

Abstract
Toll-like receptors (TLRs) are master regulators of innate immunity and play an integral role in the activation of the inflammatory response during infections. In addition, TLRs influence the body’s response to numerous forms of injury. Recent data have shown that TLRs play a modulating role in ischemic brain damage after stroke. Interestingly, their stimulation prior to ischemia induces a tolerant state that is neuroprotective. This phenomenon, referred to as TLR preconditioning, is the result of reprogramming of the TLR response to ischemic injury. This review addresses the role of TLRs in brain ischemia and the activation of endogenous neuroprotective pathways in the setting of preconditioning. We highlight the protective role of the interferon-related response and the potential site of action for TLR preconditioning involving the blood-brain-barrier. Pharmacological modulation of TLR activation to promote protection against stroke is a promising approach for the development of prophylactic and acute therapies targeting ischemic brain injury.
Keywords: Cerebral ischemia, Ischemic brain injury, Preconditioning, Stroke, Toll-like receptors
INTRODUCTION
The brain is a tissue with high oxygen demand. It represents only 2% of the total body weight yet requires 20% of the total body oxygen consumption (1). The majority of oxygen is needed to generate sufficient energy (ATP) to maintain electrochemical gradients and vital neuronal functions. Therefore, restriction of a cerebral blood vessel due to embolism, thrombosis or hypoperfusion, which limits or stops oxygen and glucose delivery to the surrounding tissue can result in serious brain damage and death.
Stroke is a leading cause of death worldwide and the major cause of disability in the western world (2). Recently, stroke fell from the third leading cause of death to fourth, as a consequence of accelerated access to medical care but with the challenge of extended disability (www.CDC.gov/datastatistics). There are 2 subtypes of stroke, hemorrhagic and ischemic, with the latter accounting for 87% of all strokes (2). The type of stroke can be quickly distinguished by medical imaging, such as computed tomography, which in turn dictates the medical management. Hemorrhagic stroke can require a range of interventions. For example, relief of high blood pressure or reduction of medications that facilitate intracranial bleeding are frequently addressed. In appropriate cases, hemorrhage may also need to be stopped with a surgical intervention.
Thrombolytic therapy is the only pharmacological treatment approved to date for ischemic stroke. The agent most commonly used is the recombinant tissue plasminogen activator (r-tPA), which converts plasminogen to plasmin, which, in turn, breaks down fibrin in blood clots. Although recent clinical trials show that the therapeutic window for r-tPA treatment may extend out to 4.5 hours in some patients, the best results are obtained when r-tPA is administered within 3 hours of stroke onset (3, 4). Advances in technology have also recently made it possible to treat ischemic stroke through the physical removal of the clot using the surgical process of endovascular revascularization. Therefore, the speed of diagnoses and treatment are critical for a favorable outcome.
A novel area of stroke research that offers therapeutic promise is the examination of inflammatory processes associated with ischemia. Toll-like receptors (TLRs) are a family of pattern recognition receptors that were initially identified for their role in the activation of innate immunity in response to the presence of exogenous microorganisms; however, TLRs also play a role in ischemic injury in the absence of infection (5). In this setting, TLRs recognize endogenous molecules released during injury. Such endogenous molecules are known as damage-associated molecular patterns (DAMPs). The binding of DAMPs to their respective receptors results in the activation of an inflammatory response that can exacerbate ischemic damage (5).
Recent data highlight the complex nature of the actions of TLRs in brain injury. A variety of studies have shown that although TLRs play an exacerbating role in the setting of ischemia, brief activation of TLRs prior to ischemia induces a state of tolerance to subsequent injury (6, 7). Whether TLR activation is detrimental or neuroprotective depends on the timing and intensity of receptor stimulation. Modulation of TLR activity to promote neuroprotective effects has great potential as a prophylactic therapy that targets ischemic brain injury.
A growing number of medical procedures have been shown to increase the risk of brain injuries. Cardiac surgery has been associated with a spectrum of brain injuries including neurological death (0.21%–2%) and stroke (1.1%–6.6%) (8). Besides stroke, cardiovascular interventions may cause long-term neuropsychological impairments, including memory loss or impaired executive functioning (9). In addition, the evaluation of patients after procedures by brain imaging may detect silent or subtle ischemic strokes. In 68% to 91% of patients undergoing transcatheter aortic valve implantation, for example, new but clinically silent lesions, predominantly caused by periprocedural emboli, were found on cerebral diffusion-weighted magnetic resonance imaging (10). Prophylactic treatment of patients who are at risk of such events could reduce the risk of ischemic injury.
This review will describe the role of TLRs in brain ischemia and how modulating TLR activation can turn on endogenous protective pathways that reduce ischemic injury. We will also discuss the potential site-of-action of TLR preconditioning shown in several recent studies that focus on the involvement of the blood-brain barrier (BBB) in TLR-induced protective effects.
TLR Structure and Signaling
Structurally, TLRs have leucine-rich repeats in the extracellular domain, and a toll/interleukin 1 receptor (TIR) intracellular domain that activates downstream signaling pathways. There are 10 TLRs identified in humans and 13 in mice (11). Expression and localization of TLRs can be cell- or tissue-dependent, although in general TLRs 1, 2, 4, 5 and 6 localize to the plasma membrane whereas others are typically located intracellularly on the surface membranes of endosomes or lysosomes (e.g. TLRs 3, 7, 8, and 9) (12). Protein or lipid structures on the surface of microbial pathogens bind TLRs expressed on the plasma membrane. Those TLRs localized on intracellular organelles recognize foreign nucleic acids thereby providing an additional level of regulation for TLR activation. It has also been suggested that different localization of the TLRs is important for discriminating between self and non-self. In this regard, Barton et al showed that TLR9 intracellular localization prevents the receptor from recognizing endogenous DNA (13).
