

Abstract
CarD is an essential mycobacterial protein that binds the RNA polymerase (RNAP) and affects the transcriptional profile of Mycobacterium smegmatis and Mycobacterium tuberculosis [6]. We predicted that CarD was directly regulating RNAP function but our prior experiments had not determined at what stage of transcription CarD was functioning and at which genes CarD interacted with the RNAP. To begin to address these open questions, we performed chromatin immunoprecipitation sequencing (ChIP-seq) to survey the distribution of CarD throughout the M. smegmatis chromosome. The distribution of RNAP subunits β and σA were also profiled. We expected that RNAP β would be present throughout transcribed regions and RNAP σA would be predominantly enriched at promoters based on work in Escherichia coli [3], however this had yet to be determined in mycobacteria. The ChIP-seq analyses revealed that CarD was never present on the genome in the absence of RNAP, was primarily associated with promoter regions, and was highly correlated with the distribution of RNAP σA. The colocalization of σA and CarD led us to propose that in vivo, CarD associates with RNAP initiation complexes at most promoters and is therefore a global regulator of transcription initiation. Here we describe in detail the data from the ChIP-seq experiments associated with the study published by Srivastava and colleagues in the Proceedings of the National Academy of Science in 2013 [5] as well as discuss the findings from this dataset in relation to both CarD and mycobacterial transcription as a whole.
The ChIP-seq data have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE48164).
Keywords: Mycobacteria, RNA polymerase, Transcription, Tuberculosis, CarD
| Specifications | |
|---|---|
| Sample and organism | Genomic DNA from Mycobacterium smegmatis mc2155 derived strains |
| Sequencer | AB SOLiD 4 system high-throughput genome sequencer |
| Data format | Raw data: sra files, normalized data: wig, SOFT, MINiML, and TXT files |
| Experimental factors | In the M. smegmatis strain that was used, the carD gene had been deleted from the native chromosomal locus and the strain instead constitutively expressed a functional C-terminal HA tagged version of CarD. The exception was the control strain that expressed an untagged HA peptide and retained the carD gene at its endogenous locus. |
| Experimental features | All M. smegmatis strains were isogenic to mc2155 and were grown at 37 °C in LB supplemented with 0.5% dextrose, 0.5% glycerol, and 0.05% Tween 80 to late log phase (ODλ600 of 1.0) before crosslinking the protein–nucleic acid complexes. |
Direct link to deposited data
The direct link for the ChIP-seq data is: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48164.
Experimental design, materials and methods
Bacterial strains and culture conditions
All M. smegmatis strains were isogenic to mc2155 and were grown at 37 °C in LB supplemented with 0.5% dextrose, 0.5% glycerol, and 0.05% Tween 80 (broth). For immunoprecipitation of CarD, RNAP β, and RNAP σA, a carD merodiploid strain was produced by integrating pMSG430smcarD-HA (constitutively expresses M. smegmatis C-terminal HA tagged CarD, kanamycin resistant) into the attB site of M. smegmatis mc2155. Allelic exchange experiments were performed with the carD merodiploid strain using a DNA donor sequence with homology to mc2155 nucleotides 6141480 to 6142268 and 6140266 to 6141010 to delete all of the carD gene except the nucleotides encoding the first 10 and last 3 amino acids from the endogenous locus, generating ΔcarD attB::tetcarD-HA [6]. For immunoprecipitation of unfused HA peptide as a control, mc2155 was transformed with pmsg431, which integrates into the attB site of the genome and constitutively expresses HA peptide. This strain was called mc2155 attB::pmsg431.
