

Abstract
Growing evidence suggests that multiple spatially, temporally, and functionally distinct pools of cyclic nucleotides exist and regulate cardiac performance, from acute myocardial contractility to chronic gene expression and cardiac structural remodeling. Cyclic nucleotide phosphodiesterases (PDEs), by hydrolyzing cAMP and cyclic GMP, regulate the amplitude, duration, and compartmentation of cyclic nucleotide–mediated signaling. In particular, PDE3 enzymes play a major role in regulating cAMP metabolism in the cardiovascular system. PDE3 inhibitors, by raising cAMP content, have acute inotropic and vasodilatory effects in treating congestive heart failure but have increased mortality in long-term therapy. PDE3A expression is downregulated in human and animal failing hearts. In vitro, inhibition of PDE3A function is associated with myocyte apoptosis through sustained induction of a transcriptional repressor ICER (inducible cAMP early repressor) and thereby inhibition of antiapoptotic molecule Bcl-2 expression. Sustained induction of ICER may also cause the change of other protein expression implicated in human and animal failing hearts. These data suggest that the downregulation of PDE3A observed in failing hearts may play a causative role in the progression of heart failure, in part, by inducing ICER and promoting cardiac myocyte dysfunction. Hence, strategies that maintain PDE3A function may represent an attractive approach to circumvent myocyte apoptosis and cardiac dysfunction.
Keywords: cAMP, PDE, ICER, heart failure, apoptosis
Cyclic AMP and cyclic cGMP are 2 critical intracellular second messengers that regulate a myriad of processes in the cardiovascular system, from short-term action in contraction/relaxation to long-term effects in cell growth/survival. For example, cAMP elevation, particularly through β-adrenergic receptor (β-AR) stimulation, has crucial positive inotropic and lusitropic effects when the cardiac demand is increased.1 However, cGMP, in most cases, exerts negative inotropic effects on the heart.2 Changes in intracellular cAMP and cGMP levels are often transient, and PDEs play very important roles in regulating the amplitude, duration, and compartmentation of cyclic nucleotide signaling. As a superfamily of enzymes, PDEs are subdivided into 11 broad families (PDE1 to PDE11) based on their tissue distribution, biochemical properties, and sensitivity to chemical inhibitors. With at least 21 genes encoding more than 50 PDE isoforms, each tissue/cell type expresses a distinct set of PDEs.3 In cardiac myocytes, multiple PDE isozymes from at least 5 different families have been described. These include PDE1, PDE2, PDE3, PDE4, and PDE5. PDE1, PDE2, and PDE3 show dual substrate specificity in vitro, whereas PDE4 and PDE5 selectively hydrolyze cAMP and cGMP, respectively. The existence of multiple PDE isoforms expressed in a single cell guarantees heterogeneous spatiotemporal patterns of cyclic nucleotide signaling and versatile cyclic nucleotide–mediated functions.
Alterations in PDE expression and activity may disturb the balance and/or localization of cyclic nucleotides and lead to pathological events. In a study of frog myocytes, for instance, PDE inhibition caused a reduction of cAMP effects on Ca2+ channels because of disruption of proper subcellular cAMP localization.4 Also, in rat neonatal cardiac myocytes, on β-AR stimulation, cAMP elevation and protein kinase A (PKA) activation occur in specific compartments proximate to the transverse tubule. Inhibition of PDEs have been shown to disturb the cAMP gradients and cause a global activation of PKA, which may reveal nonspecific and toxic effects.5 Depletion of PDE4D expression, through PDE4D gene knock out, has been shown to cause PKA hyperphosphorylation of RyR2, Ca2+ leakage, and cardiac dysfunction.6 Moreover, chronic inhibition of PDE4 activity by rolipram has been shown to induce exercise-triggered arrhythmias in these mice.6 Several clinical trials have also linked chronic administration of PDE3 inhibitors to increased mortality, primarily resulting from arrhythmia and sudden death.7,8 Taken together, these findings strongly suggest that downregulation of PDE expression in disease states, or chronic inhibition of PDE activity, may impair physiological responses while promoting pathological responses. This review highlights the role of cAMP-hydrolyzing PDE3 in the cardiac muscle, the potential cellular and molecular mechanisms of adverse cardiac effects of chronic PDE3 inhibition, as well as PDE3 regulation and function in the development of heart failure.
Multiple Structurally and Functionally Distinct PDEs in the Heart
At least 5 different PDE families, PDE1, PDE2, PDE3, PDE4, and PDE5, have been described in the heart. Among them, PDE1 to PDE4 are all able to hydrolyze cAMP in vitro, whereas PDE5 exclusively hydrolyzes cGMP. The relative contribution of each PDE family likely varies among species, developmental stages, myocyte types (atrial versus ventricular), and the cellular conditions (such as basal versus stimulated, normal versus diseased). It has not been determined whether these isoforms are attributable to cardiac myocytes specifically or to other cardiac cell types, because most of these studies used whole hearts. Individual PDEs may regulate distinct cellular functions by selectively coupling to different cAMP pools (Table). The expression and function of the newly cloned PDE families (PDE7 to PDE11) in the heart have not been well documented, likely because of the absence of selective inhibitors and lack of antibodies.
Table 1.
cAMP-Hydrolyzing PDEs in the Heart
| PDE Family Substrates (in vitro) |
Isoform* | Potential Function |
|---|---|---|
| PDE1 cAMP, cGMP |
PDE1A PDE1C |
Unknown |
| PDE2 cAMP, cGMP |
PDE2A | |
| PDE3 cAMP, cGMP |
PDE3A PDE3B |
|
| PDE4 cAMP |
PDE4A–D |
|
May vary between species, myocyte types, and cellular conditions.
cAMP-Hydrolyzing PDEs in the Heart
Ca2+/calmodulin-stimulated PDEs (PDE1 family) constitute a large family of enzymes and are encoded by 3 distinct genes: PDE1A, PDE1B, and PDE1C. Multiple N-terminal or C-terminal splice variants have also been identified for each gene. In vitro, PDE1A and PDE1B isozymes have much higher affinity for cGMP than cAMP; however, PDE1C isozymes hydrolyze both cAMP and cGMP with high affinity. In vivo, inhibition of PDE1A has been shown to preferentially elevate cGMP but not cAMP.9 Inhibition of PDE1C is, however, able to elevate intracellular cAMP inside of cells, although it is still not clear whether PDE1C is able to regulate cGMP in vivo.10,11 PDE1 activity, represented by Ca2+/calmodulin-stimulated cAMP-hydrolyzing activity, has been detected in the cytosolic fraction of human myocardium.12 Furthermore, it has been demonstrated that, in the absence of Ca2+, the myocardial cytosolic cAMP-PDE activity is mainly contributed by PDE3, whereas, in the presence of Ca2+, the cAMP-PDE activity is largely from PDE1. This suggests that PDE1 and PDE3 may differentially contribute to myocardial cAMP hydrolysis under basal and Ca2+-stimulated conditions. It has also been reported that PDE1 represents the major cGMP-hydrolyzing activity in the human heart.13 However, the abovementioned studies did not determine the identity of PDE1 isoform(s) expressed in human myocardium. In addition to human myocardium, the expression of PDE1A has been described in canine hearts.14,15 In rat hearts, PDE1 activity has been shown to represent a major cGMP-hydrolytic activity in the first peak eluting from the anion-exchange column, which is contributed by both PDE1A (50%) and PDE1C (10%), respectively.16 PDE1C expression level has also been found to be induced in 7-oxo-prostacyclin–preconditioned rat hearts.17 All of these abovementioned studies support the fact that PDE1 activity exists in whole hearts or ventricular tissues; however, an early study from Bode et al disputes the possibility for the expression of PDE1 in rat cardiac myocytes.18 For example, the report by the authors reveals predominant PDE1 activity in a nonmyocyte soluble fraction of rat ventricular tissue rather than cardiac myocytes, although this study could not rule out the possibility of an existing particulate PDE1 in cardiac myocytes.18 Immunofluorescent staining has shown that most PDE1A in the rat heart is in the smooth muscle cells of muscular arteries.16 These data together suggest that PDE1 might be expressed in noncardiac myocytes in the rat heart. Future studies are required to further characterize the individual PDE1 isoforms and cell types in the heart from various species.
Both PDE2 and PDE3 are able to hydrolyze cAMP and cGMP with high affinities. PDE2 family members are often referred to as cGMP-stimulated PDEs because binding of cGMP to the N-terminal GAF domains (cGMP-binding phosphodiesterase, Anabaena adenylyl cycles Escherichia coli FhCAs) of PDE2 greatly stimulates its catalytic activity.3 In contrast, the Vmax of PDE3 for hydrolyzing cAMP is ≈10-fold greater than that for cGMP. Thus, cGMP behaves as a competitive inhibitor of cAMP hydrolysis because of the high affinity and the low rate of cGMP hydrolysis.19 For this reason, PDE3 isozymes are also called cGMP-inhibited PDEs. The cGMP-dependent regulation of cAMP-hydrolyzing activities by PDE2 and PDE3 represent important mechanisms by which the cGMP signal regulates cAMP response in several different cell types. For example, atrial natriuretic peptide inhibits adrenocorticotropic hormone–induced aldosterone production through cGMP-dependent activation of PDE2 and thereby attenuates adrenocorticotropic hormone–stimulated cAMP signaling.20 In vascular smooth muscle cells (VSMCs), nitric oxide (NO) activates PKA signaling through cGMP-dependent inhibition of PDE3 and elevation of cAMP, which is believed to mediate a variety of NO effects on VSMCs, such as endothelium-dependent smooth muscle relaxation,21,22 the inhibitory effects of NO on VSMC growth,23 and inflammatory response.24 Similarly, NO inhibition of platelet aggregation occurs, at least in part, through inhibiting PDE3 and increasing cAMP.25,26 However, in cardiac myocytes, it appears that the roles of PDE2 and PDE3 in cGMP-dependent regulation of cAMP and cardiac functions, in the case of the cardiac L-type Ca2+ channel current Ica, vary in different species and the source of myocytes (ventricular versus atrial).2,27 Studies of frog ventricular myocytes,28,29 rat ventricular myocytes,30 and human atrial myocytes31 have demonstrated that cGMP activates cGMP-stimulated PDEs (PDE2), decreases cAMP accumulation, and attenuates cAMP-stimulated Ica. However, it has also been reported that cGMP inhibits PDE3, increases cAMP, and promotes cAMP-stimulated Ica in isolated human atrial myocytes.32 PDE2 and PDE3 may also differentially regulate Ica in response to different concentrations of NO donors in the same frog ventricular myocytes.29 For instance, PDE3 plays a major role in potentiation of cAMP-stimulated Ica when the concentrations of NO donors (such as 3-morpholinosydnonimine and sodium nitroprusside) are in the nanomolar range (low),29 whereas PDE2 plays a major role in attenuation of cAMP-stimulated Ica when the concentrations of NO donors are in the micromolar range (high).29 These are consistent with the fact that the Ki value for cGMP inhibition of PDE3 is much lower than the Ka value for cGMP activation of PDE2.25,33–35 Moreover, cGMP has been shown to activate PKG and inhibit cAMP-stimulated Ica independent of PDE in several types of mammalian ventricular myocytes such as rat, rabbit, and human.27,36 Therefore, cGMP-dependent regulation of cAMP signaling and myocyte function appears species and myocyte specific, which is probably determined by the relative expression levels of PDE2 versus PDE3, the cGMP concentrations, and/or the compartmentation of PDEs and cGMP.