Upon binding cognate ligands, TLRs form homodimers or heterodimers with or without the help of co-receptors or other accessory molecules (14), and activate signaling pathways that result in the expression of proinflammatory mediators and anti-microbial effector molecules (15–17) (Fig. 1). The binding of the adaptor molecules containing the structurally conserved toll/interleukin-1 receptor (TIR) domain activates intracellular signaling pathway. Four adaptors molecules have been described: the myeloid differentiation factor-88 (MyD88); MyD88 adaptor-like protein (MAL/TIRAP); TIR domain-containing adaptor protein inducing interferon-β (TRIF); and the TRIF-related adaptor molecule (TRAM). Recruitment of these adaptor molecules activates downstream kinases and transcription factors that regulate inflammatory and antiviral responses (18).
Figure 1.

Toll-like receptor (TLR) signaling pathways. Once activated by their cognate ligands, TLRs bind their respective adaptor molecules in the cytoplasm to initiate intracellular signaling pathways. This results in the activation of other mediators ultimately resulting in the expression of pro- or anti-inflammatory molecules. Specifically, the activation of IKK complex results in the release of nuclear factor-κ B (NF-κB) that translocates to the nucleus to initiate the expression of proinflammatory molecules. Instead, the phosphorylation of interferon regulatory factor 3 (IRF3) and interferon regulatory factor (IRF7) results in the induction of anti-inflammatory molecules. Abbreviations: IFNα, -β, interferon-α, -β; IKK, Iκb kinase; IL1β, -6, -10, interleukin-1β, -6, -10; IRAK, IL-1 receptor-associated kinase; IRF3, interferon regulatory factor 3; MAL/TIRAP, MyD88 adaptor-like protein; MYD88, myeloid differentiation factor-88; RIP1, receptor interacting protein 1; TIR, toll-interleukin 1 receptor; TBK, TANK-binding kinase; TGFβ, transforming growth factor β; TNF, tumor necrosis factor; TRAF3, -6, tumor necrosis factor receptor-associated factor 3, -6; TRAM, TRIF-related adaptor molecule; TRIF, TIR domain-containing adaptor protein inducing interferon-β.
MyD88 is recruited by all TLRs except TLR3, which exclusively signals through TRIF. MAL/TIRAP is a bridging adaptor necessary for TLR1/2/4 and 6 binding to MyD88. TLR4 is the only TLR able to bind both MyD88 and TRIF. The association of MyD88 to the cytoplasmic tail of the stimulated TLRs recruits IL-1 receptor-associated kinases (IRAKs). Specifically, IRAK4 is recruited by MyD88 and activates other IRAK family members, such as IRAK1, which signal the activation of mitogen-activated protein kinase and nuclear factor-κ B (NF-κB) pathways. In particular, IRAK1 activation results in the degradation of the NF-κB inhibitor IκB-α, which permits the translocation of NF-κB to the nucleus to initiate the transcription of inflammatory cytokines such as tumor necrosis factor (TNF), interleukin 6 (IL-6) and pro-interleukin-1β (15, 16) (Fig. 1).
Alternatively, the activation of the MyD88-independent pathway, through the recruitment of TRIF, induces anti-inflammatory molecules (e.g. interleukin-10 and transforming growth factor-β), and anti-viral type I interferon (IFN)-associated molecules including IFNβ. TRIF is recruited directly by TLR3 and through the adaptor protein TRAM by TLR4 and mediates the activation of tumor necrosis factor receptor associated factor 3 (TRAF3). TRAF3 then activates the complex TANK-binding kinase/ IκB kinase (TBK1/IKKi) that phosphorylates IFN regulatory factor 3 (IRF3), which results in IRF3 translocation into the nucleus and subsequent induction of type I IFNS (IFNα/β). TRIF can also lead to NF-κB activation through the activation of TRAF6 and receptor interacting protein 1 (RIP1) (15, 16), (Fig. 1).
TLR7, TLR8 and TLR9 are able to activate both IRF7 and NF-κB that regulate the production of type I IFNs and inflammatory cytokines, respectively, in a MyD88-dependent manner. The production of type I IFNs is induced predominantly by intracellular TLRs (TLR3, 7, 8 and 9) (19).
TLR Ligands
TLRs can be activated by specific motifs expressed on microorganisms such as bacteria, fungi, parasites and viruses known as pathogen-associated molecular patterns. This class of molecules includes lipopolysaccharide (LPS), a cell wall component of gram-negative bacteria that is a well-known ligand for TLR4. Other bacterial cell wall components, such as lipoteichoic acid and peptidoglycan, are specifically recognized by TLR2. The bacteria flagella protein flagellin is a potent activator of innate immune response through the binding of TLR5. Some TLRs are instead specifically designed to detect nucleic acids. TLR3 binds double-stranded RNA, TLR7 single-stranded RNA, and TLR9 unmethylated cytosine-guanosine-containing DNA oligonucleotides (CpG ODNs) (15).
In addition to the recognition of exogenous pathogens during infection, TLRs act as sentinels of tissue damage and activate a “sterile inflammation” response through the binding of endogenous ligands. DAMPs, released or modified after injury include the extracellular matrix fibronectin, hyaluronan or heparin sulfate (Fig. 2). Additionally, DAMPs include molecules found in the intracellular compartment such as ATP, heat shock proteins and uric acid, and molecules of nuclear origin such as high mobility group box 1 (HMGB1), dsRNA, ssRNA, DNA, microRNA (20) (Fig. 2).
Figure 2.