Chromatin immunoprecipitation
Cultures of M. smegmatis ΔcarD attB::tetcarD-HA and mc2155 attB::pmsg431 strains were grown to late log phase (ODλ600 = ~ 1) before adding a final concentration of 2% formaldehyde and shaking at room temperature for 30 min to crosslink DNA and proteins. The crosslinking was quenched by the addition of 0.25 ml of 2.5 M glycine per 5 ml of culture and incubated 5 min at 25 °C with shaking. 5 ml (~ 2.5 × 109 mycobacterial cells) of each culture was then collected by centrifugation. The cells were washed once with TE and resuspended in 100 μl of TE supplemented with Roche Complete protease inhibitor cocktail. The cell suspension was lysed using a Covaris Focused-Ultrasonicator so that the genomic DNA was sheared into ~ 100 base pair (bp) fragments, as assessed by DNA gel electrophoresis. The use of the Covaris Focused-Ultrasonicator was critical for this step and other sonicator systems were unable to yield a comparable consistency and homogeneity of DNA fragment distribution. The cell debris was spun down and the lysate was added to 400 μl ChIP lysis buffer (50 mM HEPES-KOH [pH 7.5], 140 mM NaCl, 1 mM EDTA, 1% Triton X-100) plus Roche Complete protease inhibitor cocktail.
Protein–nucleic acid complexes containing CarD-HA were immunoprecipitated from the M. smegmatis mc2155 ΔcarD attB::tetcarD-HA strain cell lysate by adding 50 μl of anti-HA agarose (Sigma). Complexes containing unfused HA were immunoprecipitated from the mc2155 attB::pmsg431 strain with the same anti-HA agarose. RNAP β and σA were immunoprecipitated from ΔcarD attB::tetcarD-HA with monoclonal antibodies specific for these subunits (Neoclone; 8RB13 for β, 2G10 for σ) immobilized on GammaBind G Sepharose (GE Healthcare Life Sciences). Each immunoprecipitation was performed in duplicate from two separate cultures, thus comprising two biological replicates. However, one of the RNAP σA samples was lost during library preparation and, therefore, there is only data for one RNAP σA replicate.
The lysates and antibodies were incubated overnight by rotating at 4 °C. The antibody matrix was washed 2 × with ChIP lysis buffer, 2 × with ChIP lysis buffer plus an additional 360 mM NaCl, 2 × with ChIP wash buffer (10 mM Tris-HCl pH 8.0, 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA), and 2 × with TE, each time by rotating for 10 min at 4 °C. Complexes that co-precipitated with the respective antibody matrix were eluted twice by adding 100 μl of ChIP elution buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% SDS), incubating for 10 min at 65 °C with agitation, spinning down the antibody matrix, and transferring the eluate to a new tube. Wash and elution buffers were all supplemented with Roche Complete protease inhibitor cocktail. To reverse the crosslinks, the eluates were incubated overnight at 65 °C. 15 μl of each sample was removed for Western blot analysis of proteins, while 100 μg/ml of proteinase K was added to the rest of each sample and incubated at 37 °C for 2 h before isolating nucleic acid by chloroform phenol extracting 2 times, ethanol precipitating and resuspending the DNA pellet in 34 μl of water.
Sequencing
Co-precipitated DNA was sequenced using an AB SOLiD 4 high-throughput genome sequencer (Life Technologies) and a 50 bp read length, which provided sufficient reads for over 100-fold coverage of the genome in each sample, wherein the M. smegmatis genome is 6,988,209 bp in length and the coverage of each sequencing reaction was over 800 Mbp. Table 1 shows the total number of reads and number of mapped reads for each sample.
Table 1.
Number of sequencing reads for each sample from the AB SOLiD 4 high-throughput genome sequencer set to a 50 bp read length.
| Sample | # of reads | # of mapped reads | % mapped reads |
|---|---|---|---|
| CarD-HA-1 | 24,988,001 | 16,452,015 | 65.84% |
| RNAP β-1 | 24,145,461 | 16,249,329 | 67.30% |
| Unfused HA-1 | 27,153,580 | 17,194,808 | 63.32% |
| CarD-HA-2 | 9,323,217 | 7,097,095 | 76.12% |
| RNAP β-2 | 12,709,226 | 9,660,422 | 76.01% |
| RNAP σA-2 | 19,596,174 | 14,868,445 | 75.87% |
| Unfused HA-2 | 11,641,903 | 8,015,559 | 68.85% |
Normalization
The DESeq method [1] was used to normalize the raw data sequence reads. Specifically, the normalized coverage (or counts) was determined by multiplying the raw (sequenced) coverage (or counts) in each sample by that sample's size factor. The size factors are determined by taking the median of the ratios of observed counts. The normalized number of sequence reads per base pair was then expressed as a log2 value. If a read mapped with equal quality at multiple loci (but not more than 3), its contribution was distributed evenly among them. For example, the sequences of the 16S, 23S, and 5S ribosomal RNA are identical in the M. smegmatis rrnA and rrnB operons. Therefore, the total number of reads for those sequences was split equally between the operons. If the number of mapping loci was higher than 3, the read was discarded. The normalized number of reads for each base pair was saved as a wig file for each sample.