PDE4 belongs to a large family of enzymes that specifically hydrolyze cAMP with high affinity. Four PDE4 genes (PDE4A, PDE4B, PDE4C, and PDE4D) have been identified, and each of them results in multiple variants through alternative promoters and/or splicing. PDE4 isozymes are found in almost all cell types, and each cell often expresses more than 1 PDE4 isozyme. The expression and function of PDE4s in hearts have not been studied extensively because PDE4 inhibitors result in minimal inotropic effects in humans. However, it has recently been reported that depletion of PDE4D gene in mice results in a progressive cardiomyopathy in senescent mice and accelerated heart failure after myocardial infarction.6 The cardiac deleterious effect of PDE4D knockout is believed to be attributable to loss of PDE4D3 from the macromolecular complex of ryanodine receptor (RyR2), a cardiac calcium channel present in the sarcoplasmic reticulum membrane.6 Loss of PDE4D3 in the RyR2 complex causes PKA hyperphosphorylation of RyR2, Ca2+ leakage, and, in turn, cardiac dysfunction.6 Furthermore, PDE4D3 has been found in human RyR2 macrocomplex and to be decreased in human failing hearts, where the RyR2 is PKA hyperphosphorylated.6 In addition, in rat neonatal cardiac myocytes, PDE4D3 and PDE4D5 are found to be recruited to membrane β2-AR via β-arrestin on isoprenaline stimulation, which may play an important role in regulating β2-AR PKA phosphorylation and G protein switching from G(s) to G(i).37 Moreover, a cAMP-responsive macromolecular complex has been found in the myocyte nuclear envelope, which contains the muscle-specific A kinase anchoring protein (mAKAP), PKA, PDE4D3, big mitogen-activated kinase 1/extracellular signal-regulated kinase 5 (BMK1/ERK5), the small GTPase Rap1, and the exchange factor Epac1.38 PDE4D3 functions as an adaptor protein that recruits Epac, activates the small GTPase Rap1, and attenuates ERK5 activation, which plays a critical role in the regulation of myocyte hypertrophy.38 PDE4, together with PDE3, provides the major cAMP-PDE activity in the heart.39– 42 However, the relative contributions might be species specific. In rat neonatal cardiac myocytes, PDE4, but not PDE3, appears to be responsible for modulating the amplitude and duration of the cAMP response to β agonists.41 This is consistent with the observations that PDE4 inhibitors enhance only β-AR agonist–induced cAMP accumulation and contractility in rats.43
PDE3 Isozymes
The PDE3 gene family contains 2 subfamilies, PDE3A and PDE3B, that are encoded by 2 different but highly related genes. At least 3 PDE3A-related proteins with different apparent molecular masses have been described.44,45 They are 136, 118, and 94 kDa and referred to as PDE3A1, PDE3A2, and PDE3A3, respectively.44 Different PDE3A proteins may be generated by excluding different amounts of 5′-coding sequences via alternative splicing, transcription promoters, or translation start sites.45,46 Although all of these PDE3A proteins may have an identical C-terminal portion that contains the PDE catalytic domain, they differ considerably in the N-terminal portion (Figure 1).45 The N-terminal hydrophobic domains, NHR1 (≈200 aa, with 6 predicted trans-membrane helices) and NHR2 (≈50 aa), are believed to be involved in targeting PDE3A subcellular localization, which are differentially distributed in distinct PDE3A proteins. For example, PDE3A1, containing both NHR1 and NHR2, is usually found in the particulate fraction. PDE3A2 (containing an intact NHR2) is found in both cytosolic and particulate fractions, and PDE3A3 (lacking both NHR1 and NHR2) is predominantly cytoplasmic.46 The 3 PDE3A isoforms may be translated from 2 mRNA transcripts: PDE3A1 (136 kDa) is derived from PDE3A1 mRNA, whereas PDE3A2 (118 kDa) and PDE3A3 (94 kDa) are the products of PDE3A2 mRNA, probably translated from different AUGs (start codons) in PDE3A2 mRNA.46 This implies that PDE3A may be regulated at both the transcriptional and posttranscriptional level, resulting in several isozymes with distinct subcellular localization and functional properties. So far, only 1 PDE3B isoform, 137-kDa PDE3B1, has been described. Despite having a divergent N-terminal region, PDE3B1 shares a nearly identical structure with PDE3A1, and it is also membrane associated.44 A more detailed description of the structural topology of PDE3 isoforms is described in a review by Shakur et al.44
Figure 1.

Structural organization of PDE3 isozymes. Three PDE3A gene products of PDE3A (PDE3A1, PDE3A2, and PDE3A3) and 1 PDE3B (PDE3B1) have been reported. The various PDE3A products differ considerably in the N-terminal portion via alternative splicing, transcription promoters, or translation start sites, while having an identical C-terminal portion that contains the PDE catalytic domain. The longest PDE3A1 contains a large N-terminal hydrophobic domain (NHR1, ≈200 aa) with 6 predicted transmembrane helices, followed by a small hydrophobic domain (NHR2, ≈50 aa) and 3 sites for phosphorylation by PKA, PKB, and PKA, respectively, between NHR1 and NHR2.
PDE3A is relatively abundant in cardiac myocytes, VSMCs, and platelets, whereas PDE3B is predominantly expressed in adipocytes, hepatocytes, and pancreatic cells.44,45 In the mammalian heart, 3 PDE3A protein products with molecular masses of ≈136, 118, and 94 kDa have been detected using PDE3A antibodies.46,47 However, it has still not been determined whether proteolysis of PDE3 accounts for the lower-molecular-mass products reported.48–50 Using human myocardium fractions, studies have found PDE3A1 (≈136 kDa) mostly in the microsomal (sarcoplasmic reticulum) fraction, PDE3A2 (≈118 kDa) in both the microsomal and cytosolic fractions, and PDE3A3 (≈94 kDa) absolutely in the cytosolic fraction of human myocardium.46,47,51 Two mRNA molecules, probably corresponding to PDE3A1 and PDE3A2/PDE3A3, have been detected by Northern blotting and RNase protection analyses.46 These observations are consistent with several early studies showing PDE3 activity present in both cardiac microsomal and cytosolic fractions in several different species.12,52,53 The functional properties of these PDE3A enzymes with distinct subcellular localizations are still not well characterized.
The existence of PDE3B1 in heart tissue has only recently been reported in mice.54 Patrucco et al demonstrate that PDE3B activity accounts for ≈30% of the total PDE3 activity in the mouse heart and that cardiac PDE3B constitutively associates with phosphoinositide 3-kinase γ (PI3Kγ).54 PI3Kγ controls L-type calcium current through its positive modulation of PDE3B.55 Because myocardial tissues contain multiple cell types (such as cardiac myocyte, cardiac fibro-blast, or cells from the vasculature), the source of each PDE3 isozyme detected in the heart is still not clear. It is possible that multiple PDE3 isozymes with distinct subcellular localizations coexist in a single cell type. For example, cytosolic PDE3A2 and PDE3A3 and membrane PDE3B1 have been detected in VSMCs.45 Taken together, these findings suggest that multiple PDE3 isoforms may be expressed in distinct cellular sites and may regulate different biological processes in the heart.
PDE3 and Heart Failure
PDE3 Inhibitors in Heart Failure: Therapeutic Effects and Mechanism of Action
PDE3-selective inhibitors such as amrinone, enoximone, and milrinone have been used clinically to acutely treat congestive heart failure.56–59 In human myocardium, PDE3 inhibitors increase the rate and magnitude of developed force as well as enhance the rate of muscle relaxation. Concurrently, in human vasculature, PDE3 inhibition reduces total peripheral and pulmonary vascular resistance and enhances coronary blood flow. Thus PDE3 inhibitors are powerful drugs for the acute treatment of the congestive heart failure because of simultaneous increased contractility of the heart and decreased resistance of blood flow through the vasculature. Such combined inotropic and vasodilatory actions make PDE3 inhibitors a more effective therapy than either inotropic or vasodilator therapy alone in improving cardiac performance in heart failure patients.60
The biochemical and molecular mechanisms by which PDE3 inhibitors reduce vascular resistance are well known. Inhibition of PDE3 enzyme(s) in VSMCs leads to cAMP elevation and PKA activation, which stimulates smooth muscle cell relaxation via PKA-dependent decrease of intra-cellular Ca2+ concentration and attenuation of myosin phosphorylation.61,62 The cardiotonic effects of PDE3 inhibitors apparently involve 2 different mechanisms of action. (1) Elevation of cAMP via PDE3 inhibition activates PKA, which results in increase in the trans-sarcolemmal influx of Ca2+, probably via phosphorylation and activation of L-type Ca2+ channel, and, in turn, triggers a much larger Ca2+ mobilization from sarcoplasmic reticulum store via RyR2.42,63– 66 In the short term, this enhances left ventricular (LV) contractile force and overall systolic function. (2) The cAMP raised by PDE3 inhibition stimulates the Ca2+ uptake through sarcoplasmic reticulum Ca2+-ATPase (SERCA2 pump), probably via PKA-dependent phosphorylation of phospholamban.63,67– 69 This event leads to LV relaxation and improves diastolic function. A strong correlation has been observed between positive inotropic response and membrane-bound PDE3 activity, suggesting the membrane-associated PDE3 may be the site of the inotropic effect of PDE3 inhibitor.40,52 The inotropic connection with PDE3 inhibitors appears to vary across species, and this discrepancy might be attributable to the species-dependent expression of the membrane-associated PDE3 isoform.40 For example, the PDE3 inhibitor imazodan elicited potent inotropic effects on LV muscles from monkeys and dogs, in which a membrane-associated PDE3 activity was identified.40 However little inotropic effect was seen in the rat, hamster, and guinea pig, in which soluble PDE3 activity was much more profound.40
Conversely, in long-term clinical trials, the early hemodynamic improvements were typically not sustained, and increased mortality attributable to arrhythmias and sudden death has often been reported. Notably, in the PROMISE (Prospective Randomized Milrinone Survival Evaluation) study, oral milrinone (40 mg daily) showed a 28% increase in mortality in patients with class III and IV heart failure. The mortality rate was 53% in patients with class IV heart failure,7 suggesting that the adverse effect of milrinone is greater in patients with more severe heart failure. Similar observations were obtained from other clinical trials with different PDE3 inhibitors (see reviews for various clinical trials8,70). Thus, PDE3 inhibitors are reserved for only the acute (rather than chronic) treatment of severe heart failure. Similar effects on increased mortality have been observed with chronic β-AR agonist therapy in heart failure patients.71–73 These results suggest that short-term increase of cAMP through PDE3 inhibition or β-AR stimulation is beneficial to patients with heart failure whose contractility is impaired, but long-term increase of cAMP is harmful.
