

Abstract
Objective:
In this study, kinetic analysis was performed to understand the functional importance of the amino terminal of the active site of previously mutated Plasmodium vivax Lactate Dehydrogenase enzyme by mimicking Toxoplasma gondii I, II, Eimeria acervulina and Eimeria tenella LDH’s.
Material and Methods:
Mutant LDH genes were amplified by PCR and 6xHistag was added to the C-terminal of the enzymes. Then LDH enzymes are overproduced as recombinant in E. coli cells, purified by Ni-NTA agarose matrix and kinetic properties were analysed.
Results:
Observing increase of Km values of mutant enzymes showed that mutations in this place caused decreasing affinity of enzyme for its substrate. However kcat values were about the same throughout all mutant proteins.
Conclusion:
Sensitivity of the studied region emphasizes the significance of this site for drug design studies for both Plasmodium and some other Apicomplexans.
Keywords: Lactate dehydrogenase, Plasmodium vivax, antimalarial drug, site-directed mutagenesis, kinetic analysis
Introduction
Malaria is an infectious disease caused by Plasmodium parasites which is transmitted to people via the bites of infected mosquito vectors belonging to the Anopheles genus. While malaria parasites spend some of their life cycles in the vectors, they need another host such as humans to complete their cycles. Five Plasmodium species cause disease in human: Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae, Plasmodium ovale and recently reported monkey malaria parasite Plasmodium knowlesi (1, 2).
Malaria shows a wide range of geographic distribution worldwide. About half of the world’s population (3.3 billion people) is at risk of malaria. Each year about 250 million people are affected and nearly 1 million deaths occur, of which 90% are in sub-Saharan Africa (3). In the next decade, malaria infections are going to rise again in many regions of Africa, Asia and South America where it is already endemic. The major cause of this increasement is the spread of resistance of the Plasmodium species to the conventional anti-malarial drugs (4). Plasmodiım falciparum is one of the most deadly species of human malaria parasites and resists all known kinds of drugs except artemisinin (5). Chloroquine resistance of the most common parasite Plasmodium vivax is increasing gradually (5). Development of resistance to currently available drugs and absence of any effective malaria vaccine has led to determining of novel drug targets (4).
Malaria parasites need high levels of energy to grow rapidly in the erythrocytes at the asexual growth stage. Absence of fully functional citric acid cycle directs parasites to gain most of the ATP energy from the glycolytic pathway for survival (6, 7). Thus, one of the first routes in the development of an antimalarial drug involves identification of vital parasite enzyme targets, and glycoloytic enzymes have been selected as the new drug target candidates (4, 8).
Lactate dehydrogenase (LDH), the terminal enzyme of the glycolytic pathway, is a vital enzyme for the Plasmodium parasites and causes oxidation of pyruvate to L-lactate and provides turnover of NAD+ required for the previous stages of glycolysis (8, 9). It has been shown that inhibition of the malarial LDH enzyme prevents the production of ATP and causes death of the Plasmodium parasites (8). Plasmodial LDH enzyme differs from its mammalian counterpart by means of kinetic parameters and also in molecular structure (10–12), so it becomes an attractive drug target candidate (10, 12–14).
Sequence of lactate dehydrogenase enzyme of Plasmodium and other Apicomplexan parasites share similar residues at the active site loop located between the 100th and 110th aminoacids. However the amino terminal end of the active site loop of lactate deyhdrogenase between Plasmodium parasites and some other Apicomplexan LDHs (Toxoplasma gondii LDH1 and LDH2, Eimeria tenella, Eimeria acervulina and Theileria parva) show amino acid exchanges (15). In this study, kinetic analyses were performed on previously mutated Plasmodium vivax LDH mutant enzymes by mimicking Toxoplasma gondii I, II, Eimeria acervulina and Eimeria tenella LDH’s to understand the structural importance of the amino terminal end amino acids of the active site loop.