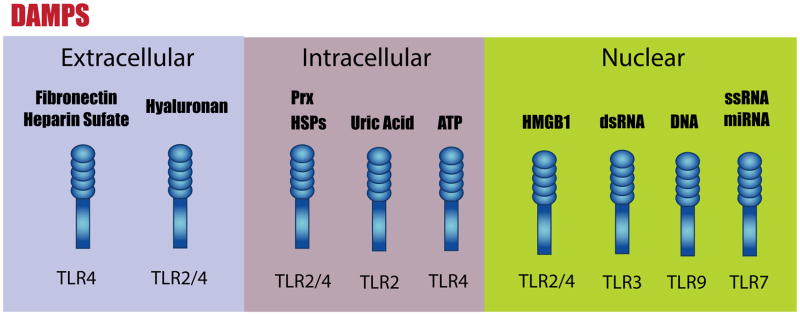
Recognition of damage-associated molecular patterns (DAMPs) by toll-like receptors (TLR)s. DAMPs are endogenous TLR ligands that are released after injury. They can have extracellular, intracellular or nuclear origin. Once released, DAMPs can bind and activate their cognate TLR. Abbreviations: HMGB1, high mobility group box 1; HSPs, heat shock proteins; Prx, peroxiredoxin protein.
The binding of either pathogen-associated molecular patterns or DAMPs to their cognate TLRs activates signaling pathways that eliminate foreign particles or host debris, respectively. This process involves the induction of inflammatory and antiviral molecules.
TLRs in Ischemic Injury
Research on innate immune responses in the generation of ischemic tissue damage has expanded greatly over the past decade. The reduction of blood flow to the brain during stroke results in an acute reaction to the lack of glucose and oxygen that causes loss of ionic balance, calcium dysregulation, acidosis and excitotoxicity (1). The subsequent cascade of events compromises the BBB, which results in infiltration of peripheral leukocytes and activation of endogenous microglia (21). Damage to cellular constituents results in the release of DAMPS (of extracellular, intracellular, and nuclear origin), which bind and activate specific TLRs (22). For example, it has been shown that the DAMP protein HMGB1, a ligand for TLR2 and TLR4, translocates from the nucleus to the cytoplasm after middle cerebral artery occlusion (MCAO) (23, 24). HMGB1 concentration is increased in serum after brain ischemia in both experimental animal models and in stroke patients (25). Interestingly, the administration of a neutralizing anti-HMGB1 antibody significantly reduces ischemic volume after MCAO, demonstrating the involvement of HMGB1 in the exacerbation of the ischemic damage (25, 26). The peroxiredoxin protein (Prx) family, which are also DAMPS, have been shown to be ubiquitous antioxidant enzymes abundantly expressed in the brain. Their expression is increased in the ischemic brain and their function is thought to be neuroprotective (27, 28). However, during cell death, Prx is released into the extracellular compartment where it functions as a DAMP and induces an inflammatory response through the activation of TLR2 and TLR4. The administration of antibodies specific for Prx immediately after experimental stroke significantly reduces infarct volume, suggesting that Prx activation of TLR signaling exacerbates cerebral ischemic injury (29).
The majority of prospective studies on the role of TLRs in brain ischemia have been conducted in rodent models, which have the advantage of advance planning, as well as access to genetic models. A fundamental question addressed in mice was whether a subpopulation of the TLR family is important in the development of ischemic damage in the brain. TLR2 and TLR4 have been extensively studied in this regard. Both receptors are expressed on mouse cortical neurons and the functional loss of either protects against ischemia (30). TLR2 is markedly upregulated in the mouse cortex after ischemia/reperfusion and TLR2 knockout mice sustained significantly less ischemic damage in the brain vs. wild type mice, thereby clearly indicating the detrimental role of TLR2 activation in brain ischemia (31, 32). On the other hand, Hua et al showed that TLR2 knockout mice had increased infarct volume and mortality compared to wild type animals, suggesting a protective role for TLR2 in ischemic injury (33). The differences in the previously mentioned studies may be explained by the different time points chosen to look at the infarct volume after MCAO. In fact, in a more recent paper, Bohacek et al showed that TLR2 deficiency has an important effect on the evolution of the ischemic volume. In particular, they found that TLR2 knockout mice have a reduced ischemic volume at early time points while they show greater ischemic volume compared to wild type at later time points, indicating that TLR2 deficiency delays evolution of ischemic brain lesion (34). Other studies involving TLR4-deficient mice also showed reduced damage compared to controls following ischemia/reperfusion (35), or permanent occlusion of the middle cerebral artery (36). Collectively, these studies indicated that TLR2 and TLR4 play a critical role in cerebral ischemic/reperfusion injury and that their activation leads to exacerbation of brain damage. Unlike TLR2 and TLR4, no role for TLR3 or TLR9 in ischemic injury has been reported (37).
Several clinical studies have examined the role of TLRs in stroke patients, including those that focus on the association of TLR4 polymorphisms with the prevalence of stroke (38, 39). Several studies have associated a significant increase in TLR2 (40) and TLR4 (40–42) on peripheral monocytes, with a poorer outcome following stroke. A subsequent study that looked at gene expression in post-stroke patients found upregulation of peripheral blood TLR4 and MyD88-independent pathways (43). In addition, higher levels of TLR7 and 8 in blood samples were associated with worse stroke outcomes, but not TLR 3 or 9 (44). The authors speculate that these TLRs, which are located in endosomes and are activated by foreign RNA, respond to insufficient clearance of “self” nucleic acids from necrotic debris. Thus, TLRs appear to be involved in ischemic injury both in experimental models and in clinical studies, making them potential targets for therapeutic manipulation.
TLRs in Neuroprotection
Recently, investigators have tested the concept of prophylactic treatment (preconditioning) as a protective mechanism against ischemic damage. This mechanism relies on the body’s response to a sub-threshold level of damage, which subsequently protects it against a more severe insult. Several preconditioning stimuli have been described, including anesthesia, pharmacological compounds, inflammatory agents, and brief ischemia (45). In the mouse, brief ischemic preconditioning (IP) involves the occlusion of the middle cerebral artery for a short time, (10–15 minutes), recovery for >1 day, followed by exposure to a subsequent middle cerebral artery occlusion (> 45 minutes). This preconditioning paradigm significantly reduces ischemic injury and has been shown to mediate this effect, at least partially through TLR4 activation. This was demonstrated in TLR4 deficient mice, which have significantly less IP-induced protection compared to wild type mice (46). Even more importantly, direct administration of low dose TLR ligands prior to stroke induces neuroprotection, further establishing the important role of these receptors in preconditioning (6, 7).