Data analysis
We first determined how well replicate samples of the distribution of a given protein correlated to each other and how well the distribution of CarD correlated to the distributions of RNAP β and RNAP σA (Table 2, Table 3). The correlations were obtained by computing the Pearson correlation of the genomic coverage profiles of each pair of samples. The coverage profiles were computed by summing the contributions of all mapped fragments, assuming they were 100 bp long, and then, in 20-bp steps along the entire genome, computing the average coverage of the surrounding 100-bp window. Table 2 shows the correlations between the individual replicates. These data showed that individual replicates for a single immunoprecipitation condition correlated highly with one another (bolded in Table 2) and indicated that the distribution of CarD-HA or RNAP β was consistent between biological replicates. This consistency between replicates allowed us to average the Pearson correlation values for each comparison to simplify the comparisons between immunoprecipitation conditions (Table 3). The correlation between the distribution of CarD-HA and the distribution of RNAP σA (bolded in Table 3) was almost as high as the correlation between the two CarD-HA replicates, indicating that the distribution of CarD-HA is very similar to that of RNAP σA.
Table 2.
Pearson correlations of the genomic coverage profiles of each pair of samples. The bolded numbers show the correlation between the distributions of individual replicates for a single immunoprecipitation condition.
| CarD-HA-1 | RNAP β-1 | Unfused HA-1 | CarD-HA-2 | RNAP β-2 | RNAP σA-1 | Unfused HA-2 | |
|---|---|---|---|---|---|---|---|
| CarD-HA-1 | 1.00 | 0.71 | 0.57 | 0.92 | 0.71 | 0.89 | 0.62 |
| RNAP β-1 | 1.00 | 0.71 | 0.63 | 0.91 | 0.60 | 0.76 | |
| Unfused HA-1 | 1.00 | 0.41 | 0.65 | 0.36 | 0.87 | ||
| CarD-HA-2 | 1.00 | 0.70 | 0.95 | 0.52 | |||
| RNAP β-2 | 1.00 | 0.66 | 0.80 | ||||
| RNAP σA-2 | 1.00 | 0.47 | |||||
| Unfused HA-2 | 1.00 |
Table 3.
Average Pearson correlations of the genomic coverage profiles for each immunoprecipitation condition examined. Each sample was done in duplicate, except σA was done once. Correlations are the average of each duplicate to one another. The bolded number shows the correlation between the distribution of CarD-HA and the distribution of RNAP σA.