Material and Methods
Homology modelling
The LDH aminoacid sequence in FASTA format of wild type Plasmodium vivax was obtained from NCBI (http://www.ncbi.nlm.nih.gov) with the accession number of 2A92_A. Amino acid changes on this template were made to obtain mutant proteins by using SWISS-MODEL, automated protein homology-modelling server (http://swissmodel.expasy.org). Constructed mutant protein models were expressed as Protein Data Bank (PDB) file format. PDB file of wild type PvLDH enzyme were downloaded from Protein Data Bank (http://www.rcsb.org/pdb) with the ID code 2A92. PDB files for every mutant protein models and also wild type one were analyzed for superimposition and visualized by Molsoft ICM-Pro 3.4–9a.
Bacterial strains, growth media, plasmid and enzymes
Escherichia coli JM105 strain {supE endA sbcB15 hsdr4 rpsL thiD (lac-proAB)F′ [traD36 proAB+ lacIq lacZDM15] was used as a host for plasmids propagation and as an expression strain. Strains were cultured in double-strength yeast-tripton (2×YT) agar plate and broth media at 37°C. Ampicillin was used in the media in appropriate amounts (100 μg/mL) for the selection and growth of transformants (16). The E. coli plasmid vector pKK223-3 (Pharmacia LKB Biotechnology AB, Uppsala, Sweden) was used for subcloning, expression and sequencing. Pfu DNA Polymerase (2.5 U/μL) was supplied by Fermentas (Lithuania), EcoRI (12 U/μL) and Pstl (10 U/μL) restriction enzymes and T4 DNA Ligase (3 U/μL) were purchased from Promega (USA).
Addition of His tag to the mutant PvLDH genes
His tag was added by PCR amplification to the C-terminal of previously mutated (17) P. vivax enzymes. Primers used for amplification complementery to the forward and reverse strands were Pv7; 5′-GCC GAC CCG GAA TTC ATG ACG CCG AAA CCC AAA ATT GTG C-3′ (EcoRI restriction site) and Pv15; 5′-TTT TCT GCA GTT AGT GAT GGT GAT GGT GAT GAA TGA GCG CCT TC ATC C-3′ (Pst1 restriction site and 6xHis tag site). PCR was performed in a total of 50 μl volume containing 5 μL 10x Pfu Buffer, 5 μL stock dNTPs (10 mL of each 10 mM dNTPS and 10 mL of dH2O), 2.5 μL of each 5′ Primer (Pv7, 20 pmol/μL) and 3′ primer (Pv13, 20 pmol/μL), 1 μL template mutant PvLDH DNA, 1 μL Pfu DNA Polymerase (2.5 U/μL) and 33 μL sterile dH2O. The PCR was carried out at 94°C for 1.5 min, 50°C for 2 min. and 72°C for 2 min. for 30 cycles. Double-strand amplicons run on a 1% crystal violet stained agarose gel for preparative purposes and a parallel PCR product obtained using the same reaction was run on the 1% ethidium bromide (EtBr) stained agarose gel for visualisation of the amplicons (18). The DNA was recovered by QIA quick Gel Extraction Kit (QIAGEN, Germany) following the electrophoresis.
Restriction digest, ligation and transformation
Both of the mutant DNA samples and pKK223-3 expression vector were digested by Eco RI and PstI restriction endonucleases. Digested vector and samples were mixed at the ratio of 1:1 and the mutant genes were ligated into digested expression vector pKK223-3. The reaction mixture contained 100 ng DNA (insert), 100 ng vector, 1 μL T4 DNA Ligase (3 U/μL), 1 μL 10X ligation buffer and XμL sterile dH20 to a final volume of 10 μL. The ligation was set up at 4°C overnight in the thermal cycler (Eppendorf). The resulting recombinant DNA plasmids were transformed into CaCl2 competent cells of E. coli JM105 according to Sambrook et al. (16) and plated onto the 2xYT agar plates supplemented with 100 μg/mL ampicillin and grown overnight at 37°C.