It is somewhat interesting that another PDE3 inhibitor, cilostazol (a quinolinone derivative), which has a much weaker positive inotropic effect than milrinone, also lacks significant adverse cardiac effects. Cilostazol (Pletal) has been used clinically to treat symptoms of intermittent claudication for decades in Japan and other Asian countries and more recently in the US. The therapeutic effects of cilostazol are mainly attributed to its vasodilatory and antiplatelet actions, via raising intracellular cAMP levels in VSMCs and platelets, respectively. An adjunct to inhibiting PDE3, cilostazol also blocks adenosine uptake, which may distinguish it from other PDE3 inhibitors such as milrinone.74 It has been reported in smooth muscle and platelets that elevated interstitial and circulating adenosine levels by cilostazol could potentiate its cAMP-raising effect through inhibiting PDE3 and, thereby, augmenting antiplatelet and vasodilatory effects of the drug74; whereas in the heart, interstitial adenosine elevation by cilostazol attenuates cAMP response through PDE3 inhibition and thus eliminates the cAMP-elicited cardiac effects.74–76 Therefore, the inhibitory effect of cilostazol on adenosine uptake may lessen its cardiac effects including the beneficial acute inotropic effects as well as the detrimental chronic effects of cAMP.
Alteration of PDE3 Expression in Failing Hearts
Because the hemodynamic effects are not sustained and mortality is increased with chronic PDE3 inhibitor therapy, changes in PDE3 expression have been investigated as a link to progressive heart failure. Several early studies using LV muscles from patients with various stages of heart failure have shown reduced positive inotropic effects of PDE3 inhibitors with the progression of heart failure.77,78 This loss of cardiac efficacy of PDE3 inhibitors has been proposed to associate with disease-related reduction of PDE3 function.77,78 Further investigation indeed revealed a 50% to 70% reduction of PDE3 activity in the LV myocardium from human hearts with idiopathic cardiomyopathy, compared with normal hearts.79 Moreover, we have recently demonstrated that the overall PDE3 activity and PDE3A protein levels are significantly reduced in LV myocardial samples from end-stage heart failure patients with both dilated cardiomyopathy and ischemic heart disease.80 However, a report by Movsesian et al demonstrates no significant alteration in PDE3 activity in the microsomal fractions of human failing hearts.81 The decrease of PDE3 expression/activity has also been observed in several animal models of heart disease. For example, PDE3 activity in LV endocardium has been shown to be decreased in pacing-induced failing canine hearts, which accompanied desensitized inotropic effects of milrinone.82 A similar pacing-induced canine heart failure model demonstrated a 50% to 59% reduction of PDE3A mRNA levels in both LV and RV of failing canine hearts.83,84 Consistent with the mRNA changes, PDE3A protein products, corresponding to microsomal 135 kDa, cytosolic 120 kDa, and cytosolic 80 kDa, were all found to be decreased in the canine failing hearts.84 Recently, we have further shown a time-dependent downregulation of PDE3A protein expression in a mouse model of heart failure induced by chronic pressure overload via thoracic aorta constriction for up to 8 weeks.80 Because excessive neurohormonal activation of the angiotensin and adrenergic systems contribute to progressive ventricular remodeling and worsening cardiac function,85,86 the expression of PDE3A was directly evaluated in rodent hearts receiving chronic angiotensin II (Ang II) or β-AR stimulation. We found that PDE3A protein is downregulated in hypertrophied rodent hearts with chronic infusion of Ang II or β-AR agonist isoproterenol (ISO).87 However, an enhancement of PDE3B activity and gene expression was reported in rat hypertrophied and failing hearts with salt-induced hypertension.88 Taken together, despite a few opposing reports of PDE3 activity/expression in failing hearts,81,88 most of the abovementioned evidence indirectly or directly indicates a downregulation of PDE3 expression/activity in failing hearts. The correlation between PDE3 downregulation and heart failure suggests that downregulation of PDE3 may play a causative role in the development of heart failure. PDE3A knockout mice (PDE3A−/−), however, did not spontaneously develop noticeable cardiac abnormalities during their normal adulthood.89 Most likely, depletion of PDE3A alone is insufficient to induce cardiac dysfunction in mice under the normal physiological circumstances, and additional cardiac stresses are necessary. For example, PDE4D−/− mice did not spontaneously develop heart failure until senescence, when the heart shows many pathological changes.6 In young PDE4D−/− mice, accelerated heart failure was observed only after applying myocardial infarction.6 Similarly, PI3Kγ−/− mice developed heart failure only in the presence of chronic pressure overload.54 Therefore, it will be interesting to challenge the PDE3A−/− mice with pathological stresses such as pressure overload by thoracic aorta constriction or myocardial infarction.
Pathological Roles of PDE3 Downregulation
PDE3 and Cardiac Myocyte Apoptosis
The progressive loss of cardiac myocytes caused by apoptosis is critical in pathological cardiac remodeling and heart failure.90,91 To determine the role of PDE3 downregulation in cardiac myocyte pathology, the effects of PDE3 inhibition on cardiac myocyte apoptosis have been evaluated in vitro.80,87 We found that 2 different PDE3 selective inhibitors, milrinone and cilostamide, significantly increased rat neonatal cardiac myocyte apoptosis similar to that induced by chronic Ang II and β-AR agonist ISO treatment.80 As for PDE3 inhibitors, selective downregulation of PDE3A expression using PDE3A antisense also induced myocyte apoptosis.80 We found that PDE3A protein and mRNA levels were significantly decreased in primary rat neonatal cardiac myocytes on chronic stimulation with Ang II (via Ang II type 1 receptor [AT1R]) or ISO (via β1-AR),80,87 consistent with PDE3 downregulation in rodent heart with chronic infusion of Ang II or ISO in vivo. Interestingly, expression of exogenous wild-type PDE3A1, but not catalytically inactive PDE3A1, prevented Ang II– and ISO-induced myocyte apoptosis,80 indicating that PDE3A downregulation is essential for Ang II– and ISO-induced cardiac myocyte apoptosis.
In rat cardiac myocytes, PDE3 and PDE4 account for the major cAMP-PDE activity,41,42 and the cAMP elevation by PDE4 inhibition is much more profound than that by PDE3 inhibition.41,80 However, we did not see increased myocyte apoptosis by inhibiting PDE4 activity with PDE4 selective inhibitor rolipram,80 suggesting that myocyte apoptosis may be regulated by a subset of cAMP molecules that are preferentially coupled to PDE3A, regardless of the intracellular cAMP levels. PDE4D, however, differentially regulates cardiac myocyte function. For example, PDE4D3, associating with the macrocomplex of RyR2, regulates PKA-dependent RyR2 phosphorylation and function. Loss of PDE4D3 from the RyR2 complex causes leakiness of RyR2 because of PKA hyperphosphorylation of RyR2, which may lead to cardiac dysfunction.6 PDE3B has been also found in the mouse heart, constitutively associating with PI3Kγ in a multimolecular complex.54 PDE3B function was significantly decreased in PI3Kγ knockout mice (PI3Kγ−/−), which develop massive myocyte necrosis after chronic pressure overload. However, it is not clear whether the myocyte death in PI3Kγ−/− mice is attributable to PDE3B dysfunction. Taken together, these observations strongly indicate that different cAMP-hydrolyzing PDEs may regulate spatially and functionally distinct cAMP pools in cardiac myocytes.
PDE3 and Cardiac Myocyte Gene Expression
Chronic reduction of PDE3 activity may lead to changes in cAMP/PKA-dependent gene expression. Transcriptional activation coupled to cAMP/PKA signaling is through nuclear activators including CREB (cAMP response element–binding protein), CREM (cAMP responsive element modulator), and ATF-1 (activating transcription factor 1). For example, it is well known that increased cAMP activates PKA, which can phosphorylate/activate the transcription activator CREB and subsequently modulate cAMP response element (CRE)-mediated gene expression.92 ICERs (inducible cAMP early repressors) are members of the CREM family, which are generated through an alternative promoter (P2) located within an intron near the 3′ end of the CREM gene (Figure 2).93,94 Four ICER isoforms (named ICER-I, ICER-Iγ, ICER-II, and ICER-IIγ) have been described and are often referred to as ICER collectively because the functional difference of distinct ICER isoforms is not noticed. ICERs are transcription-ally induced by CREB via a CRE-binding sequence in the ICER promoter.95 ICER proteins contain DNA-binding and leucine zipper domains but lack the N-terminal transactivation domain, which renders them endogenous inhibitors of gene transcription driven by its cognates such as CREB and ATF-1 (Figure 2).93–95 For example, Tomita et al were first to show that ICER is induced by various agonists in the adult hearts and cultured neonatal cardiac myocytes.96 And induction of ICER in cardiac myocytes attenuates myocyte hypertrophic growth and promotes myocyte apoptosis by suppressing survival protein Bcl-2 expression.96 These findings support an important role of ICER as an inhibitor in CRE-mediated hypertrophic and survival signaling.97 In addition to competing with and thereby repressing CREB-mediated gene transcription, ICER may also inhibit functions of other transcription factors. For example, it has been shown that ICER forms a complex with NFAT (nuclear factor at activated T cells) and inhibits NFAT-dependent gene expression in T lymphocytes.98
Figure 2.
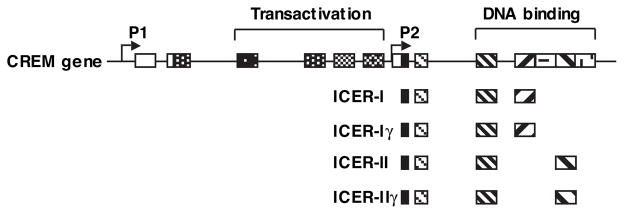
Schematic diagram showing the organization of CREM gene and ICER proteins. Line indicates intron; box, exon; open box, noncoding region; filled box, coding region; P1 and P2, promoters.
