

Abstract
Transcription factors (TFs) bind to DNA and regulate the transcription of nearby genes. However, only a small fraction of TF binding sites have such regulatory effects. Here we search for the predictors of functional binding sites by carrying out a systematic computational screening of a variety of contextual factors (histone modifications, nuclear lamin-bindings, and cofactor bindings). We used regression analysis to test if contextual factors are associated with upregulation or downregulation of neighboring genes following the induction or knockdown of the 9 TFs in mouse embryonic stem (ES) cells. Functional TF binding sites appeared to be either active (i.e., bound by P300, CHD7, mediator, cohesin, and SWI/SNF) or repressed (i.e., with H3K27me3 histone marks and bound by Polycomb factors). Active binding sites mediated the downregulation of nearby genes upon knocking down the activating TFs or inducing repressors. Repressed TF binding sites mediated the upregulation of nearby genes (e.g., poised developmental regulators) upon inducing TFs. In addition, repressed binding sites mediated repressive effects of TFs, identified by the downregulation of target genes after the induction of TFs or by the upregulation of target genes after the knockdown of TFs. The contextual factors associated with functions of DNA-bound TFs were used to improve the identification of candidate target genes regulated by TFs.
Key words: : chromatin modification, cis-regulatory module, embryonic stem cells, enhancer, target genes, transcription factor binding site
1. Introduction
Transcription factors (TFs) bind to DNA and regulate the expression of nearby genes (i.e., target genes of TFs). Locations of TF binding sites are identified at the genome scale using chromatin immunoprecipitation (ChIP) followed by sequencing (ChIP-seq) or hybridization to microarrays (ChIP-chip) (Buck and Lieb, 2004; Chen et al., 2008; Marson et al., 2008). However, the majority of binding sites are not involved in the regulation of expression of targets (Sharov et al., 2008; Yu et al., 2011). A novel method STARR-seq (Arnold et al., 2013) helped to identify functional enhancers in Drosophila. However, enhancers were tested outside of their genomic contexts, and this method has not been used for mammals. Thus, it is important to identify additional factors (e.g., histone modifications and binding of cofactors) that control the functionality of DNA-bound TFs. For example, mammalian enhancers are characterized by H3K4me1 chromatin marks (Barski et al., 2007; Heintzman et al., 2007; Creyghton et al., 2010; Rye et al., 2011). In addition, enhancers that are active in a specific cell type are bound by P300 and CHD7 (Visel et al., 2009; Schnetz et al., 2010) and mediator and cohesin complexes (Kagey et al., 2010), and enriched in histone acetylation (Creyghton et al., 2010). Enhancers near silent genes often have H3K27me3 histone marks and were called “poised enhancers,” because they are associated with target genes that are ready for activation (Rada-Iglesias et al., 2011; Zentner et al., 2011; Xiao et al., 2012).
Current analyses of the function of regulatory DNA regions are usually based on the comparison of chromatin modifications, cofactor bindings, and “static” gene expression profiles in various cell types (Creyghton et al., 2010; Kagey et al., 2010; Rada-Iglesias et al., 2011). Obviously, cell types differ in the expression of hundreds of TFs, and therefore the differences in the expression of downstream genes cannot be attributed to a specific TF. To analyze the role of histone modifications and cofactors in modulating the effect of TF binding to DNA on gene expression, it is necessary to quantify the effect of a large number of potential factors with multivariate statistics, which may help to discriminate causal relationships from mere correlations. So far, only one study applied multivariate statistics to discriminate between enhancers bound by Oct4, Sox2, and Nanog (OSN) in embryonic stem (ES) cells from promoter-like DNA segments bound by Myc and Mycn (Chen et al., 2012). This study showed the importance of p300, H3K4me1, Med12, and Nipbl as an epigenetic context for OSN functions. The limitation of the study is that it did not discriminate between the activating and repressing roles of TFs, and it was applied to only one kind of active enhancers.