| HA | CarD-HA | RNAP β | RNAP σA | |
|---|---|---|---|---|
| HA | 0.934 | 0.530 | 0.730 | 0.417 |
| CarD-HA | 0.962 | 0.687 | 0.919 | |
| RNAP β | 0.954 | 0.629 | ||
| RNAP σA | 1.000 |
To directly compare the genome distributions of CarD-HA, RNAP β, and RNAP σA, the reads per base pair from the unfused HA peptide sample served as the background control and were subtracted from the other datasets. The rationale for this control was that as a non-DNA binding protein, the HA peptide should be diffusely localized throughout the cell and serve as a readout for the background levels of nonspecific crosslinking to the DNA. The normalized, background-corrected log2 reads per base pair were then smoothed over a 20-bp window and RNAP σA and CarD-HA peaks were identified as described previously [3], [4]. Briefly, maxima and minima were assigned as inflection points where the values ± 10 bp were both lower or both higher, respectively. Maxima within 20 bp were merged with the peak location assigned to the maximum with the highest absolute signal value. Adjacent minima were merged analogously. To assess the statistical significance, peaks were divided into 0.1 interval bins of peak heights with a lower cutoff of peak height of 0.4 log2 reads per base pair. Starting with the lowest bin, we then calculated the distance of each peak to the nearest gene start and compared these distances to those computed using genome coordinates arbitrarily rotated 1 × 106 bp around the M. smegmatis genome. Using the Wilcoxon–Mann–Whitney ranksum test for nonsimilarity of distributions [3], [4], RNAP σA peaks in the 1.1–1.2 peak-height bins and CarD-HA peaks in the 0.5–0.6 peak-height bins were statistically significant (P values for similarity of the distributions < 0.0001). In other words, for each peak-height bin, the two lists of peak-to-gene start distances (actual and rotated by 1 × 106 bp) were tested for whether they were from different populations using the Wilcoxon–Mann–Whitney ranksum test. Peaks in the lowest peak-height bin for which peak-to-gene start distances differed from random with a P value of < 0.0001 plus all peaks with greater heights were then used as the statistically significant peaks. We then identified RNAP σA peaks associated with each gene as the closest RNAP σA peak upstream from the gene start and CarD-HA peaks associated with the RNAP σA peaks as the closest CarD-HA peak to each selected RNAP σA peak. To calculate average ChIP signals for the aggregate profiles (Fig. 1), we selected a subset of 62 genes meeting the following criteria: (i) ≥ 300 bp in gene length, (ii) average RNAP log2 ChIP signal ≥ 1.6/bp, (iii) associated with an RNAP σA peak with log2 ChIP signal ≥ 3/bp, (iv) absence of other RNAP σA peaks within 500 bp upstream or 1000 bp downstream of the associated RNAP σA peak, (v) absence of an oppositely oriented gene with an average RNAP β log2 ChIP signal ≥ 1 upstream from the gene (because an oppositely oriented gene could create a divergent promoter region with potential for overlapping RNAP σA and CarD-HA ChIP signals), and (vi) absence of an upstream gene with average RNAP log2 ChIP signal > 0 within 100 bp upstream from the gene (because such an arrangement would indicate the gene is an internal member of an operon).
Fig. 1.
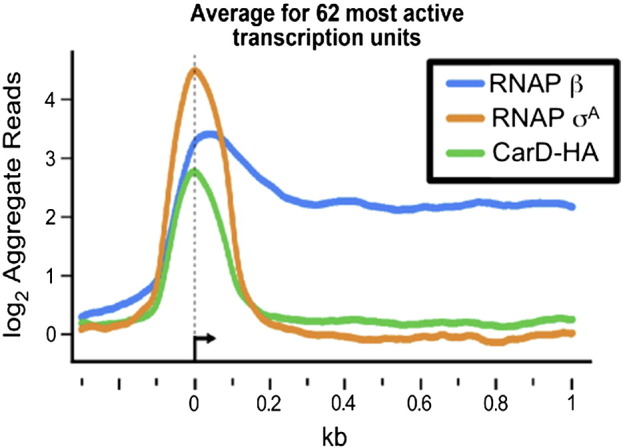
Normalized log2 of ChIP-seq reads from M. smegmatis DNA co-immunoprecipitated with RNAP β, RNAP σA, or CarD-HA. Protein–DNA complexes containing CarD-HA, RNAP β, and RNAP σA were immunoprecipitated from M. smegmatis lysates. The co-precipitated DNA was sequenced, and the number of sequence reads per bp was normalized to total reads per sample and expressed as a log2 value. Normalized reads per base pair from DNA precipitated from cells expressing only the HA epitope were used as background and subtracted from the other samples. Shown are the aggregate profiles averaged over 62 highly active transcription units with the 0 designating the estimated transcriptional start sites. The 62 transcription units were selected on the basis of high signal and isolation from surrounding transcription units.