With this knowledge, the effects of PDE3 inhibition on CREB activation, ICER induction, and Bcl-2 expression in cardiac myocytes have been examined. We found that PDE3 inhibitors milrinone and cilostamide or PDE3A downregulation by its antisense stimulated a sustained CREB activation, ICER induction, and Bcl-2 reduction.80,87 Ang II and ISO also stimulated CREB activation, ICER induction, and BCL-2 reduction, which were prevented by expressing wild-type PDE3A1, consistent with observations in myocyte apoptosis.80,87 The role of CREB activation and ICER induction in Ang II, ISO, or PDE3 reduction-mediated myocyte apoptosis was further confirmed by the observations that myocyte apoptosis is prevented by blocking CREB activation and ICER expression through overexpressing dominant negative CREB or ICER antisense, respectively.80,87 Taken together, these results indicate that CREB activation, ICER induction, and Bcl-2 reduction may represent an important mechanism for PDE3 reduction–mediated myocyte apoptosis. Conversely, altering PDE4 activity through PDE4-selective inhibitor or overexpressing PDE4D3 had no significant effects on CREB activation, ICER induction, Bcl-2 expression,80 which was consistent with observations from cardiac myocyte apoptosis. These findings again suggest that PDE3 and PDE4 may regulate different cAMP/PKA signaling pathways, which have distinct effects on CREB activation, ICER induction, Bcl-2 expression, and myocyte apoptosis. Besides Bcl-2, induction of ICER may change the expression of other genes that are implicated in heart failure. For example, via a microarray analysis, we have seen that sustained expression of exogenous ICER can cause dysregulation of a number of genes that have been reported in failing hearts and that are involved in different aspects of cardiac pathologies. These include structural proteins, ion channels/transporters, signaling molecules, and transcription factors/regulators (Yan C, Abe J, Ding B, Liu W, unpublished data). We further confirmed that the expression of SERCA2 was downregulated by Ang II, ISO, and PDE3 inhibitors and that the ISO-induced downregulation of SERCA2 was prevented by expressing exogenous PDE3A or antisense ICER (Yan C, Abe J, Ding, B, Liu W, unpublished data). SERCA2 plays a critical role in calcium cycling and the beat-to-beat function of the heart. SERCA2 is decreased in animal and human failing hearts, which is associated with impaired cardiac function.99–101
The PDE3-ICER Positive-Feedback Loop and Sustained ICER Expression
Temporally restricted gene expression is critical for many biological processes. Under physiological conditions, ICER expression is a transient phenomenon because ICER is able to block its own transcription by competing with CREB in binding CRE sequences in the ICER promoter.95,102 ICER protein levels are decreased by proteasome-dependent degradation.103 This transient expression of ICER is physiologically important for negative-feedback regulation of CREB-dependent gene expression and allows the gene expression to be activated in a cyclic fashion (Figure 3). For example, CREB-regulated genes are rapidly induced by binding of phosphorylated CREB (p-CREB, activated) to CRE in the promoter regions and then activating transcription, which also includes ICER. Induction of ICER, in turn, leads to termination of mRNA accumulation of CREB-dependent genes as well as ICER itself. The clearance of ICER allows CREB-dependent gene expression to be activated repeatedly. The p-CREB/ICER balanced regulation of gene expression was first described to be essential for circadian rhythm in pineal gland104,105 and lately has also been shown to play critical roles in many other types of neuronal cells and nonneuronal systems.106 In contrast, sustained elevation of ICER levels may cause persistent suppression of CREB-regulated gene expression and thus lead to pathological consequences (Figure 3). For example, it has been shown that expressing exogenous ICER induced cell death in neurons102 and cardiac myocytes.80,96 We have further shown that chronic stimulation with Ang II and β-AR agonist ISO caused sustained ICER induction through PKA-dependent stabilization of ICER protein, which is essential for Ang II– and ISO-induced myocyte apoptosis.80,87 Our observations that myocyte apoptosis can be prevented by blocking ICER expression using ICER antisense 24 hours after ICER has been induced87 further support the notion that persistent but not transient ICER induction is critical for promoting cardiac myocyte apoptosis.
Figure 3.

The role of transient versus sustained induction of ICER. Transcription activator CREB is activated on phosphorylation at serine 133. Phosphorylated CREB (p-CREB) binds to CRE in the promoter region and activates transcription. ICER is also transcriptionally induced by p-CREB via a CRE-binding sequence in the ICER promoter. Induction of ICER, in turn, blocks CREB-mediated gene transcription, including ICER itself, by competing with CREB in binding CRE sequences. ICER proteins are subjected to proteasome-dependent degradation. Reduction of ICER allows cAMP-inducible gene expression to be activated repeatedly. Therefore, ICER induction is a transient phenomenon. This transient expression of ICER is physiologically important for negative-feedback regulation of CREB-dependent gene expression and allows CREB-dependent genes to be activated in a cyclic fashion. In contrast, sustained induction of ICER may cause persistent suppression of CREB-regulated gene expression and thus lead to pathological consequences.
We have discovered that the sustained induction of ICER is maintained by a positive-feedback loop (PDE3A-ICER positive-feedback loop) in which PDE3A is a key mediator (Figure 4).87,107 The initiation of ICER expression depends on CREB activation, likely via CREB-dependent ICER gene transcription. ICER represses PDE3A gene transcription by interacting with the PDE3A promoter.87 PDE3A downregulation activates PKA via increased local cAMP. PKA activation leads to ICER protein elevation by stabilization of ICER protein.87 This constitutes an autoregulatory positive-feedback loop (Figure 4). Ang II, via AT1R, can initiate this PDE3A-ICER positive-feedback loop by the activation of the classical PKC and CREB pathways. It is noteworthy that Ang II stimulation of AT1R, PKC, and CREB is critical only for the initiation, not the continuation, of the feedback loop.87 This explains why AT1R antagonists (such as losartan) cannot terminate the feedback loop once it is established.87 β-AR stimulation by ISO (via β1-AR) initiates the feedback loop through activation of Ca2+/calmodulin kinase II (Van C, Abe J, Ding B, Liu W, unpublished data) and CREB.87 PDE3 inhibitors can also induce this positive-feedback loop.87 We have demonstrated that this positive-feedback regulatory mechanism is essential for sustained ICER induction and subsequent cardiomyocyte apoptosis induced by Ang II, ISO, and PDE3 inhibitors (Figure 4).87 It appears that this feedback mechanism is not limited only to neonatal cardiomyocytes but also occurs in adult cardiomyocytes. For example, we have confirmed that in adult rat cardiac myocytes, Ang II–induced sustained ICER induction, Bcl-2 reduction, and myocyte apoptosis can be attenuated by preventing the PDE3A-ICER feedback loop via targeting 3 key molecular players in the feedback loop (PDE3A, PKA, and ICER), through overexpressing wild-type PDE3A, the endogenous PKA inhibitor (PKI), and antisense ICER (Figure 5).87 More importantly, the reciprocal and concomitant changes of PDE3A and ICER were also observed in adult human and mouse failing hearts, as well as adult rodent hearts with chronic infusion of Ang II or ISO.80,87 These findings strongly suggest that the positive-feedback loop regulated by PDE3A and ICER is pathologically important in adult hearts.
Figure 4.
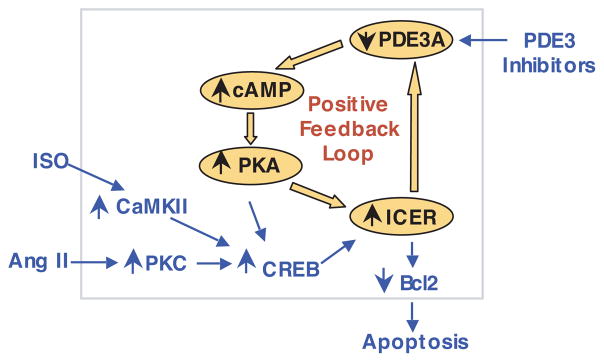
Schematic diagram showing the PDE3A-ICER positive-feedback loop stimulated by Ang II, ISO, and PDE3 inhibitors, leading to myocyte apoptosis. With respect to Ang II, activation of PKC and CREB by Ang II via AT1R initiates the PDE3A-ICER feedback loop probably by CREB-dependent ICER gene transcription (shown in blue). Induction of ICER leads to a reduction of PDE3A expression, and reduction of PDE3A leads to ICER elevation because of PKA-dependent ICER stabilization that prevents ICER degradation; this constitutes the positive PDE3A-ICER feedback loop (shown in orange) and allows a persistent induction of ICER, which plays a key role in cardiac myocyte apoptosis. CaMKII indicates Ca2+/calmodulin kinase II. Adapted from Ding et al.87
Figure 5.

Role of PDE3-ICER feedback loop in the regulation of rat adult cardiomyocyte apoptosis. Rat adult cardiac myocytes were transduced with 100 multiplicities of infection of Ad-LacZ, Ad-PDE3A1, Ad-PKI, or Ad-AS-ICER, followed by the treatment with vehicle or Ang II (200 nmol/L) for 48 hours. A, Western blots showing the expression levels of ICER and Bcl2. B, Microscopic images of cardiomyocytes stained with TUNEL (left panels) and DAPI (4′,6′-diamido-2-phenylindole) (right panels). Arrows point to apoptotic cells showing positive nuclear TUNEL staining. C, Quantitative results of TUNEL experiments. Data are means±SEM (n=3). *P<0.vs Ang II+Ad-LacZ. Adapted from Ding et al.87
The fact that expressing exogenous PDE3A blocked the PDE3A-ICER positive-feedback loop and cardiac myocyte apoptosis stimulated by Ang II and β-AR agonist87 suggests that maintaining PDE3A function is critical and that PDE3A gene therapy may represent a strategy to prevent the pathological effects from chronic neurohormonal stimulation. Therefore, it is necessary to further determine whether cardiac-specific expression of exogenous PDE3A is able to protect the heart from cardiac damage and dysfunction induced by various stresses in vivo. In addition, it is also critical to define other regulators and signaling pathways that positively regulate PDE3A expression, which may lead to new therapeutic agents that are capable of maintaining PDE3A function, abrogating the pathogenic PDE3A-ICER positive-feedback loop, and preventing cardiac dysfunction.
The Inhibition of PDE3A-ICER Feedback Loop: Role of Insulin-Like Growth Factor-1–Mediated ERK5 Activation
It has been proposed that the inhibition of cardiomyocyte apoptosis could prevent or slow cardiac failure progression. One of the important cardiomyocyte survival factors is insulin-like growth factor-1 (IGF-1).108–110 Mehrhof et al have reported that IGF-1 activates Akt and ERK1/2 and inhibits apoptosis.111 Although the importance of IGF-1/PI3-K activation in cell survival is well established,112,113 the specific role of Akt in heart failure has become more complicated by several recently published studies.114–116 For example, Nagoshi et al have reported that ischemia/reperfusion injury is increased in myristoylated Akt Tg (myr-Akt-Tg) mice because of feedback inhibition of insulin receptor substrate-1/PI3K signaling and suggested an Akt-independent cardioprotective pathway in IGF-1 signaling may be operative.114 Interestingly, the activity of serum- and glucocorticoid-regulated kinase-1 (SGK-1),117 a downstream substrate of ERK5 that promotes myocyte survival in vitro,118 was downregulated in myr-Akt-Tg mice.114 ERK5/BMK1 is a member of the MAP kinase family. The upstream kinase that phosphorylates ERK5 has been identified as MEK5.119,120 Like many MAP kinase family members, ERK5 plays a significant role in cell growth and differentiation, although emerging evidence suggests unique functional characteristics. Activation of ERK5 is documented to have an antiapoptotic effect in neuronal and endothelial cells.121,122 ERK5 knockout mice have impaired cardiac and vascular development.123 The transgenic mice with cardiac-specific expression of the constitutively active MEK5α revealed accelerated LV function recovery after ischemia/reperfusion.124 The results from these aforementioned studies led us to hypothesize that IGF-1–mediated ERK5 activation may be acting as the Akt-independent cardioprotective signaling pathway proposed by Nagoshi et al.
To determine the physiological relevance of ERK5 activation in regulating the positive-feedback loop of PDE3A and ICER, we investigated the pressure overload or doxorubicin-induced change of PDE3A and ICER expression, myocyte apoptosis, and LV function in transgenic mice with cardiac-specific expression of the constitutively active form of MEK5α (CA-MEK5α) and in normal control mice. We found that pressure overload or doxorubicin caused significant PDE3A reduction and ICER induction in normal control mice, which was prevented in the CA-MEK5α transgenic mice. In addition, myocyte apoptosis and LV developed pressure (an indication of LV function) induced by pressure overload or doxorubicin were also prevented in CA-MEK5α transgenic mice.125 These data suggest that blocking the progression of PDE3A-ICER feedback loop by ERK5 activation may inhibit myocytes apoptosis and thereby prevent cardiac dysfunction.