Here we have used published data sets on gene expression changes after induction or knockdown of TFs in mouse ES cells (Nishiyama et al., 2009; Correa-Cerro et al., 2011; Nishiyama et al., 2013) and systematically analyzed the associations between the functionality of binding sites of nine TFs with high, low, and intermediate expression and their chromatin and cofactor profiles. Unlike the “static” gene expression profiles used in the previous studies, “dynamic” changes of gene expression profiles capture short-term responses of target genes to a specific TF, and thus they are more relevant for studying the effect of chromatin modifications and cofactors at TF binding sites. This method allowed us to discriminate between activating and repressing effects of TFs.
2. Methods
Genome coordinates of TF binding sites are taken from the original articles (Supplementary Table S1; Supplementary Material is available online at www.liebertpub.com/cmb) and converted to the mm9 version of mouse genome using the UCSC LiftOver tool. Binding sites of TFs are associated with the nearest transcription start site (TSS) of a gene with symbol (excluding RIKEN clones, hypothetical genes, and gene models) within 100 kb maximum distance. If no gene with a symbol is found, then binding site is associated with the nearest other gene within 100 kb. The location of the main TSS for 17,412 nonredundant genes is taken from Mikkelsen et al. (2007) and for 5,101 other genes is identified using CisView (Sharov et al., 2006). Whole-genome data on contextual factors are taken from ChIP-seq data (Supplementary Table S1). The abundance of ChIP-seq reads of contextual factors is estimated within a 1 kb region centered at each TF binding site and normalized to the average density in the genome. Data from multiple data sources for H3K9me3 and H3K27me3 are then averaged. Binding of DNA to Lamin B1 is assessed by the median log ratio of DAM-ID signals for all probes in the tiling arrays within a 1 kb region centered at each TF binding site (Kim et al., 2011).
To identify contextual factors associated with functional TF binding sites, the change of gene expression of each target gene after manipulation of a certain TF is regressed against the abundance of each factor at TF binding sites. Response of genes to the induction of TFs is measured as a log ratio of gene expression in cells cultured in the absence of doxycycline (dox− condition: a transgenic TF is induced) versus cells cultured in the presence of doxycycline (dox+ condition: a transgenic TF is silent) (Nishiyama et al., 2009; Correa-Cerro et al., 2011). Response of genes to the knockdown of TFs is measured as a log ratio of gene expression in cells transfected with shRNA against specific TF versus median expression (Nishiyama et al., 2013). Based on preliminary analysis with different transformations (log, square root, and none), the following transformations of variables are selected: log[abs(xi)], where xi is the log ratio of expression change of target gene i, and log(qi + 0.5), where qi is the enrichment ratio of a contextual factor at TF binding sites near gene i compared with the genome-wide abundance of this factor. Transformation of variables helps to stabilize the error variance. In the case of multiple binding sites per target gene, we use three alternative methods of calculating the abundance of each contextual factor: (1) average abundance of factor in all TF binding sites associated with target gene; (2) abundance in the TF binding site closest to the TSS of target gene; and (3) highest abundance of this factor among TF binding sites associated with target gene. Also, we use four additional methods of analysis: (4) rank-correlation applied to the average abundance of factor in all TF binding sites associated with target gene (i.e., it is similar to method 1 but use rank correlation); (5–7) multivariate regression is similar to methods 1–3 but the distance from binding sites of TFs to TSS of targets is used as the second independent variable. Distance from TF binding sites to TSS, d, is transformed: exp(−cd), where coefficient c = 0.0001 (it is selected based on preliminary tests). The analysis is done separately for upregulated and downregulated target genes (change >1.2-fold), because positive and negative effects on gene expression may be associated with different molecular mechanisms. The strength of such relationships is assessed with z-value: z = b/SE(b), where b is regression coefficient and SE(b) is its standard error. z-Values obtained by seven different methods of analyses matched closely (Supplementary Tables S2 and S3). Statistical significance is determined using random permutations of gene expression responses within each subset of analyzed target genes. To account for possible effects of multiple hypotheses testing, we use the false discovery rate criterion (FDR < 0.05) to call effects significant. FDR is estimated directly from the results of permutation analysis. In Figure 2, we use average z-values obtained with seven methods of analysis and mark the effect as significant if FDR is <0.05 for all methods.