Because TLR stimulation is evident following stroke, low dose TLR activation prior to ischemia has the potential to alter the subsequent stroke-induced TLR response. Indeed, in mouse models, a low dose of TLR2, TLR3, TLR4, TLR7 or TLR9 ligand reduces infarct size when administered prior to stroke (47–51). This state of neuroprotection induced by TLR preconditioning is dose-and time-dependent. For example, preconditioning with the TLR4 ligand LPS, the TLR7 ligand Gardiquimod (GDQ), or the TLR 9 ligand CpG, requires 1 day to develop, lasts approximately 7 days, and is gone by 14 days (52). Thus, the identification of the pathways of protection induced by TLR preconditioning is important to define the mechanisms of endogenous neuroprotection that can lead to the discovery of new potential targets for stroke therapy.
We have performed comparative microarray analysis to determine genomic changes associated with preconditioning-induced neuroprotection. We compared genomic alterations in the brains of mice preconditioned with the TLR4 ligand LPS, the TLR9 ligand CpG, or IP to define conserved transcriptional changes between these 3 preconditioning paradigms. We found that a subset of genes is specifically induced in response to exposure to ischemia after each preconditioning paradigm but is not evident in the genomic profile of brains that were subjected to stroke without preconditioning. The analysis of the promoter region of these shared genes revealed that each contained sequences required for IRF-mediated transcription (53). To define the relevance of IRF in preconditioning-induced protection better, mice deficient for IRF3 or IRF7 were preconditioned with LPS, CpG or IP prior to stroke. It was found that both IRF transcription factors are required for LPS and CpG preconditioning, whereas there was only a partial effect in IP (53). These data indicate that TLR4 and TLR9 preconditioning signal through IRF3 and IRF7 to induce protection. Our data in IP support a partial dependency on IRF3 and IRF7 because protection was attenuated but not lost completely in this model. Our data are consistent with the work by Pradillo discussed above that showed partial attenuation of IP in TLR4-deficient mice (46). Taken together, these data indicate that each of the 3 preconditioning stimuli induces an IFN-related response that is important for neuroprotection.
The importance of IFN to preconditioning-induce protection has been further established in studies demonstrating that LPS preconditioning is associated with increased levels of IFN-β in the brain compared to animals who were not preconditioned prior to exposure to ischemia (54) or traumatic brain injury (55). Importantly, exogenous intracerebroventricular administration of IFN-β immediately before and after ischemia decreases the volume of ischemic damage (54). In vitro treatment with IFN is able to increase neuronal survival following exposure to an excitotoxic insult, suggesting that IFN can provide direct protection to neurons undergoing excitotoxic injury (56).
Tumor necrosis factor is another important factor in TLR preconditioning. Although TNF contributes to the activation of damaging events following stroke (e.g. microglial and vascular endothelial activation, upregulation of cyclooxygenase and induction of apoptotic signaling pathways [57, 58]), both LPS and CpG preconditioning require TNF to induce protection. This has been demonstrated by the loss of neuroprotection by LPS or CpG preconditioning in TNF-deficient mice (50, 52). Preconditioning by administration of LPS causes an increase in systemic TNF levels within hours after injection (52). We have speculated that the LPS-induced increase in TNF levels prior to stroke may be an early signal that primes the brain against subsequent ischemic injury by limiting the subsequent harmful TNF signaling in the setting of ischemia (52). As discussed above, LPS (TLR4) and CpG (TLR9) preconditioning require TNF, IRF3 and IRF7; however, neither require type I IFN receptor (IFNAR) signaling (48), suggesting that TLR signaling directly activates IRF transcription factors independent of the IFNAR. In contrast, studies using the TLR7 ligand Gardiquimod (GDQ) or the TLR3 ligand Poly I:C demonstrated that both require IFNAR (48, 59). Thus, the specific mechanism of protection may be dependent on the TLR ligand; however, there is a common link to IFN and IFN-related transcription that appears to be essential.
To summarize, stroke induces the release of DAMPs, which activate TLRs. In a non-preconditioned animal the TLR activation by DAMPs results in an inflammatory response that increases ischemic damage. Instead, in a preconditioned animal, the TLRs response to the presence of DAMPs has been reprogrammed by the previous exposure to a TLR ligand. As a consequence, the TLRs response initiates signaling dominated by IRF-IFN, resulting in reduced ischemic damage (Fig. 3).
Figure 3.

Model of toll-like receptor (TLR) preconditioning-induced protection. Strokes cause the release of damage-associated molecular patterns (DAMPs). Once released, the DAMPs bind and activate TLRs. Non-preconditioned TLRs respond to DAMPs by activating an inflammatory response that increases ischemic damage. In preconditioned TLR the response to the presence of DAMPs has been reprogrammed by previous exposure to a TLR ligand. Consequently, they activate an interferon regulatory factor (IRF)-mediated response that is associated with neuroprotection. HMGB1, high mobility group box 1; HSPs, heat shock proteins; Prx, peroxiredoxin protein.
Target of TLR Preconditioning
Although the signaling pathways involved in the TLR preconditioning-induce neuroprotection are beginning to be deciphered, little is known about the site-of-action of TLR-mediated protection. TLRs are expressed by a range of cells. Systemically, TLRs are generally expressed by antigen-presenting cells including B cells, dendritic cells, monocytes, macrophages in the blood, epithelial and endothelial cells and lymphoid tissues. In the brain, TLRs are constitutively expressed by microglia and astrocytes, but other brain cells (e.g. brain endothelial cells, oligodendrocytes and neurons) also express TLRs (60, 61). Thus, cells in the brain and periphery are potential targets of TLR ligand stimulation and these cells appear to play a role in TLR preconditioning-induced protection.
