j.febslet.2004.03.120. [DOI] [PubMed] [Google Scholar]
- 21.Ren J, Samson WK, Sowers JR. Insulin-like growth factor I as a cardiac hormone: physiological and pathophysiological implications in heart disease. J Mol Cell Cardiol. 1999;31:2049–2061. doi: 10.1006/jmcc.1999.1036. [DOI] [PubMed] [Google Scholar]
- 22.Buerke M, Murohara T, Skurk C, Nuss C, Tomaselli K, Lefer AM. Cardioprotective effect of insulin-like growth factor I in myocardial ischemia followed by reperfusion. Proc Natl Acad Sci U S A. 1995;92:8031–8035. doi: 10.1073/pnas.92.17.8031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anversa P, Kajstura J, Cheng W, Reiss K, Cigola E, Olivetti G. Insulin-like growth factor-1 and myocyte growth: the danger of a dogma. Part I. Postnatal myocardial development: normal growth. Cardiovasc Res. 1996;32:219–225. doi: 10.1016/0008-6363(96)00035-1. [DOI] [PubMed] [Google Scholar]
- 24.Reiss K, Cheng W, Ferber A, Kajstura J, Li P, Li B, Olivetti G, Homcy CJ, Baserga R, Anversa P. Overexpression of insulin-like growth factor-1 in the heart is coupled with myocyte proliferation in transgenic mice. Proc Natl Acad Sci U S A. 1996;93:8630–8635. doi: 10.1073/pnas.93.16.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehrhof FB, Muller FU, Bergmann MW, Li P, Wang Y, Schmitz W, Dietz R, von Harsdorf R. In cardiomyocyte hypoxia, insulin-like growth factor-I-induced antiapoptotic signaling requires phosphatidylinositol-3-OH-kinase-dependent and mitogen-activated protein kinase-dependent activation of the transcription factor cAMP response element-binding protein. Circulation. 2001;104:2088–2094. doi: 10.1161/hc4201.097133. [DOI] [PubMed] [Google Scholar]
- 26.Welch S, Plank D, Witt S, Glascock B, Schaefer E, Chimenti S, Andreoli AM, Limana F, Leri A, Kajstura J, Anversa P, Sussman MA. Cardiac-specific IGF-1 expression attenuates dilated cardiomyopathy in tropomodulin-overexpressing transgenic mice. Circ Res. 2002;90:641–648. doi: 10.1161/01.res.0000013780.77774.75. [DOI] [PubMed] [Google Scholar]
- 27.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 28.Kajstura J, Fiordaliso F, Andreoli AM, Li B, Chimenti S, Medow MS, Limana F, Nadal-Ginard B, Leri A, Anversa P. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes. 2001;50:1414–1424. doi: 10.2337/diabetes.50.6.1414. [DOI] [PubMed] [Google Scholar]
- 29.Nagoshi T, Matsui T, Aoyama T, Leri A, Anversa P, Li L, Ogawa W, del Monte F, Gwathmey JK, Grazette L, Hemmings BA, Kass DA, Champion HC, Rosenzweig A. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J Clin Invest. 2005;115:2128–2138. doi: 10.1172/JCI23073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Neill BT, Abel ED. Akt1 in the cardiovascular system: friend or foe? J Clin Invest. 2005;115:2059–2064. doi: 10.1172/JCI25900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pi X, Yan C, Berk BC. Big mitogen-activated protein kinase (BMK1)/ERK5 protects endothelial cells from apoptosis. Circ Res. 2004;94:362–369. doi: 10.1161/01.RES.0000112406.27800.6F. [DOI] [PubMed] [Google Scholar]
- 33.Kato Y, Zhao M, Morikawa A, Sugiyama T, Chakravortty D, Koide N, Yoshida T, Tapping RI, Yang Y, Yokochi T, Lee JD. Big mitogen-activated kinase regulates multiple members of the MEF2 protein family. J Biol Chem. 2000;275:18534–18540. doi: 10.1074/jbc.M001573200. [DOI] [PubMed] [Google Scholar]
- 34.Molkentin JD, Black BL, Martin JF, Olson EN. Mutational analysis of the DNA binding, dimerization, and transcriptional activation domains of MEF2C. Mol Cell Biol. 1996;16:2627–2636. doi: 10.1128/mcb.16.6.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yehia G, Schlotter F, Razavi R, Alessandrini A, Molina CA. Mitogen-activated protein kinase phosphorylates and targets inducible cAMP early repressor to ubiquitin-mediated destruction. J Biol Chem. 2001;276:35272–35279. doi: 10.1074/jbc.M105404200. [DOI] [PubMed] [Google Scholar]
- 36.Ding B, Price RL, Goldsmith EC, Borg TK, Yan X, Douglas PS, Weinberg EO, Bartunek J, Thielen T, Didenko VV, Lorell BH. Left ventricular hypertrophy in ascending aortic stenosis mice: anoikis and the progression to early failure. Circulation. 2000;101:2854–2862. doi: 10.1161/01.cir.101.24.2854. [DOI] [PubMed] [Google Scholar]
- 37.Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard M, Force TL, Franke TF, Hajjar RJ, Rosenzweig A. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;104:330–335. doi: 10.1161/01.cir.104.3.330. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.