FIG. 2.

Correlation analysis of the response of target genes to manipulations of transcription factors (TFs) and presence of chromatin modifications and cofactors at TF binding sites. Correlation patterns reveal two kinds of functional TF binding sites: (1) active sites (marked “A” at right) are bound by P300, CHD7, mediator (Med1, Med12), cohesin (Smc1, Nipbl, Rad21), and SWI/SNF (Brg1), and (2) repressed sites (marked “R” at right) carry repressive histone marks H3K27me3 and are bound by Polycomb TFs (Ezh2, Suz12, Mtf2, and Ring1b). Color corresponds to the average z-value from seven types of analysis, including three algorithms for estimating factor abundance, rank correlation, and multivariate regression; statistical significance is evaluated using false discovery rate (FDR) estimated from permutations of gene expression change. Effects marked as significant satisfy condition FDR ≤ 0.05 in all 7 methods of analysis.
The expected proportion of false positives (EPFP) for a target gene with rank i in the list of all genes ordered by increasing (or decreasing) log ratio of expression response to the manipulation of a TF is estimated using the following equation:
 |
where Q is the proportion of targets (i.e., genes with TF binding sites) within the “control” set of genes that have limited change of expression (here, <1.2-fold change), and Pj is the proportion of targets among genes with rank ≤j. The minimum operator in Equation 1 ensures the monotonic property of the EPFP (Sharov et al., 2008). If the abundance of contextual factors at TF binding sites is significantly correlated with gene expression change after the manipulation of TFs (see Fig. 2), we use these factors to narrow down the set of TF binding sites, hoping to find more significant targets regulated by TFs. The modified EPFP* for a target gene with rank i is estimated:
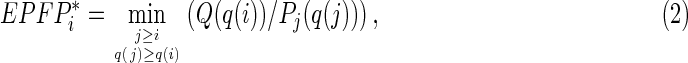 |
where q(i) is the enrichment ratio of a contextual factor at the TF binding site(s) near gene i; Q(q(i)) is the proportion of targets within the “control” set of genes (see above) with factor enrichment ratio >q(i), and Pj(q(j)) is the proportion of targets among genes with rank ≤j and enrichment ratio >q(j). Equation 2 is derived using the same logic as Equation 1; the only difference is that the set of target genes is not fixed but instead depends on the abundance of contextual factors. The minimum is found by varying two parameters: the threshold of fold change after the induction of TF (gene rank j), and the enrichment ratio of sequence reads, q(j). Because q(j) depends on j, the minimization is one-dimensional.
3. Findings and Discussion
If a chromatin modification or cofactor (in short, contextual factors) assists a TF in regulating the expression of its target genes, then it is reasonable to expect that the magnitude of expression change among target genes after the induction (or knockdown) of the TF correlates with the abundance of contextual factors at TF binding sites near target genes. Thus, we used regression analysis to study the association between the response of target genes and the abundance of contextual factors at TF binding sites after manipulation of 9 TFs in ES cells (Fig. 1). These 9 TFs were selected as representatives of genes expressed highly (Pou5f1, Sox2, and Nanog), genes expressed at the intermediate level (Klf4, Tcf3, and Tcfcp2l1), and silent genes (Cdx2, Myod1, and Sfpi1). We analyzed data sets of 12 manipulations of these TFs: overexpression of all 9 TFs in doxycycline-regulatable transgenic cell lines (Nishiyama et al., 2009) and knockdown of 3 highly expressed TFs via shRNA (Nishiyama et al., 2013). For example, the response of target genes to the knockdown of Nanog correlated positively with the enrichment of P300 at the binding sites of Nanog (Fig. 1). The analysis was repeated using nonparametric correlation (Spearman) and multivariate regression analysis to model the change of gene expression versus two independent variables: (1) abundance of contextual factors at TF binding sites and (2) distance from TF binding sites to the TSS of targets. Results (z-values) obtained with 7 different methods of analysis (see Methods) for 648 combinations of factors ([12 TF manipulations] × [27 contextual factors] × [2 directions of gene expression changes]) appeared consistent (r > 0.9 for all pair-wise comparisons) (Supplementary Table S2). Statistical significance was assessed using permutation test and false discovery rate (FDR < 0.05).