We have further studied the role and molecular mechanism of IGF-1/ERK5 signaling in regulation of the PDE3A-ICER feedback loop using cardiac myocytes in vitro. We found that IGF-1 significantly increased ERK5 activation as well as PDE3A expression in rat neonatal cardiac myocytes.125 ERK5 activation was necessary for this IGF-1–mediated PDE3A expression.125 In addition, activation of ERK5 by CA-MEK5α inhibited β-AR agonist, ISO-mediated PDE3A reduction and ICER induction.125 Consistent with the effects on PDE3A and ICER expression, IGF-1 blocked the ISO-induced cardiac myocyte apoptosis, which is dependent on ERK5 activation.125 These data implicated IGF-1–mediated ERK5 activation in inhibiting the ISO-mediated PDE3A-ICER feedback loop and thereby cardiac myocyte apoptosis. We also discovered that transcription factor myocyte-enhancer factor-2 (MEF2), a downstream substrate of ERK5, played a critical role in ERK5-mediated inhibition of the PDE3-ICER feedback loop and myocyte apoptosis.125 Based on sequence analysis, there are putative MEF2 consensus sites in the in the proximal 3-kb 5′ upstream flanking region of the human PDE3A gene promoter.87 These data suggest that activation of MEF2 by ERK5 may directly act on the PDE3A promoter to prevent the downregulation of PDE3A expression by ICER. Further studies are required to clarify the regulatory mechanism of PDE3A promoter by ERK5/MEF2.
As discussed above, PKA-dependent stabilization of ICER protein is critical for maintaining the PDE3A-ICER feedback loop (Figure 4). ICER stability is regulated by proteasome-dependent degradation.103 cAMP has been shown to attenuate ICER protein ubiquitination and degradation probably via a cAMP-dependent inhibition of MAP kinase ERK1/2.126 It has been shown that ERK1/2 physically interacts with ICER and mediates the phosphorylation of ICER on a critical serine residue (Ser-41) in mouse pituitary tumor AtT20 cells that promotes ICER degradation.126 In this report, the involvement of MEK1/2 and ERK1/2 was proposed because inhibitor PD98059 (initially defined as a MEK1/2 inhibitor) inhibited epidermal growth factor–induced ICER phosphorylation and degradation. However, recent studies indicate that PD98059 also effectively inhibits MEK5 and ERK5 pathway.127 Therefore, epidermal growth factor–induced phosphorylation of ICER is possible, in part or entirely, through the MEK5/ERK5 pathway. In fact, we found that activation of ERK5 significantly decreased forskolin-enhanced ICER stability (Yan C, Abe J, Ding B, unpublished data, 2006).
Taken together, our results suggest 2 potential mechanisms by which ERK5 activation intervenes PDE3A-ICER feedback loop: (1) blocking PDE3A downregulation via MEF2-mediated regulation of PDE3A promoter activity and (2) promoting ICER degradation via ERK5 phosphorylation of ICER. Further investigation will be required to define the precise mechanisms of ERK5 activation in antagonizing PDE3A-ICER feedback loop.
Conclusion
Multiple distinct PDE isozymes coexisting in cardiac myocytes concertedly and differentially regulate the temporal, spatial, and kinetic properties of cyclic nucleotide responses. Precise time, duration, and location of cyclic nucleotide elevation are critical for specifically regulating signaling pathways that result in physiological consequences. Long-term alteration of PDE activity/expression, which is vital for normal physiological function, may bring about undesired toxic effects and pathological consequences. Using PDE3 as an example, PDE3A expression is significantly downregulated in failing hearts because of a variety of etiologies. Chronic inhibition of PDE3 activity by PDE3 inhibitors in vitro induces sustained expression of transcription repressor ICER and thereby cardiac myocyte apoptosis and SERCA2 downregulation. These findings suggest that PDE3A reduction observed in failing hearts may play a causative role in the development of heart failure. Sustained ICER induction is permitted by an autoregulatory positive-feedback loop (PDE3A-ICER positive-feedback loop) in which PDE3A and ICER are key mediators. Pathological stresses (such as chronic pressure overload) and neurohormonal overactivation promote, whereas cardiac protective signaling IGF1/ERK5 attenuates, the PDE3A-ICER feedback loop (Figure 6). Sustained ICER expression resulting from the feedback loop is not only essential for cardiac myocyte apoptosis but may also contribute to other protein alteration and cardiac malfunction associated with heart failure (such as contractile dysfunction attributable to SERCA2) (Figure 6). Maintaining PDE3A function may abrogate the pathogenic PDE3A-ICER positive-feedback loop and prevent cardiac myocyte apoptosis and cardiac dysfunction.
Figure 6.

Proposed model showing the regulation and function the PDE3A-ICER feedback loop. Pathological stimuli (such as chronic pressure overload and neurohormonal overactivation) promote the PDE3A-ICER feedback loop via initial activation of CREB, whereas cardiac protectors (such as IGF-1) attenuate it through activation of ERK5. The persistent ICER induction resulting from the PDE3A-ICER feedback loop dysregulates the expression of a number of proteins (such as downregulation of Bcl-2 and SERCA2) and may, in turn, lead to cardiac myocyte death and contractile dysfunction, which is associated with impaired cardiac function.
Acknowledgments
Sources of Funding
This work was supported by American Heart Association Grant 0455847T (to C.Y.) and NIH grants HL077789 (to C.Y.), HL-66919 (to J.A.), HL-077789 (to J.A.), and GM-071485 (to J.A.).
Footnotes
Disclosures
None.
References
- 1.Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003;93:896–906. doi: 10.1161/01.RES.0000102042.83024.CA. [DOI] [PubMed] [Google Scholar]
- 2.Lohmann SM, Fischmeister R, Walter U. Signal transduction by cGMP in heart. Basic Res Cardiol. 1991;86:503–514. doi: 10.1007/BF02190700. [DOI] [PubMed] [Google Scholar]
- 3.Soderling SH, Beavo JA. Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functions. Curr Opin Cell Biol. 2000;12:174–179. doi: 10.1016/s0955-0674(99)00073-3. [DOI] [PubMed] [Google Scholar]
- 4.Jurevicius J, Fischmeister R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by beta-adrenergic agonists. Proc Natl Acad Sci U S A. 1996;93:295–299. doi: 10.1073/pnas.93.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaccolo M, Pozzan T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 2002;295:1711–1715. doi: 10.1126/science.1069982. [DOI] [PubMed] [Google Scholar]
- 6.Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Packer M, Carver JR, Rodeheffer RJ, Ivanhoe RJ, DiBianco R, Zeldis SM, Hendrix GH, Bommer WJ, Elkayam U, Kukin ML, Mallis GI, Sollano JA, Shannon JD, Tandon PK, DeMets DL, et al. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N Engl J Med. 1991;325:1468–1475. doi: 10.1056/NEJM199111213252103. [DOI] [PubMed] [Google Scholar]
- 8.Amsallem E, Kasparian C, Haddour G, Boissel JP, Nony P. Phosphodiesterase III inhibitors for heart failure. Cochrane Database Syst Rev. 2005;25:CD002230. doi: 10.1002/14651858.CD002230.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagel DJ, Aizawa T, Jeon KI, Liu W, Mohan A, Wei H, Miano JM, Florio VA, Gao P, Korshunov VA, Berk BC, Yan C. Role of nuclear Ca2+/calmodulin-stimulated phosphodiesterase 1A in vascular smooth muscle cell growth and survival. Circ Res. 2006;98:777–784. doi: 10.1161/01.RES.0000215576.27615.fd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han P, Werber J, Surana M, Fleischer N, Michaeli T. The calcium/calmodulin-dependent phosphodiesterase PDE1C down-regulates glucose-induced insulin secretion. J Biol Chem. 1999;274:22337–22344. doi: 10.1074/jbc.274.32.22337. [DOI] [PubMed] [Google Scholar]
- 11.Rybalkin SD, Rybalkina I, Beavo JA, Bornfeldt KE. Cyclic nucleotide phosphodiesterase 1C promotes human arterial smooth muscle cell proliferation. Circ Res. 2002;90:151–157. doi: 10.1161/hh0202.104108. [DOI] [PubMed] [Google Scholar]
- 12.Hambleton R, Krall J, Tikishvili E, Honeggar M, Ahmad F, Manganiello VC, Movsesian MA. Isoforms of cyclic nucleotide phosphodiesterase PDE3 and their contribution to camp hydrolytic activity in subcellular fractions of human myocardium. J Biol Chem. 2005;280:39168–39174. doi: 10.1074/jbc.M506760200. [DOI] [PubMed] [Google Scholar]
- 13.Wallis RM, Corbin JD, Francis SH, Ellis P. Tissue distribution of phosphodiesterase families and the effects of sildenafil on tissue cyclic nucleotides, platelet function, and the contractile responses of trabeculae carneae and aortic rings in vitro. Am J Cardiol. 1999;83:3C–12C. doi: 10.1016/s0002-9149(99)00042-9. [DOI] [PubMed] [Google Scholar]
- 14.Yanaka N, Kurosawa Y, Minami K, Kawai E, Omori K. cGMP-phosphodiesterase activity is up-regulated in response to pressure overload of rat ventricles. Biosci Biotechnol Biochem. 2003;67:973–979. doi: 10.1271/bbb.67.973. [DOI] [PubMed] [Google Scholar]
- 15.Clapham JC, Wilderspin AF. Cloning of dog heart PDE1A—a first detailed characterization at the molecular level in this species. Gene. 2001;268:165–171. doi: 10.1016/s0378-1119(01)00413-9. [DOI] [PubMed] [Google Scholar]
- 16.Sonnenburg WK, Rybalkin SD, Bornfeldt KE, Kwak KS, Rybalkina IG, Beavo JA. Identification, quantitation, and cellular localization of PDE1 calmodulin-stimulated cyclic nucleotide phosphodiesterases. Methods. 1998;14:3–19. doi: 10.1006/meth.1997.0561. [DOI] [PubMed] [Google Scholar]
- 17.Kostic MM, Erdogan S, Rena G, Borchert G, Hoch B, Bartel S, Scotland G, Huston E, Houslay MD, Krause EG. Altered expression of PDE1 and PDE4 cyclic nucleotide phosphodiesterase isoforms in 7-oxo-prostacyclin-preconditioned rat heart. J Mol Cell Cardiol. 1997;29:3135–3146. doi: 10.1006/jmcc.1997.0544. [DOI] [PubMed] [Google Scholar]