Studies involving peripheral viral infection showed that systemic administration of TLR ligands affects gene expression in the brain, and that this effect is not necessarily associated with the TLR ligand crossing the BBB (62). For example, blood plasma collected 3 hours following intraperitoneal injection of Poly I:C and subsequently injected into naïve animals induced an inflammatory response in the brain, although negligible amounts of PolyI:C were detected in the blood. The authors postulated that Poly I:C, rather than crossing the BBB and directly affecting cells of the brain parenchyma, is likely to affect brain gene expression via indirect mechanisms (62).
In the context of preconditioning, it is still unclear whether the effect of preconditioning requires direct or indirect stimulation of the brain. To investigate this, we generated TLR9 bone-marrow chimeric mice that exhibited TLR9 expression only on hematopoietic cells or only on parenchymal cells but not both. We found that neuroprotection by CpG preconditioning is absent when mice were deficient in TLR9 in either hematopoietic or parenchymal cells (63). These data suggest that TLR9-induced neuroprotection requires the expression of TLR9 in both cellular compartments and that there is an important interaction between hematopoietic and parenchymal cells. Because endothelial cells and hematopoietic cells express TLRs, systemic administration of TLR ligands directly exposes both the endothelium and the circulating hematopoietic cells to the ligand potentially altering the interaction of circulating cells to the activated endothelium. This interaction may activate signaling pathways that propagate through the brain resulting in altered gene expression and reprogramming (64).
It cannot be excluded that TLR ligands could cross the BBB and have a direct effect on brain cells. In vitro studies with cortical neuronal cultures showed that TLR ligands induce protection against oxygen glucose deprivation (49, 50, 52), suggesting that TLR ligands directly protect neurons against injury. We also found that intranasal administration of CpG to mice protects against ischemic damage in a model of stroke, further suggesting that CpG may have a direct protective effect on brain cells (63).
BBB Involvement in TLR Preconditioning
The BBB is the physical interface between the blood and the brain and consists of a selective barrier formed by endothelial cells lining the cerebral microvessels. The integrity of the BBB is due to the presence of tight junction proteins between adjacent endothelial cells that strongly limits the passage of molecules between the vascular and brain compartment. BBB endothelial cells are in close association with the astrocytes and microglia of the brain parenchyma resulting in a complex network of cellular interactions that is essential for the maintenance of the nervous system microenvironment. Thus, the endothelial cells of the BBB are in a prime location to transmit signals from systemically administered TLR ligands.
We have recently shown that TLR preconditioning can directly affect the BBB. We used an in vitro BBB model consisting of a co-culture of primary brain microvascular endothelial cells and primary mixed astrocytes/microglial cells. This in vitro BBB system retains the features of a functional BBB, such as a high trans-endothelial electrical resistance (TEER), which is the resistance of the endothelial cells to the passage of ions from the upper to the lower compartment, and low permeability. Using this model we evaluated the effect of preconditioning with Poly-ICLC (carboxymethylcellulose, polyinosinic-polycytidylic acid, and poly-L-lysine double-stranded RNA), a ligand for TLR3 and for the cytoplasmic receptors melanoma differentiation associated gene-5 (MDA5) and retinoic acid-inducible gene I (RIGI), against modeled ischemia, which consisted of 5 hours of oxygen-glucose deprivation (OGD). Poly-ICLC was administered 24 hours prior to OGD. OGD caused a drastic reduction of TEER and a significant increase in permeability (Fig. 4). Poly-ICLC preconditioning preserved BBB function against OGD, as shown by the amelioration of OGD-induced effects on TEER and permeability of the in vitro BBB system treated with Poly-ICLC compared to vehicle-treated cells (Fig. 4A) (59). In addition, Poly-ICLC preconditioning also preserved in vitro BBB integrity as shown by the maintenance of the tight junction proteins (59).
Figure 4.

Poly-ICLC (carboxymethylcellulose, polyinosinic-polycytidylic acid, and poly-L-lysine double-stranded RNA) preconditioning effect in the in vitro blood-brain barrier (BBB) model. (A–C) Poly-ICLC preconditioning prevented the reduction of trans-endothelial electrical resistance (TEER) and the increase of permeability induced by oxygen-glucose deprivation (OGD) when wild type (WT) cells were used in the co-culture system (A), while it was not effective when interferon-β (IFNβ)−/− glial cells (B), or type I interferon receptor-deficient (IFNAR−/−) endothelial cells were used in the co-culture (C). Data are reported as mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control (CTR); °p < 0.05, °°p < 0.01 vs. OGD. Adapted from (59).
Interestingly, as in the in vivo model, Poly-ICLC-induced BBB protection was dependent on signaling through the IFNAR. In fact, we found that Poly-ICLC induced astrocyte/microglial cell production of IFNβ prior to OGD (59). This induction is required for the Poly-ICLC-induced protective effect, as shown by the lack of protection when IFNβ-deficient glial cells were used in the co-culture (Fig. 4B). We further showed that Poly-ICLC preconditioning is not effective in inducing protection when type I IFN receptor-deficient endothelial cells were used in the co-culture (Fig. 4C). Based on these results, we hypothesized that systemically administered Poly-ICLC is able to cross the BBB endothelial cells, reach the glial cells and stimulate them to produce IFNβ. This IFNβ is then able to bind and signal through its receptor (IFNAR) expressed on the abluminal side of the BBB endothelial cells to stabilize the BBB and confer protection. This is the first in vitro demonstration that poly-ICLC preconditioning attenuates ischemia-induced BBB dysfunction. Importantly, this mechanism can be an important feature of TLR-mediated neuroprotection and highlights the therapeutic potential of targeting BBB signaling pathways to protect the brain against stroke.