FIG. 1.

Association between the enrichment of P300 at Nanog binding sites and response of target genes to the knockdown of Nanog.
We detected 88 positive and 87 negative associations between contextual factors at TF binding sites and their regulatory functions that were significant for all 7 tested methods of analysis (Fig. 2 and Supplementary Table S3). These results indicate that functional TF binding sites can be roughly classified into two categories: (1) active sites that are also bound by P300, Chd7, mediator (Med1, Med12), cohesin (Smc1, Nipbl, Rad21), and SWI/SNF (Brg1), and (2) repressed sites that carry repressive histone marks H3K27me3 and are bound by Polycomb TFs (Ezh2, Suz12, Mtf2, and Ring1b). In addition, repressed sites were negatively associated with binding of Med1, Med12, Nipb1, histone acetylation (H3K9ac, H3K27ac), and histone methylation (H3K4me3). Associations found between regulatory functions of TF binding sites and the abundance of contextual factors were not mediated by the distance to TSS, because the effects remained significant, even if distance to TSS was used as a covariate (Supplementary Table S3). The main advantage of our approach compared with other methods for identifying predictors of functional binding sites is in the use of experiments on TF manipulation instead of comparing chromatin profiles in distinct cell types (Creyghton et al., 2010; Kagey et al., 2010; Rada-Iglesias et al., 2011) or in enhancers and promoter-like regions (Chen et al., 2012). Thus, our results are TF-specific and represent both activating and repressing roles of TFs: the proposed algorithm of analysis is unique in accounting for quantitative change of expression of individual target genes after manipulation of each of the 9 TFs and in using the distance from TSS as a covariate.
These results are mostly consistent with previous reports on the role of cofactors and chromatin modifications in the function of enhancers (Visel et al., 2009; Creyghton et al., 2010; Schnetz et al., 2010; Chen et al., 2012). In ES cells, P300 is recruited by Pou5f1, Sox2, and Nanog (Chen et al., 2008), and controls the differentiation of ES cells (Zhong and Jin, 2009). The mediator connects enhancers with the proximal promoter, and cohesin locks the enhancer with the promoter (Kagey et al., 2010; Goke et al., 2012). Brg1 (a component of the SWI/SNF complex) activates enhancers bound by Stat3 (Ho et al., 2011), although our analysis indicates that it is also present at functional binding sites of Pou5f1 and Sox2 (Fig. 2). The presence of H3K27me3 chromatin marks at repressed TF binding sites is consistent with the description of “poised enhancers” (Rada-Iglesias et al., 2011; Zentner et al., 2011). These histone marks are known to be established and maintained by Polycomb TFs (Chamberlain et al., 2008; Walker et al., 2010). Two classes of functional TF binding sites (active and repressed) possibly represent dynamic states of DNA regulatory regions controlled by various TFs. For example, some developmental enhancers are repressed by pluripotency factors—Pou5f1, Sox2, and Nanog (Goke et al., 2012). However, testing this hypothesis is beyond the scope of this article.