- 18.Bode DC, Kanter JR, Brunton LL. Cellular distribution of phosphodiesterase isoforms in rat cardiac tissue. Circ Res. 1991;68:1070–1079. doi: 10.1161/01.res.68.4.1070. [DOI] [PubMed] [Google Scholar]
- 19.Leroy MJ, Degerman E, Taira M, Murata T, Wang LH, Movsesian MA, Meacci E, Manganiello VC. Characterization of two recombinant PDE3 (cGMP-inhibited cyclic nucleotide phosphodiesterase) isoforms, RcGIP1 and HcGIP2, expressed in NIH 3006 murine fibroblasts and Sf9 insect cells. Biochemistry. 1996;35:10194–10202. doi: 10.1021/bi952711t. [DOI] [PubMed] [Google Scholar]
- 20.MacFarland RT, Zelus BD, Beavo JA. High concentrations of a cGMP-stimulated phosphodiesterase mediate ANP-induced decreases in cAMP and steroidogenesis in adrenal glomerulosa cells. J Biol Chem. 1991;266:136–142. [PubMed] [Google Scholar]
- 21.Lugnier C, Komas N. Modulation of vascular cyclic nucleotide phosphodiesterases by cyclic GMP: role in vasodilatation. Eur Heart J. 1993;14(suppl I):141–148. [PubMed] [Google Scholar]
- 22.Komas N, Lugnier C, Stoclet JC. Endothelium-dependent and independent relaxation of the rat aorta by cyclic nucleotide phosphodiesterase inhibitors. Br J Pharmacol. 1991;104:495–503. doi: 10.1111/j.1476-5381.1991.tb12457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Osinski MT, Rauch BH, Schror K. Antimitogenic actions of organic nitrates are potentiated by sildenafil and mediated via activation of protein kinase A. Mol Pharmacol. 2001;59:1044–1050. doi: 10.1124/mol.59.5.1044. [DOI] [PubMed] [Google Scholar]
- 24.Aizawa T, Wei H, Miano JM, Abe J, Berk BC, Yan C. Role of phosphodiesterase 3 in NO/cGMP-mediated antiinflammatory effects in vascular smooth muscle cells. Circ Res. 2003;93:406– 413. doi: 10.1161/01.RES.0000091074.33584.F0. [DOI] [PubMed] [Google Scholar]
- 25.Maurice DH, Haslam RJ. Molecular basis of the synergistic inhibition of platelet function by nitrovasodilators and activators of adenylate cyclase: inhibition of cyclic AMP breakdown by cyclic GMP. Mol Pharmacol. 1990;37:671– 681. [PubMed] [Google Scholar]
- 26.Bowen R, Haslam RJ. Effects of nitrovasodilators on platelet cyclic nucleotide levels in rabbit blood: role for cyclic AMP in synergistic inhibition of platelet function by SIN-1 and prostaglandin E1. J Cardiovasc Pharmacol. 1991;17:424– 433. doi: 10.1097/00005344-199103000-00011. [DOI] [PubMed] [Google Scholar]
- 27.Fischmeister R, Castro L, Abi-Gerges A, Rochais F, Vandecasteele G. Species- and tissue-dependent effects of NO and cyclic GMP on cardiac ion channels. Comp Biochem Physiol A Mol Integr Physiol. 2005;142:136–143. doi: 10.1016/j.cbpb.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 28.Dittrich M, Jurevicius J, Georget M, Rochais F, Fleischmann B, Hescheler J, Fischmeister R. Local response of L-type Ca(2+) current to nitric oxide in frog ventricular myocytes. J Physiol. 2001;534:109–121. doi: 10.1111/j.1469-7793.2001.00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mery PF, Pavoine C, Belhassen L, Pecker F, Fischmeister R. Nitric oxide regulates cardiac Ca2+ current. Involvement of cGMP-inhibited and cGMP-stimulated phosphodiesterases through guanylyl cyclase activation. J Biol Chem. 1993;268:26286–26295. [PubMed] [Google Scholar]
- 30.Mongillo M, Tocchetti CG, Terrin A, Lissandron V, Cheung YF, Dostmann WR, Pozzan T, Kass DA, Paolocci N, Houslay MD, Zaccolo M. Compartmentalized phosphodiesterase-2 activity blunts {beta}-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ Res. 2006;98:226–234. doi: 10.1161/01.RES.0000200178.34179.93. [DOI] [PubMed] [Google Scholar]
- 31.Rivet-Bastide M, Vandecasteele G, Hatem S, Verde I, Benardeau A, Mercadier JJ, Fischmeister R. cGMP-stimulated cyclic nucleotide phosphodiesterase regulates the basal calcium current in human atrial myocytes. J Clin Invest. 1997;99:2710–2718. doi: 10.1172/JCI119460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirstein M, Rivet-Bastide M, Hatem S, Benardeau A, Mercadier JJ, Fischmeister R. Nitric oxide regulates the calcium current in isolated human atrial myocytes. J Clin Invest. 1995;95:794– 802. doi: 10.1172/JCI117729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Degerman E, Belfrage P, Manganiello VC. Structure, localization, and regulation of cGMP-inhibited phosphodiesterase (PDE3) J Biol Chem. 1997;272:6823– 6826. doi: 10.1074/jbc.272.11.6823. [DOI] [PubMed] [Google Scholar]
- 34.Beavo JA, Hardman JG, Sutherland EW. Stimulation of adenosine 3′,5′-monophosphate hydrolysis by guanosine 3′,5′-monophosphate. J Biol Chem. 1971;246:3841–3846. [PubMed] [Google Scholar]
- 35.Wada H, Manganiello VC, Osborne JC., Jr Analysis of the kinetics of cyclic AMP hydrolysis by the cyclic GMP-stimulated cyclic nucleotide phosphodiesterase. J Biol Chem. 1987;262:13938–13945. [PubMed] [Google Scholar]
- 36.Mery PF, Lohmann SM, Walter U, Fischmeister R. Ca2+ current is regulated by cyclic GMP-dependent protein kinase in mammalian cardiac myocytes. Proc Natl Acad Sci U S A. 1991;88:1197–1201. doi: 10.1073/pnas.88.4.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ, Houslay MD. beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi. Proc Natl Acad Sci U S A. 2003;100:940–945. doi: 10.1073/pnas.262787199. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature. 2005;437:574–578. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weishaar RE, Kobylarz-Singer DC, Kaplan HR. Subclasses of cyclic AMP phosphodiesterase in cardiac muscle. J Mol Cell Cardiol. 1987;19:1025–1036. doi: 10.1016/s0022-2828(87)80574-6. [DOI] [PubMed] [Google Scholar]
- 40.Weishaar RE, Kobylarz-Singer DC, Steffen RP, Kaplan HR. Subclasses of cyclic AMP-specific phosphodiesterase in left ventricular muscle and their involvement in regulating myocardial contractility. Circ Res. 1987;61:539–547. doi: 10.1161/01.res.61.4.539. [DOI] [PubMed] [Google Scholar]
- 41.Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, Huston E, Hannawacker A, Lohse MJ, Pozzan T, Houslay MD, Zaccolo M. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res. 2004;95:67–75. doi: 10.1161/01.RES.0000134629.84732.11. [DOI] [PubMed] [Google Scholar]
- 42.Rochais F, Abi-Gerges A, Horner K, Lefebvre F, Cooper DM, Conti M, Fischmeister R, Vandecasteele G. A specific pattern of phosphodies-terases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ Res. 2006;98:1081–1088. doi: 10.1161/01.RES.0000218493.09370.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katano Y, Endoh M. Differential effects of Ro 20–1724 and isobutyl-methylxanthine on the basal force of contraction and beta-adrenoceptor-mediated response in the rat ventricular myocardium. Biochem Biophys Res Commun. 1990;167:123–129. doi: 10.1016/0006-291x(90)91739-f. [DOI] [PubMed] [Google Scholar]
- 44.Shakur Y, Holst LS, Landstrom TR, Movsesian M, Degerman E, Manganiello V. Regulation and function of the cyclic nucleotide phos-phodiesterase (PDE3) gene family. Prog Nucleic Acid Res Mol Biol. 2001;66:241–277. doi: 10.1016/s0079-6603(00)66031-2. [DOI] [PubMed] [Google Scholar]
- 45.Choi YH, Ekholm D, Krall J, Ahmad F, Degerman E, Manganiello VC, Movsesian MA. Identification of a novel isoform of the cyclic-nucleotide phosphodiesterase PDE3A expressed in vascular smooth-muscle myocytes. Biochem J. 2001;353:41–50. [PMC free article] [PubMed] [Google Scholar]
- 46.Wechsler J, Choi YH, Krall J, Ahmad F, Manganiello VC, Movsesian MA. Isoforms of cyclic nucleotide phosphodiesterase PDE3A in cardiac myocytes. J Biol Chem. 2002;277:38072–38078. doi: 10.1074/jbc.M203647200. [DOI] [PubMed] [Google Scholar]
- 47.Smith CJ, Krall J, Manganiello VC, Movsesian MA. Cytosolic and sarcoplasmic reticulum-associated low Km, cGMP-inhibited cAMP phosphodiesterase in mammalian myocardium. Biochem Biophys Res Commun. 1993;190:516–521. doi: 10.1006/bbrc.1993.1078. [DOI] [PubMed] [Google Scholar]
- 48.Grant PG, Colman RW. Purification and characterization of a human platelet cyclic nucleotide phosphodiesterase. Biochemistry. 1984;23:1801–1807. doi: 10.1021/bi00303a034. [DOI] [PubMed] [Google Scholar]
- 49.Harrison SA, Reifsnyder DH, Gallis B, Cadd GG, Beavo JA. Isolation and characterization of bovine cardiac muscle cGMP-inhibited phosphodiesterase: a receptor for new cardiotonic drugs. Mol Pharmacol. 1986;29:506–514. [PubMed] [Google Scholar]
- 50.Degerman E, Belfrage P, Newman AH, Rice KC, Manganiello VC. Purification of the putative hormone-sensitive cyclic AMP phosphodiesterase from rat adipose tissue using a derivative of cilostamide as a novel affinity ligand. J Biol Chem. 1987;262:5797–5807. [PubMed] [Google Scholar]
- 51.Movsesian MA. PDE3 cyclic nucleotide phosphodiesterases and the compartmentation of cyclic nucleotide-mediated signalling in cardiac myocytes. Basic Res Cardiol. 2002;97(suppl 1):I83–I90. doi: 10.1007/s003950200035. [DOI] [PubMed] [Google Scholar]
- 52.Kauffman RF, Crowe VG, Utterback BG, Robertson DW. LY195115: a potent, selective inhibitor of cyclic nucleotide phosphodiesterase located in the sarcoplasmic reticulum. Mol Pharmacol. 1986;30:609– 616. [PubMed] [Google Scholar]
- 53.Kithas PA, Artman M, Thompson WJ, Strada SJ. Subcellular distribution of high-affinity type IV cyclic AMP phosphodiesterase activity in rabbit ventricular myocardium: relations to the effects of cardiotonic drugs. Circ Res. 1988;62:782–789. doi: 10.1161/01.res.62.4.782. [DOI] [PubMed] [Google Scholar]
- 54.Patrucco E, Notte A, Barberis L, Selvetella G, Maffei A, Brancaccio M, Marengo S, Russo G, Azzolino O, Rybalkin SD, Silengo L, Altruda F, Wetzker R, Wymann MP, Lembo G, Hirsch E. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell. 2004;118:375–387. doi: 10.1016/j.cell.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 55.Marcantoni A, Levi RC, Gallo MP, Hirsch E, Alloatti G. Phosphoino-sitide 3-kinasegamma (PI3Kgamma) controls L-type calcium current (ICa,L) through its positive modulation of type-3 phosphodiesterase (PDE3) J Cell Physiol. 2006;206:329–336. doi: 10.1002/jcp.20467. [DOI] [PubMed] [Google Scholar]