DNA sequencing
After transformation, the resultant colonies were screened by colony PCR for plasmid inserts using Pv7 and Pv15 primers. Plasmid DNAs were purifed from the positive colonies using QIAprep Spin Miniprep Kit (QIAGEN) and then sequenced by Iontek (Turkey) using internal primers to show the addition of the 6-His tag to the C terminal of the mutant genes.
All general methods were applied according to Sambrook et al. (16) unless otherwise stated.
Purification of the His tagged mutant PvLDH enzymes
Starting culture of E. coli JM105 containing recombinant plasmids were inoculated into yeast-tripton (2×YT) broth containing ampicillin (100 μg/mL) and grown at 37°C overnight. The culture was inoculated (2 mL) into 500 mL growth medium and the cultivation was performed until OD600 reached 0.6. Expressions of mutant PvLDH enzymes were induced by the addition of isopropyl-β-D-thiogalactopyranoside (IPTG) with 1 mM final concentration and incubated overnight at 37°C while shaking at 200 rpm. Subsequently, protein purification was performed according to Shoemark et al. (19).
Following the incubation, cells were harvested by centrifugation at 5000 rpm for 15 min at 4°C and the pellet was suspended in 6 mL lysis buffer (50 mM Na2HPO4, 300 mM NaCl, 20 mM imidazole, 10% gycerol, pH 7.5). The cells were sonicated at 60% Amp. (9×10s pulses with 30s intervals) on ice and centrifuged at 14.000 rpm for 1 hour at 4°C to remove cell debris. Supernatant was collected and used in the following applications. Ni-NTA agarose matrix was equilibrated with the lysis buffer prior to loading the sample. The supernatant was applied to the column and flow through was collected. The column was washed with 40 mL wash buffer (50 mM Na2HPO4, 300 mM NaCl, 20 mM imidazole, 10% gycerol, pH 7.5) which is the same as lysis buffer and fractions were collected. Proteins bound to the column were eluted using 10x of 1 mL elution buffer (50mM Na2HPO4, 300 mM NaCl, 250 mM imidazole, 10% gycerol, pH 7.5) (19). The purity of protein fractions were checked by both SDS-PAGE (20) and by scanning the protein at 260–320 nm by UV-visible spectrophotometer. Protein concentration was determined by using the absorbance value obtained at 280 nm by UV-visible spectrophotometer.
Western blot analysis of mutant protiens
The primary antibody that was raised against active P. falciparum LDH in New Zealand white rabbit was used in the assays (13). The secondary antibody, goat anti-rabbit IgG, was purchased from the Bio-Rad, UK. Protein samples were run on the SDS-polyacrylamide gel prior to blotting (20). Proteins were transferred to nitrocellulose membranes electrophoretically after the SDS-PAGE for western blotting. The membrane was blocked in PBS Tween 20 containing 5% skimmed milk powder overnight at 4°C. After the blocking, the membrane was washed in PBS Tween 20 for 10 minutes and then incubated in the blocking buffer with 1:1000 diluted primary antibody at room temperature for 1 hour on a shaker. After the incubation the membrane was washed three times in PBS tween 20. The goat anti-rabbit IgG conjugated to peroxidase was diluted 1:12000 in 5% PBS Tween 20, and the washed membrane was incubated in this solution at room temperature for 1 h. Later, the membrane was washed twice in PBS Tween 20, once in PBS, and once in H2O. Finally, the filter was incubated in 60 mg 3.3 diaminobenzidine (DAB), 100 mL PBS, 100 mL H2O2 for 30 seconds and rinsed in H2O (13).