The RNAP β, CarD-HA, and RNAP σA signals from the 62 genes were then averaged using the distance from the center of the associated σA peaks to align the genes (Fig. 1). For the gene alignments, the distance from the center of the associated σA peak served as a proxy for the transcriptional start site, since most transcriptional start sites are not mapped in M. smegmatis. This analysis showed that whereas RNAP β was found throughout transcribed regions of the genome, CarD-HA and RNAP σA were primarily associated with promoter regions. These data matched the high correlation calculated for the distribution of CarD-HA and RNAP σA (Table 2, Table 3). Levels of both CarD-HA and RNAP σA dropped off immediately following the promoter sequences, suggesting that these proteins are lost from the RNAP elongating complex after transcription initiation. The colocalization of RNAP σA and CarD-HA led us to propose that in vivo, CarD associates with RNAP initiation complexes at most promoters and is therefore a global regulator of transcription initiation. Further analysis of the dataset also revealed that CarD was never present on the genome in the absence of RNAP, suggesting that it may be targeted to the genome through its interaction with RNAP.
Discussion
CarD modulates transcription through its direct interaction with RNAP [6], [7]. To determine at which stage of the transcription cycle (initiation, elongation, or termination) CarD acts, we used ChIP-seq [2] to survey the distribution of CarD throughout the M. smegmatis chromosome. Our data shows that CarD is localized to promoters throughout the M. smegmatis genome, indicating that CarD functions during transcription initiation. Despite the previous finding that CarD has sequence non-specific DNA binding activity [5], the ChIP-seq experiments also revealed that CarD was never present on the genome in the absence of RNAP β or RNAP σA, suggesting that CarD is targeted to the genome through its interaction with RNAP. The ChIP-seq data for the distribution of RNAP σA also serves as a map of potential promoter elements throughout the M. smegmatis genome, which has never before been experimentally examined.
The ChIP-seq experimental dataset has also raised a number of new questions. Compilation of the ChIP-seq data and previous microarray expression profiling analyses [6] indicates that CarD is broadly distributed on promoters of most transcription units regardless of whether they were deregulated during CarD depletion. This brings into question whether CarD activity exhibits promoter specificity. There is also the striking correlation between the distributions of CarD and RNAP σA on the genome, despite the fact that no direct interaction between these proteins has been reported. The factors contributing to the enrichment of CarD at RNAP σ containing holoenzymes as opposed to elongating RNAP core complexes remain unknown and will be a topic of future study. All together, results from these experiments have provided invaluable information that will help direct the ongoing efforts in determining the mechanism of transcription regulation by CarD. In addition, this work serves as a framework for further investigations into RNAP function in mycobacteria.
Acknowledgments
The authors thank the Genomics Core Laboratory (GCL) at Memorial Sloan Kettering Cancer Center (MSKCC) for performing the next-generation sequencing for ChIP-seq experiments. The GCL is supported by the cancer center core grant P30 CA008748.
References
- 1.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson D.S., Mortazavi A., Myers R.M., Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 3.Mooney R.A., Davis S.E., Peters J.M., Rowland J.L., Ansari A.Z., Landick R. Regulator trafficking on bacterial transcription units in vivo. Mol. Cell. 2009;33:97–108. doi: 10.1016/j.molcel.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reppas N.B., Wade J.T., Church G.M., Struhl K. The transition between transcriptional initiation and elongation in E. coli is highly variable and often rate limiting. Mol. Cell. 2006;24:747–757. doi: 10.1016/j.molcel.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 5.Srivastava D.B., Leon K., Osmundson J., Garner A.L., Weiss L.A., Westblade L.F., Glickman M.S., Landick R., Darst S.A., Stallings C.L., Campbell E.A. Structure and function of CarD, an essential mycobacterial transcription factor. Proc. Natl. Acad. Sci. U. S. A. 2013;110:12619–12624. doi: 10.1073/pnas.1308270110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stallings C.L., Stephanou N.C., Chu L., Hochschild A., Nickels B.E., Glickman M.S. CarD is an essential regulator of rRNA transcription required for Mycobacterium tuberculosis persistence. Cell. 2009;138:146–159. doi: 10.1016/j.cell.2009.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weiss L.A., Harrison P.G., Nickels B.E., Glickman M.S., Campbell E.A., Darst S.A., Stallings C.L. Interaction of CarD with RNA polymerase mediates Mycobacterium tuberculosis viability, rifampin resistance, and pathogenesis. J. Bacteriol. 2012;194:5621–5631. doi: 10.1128/JB.00879-12. [DOI] [PMC free article] [PubMed] [Google Scholar]