- 56.Benotti JR, Grossman W, Braunwald E, Davolos DD, Alousi AA. Hemodynamic assessment of amrinone. A new inotropic agent. N Engl J Med. 1978;299:1373–1377. doi: 10.1056/NEJM197812212992501. [DOI] [PubMed] [Google Scholar]
- 57.Baim DS, McDowell AV, Cherniles J, Monrad ES, Parker JA, Edelson J, Braunwald E, Grossman W. Evaluation of a new bipyridine inotropic agent—milrinone—in patients with severe congestive heart failure. N Engl J Med. 1983;309:748–756. doi: 10.1056/NEJM198309293091302. [DOI] [PubMed] [Google Scholar]
- 58.Uretsky BF, Generalovich T, Reddy PS, Spangenberg RB, Follansbee WP. The acute hemodynamic effects of a new agent, MDL 17,043, in the treatment of congestive heart failure. Circulation. 1983;67:823– 828. doi: 10.1161/01.cir.67.4.823. [DOI] [PubMed] [Google Scholar]
- 59.Jaski BE, Fifer MA, Wright RF, Braunwald E, Colucci WS. Positive inotropic and vasodilator actions of milrinone in patients with severe congestive heart failure. Dose-response relationships and comparison to nitroprusside. J Clin Invest. 1985;75:643– 649. doi: 10.1172/JCI111742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parnley WW, Chatterjee K. Combined vasodilator and inotropic therapy: a new approach in the treatment of heart failure. In: Mason DT, editor. Advances in Health Disease. New York: Grune and Stratton; 1977. pp. 45–47. [Google Scholar]
- 61.McDaniel NL, Rembold CM, Murphy RA. Cyclic nucleotide dependent relaxation in vascular smooth muscle. Can J Physiol Pharmacol. 1994;72:1380–1385. doi: 10.1139/y94-199. [DOI] [PubMed] [Google Scholar]
- 62.Lincoln TM, Cornwell TL. Towards an understanding of the mechanism of action of cyclic AMP and cyclic GMP in smooth muscle relaxation. Blood Vessels. 1991;28:129–137. doi: 10.1159/000158852. [DOI] [PubMed] [Google Scholar]
- 63.Malecot CO, Bers DM, Katzung BG. Biphasic contractions induced by milrinone at low temperature in ferret ventricular muscle: role of the sarcoplasmic reticulum and transmembrane calcium influx. Circ Res. 1986;59:151–162. doi: 10.1161/01.res.59.2.151. [DOI] [PubMed] [Google Scholar]
- 64.Vandecasteele G, Verde I, Rucker-Martin C, Donzeau-Gouge P, Fischmeister R. Cyclic GMP regulation of the L-type Ca(2+) channel current in human atrial myocytes. J Physiol. 2001;533:329–340. doi: 10.1111/j.1469-7793.2001.0329a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Verde I, Vandecasteele G, Lezoualc’h F, Fischmeister R. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br J Pharmacol. 1999;127:65–74. doi: 10.1038/sj.bjp.0702506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jurevicius J, Skeberdis VA, Fischmeister R. Role of cyclic nucleotide phosphodiesterase isoforms in cAMP compartmentation following beta2-adrenergic stimulation of ICa, L in frog ventricular myocytes. J Physiol. 2003;551:239–252. doi: 10.1113/jphysiol.2003.045211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gaide MS, Fitterman WS, Wiggins JR, Myerburg RJ, Cameron JS, Bassett AL. Amrinone relaxes potassium-induced contracture of failing right ventricular muscle of cats. J Cardiovasc Pharmacol. 1983;5:335–340. doi: 10.1097/00005344-198303000-00028. [DOI] [PubMed] [Google Scholar]
- 68.Gwathmey JK, Morgan JP. The effects of milrinone and piroximone on intracellular calcium handling in working myocardium from the ferret. Br J Pharmacol. 1985;85:97–108. doi: 10.1111/j.1476-5381.1985.tb08835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yano M, Kohno M, Ohkusa T, Mochizuki M, Yamada J, Hisaoka T, Ono K, Tanigawa T, Kobayashi S, Matsuzaki M. Effect of milrinone on left ventricular relaxation and Ca(2+) uptake function of cardiac sarcoplasmic reticulum. Am J Physiol Heart Circ Physiol. 2000;279:H1898–H1905. doi: 10.1152/ajpheart.2000.279.4.H1898. [DOI] [PubMed] [Google Scholar]
- 70.Movsesian MA, Alharethi R. Inhibitors of cyclic nucleotide phosphodi-esterase PDE3 as adjunct therapy for dilated cardiomyopathy. Expert Opin Investig Drugs. 2002;11:1529–1536. doi: 10.1517/13543784.11.11.1529. [DOI] [PubMed] [Google Scholar]
- 71.Xamoterol in severe heart failure. The Xamoterol in Severe Heart Failure Study Group. Lancet. 1990;336:1– 6. [PubMed] [Google Scholar]
- 72.Oliva F, Latini R, Politi A, Staszewsky L, Maggioni AP, Nicolis E, Mauri F. Intermittent 6-month low-dose dobutamine infusion in severe heart failure: DICE multicenter trial. Am Heart J. 1999;138:247–253. doi: 10.1016/s0002-8703(99)70108-0. [DOI] [PubMed] [Google Scholar]
- 73.O’Connor CM, Gattis WA, Uretsky BF, Adams KF, Jr, McNulty SE, Grossman SH, McKenna WJ, Zannad F, Swedberg K, Gheorghiade M, Califf RM. Continuous intravenous dobutamine is associated with an increased risk of death in patients with advanced heart failure: insights from the Flolan International Randomized Survival Trial (FIRST) Am Heart J. 1999;138:78– 86. doi: 10.1016/s0002-8703(99)70250-4. [DOI] [PubMed] [Google Scholar]
- 74.Liu Y, Shakur Y, Yoshitake M, Kambayashi Ji J. Cilostazol (pletal): a dual inhibitor of cyclic nucleotide phosphodiesterase type 3 and adenosine uptake. Cardiovasc Drug Rev. 2001;19:369–386. doi: 10.1111/j.1527-3466.2001.tb00076.x. [DOI] [PubMed] [Google Scholar]
- 75.Wang S, Cone J, Fong M, Yoshitake M, Kambayashi J, Liu Y. Interplay between inhibition of adenosine uptake and phosphodiesterase type 3 on cardiac function by cilostazol, an agent to treat intermittent claudication. J Cardiovasc Pharmacol. 2001;38:775–783. doi: 10.1097/00005344-200111000-00014. [DOI] [PubMed] [Google Scholar]
- 76.Liu Y, Fong M, Cone J, Wang S, Yoshitake M, Kambayashi J. Inhibition of adenosine uptake and augmentation of ischemia-induced increase of interstitial adenosine by cilostazol, an agent to treat intermittent claudication. J Cardiovasc Pharmacol. 2000;36:351–360. doi: 10.1097/00005344-200009000-00011. [DOI] [PubMed] [Google Scholar]
- 77.Bohm M, Diet F, Kemkes B, Erdmann E. Enhancement of the effectiveness of milrinone to increase force of contraction by stimulation of cardiac beta-adrenoceptors in the failing human heart. Klin Wochenschr. 1988;66:957–962. doi: 10.1007/BF01738110. [DOI] [PubMed] [Google Scholar]
- 78.Feldman MD, Copelas L, Gwathmey JK, Phillips P, Warren SE, Schoen FJ, Grossman W, Morgan JP. Deficient production of cyclic AMP: pharmacologic evidence of an important cause of contractile dysfunction in patients with end-stage heart failure. Circulation. 1987;75:331–339. doi: 10.1161/01.cir.75.2.331. [DOI] [PubMed] [Google Scholar]
- 79.Silver PJ, Allen P, Etzler JH, Hamel LT, Bentley RG, Pagani ED. Cellular distribution and pharmacological sensitivity of low Km cyclic nucleotide phosphodiesterase isozymes in human cardiac muscle from normal and cardiomyopathic subjects. Second Messengers Phospho-proteins. 1990;13:13–25. [PubMed] [Google Scholar]
- 80.Ding B, Abe J, Wei H, Huang Q, Walsh RA, Molina CA, Zhao A, Sadoshima J, Blaxall BC, Berk BC, Yan C. Functional role of phospho-diesterase 3 in cardiomyocyte apoptosis: implication in heart failure. Circulation. 2005;111:2469–2476. doi: 10.1161/01.CIR.0000165128.39715.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Movsesian MA, Smith CJ, Krall J, Bristow MR, Manganiello VC. Sarcoplasmic reticulum-associated cyclic adenosine 5′-monophosphate phosphodiesterase activity in normal and failing human hearts. J Clin Invest. 1991;88:15–19. doi: 10.1172/JCI115272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sato N, Asai K, Okumura S, Takagi G, Shannon RP, Fujita-Yamaguchi Y, Ishikawa Y, Vatner SF, Vatner DE. Mechanisms of desensitization to a PDE inhibitor (milrinone) in conscious dogs with heart failure. Am J Physiol. 1999;276:H1699–H1705. doi: 10.1152/ajpheart.1999.276.5.H1699. [DOI] [PubMed] [Google Scholar]
- 83.Smith CJ, Huang R, Sun D, Ricketts S, Hoegler C, Ding JZ, Moggio RA, Hintze TH. Development of decompensated dilated cardiomyopathy is associated with decreased gene expression and activity of the milrinone-sensitive cAMP phosphodiesterase PDE3A. Circulation. 1997;96:3116–3123. doi: 10.1161/01.cir.96.9.3116. [DOI] [PubMed] [Google Scholar]
- 84.Smith CJ, He J, Ricketts SG, Ding JZ, Moggio RA, Hintze TH. Down-regulation of right ventricular phosphodiesterase PDE-3A mRNA and protein before the development of canine heart failure. Cell Biochem Biophys. 1998;29:67– 88. doi: 10.1007/BF02737829. [DOI] [PubMed] [Google Scholar]
- 85.Sharma R, Anker SD. Immune and neurohormonal pathways in chronic heart failure. Congest Heart Fail. 2002;8:23–28. 48. doi: 10.1111/j.1527-5299.2002.00724.x. [DOI] [PubMed] [Google Scholar]
- 86.Chatterjee K. Congestive heart failure: what should be the initial therapy and why? Am J Cardiovasc Drugs. 2002;2:1– 6. doi: 10.2165/00129784-200202010-00001. [DOI] [PubMed] [Google Scholar]
- 87.Ding B, Abe J, Wei H, Xu H, Che W, Aizawa T, Liu W, Molina CA, Sadoshima J, Blaxall BC, Berk BC, Yan C. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc Natl Acad Sci U S A. 2005;102:14771–14776. doi: 10.1073/pnas.0506489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takahashi K, Osanai T, Nakano T, Wakui M, Okumura K. Enhanced activities and gene expression of phosphodiesterase types 3 and 4 in pressure-induced congestive heart failure. Heart Vessels. 2002;16:249–256. doi: 10.1007/s003800200032. [DOI] [PubMed] [Google Scholar]
- 89.Masciarelli S, Horner K, Liu C, Park SH, Hinckley M, Hockman S, Nedachi T, Jin C, Conti M, Manganiello V. Cyclic nucleotide phospho-diesterase 3A-deficient mice as a model of female infertility. J Clin Invest. 2004;114:196–205. doi: 10.1172/JCI21804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kang PM, Izumo S. Apoptosis and heart failure: a critical review of the literature. Circ Res. 2000;86:1107–1113. doi: 10.1161/01.res.86.11.1107. [DOI] [PubMed] [Google Scholar]