Conclusions
TLRs are surveillance receptors that have important roles in detection and removal of pathogens. In addition, evidence from experimental models showed that TLRs play a key role in initiating events that contribute to ischemic damage. Importantly, preconditioning that results in the modulation of TLR activation confers a neuroprotective state that is tolerant against ischemic injury. Modulation of TLR activation by preconditioning offers great promise as prophylactic therapy against ischemic injury. In general, preconditioning is a powerful strategy that can be used to protect the brain against injury. This concept has been applied in the clinic using remote ischemic preconditioning wherein exposure to short sublethal ischemia and reperfusion in one organ is used to induce ischemic tolerance in a distant organ (65). Encouraging clinical trials showed that remote ischemic preconditioning applied in different clinical settings (pediatric surgery, coronary bypass, abdominal aneurysm repair and coronary angioplasty) protects cardiac tissue, as measured by reduced troponin release, and also preserves kidney function (65). Interestingly, in one of these studies where limb preconditioning prior to cardiac surgery was used to protect the heart they also found reduced cerebral ischemic events at 6 months following surgery, suggesting that the protective effect extends to the brain (66). In a more recent study, Meng et al found that 5 brief cycles of bilateral upper arm ischemic preconditioning (BAIPC) performed twice daily over 300 consecutive days reduced the occurrence of stroke in patients with intracranial arterial stenosis (67). This suggested that BAIPC could be effective in ameliorating abnormal cerebral perfusion and reducing the recurrence of stroke in high-risk patients. The development of pharmacological agents would be preferable to long and uncomfortable protocols like BAIPC. Thus, TLR agonists may be considered a viable approach because they can be administered easily through simple injection in particular conditions before surgery as prophylactic treatments against stroke.
The promise of using TLR agonists as prophylaxis against brain ischemic injury has been confirmed in a non-human primate model of stroke (68). Administration of a TLR9 ligand 3 days prior to experimentally induced stroke significantly reduced ischemic damage. This study represents the first to show that pharmacological preconditioning induces protection against ischemic injury in a clinically relevant model and represents an important proof-of-concept justifying continued development of TLR agonists for prophylactic treatment against stroke.
Some TLR agonists have been approved by the FDA for use in humans (69), making clinical translation more plausible. One such agonist is the bacillus Calmette-Guérin. This is an attenuated strain of Mycobacterium bovis, an agonist for TLR2 and TLR4, used as a vaccine against tuberculosis as well as an adjuvant in the immunotherapy of in situ bladder carcinoma. The second is a derivative of Salmonella Minnesota endotoxin, the monophosphoryl lipid A recognizing TLR4. It is included in the formulation of Cervarix, a vaccine against human papillomavirus. The third is the TLR7 agonist imiquimod, a synthetic imidazoquinoline employed for actinic keratosis, superficial basal cell carcinoma, and external genital warts. These findings demonstrate that TLR agonists have a favorable safety profile in humans and suggest that they could be good candidates for development as therapies against stroke.
The study of TLR preconditioning in the context of stroke has 2 important consequences. The first is to increase our knowledge on the features of these compounds that may be helpful for the development of prophylactic therapies that can be used in selected patient population at high risk of stroke or peri-operative ischemic injury. The second is to understand the molecular basis of endogenous neuroprotection. The understanding of the mechanisms associated with the protective state can lead to the development of acute approaches aimed at enhancing endogenous neuroprotection following stroke. The expansion of the clinical window of drug efficacy is critical for stroke treatment and remains a major goal into the foreseeable future.
Acknowledgments
This work was supported by funding from the National Institute of Neurological Disorders and Stroke NS062381, NS050567 and NS064953.
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- 1.Doyle KP, Simon RP, Stenzel-Poore MP. Mechanisms of ischemic brain damage. Neuropharmacology. 2008;55:310–18. doi: 10.1016/j.neuropharm.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carpenter CR, Keim SM, Milne WK, et al. Thrombolytic therapy for acute ischemic stroke beyond three hours. J Emerg Med. 2011;40:82–92. doi: 10.1016/j.jemermed.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shobha N, Buchan AM, Hill MD. Thrombolysis at 3–4. 5 hours after acute ischemic stroke onset--evidence from the Canadian Alteplase for Stroke Effectiveness Study (CASES) registry. Cerebrovasc Dis. 2011;31:223–28. doi: 10.1159/000321893. [DOI] [PubMed] [Google Scholar]
- 5.Shichita T, Ago T, Kamouchi M, et al. Novel therapeutic strategies targeting innate immune responses and early inflammation after stroke. J Neurochem. 2012;123 (Suppl 2):29–38. doi: 10.1111/j.1471-4159.2012.07941.x. [DOI] [PubMed] [Google Scholar]
- 6.Marsh BJ, Williams-Karnesky RL, Stenzel-Poore MP. Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience. 2009;158:1007–20. doi: 10.1016/j.neuroscience.2008.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vartanian KB, Stenzel-Poore MP. Toll-like receptor tolerance as a mechanism for neuroprotection. Transl Stroke Res. 2010;1:252–60. doi: 10.1007/s12975-010-0033-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barber PA, Hach S, Tippett LJ, et al. Cerebral ischemic lesions on diffusion-weighted imaging are associated with neurocognitive decline after cardiac surgery. Stroke. 2008;39:1427–33. doi: 10.1161/STROKEAHA.107.502989. [DOI] [PubMed] [Google Scholar]