Our data from experiments on TF manipulation provided a unique opportunity to explore the mechanisms of repressive effects of TFs on their target genes, which were inferred from the repressive effects from the downregulation of target genes after the induction of TFs or from the upregulation of target genes after the knockdown of TFs. The majority of these effects (e.g., downregulation of target genes upon the overexpression of Klf4 and Tcfcp2l1, and the upregulation of target genes after the knockdown of Nanog, Sox2, and Pou5f1) were mediated by the repressed TF binding sites (Fig. 2). By contrast, the downregulation of target genes after the overexpression of Tcf3 and Pou5f1 was mediated by the active binding sites of these TFs. Involvement of both active and repressed sites in the repressive action of TFs seems to be associated with the phase of target gene repression. If repression has not started yet (e.g., before the overexpression of Tcf3 and Pou5f1), then the TF binding sites are still in the active state. But, if repression is under way (e.g., before the overexpression of Klf4, and Tcfcp2l1), then corresponding TF binding sites would be in the repressed state. Note that at normal or reduced levels of expression, Pou5f1 activates the expression of target genes in ES cells, but if artificially overexpressed, it then turns into a repressor (Aiba et al., 2009; Niwa et al., 2000).
Next, we used contextual factors at TF binding sites to identify target genes regulated by TFs, which are defined as genes that (1) have a TF binding site and (2) change their expression in response to TF manipulation (Loh et al., 2006; Johnson et al., 2008; Lee et al., 2010). However, the overlap between the set of genes with TF binding sites and the set of genes that responded to TF manipulations may include many false positives (i.e., genes with nonfunctional TF binding sites that responded to TF manipulation via alternative regulatory pathways). To address this issue, we evaluated the EPFP by comparing the proportion of genes with TF binding sites (i.e., targets) within two subsets of genes: (1) genes that responded to TF manipulation, and (2) “control” genes that did not change their expression (Sharov et al., 2008). If the percent of targets in these subsets of genes is 25% and 5%, respectively, then out of 25% targets in the set (1) 5% are expected to be false positives, because the “control” set of genes includes 5% targets. Thus, the EPFP equals 0.05/0.25 (i.e., 0.2). By varying the threshold of fold change (i.e., higher or lower than 2-fold), we can find conditions when EPFP = 0.3 (threshold used here). The EPFP measure is similar to the FDR statistical criterion, and in our previous publications we called it “FDR” (Sharov et al., 2008). However, because it differs in both underlying assumptions (EPFP compares the frequency of target genes between test and control subsets of genes, whereas no control is available for FDR estimation) and estimation methods (FDR method evaluates the p-value for each individual hypothesis, whereas EPFP is not derived from p-values, because each gene is unique and has no replications), we have chosen a different name here.
In this article, we further improved the EPFP estimates by using additional information on contextual factors at TF binding sites (see Methods). For example, using the previous EPFP statistics (EPFP ≤ 0.3 and fold change ≥ 1.5), we found only 28 target genes for Tcf3 overexpression, and no target genes for other TFs manipulation. However, using the new EPFP* statistics (i.e., after incorporating data on the abundance of contextual factors), we detected a large number of TF-regulated target genes (N = 4,455) with the same statistical criteria (Fig. 3b). Our lists of positively regulated targets of pluripotency factors—Pou5f1, Sox2, and Nanog (i.e., targets that were downregulated after the knockdown of these TFs)—substantially increased compared with previous publications (Loh et al., 2006; Matoba et al., 2006; Sharov et al., 2008) (Supplementary Table S3). In addition, we found sets of target genes that were negatively regulated by pluripotency factors (i.e., targets that were upregulated after the knockdown of TFs). Negative regulation was difficult to detect, because repressed binding sites of Pou5f1, Sox2, and Nanog are strongly diluted among all binding sites. However, the new EPFP* statistics with the additional predictors, such as H3K27me3 histone marks and binding of Polycomb factors, reliably detected these effects. Lists of targets regulated by Klf4 and Tcfcp2l1 were compiled here for the first time, whereas lists of the regulated targets of Tcf3, Cdx2, Myod1, and Sfpi1 were updated as compared with the previous reports (Cole et al., 2008; Nishiyama et al., 2009; Cao et al., 2010; Heinz et al., 2010). Cdx2, Myod1, and Sfpi1 mostly positively regulated their targets (Fig. 3b). By contrast, Tcf3, Klf4, and Tcfcp2l1 affected the expression of their target genes both positively and negatively. The repressive functions of Nanog and Klf4 have been suggested previously based on their association with repressive protein complexes (Liang et al., 2008; van den Berg et al., 2010) or analysis of transcription regulation of individual genes (Liu et al., 2005; Addis et al., 2010). Our data, however, provide the first evidence of their repressive effects based on the genome-wide analysis of TF manipulation experiments.