- 91.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–1141. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 92.Yamamoto KK, Gonzalez GA, Biggs WH, 3rd, Montminy MR. Phos-phorylation-induced binding and transcriptional efficacy of nuclear factor CREB. Nature. 1988;334:494– 498. doi: 10.1038/334494a0. [DOI] [PubMed] [Google Scholar]
- 93.Molina CA, Foulkes NS, Lalli E, Sassone-Corsi P. Inducibility and negative autoregulation of CREM: an alternative promoter directs the expression of ICER, an early response repressor. Cell. 1993;75:875– 886. doi: 10.1016/0092-8674(93)90532-u. [DOI] [PubMed] [Google Scholar]
- 94.Stehle JH, Foulkes NS, Molina CA, Simonneaux V, Pevet P, Sassone-Corsi P. Adrenergic signals direct rhythmic expression of transcriptional repressor CREM in the pineal gland. Nature. 1993;365:314–320. doi: 10.1038/365314a0. [DOI] [PubMed] [Google Scholar]
- 95.Mioduszewska B, Jaworski J, Kaczmarek L. Inducible cAMP early repressor (ICER) in the nervous system—a transcriptional regulator of neuronal plasticity and programmed cell death. J Neurochem. 2003;87:1313–1320. doi: 10.1046/j.1471-4159.2003.02116.x. [DOI] [PubMed] [Google Scholar]
- 96.Tomita H, Nazmy M, Kajimoto K, Yehia G, Molina CA, Sadoshima J. Inducible cAMP early repressor (ICER) is a negative-feedback regulator of cardiac hypertrophy and an important mediator of cardiac myocyte apoptosis in response to beta-adrenergic receptor stimulation. Circ Res. 2003;93:12–22. doi: 10.1161/01.RES.0000079794.57578.F1. [DOI] [PubMed] [Google Scholar]
- 97.Sussman MA. ICER-capades: putting cardiac cyclic AMP signaling “on ice”. Circ Res. 2003;93:6– 8. doi: 10.1161/01.RES.0000082770.81721.44. [DOI] [PubMed] [Google Scholar]
- 98.Bodor J, Bodorova J, Gress RE. Suppression of T cell function: a potential role for transcriptional repressor ICER. J Leukoc Biol. 2000;67:774–779. doi: 10.1002/jlb.67.6.774. [DOI] [PubMed] [Google Scholar]
- 99.Houser SR, Piacentino V, 3rd, Weisser J. Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol. 2000;32:1595–1607. doi: 10.1006/jmcc.2000.1206. [DOI] [PubMed] [Google Scholar]
- 100.del Monte F, Hajjar RJ. Targeting calcium cycling proteins in heart failure through gene transfer. J Physiol. 2003;546:49– 61. doi: 10.1113/jphysiol.2002.026732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Frank KF, Bolck B, Brixius K, Kranias EG, Schwinger RH. Modulation of SERCA: implications for the failing human heart. Basic Res Cardiol. 2002;97(suppl 1):I72–I78. doi: 10.1007/s003950200033. [DOI] [PubMed] [Google Scholar]
- 102.Jaworski J, Mioduszewska B, Sanchez-Capelo A, Figiel I, Habas A, Gozdz A, Proszynski T, Hetman M, Mallet J, Kaczmarek L. Inducible cAMP early repressor, an endogenous antagonist of cAMP responsive element-binding protein, evokes neuronal apoptosis in vitro. J Neurosci. 2003;23:4519– 4526. doi: 10.1523/JNEUROSCI.23-11-04519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Folco EJ, Koren G. Degradation of the inducible cAMP early repressor (ICER) by the ubiquitin-proteasome pathway. Biochem J. 1997;328(pt 1):37– 43. doi: 10.1042/bj3280037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maronde E, Wicht H, Tasken K, Genieser HG, Dehghani F, Olcese J, Korf HW. CREB phosphorylation and melatonin biosynthesis in the rat pineal gland: involvement of cyclic AMP dependent protein kinase type II. J Pineal Res. 1999;27:170–182. doi: 10.1111/j.1600-079x.1999.tb00613.x. [DOI] [PubMed] [Google Scholar]
- 105.Pfeffer M, Maronde E, Korf HW, Stehle JH. Antisense experiments reveal molecular details on mechanisms of ICER suppressing cAMP-inducible genes in rat pinealocytes. J Pineal Res. 2000;29:24–33. doi: 10.1034/j.1600-079x.2000.290104.x. [DOI] [PubMed] [Google Scholar]
- 106.Stehle JH, von Gall C, Korf HW. Analysis of cell signalling in the rodent pineal gland deciphers regulators of dynamic transcription in neural/endocrine cells. Eur J Neurosci. 2001;14:1–9. doi: 10.1046/j.0953-816x.2001.01627.x. [DOI] [PubMed] [Google Scholar]
- 107.Brunton LL. A positive feedback loop contributes to the deleterious effects of angiotensin. Proc Natl Acad Sci U S A. 2005;102:14483–14484. doi: 10.1073/pnas.0507070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Buerke M, Murohara T, Skurk C, Nuss C, Tomaselli K, Lefer AM. Cardioprotective effect of insulin-like growth factor I in myocardial ischemia followed by reperfusion. Proc Natl Acad Sci U S A. 1995;92:8031– 8035. doi: 10.1073/pnas.92.17.8031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Anversa P, Kajstura J, Cheng W, Reiss K, Cigola E, Olivetti G. Insulin-like growth factor-1 and myocyte growth: the danger of a dogma. Part I. Postnatal myocardial development: normal growth. Cardiovasc Res. 1996;32:219–225. doi: 10.1016/0008-6363(96)00035-1. [DOI] [PubMed] [Google Scholar]
- 110.Reiss K, Cheng W, Ferber A, Kajstura J, Li P, Li B, Olivetti G, Homcy CJ, Baserga R, Anversa P. Overexpression of insulin-like growth factor-1 in the heart is coupled with myocyte proliferation in transgenic mice. Proc Natl Acad Sci U S A. 1996;93:8630– 8635. doi: 10.1073/pnas.93.16.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mehrhof FB, Muller FU, Bergmann MW, Li P, Wang Y, Schmitz W, Dietz R, von Harsdorf R. In cardiomyocyte hypoxia, insulin-like growth factor-I-induced antiapoptotic signaling requires phosphatidylinositol-3-OH-kinase-dependent and mitogen-activated protein kinase-dependent activation of the transcription factor cAMP response element-binding protein. Circulation. 2001;104:2088–2094. doi: 10.1161/hc4201.097133. [DOI] [PubMed] [Google Scholar]
- 112.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661– 665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 113.Kajstura J, Fiordaliso F, Andreoli AM, Li B, Chimenti S, Medow MS, Limana F, Nadal-Ginard B, Leri A, Anversa P. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes. 2001;50:1414–1424. doi: 10.2337/diabetes.50.6.1414. [DOI] [PubMed] [Google Scholar]
- 114.Nagoshi T, Matsui T, Aoyama T, Leri A, Anversa P, Li L, Ogawa W, del Monte F, Gwathmey JK, Grazette L, Hemmings BA, Kass DA, Champion HC, Rosenzweig A. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J Clin Invest. 2005;115:2128–2138. doi: 10.1172/JCI23073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angio-genesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.O’Neill BT, Abel ED. Akt1 in the cardiovascular system: friend or foe? J Clin Invest. 2005;115:2059–2064. doi: 10.1172/JCI25900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hayashi M, Tapping RI, Chao TH, Lo JF, King CC, Yang Y, Lee JD. BMK1 mediates growth factor-induced cell proliferation through direct cellular activation of serum and glucocorticoid-inducible kinase. J Biol Chem. 2001;276:8631– 8634. doi: 10.1074/jbc.C000838200. [DOI] [PubMed] [Google Scholar]
- 118.Aoyama T, Matsui T, Novikov M, Park J, Hemmings B, Rosenzweig A. Serum and glucocorticoid-responsive kinase-1 regulates cardiomyocyte survival and hypertrophic response. Circulation. 2005;111:1652–1659. doi: 10.1161/01.CIR.0000160352.58142.06. [DOI] [PubMed] [Google Scholar]
- 119.Zhou G, Bao ZQ, Dixon JE. Components of a new human protein kinase signal transduction pathway. J Biol Chem. 1995;270:12665–12669. doi: 10.1074/jbc.270.21.12665. [DOI] [PubMed] [Google Scholar]
- 120.Lee JD, Ulevitch RJ, Han J. Primary structure of BMK1: a new mammalian MAP kinase. Biochem Biophys Res Commun. 1995;213:715–724. doi: 10.1006/bbrc.1995.2189. [DOI] [PubMed] [Google Scholar]
- 121.Suzaki Y, Yoshizumi M, Kagami S, Koyama AH, Taketani Y, Houchi H, Tsuchiya K, Takeda E, Tamaki T. Hydrogen peroxide stimulates c-Src-mediated big mitogen-activated protein kinase 1 (BMK1) and the MEF2C signaling pathway in PC12 cells: potential role in cell survival following oxidative insults. J Biol Chem. 2002;277:9614–9621. doi: 10.1074/jbc.M111790200. [DOI] [PubMed] [Google Scholar]
- 122.Pi X, Yan C, Berk BC. Big mitogen-activated protein kinase (BMK1)/ERK5 protects endothelial cells from apoptosis. Circ Res. 2004;94:362–369. doi: 10.1161/01.RES.0000112406.27800.6F. [DOI] [PubMed] [Google Scholar]
- 123.Regan CP, Li W, Boucher DM, Spatz S, Su MS, Kuida K. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc Natl Acad Sci U S A. 2002;99:9248–9253. doi: 10.1073/pnas.142293999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cameron SJ, Itoh S, Baines CP, Zhang C, Ohta S, Che W, Glassman M, Lee JD, Yan C, Yang J, Abe J. Activation of big MAP kinase 1 (BMK1/ERK5) inhibits cardiac injury after myocardial ischemia and reperfusion. FEBS Lett. 2004;566:255–260. doi: 10.1016/j.febslet.2004.03.120. [DOI] [PubMed] [Google Scholar]
- 125.Ding B, Yan C, Molina CA, Blaxall BC, Abe J. Effects of insulin growth factor (ICF-1)-induced ERK5 activation on a novel positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER)-mediated apoptosis: Akt independent cell survival pathways. Circulation. 2005;112(suppl II):II-219. [Google Scholar]
- 126.Yehia G, Schlotter F, Razavi R, Alessandrini A, Molina CA. Mitogen-activated protein kinase phosphorylates and targets inducible cAMP early repressor to ubiquitin-mediated destruction. J Biol Chem. 2001;276:35272–35279. doi: 10.1074/jbc.M105404200. [DOI] [PubMed] [Google Scholar]
- 127.Kamakura S, Moriguchi T, Nishida E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J Biol Chem. 1999;274:26563–26571. doi: 10.1074/jbc.274.37.26563. [DOI] [PubMed] [Google Scholar]
- 128.Xiang Y, Naro F, Zoudilova M, Jin SL, Conti M, Kobilka B. Phospho-diesterase 4D is required for beta2 adrenoceptor subtype-specific signaling in cardiac myocytes. Proc Natl Acad Sci U S A. 2005;102:909–914. doi: 10.1073/pnas.0405263102. [DOI] [PMC free article] [PubMed] [Google Scholar]