- 9.Adams HPJ. Ischemic cerebrovascular complications of cardiac procedures. Circulation. 2010;121:846–47. doi: 10.1161/CIR.0b013e3181d4c53b. [DOI] [PubMed] [Google Scholar]
- 10.Kahlert P, Al-Rashid F, Dottger P, et al. Cerebral embolization during transcatheter aortic valve implantation: a transcranial Doppler study. Circulation. 2012;126:1245–55. doi: 10.1161/CIRCULATIONAHA.112.092544. [DOI] [PubMed] [Google Scholar]
- 11.Mallard C. Innate immune regulation by toll-like receptors in the brain. ISRN Neurol. 2012;2012:701950. doi: 10.5402/2012/701950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 13.Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol. 2006;7:49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 14.Song DH, Lee JO. Sensing of microbial molecular patterns by Toll-like receptors. Immunol Rev. 2012;250:216–29. doi: 10.1111/j.1600-065X.2012.01167.x. [DOI] [PubMed] [Google Scholar]
- 15.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 16.Ostuni R, Zanoni I, Granucci F. Deciphering the complexity of Toll-like receptor signaling. Cell Mol Life Sci. 2010;67:4109–34. doi: 10.1007/s00018-010-0464-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qian C, Cao X. Regulation of Toll-like receptor signaling pathways in innate immune responses. Ann N Y Acad Sci. 2013;1283:67–74. doi: 10.1111/j.1749-6632.2012.06786.x. [DOI] [PubMed] [Google Scholar]
- 18.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 19.Brown J, Wang H, Hajishengallis GN, et al. TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J Dent Res. 2011;90:417–27. doi: 10.1177/0022034510381264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010;2010 doi: 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanisch UK, Johnson TV, Kipnis J. Toll-like receptors: roles in neuroprotection? Trends Neurosci. 2008;31:176–82. doi: 10.1016/j.tins.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Qiu J, Nishimura M, Wang Y, et al. Early release of HMGB-1 from neurons after the onset of brain ischemia. J Cereb Blood Flow Metab. 2008;28:927–38. doi: 10.1038/sj.jcbfm.9600582. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, Takahashi HK, Liu K, et al. Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke. 2011;42:1420–28. doi: 10.1161/STROKEAHA.110.598334. [DOI] [PubMed] [Google Scholar]
- 25.Muhammad S, Barakat W, Stoyanov S, et al. The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci. 2008;28:12023–31. doi: 10.1523/JNEUROSCI.2435-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu K, Mori S, Takahashi HK, et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007;21:3904–16. doi: 10.1096/fj.07-8770com. [DOI] [PubMed] [Google Scholar]
- 27.Patenaude A, Murthy MR, Mirault ME. Emerging roles of thioredoxin cycle enzymes in the central nervous system. Cell Mol Life Sci. 2005;62:1063–80. doi: 10.1007/s00018-005-4541-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rashidian J, Rousseaux MW, Venderova K, et al. Essential role of cytoplasmic cdk5 and Prx2 in multiple ischemic injury models, in vivo. J Neurosci. 2009;29:12497–505. doi: 10.1523/JNEUROSCI.3892-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shichita T, Hasegawa E, Kimura A, et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med. 2012;18:911–17. doi: 10.1038/nm.2749. [DOI] [PubMed] [Google Scholar]
- 30.Tang SC, Arumugam TV, Xu X, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci. 2007;104:13798–803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lehnardt S, Lehmann S, Kaul D, et al. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol. 2007;190:28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 32.Ziegler G, Harhausen D, Schepers C, et al. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun. 2007;359:574–79. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]
- 33.Hua F, Ma J, Ha T, et al. Differential roles of TLR2 and TLR4 in acute focal cerebral ischemia/reperfusion injury in mice. Brain Res. 2009;1262:100–108. doi: 10.1016/j.brainres.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bohacek I, Cordeau P, Lalancette-Hebert M, et al. Toll-like receptor 2 deficiency leads to delayed exacerbation of ischemic injury. J Neuroinflammation. 2012;9:191. doi: 10.1186/1742-2094-9-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao CX, Yang QW, Lv FL, et al. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem Biophys Res Commun. 2007;353:509–14. doi: 10.1016/j.bbrc.2006.12.057. [DOI] [PubMed] [Google Scholar]
- 36.Caso JR, Pradillo JM, Hurtado O, et al. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- 37.Hyakkoku K, Hamanaka J, Tsuruma K, et al. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience. 2010;171:258–67. doi: 10.1016/j.neuroscience.2010.08.054. [DOI] [PubMed] [Google Scholar]
- 38.Reismann P, Lichy C, Rudofsky G, et al. Lack of association between polymorphisms of the toll-like receptor 4 gene and cerebral ischemia. J Neurol. 2004;251:853–58. doi: 10.1007/s00415-004-0447-7. [DOI] [PubMed] [Google Scholar]
- 39.Lin YC, Chang YM, Yu JM, et al. Toll-like receptor 4 gene C119A but not Asp299Gly polymorphism is associated with ischemic stroke among ethnic Chinese in Taiwan. Atherosclerosis. 2005;180:305–309. doi: 10.1016/j.atherosclerosis.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 40.Brea D, Blanco M, Ramos-Cabrer P, et al. Toll-like receptors 2 and 4 in ischemic stroke: outcome and therapeutic values. J Cereb Blood Flow Metab. 2011;31:1424–31. doi: 10.1038/jcbfm.2010.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Urra X, Villamor N, Amaro S, et al. Monocyte subtypes predict clinical course and prognosis in human stroke. J Cereb Blood Flow Metab. 2009;29:994–1002. doi: 10.1038/jcbfm.2009.25. [DOI] [PubMed] [Google Scholar]
- 42.Yang QW, Li JC, Lu FL, et al. Upregulated expression of toll-like receptor 4 in monocytes correlates with severity of acute cerebral infarction. J Cereb Blood Flow Metab. 2008;28:1588–96. doi: 10.1038/jcbfm.2008.50. [DOI] [PubMed] [Google Scholar]
- 43.Wu D, Lee YC, Liu HC, et al. Identification of TLR downstream pathways in stroke patients. Clin Biochem. 2013;46:1058–64. doi: 10.1016/j.clinbiochem.2013.05.059. [DOI] [PubMed] [Google Scholar]