FIG. 3.

Number of target genes upregulated and downregulated after manipulation of transcription factors, as identified using criteria: expected proportion of false positives ≤0.3 and change >1.5-fold. See Supplementary Table S4 for lists of regulated target genes.
4. Conclusions
In summary, we have developed methods for systematic analysis of contextual factors associated with functional TF binding sites. These methods integrate data on the localization of TFs and chromatin modifications (ChIP-seq) with data on gene expression change after manipulation of TFs and facilitate identification of target genes regulated by TFs. In particular, we have identified contextual factors associated with active and repressed states of TF binding sites. The use of TF manipulations has also allowed us to characterize repressive effects of TFs, which appeared to be mediated by TF binding sites in repressed states.
Supplementary Material
Acknowledgments
This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute on Aging. We thank current and past lab members for discussion and contribution to data published previously and integrated into the meta-analysis in this article.
Author Disclosure Statement
No competing financial interests exist.
References
- Addis R.C., Prasad M.K., Yochem R.L., et al. . 2010. OCT3/4 regulates transcription of histone deacetylase 4 (Hdac4) in mouse embryonic stem cells. J. Cell Biochem. 111, 391–401 [DOI] [PubMed] [Google Scholar]
- Aiba K., Nedorezov T., Piao Y., et al. . 2009. Defining developmental potency and cell lineage trajectories by expression profiling of differentiating mouse embryonic stem cells. DNA Res. 16, 73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold C.D., Gerlach D., Stelzer C., et al. . 2013. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339, 1074–1077 [DOI] [PubMed] [Google Scholar]
- Barski A., Cuddapah S., Cui K., et al. . 2007. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 [DOI] [PubMed] [Google Scholar]
- Buck M.J., and Lieb J.D.2004. ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics 83, 349–360 [DOI] [PubMed] [Google Scholar]
- Cao Y., Yao Z., Sarkar D., et al. . 2010. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev. Cell 18, 662–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain S.J., Yee D., and Magnuson T.2008. Polycomb repressive complex 2 is dispensable for maintenance of embryonic stem cell pluripotency. Stem Cells 26, 1496–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.Y., Morris Q., and Mitchell J.A.2012. Enhancer identification in mouse embryonic stem cells using integrative modeling of chromatin and genomic features. BMC Genomics 13, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Xu H., Yuan P., et al. . 2008. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117 [DOI] [PubMed] [Google Scholar]
- Cole M.F., Johnstone S.E., Newman J.J., et al. . 2008. Tcf3 is an integral component of the core regulatory circuitry of embryonic stem cells. Genes Dev. 22, 746–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa-Cerro L.S., Piao Y., Sharov A.A., et al. . 2011. Generation of mouse ES cell lines engineered for the forced induction of transcription factors. Sci. Rep. 1, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton M.P., Cheng A.W., Welstead G.G., et al. . 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 107, 21931–21936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goke J., Jung M., Behrens S., et al. . 2012. Combinatorial binding in human and mouse embryonic stem cells identifies conserved enhancers active in early embryonic development. PLoS Comput. Biol. 7, e1002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman N.D., Stuart R.K., Hon G., et al. . 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318 [DOI] [PubMed] [Google Scholar]