Kinetic analysis of the mutant PvLDH enzymes
Molecular masses and extinction coefficients of mutant PvLDHs were calculated by using web based the “Peptide Property Calculator” (http://www.basic.northwestern.edu/biotools/proteincalc.html) programme. Steady-State kinetic parameters of the mutant enzymes were determined by following the rate of absorbance change at 340 nm using the UNICAM UV/Visible spectrophometer. Enzyme assays were performed in 50 mM Trizma Base/KCI kinetic buffer, pH 7.5, containing 200 μM NADH at different concentrations of pyruvate (20 μM, 60 μM, 180 μM, 340 μM, 1.6 mM, 4.8 mM, 14.48 mM, 20.4 mM, 29 mM) at 25°C. Data were used to determine Km and Vmax values and using Grafit 3.0 and then the values were used to calculate kcat (21). Kinetic analysis was performed without removing 6xHis tag from the N-terminal of the enzyme which had no effect on the kinetic constants (22).
Results
Sequence alignment of PvLDH to other target LDHs
The N terminal sequence of the active site loop placed between positions 100–108 of wild type PvLDH enzyme was compared to the same region from the previously mutated PvLDH enzymes (Table 1) (17). Alignment results indicated the terminal sequence of the active site of PvLDH is slightly different from other Apicomplexans LDHs which differ from 2 or 3 residues. Amino acid exchanges made on the PvLDH in the previous study to mimick Toxoplasma gondii I LDH (TgILDH) were F100L, A103V and S108P; for Toxoplasma gondii II LDH (TgIILDH) F100L and A103V, for Eimeria tenella LDH (EtLDH) F100I and P105A; for Eimeria acervulina LDH (EaLDH) were F100I and A103I (17).
Table 1.
Wild type PvLDH and mutated Apicomlexan LDH amino acid sequences of the amino terminal end of active site loop. PvLDH (Plasmodium vivax LDH), TgILDH (Toxoplasma gondii I LDH), TgIILDH (Toxoplasma gondii II LDH), EtLDH (Eimeria tenella LDH) and EaLDH (Eimeria acervulina LDH)
| 100 | 108 | ||
|---|---|---|---|
| PvLDH | F T K A | - | P G K S |
| TgILDH | L T K V | - | P G K P |
| TgIILDH | L T K V | - | P G K S |
| EtLDH | I T K A | - | A G K S |
| EaLDH | I T K I | - | P G K S |
Addition of His tag to the mutant PvLDH genes
Amplification of the mutant LDHs were performed by using specific primers that had a His tag to enable purification of the protein by affinity purification and amplicons run on a 1% Ethidium Bromide (EtBr) stained agarose gel to confirm the presence of the correct band (Figure 1). EtBr stained gel was only used to identify the molecular weight of the amplified genes. Gel result indicated the amplicons were about 1 kb pair long. This result confirmed the amplication of the desired gene product.
Figure 1.
PCR amplicon run on EtBr stained agarose gel. 1: Marker; 2: 6×His tagged mutant gene
Restriction digest, ligation, transformation and DNA sequencing
After recovery and restriction digest of the mutant genes, they were ligated into the E. coli expression vector pKK223-3 and were transformed into CaCl2 competent cells of E. coli JM105. Colony PCR were used for pre-identification of plasmid containing positive colonies. Recombinant plasmids were isolated from the positive ones and sequenced using plasmid specific primers. Sequencing showed presence of the addition of the 6-His tag to the C terminal of each mutant PvLDH enzymes.
Purification of the mutant PvLDH proteins
Mutant proteins expressed in E. coli were purified by gradient flow affinity chromatography using Ni-NTA agarose beats that have affinity to 6xHis tags of mutant proteins. According to SDS-PAGE analysis of flow through, washing and elution samples and enzyme activity measurements, purity and amount (6 mg/mL) of enzymes were found to be sufficient for kinetic analysis (Figure 2).
Figure 2.