- 44.Brea D, Sobrino T, Rodriguez-Yanez M, et al. Toll-like receptors 7 and 8 expression is associated with poor outcome and greater inflammatory response in acute ischemic stroke. Clin Immunol. 2011;139:193–98. doi: 10.1016/j.clim.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 45.Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8:398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pradillo JM, Fernandez-Lopez D, Garcia-Yebenes I, et al. Toll-like receptor 4 is involved in neuroprotection afforded by ischemic preconditioning. J Neurochem. 2009;109:287–94. doi: 10.1111/j.1471-4159.2009.05972.x. [DOI] [PubMed] [Google Scholar]
- 47.Hua F, Ma J, Ha T, et al. Preconditioning with a TLR2 specific ligand increases resistance to cerebral ischemia/reperfusion injury. J Neuroimmunol. 2008;199:75–82. doi: 10.1016/j.jneuroim.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leung PY, Stevens SL, Packard AE, et al. Toll-like receptor 7 preconditioning induces robust neuroprotection against stroke by a novel type I interferon-mediated mechanism. Stroke. 2012;43:1383–89. doi: 10.1161/STROKEAHA.111.641522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Packard AE, Hedges JC, Bahjat FR, et al. Poly-IC preconditioning protects against cerebral and renal ischemia-reperfusion injury. J Cereb Blood Flow Metab. 2012;32:242–47. doi: 10.1038/jcbfm.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stevens SL, Ciesielski TM, Marsh BJ, et al. Toll-like receptor 9: a new target of ischemic preconditioning in the brain. J Cereb Blood Flow Metab. 2008;28:1040–47. doi: 10.1038/sj.jcbfm.9600606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tasaki K, Ruetzler CA, Ohtsuki T, et al. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 1997;748:267–70. doi: 10.1016/s0006-8993(96)01383-2. [DOI] [PubMed] [Google Scholar]
- 52.Rosenzweig HL, Minami M, Lessov NS, et al. Endotoxin preconditioning protects against the cytotoxic effects of TNFalpha after stroke: a novel role for TNFalpha in LPS-ischemic tolerance. J Cereb Blood Flow Metab. 2007;27:1663–74. doi: 10.1038/sj.jcbfm.9600464. [DOI] [PubMed] [Google Scholar]
- 53.Stevens SL, Leung PY, Vartanian KB, et al. Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. Journal of Neuroscience. 2011;31:8456–63. doi: 10.1523/JNEUROSCI.0821-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marsh B, Stevens SL, Packard AE, et al. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci. 2009;29:9839–49. doi: 10.1523/JNEUROSCI.2496-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Longhi L, Gesuete R, Perego C, et al. Long-lasting protection in brain trauma by endotoxin preconditioning. J Cereb Blood Flow Metab. 2011;31:1919–29. doi: 10.1038/jcbfm.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pappas DJ, Gabatto PA, Oksenberg D, et al. Transcriptional expression patterns triggered by chemically distinct neuroprotective molecules. Neuroscience. 2012;226:10–20. doi: 10.1016/j.neuroscience.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hallenbeck JM. The many faces of tumor necrosis factor in stroke. Nat Med. 2002;8:1363. doi: 10.1038/nm1202-1363. [DOI] [PubMed] [Google Scholar]
- 58.Muppidi JR, Tschopp J, Siegel RM. Life and death decisions: Secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity. 2004;21:461–65. doi: 10.1016/j.immuni.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 59.Gesuete R, Packard AE, Vartanian KB, et al. Poly-ICLC preconditioning protects the blood-brain barrier against ischemic injury in vitro through type I interferon signaling. J Neurochem. 2012;123 (Suppl 2):75–85. doi: 10.1111/j.1471-4159.2012.07946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bsibsi M, Ravid R, Gveric D, et al. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–21. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 61.Constantin D, Cordenier A, Robinson K, et al. Neisseria meningitidis-induced death of cerebrovascular endothelium: mechanisms triggering transcriptional activation of inducible nitric oxide synthase. J Neurochem. 2004;89:1166–74. doi: 10.1111/j.1471-4159.2004.02393.x. [DOI] [PubMed] [Google Scholar]
- 62.Fil D, Borysiewicz E, Konat GW. A broad upregulation of cerebral chemokine genes by peripherally-generated inflammatory mediators. Metab Brain Dis. 2011;26:49–59. doi: 10.1007/s11011-010-9231-9. [DOI] [PubMed] [Google Scholar]
- 63.Packard AE, Leung PY, Vartanian KB, et al. TLR9 bone marrow chimeric mice define a role for cerebral TNF in neuroprotection induced by CpG preconditioning. J Cereb Blood Flow Metab. 2012;32:2193–2200. doi: 10.1038/jcbfm.2012.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quan N, Banks WA. Brain-immune communication pathways. Brain Behav Immun. 2007;21:727–35. doi: 10.1016/j.bbi.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 65.Veighey K, Macallister RJ. Clinical applications of remote ischemic preconditioning. Cardiol Res Pract. 2012;2012:620681. doi: 10.1155/2012/620681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoole SP, Heck PM, Sharples L, et al. Cardiac Remote Ischemic Preconditioning in Coronary Stenting (CRISP Stent) Study: a prospective, randomized control trial. Circulation. 2009;119:820–27. doi: 10.1161/CIRCULATIONAHA.108.809723. [DOI] [PubMed] [Google Scholar]
- 67.Meng R, Asmaro K, Meng L, et al. Upper limb ischemic preconditioning prevents recurrent stroke in intracranial arterial stenosis. Neurology. 2012;79:1853–61. doi: 10.1212/WNL.0b013e318271f76a. [DOI] [PubMed] [Google Scholar]
- 68.Bahjat FR, Williams-Karnesky RL, Kohama SG, et al. Proof of concept: pharmacological preconditioning with a Toll-like receptor agonist protects against cerebrovascular injury in a primate model of stroke. J Cereb Blood Flow Metab. 2011;31:1229–42. doi: 10.1038/jcbfm.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vacchelli E, Galluzzi L, Eggermont A, et al. Trial watch: FDA-approved Toll-like receptor agonists for cancer therapy. Oncoimmunology. 2012;1:894–907. doi: 10.4161/onci.20931. [DOI] [PMC free article] [PubMed] [Google Scholar]