- Heinz S., Benner C., Spann N., et al. . 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L., Miller E.L., Ronan J.L., et al. . 2011. esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat. Cell Biol. 13, 903–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R., Teh C.H., Kunarso G., et al. . 2008. REST regulates distinct transcriptional networks in embryonic and neural stem cells. PLoS Biol. 6, e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey M.H., Newman J.J., Bilodeau S., et al. . 2010. Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Sharov A.A., McDole K., et al. . 2011. Mouse B-type lamins are required for proper organogenesis but not by embryonic stem cells. Science 334, 1706–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.H., Li M., Michalowski A.M., et al. . 2010. A genomewide study identifies the Wnt signaling pathway as a major target of p53 in murine embryonic stem cells. Proc. Natl. Acad. Sci. USA 107, 69–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J., Wan M., Zhang Y., et al. . 2008. Nanog and Oct4 associate with unique transcriptional repression complexes in embryonic stem cells. Nat. Cell Biol. 10, 731–739 [DOI] [PubMed] [Google Scholar]
- Liu Y., Sinha S., McDonald O.G., et al. . 2005. Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J. Biol. Chem. 280, 9719–9727 [DOI] [PubMed] [Google Scholar]
- Loh Y.H., Wu Q., Chew J.L., et al. . 2006. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 38, 431–440 [DOI] [PubMed] [Google Scholar]
- Marson A., Levine S.S., Cole M.F., et al. . 2008. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 134, 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba R., Niwa H., Masui S., et al. . 2006. Dissecting Oct3/4-regulated gene networks in embryonic stem cells by expression profiling. PLoS One 1, e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen T.S., Ku M., Jaffe D.B., et al. . 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A., Sharov A.A., Piao Y., et al. . 2013. Systematic repression of transcription factors reveals limited patterns of gene expression changes in ES cells. Sci. Rep. 3, 1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A., Xin L., Sharov A.A., et al. . 2009. Uncovering early response of gene regulatory networks in ES cells by systematic induction of transcription factors. Cell Stem Cells 5, 420–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H., Miyazaki J., and Smith A.G.2000. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat. Genet. 24, 372–376 [DOI] [PubMed] [Google Scholar]
- Rada-Iglesias A., Bajpai R., Swigut T., et al. . 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rye M., Saetrom P., Handstad T., and Drablos F.2011. Clustered ChIP-Seq-defined transcription factor binding sites and histone modifications map distinct classes of regulatory elements. BMC Biol. 9, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnetz M.P., Handoko L., Akhtar-Zaidi B., et al. . 2010. CHD7 targets active gene enhancer elements to modulate ES cell-specific gene expression. PLoS Genet. 6, e1001023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharov A.A., Dudekula D.B., and Ko M.S.2006. CisView: a browser and database of cis-regulatory modules predicted in the mouse genome. DNA Res. 13, 123–134 [DOI] [PubMed] [Google Scholar]
- Sharov A.A., Masui S., Sharova L.V., et al. . 2008. Identification of Pou5f1, Sox2, and Nanog downstream target genes with statistical confidence by applying a novel algorithm to time course microarray and genome-wide chromatin immunoprecipitation data. BMC Genomics 9, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg D.L., Snoek T., Mullin N.P., et al. . 2010. An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell 6, 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A., Blow M.J., Li Z., et al. . 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker E., Chang W.Y., Hunkapiller J., et al. . 2010. Polycomb-like 2 associates with PRC2 and regulates transcriptional networks during mouse embryonic stem cell self-renewal and differentiation. Cell Stem Cell 6, 153–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S., Xie D., Cao X., et al. . 2012. Comparative epigenomic annotation of regulatory DNA. Cell 149, 1381–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.B., Johnson R., Kunarso G., and Stanton L.W.2011. Coassembly of REST and its cofactors at sites of gene repression in embryonic stem cells. Genome Res. 21, 1284–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner G.E., Tesar P.J., and Scacheri P.C.2011. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 21, 1273–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X., and Jin Y.2009. Critical roles of coactivator p300 in mouse embryonic stem cell differentiation and Nanog expression. J. Biol. Chem. 284, 9168–9175 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.