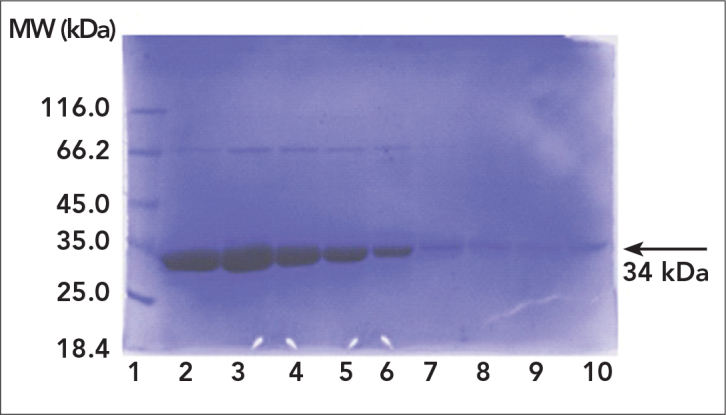
SDS-PAGE analysis of purified mutant protein. 1: Marker; 2–10: Elutions
Western blot analysis of mutant protiens
Due to gene expression PvLDH is weak and not detected by SDS-PAGE analysis prior to purification, expression of mutant proteins were shown by Western blot analysis after the enzyme purification step. Elution 7 from every purified mutant proteins were subjected to Western blotting. Analysis results showed that mutant enzymes were at the correct size with high purity on the membrane (Figure 3).
Figure 3.
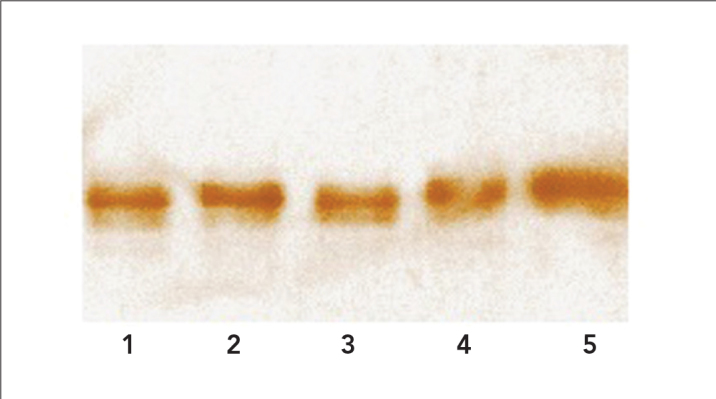
Western blot analysis of all mutant proteins. 1: PvLM3Tg1; 2: PvLM2Tg2; 3: PvLM3Et; 4: PvLM2Ea; 5: Wild type P. vivax LDH
Kinetic analysis of mutant proteins
After the affinity purification, eluted samples possessing the most pure protein samples were selected and used in the kinetic measurements. Kinetic measurements of mutant proteins were performed by using nine different pyruvate concentrations. 200 μM NADH was used as cofactor. Analysis was performed in a total of 1 mL kinetic buffer containing 50mM trizma base and 50mM KCI. Km and Vmax values of mutant enzymes were identified by Grafit 3.0. The kcat value was calculated by using these results and the amount of the enzyme used in the reaction. The resultant parameters are presented in Table 2.
Table 2.
Steady-state kinetic data for mutant enzymes. PvLDH (wild type Plasmodium vivax LDH), PvLM3Tg1 (Toxoplasma gondii I LDH), PvLM2Tg2 (Toxoplasma gondii II LDH), PvLM3Et (Eimeria tenella LDH) and PvLM2Ea (Eimeria acervulina LDH)
| Km, pyruvate (mM) | kcat, pyruvate (s−1) | Vmax | Standart Errors, | Vmax | |
|---|---|---|---|---|---|
| PvLDH | 0.0378 | 31.7 | 0.1413 | 0.0038 | (24) |
| PvLM3Tg1 | 0.1294 | 30.6 | 0.1506 | 0.0047 | |
| PvLM2Tg2 | 0.0811 | 29.8 | 0.1671 | 0.0034 | |
| PvLM3Et | 0.1665 | 38.15 | 0.1420 | 0.0049 | |
| PvLM2Ea | 0.1865 | 40.64 | 0.1653 | 0.0044 |
Homology modelling
Mutant protein sequences were obtained by exchanging the desired aminoacids on the FASTA sequence of the wild type enzyme prior to the modelling study. Homology modelling of mutant types of the wild type enzyme were done using SWISS-MODEL, automated protein homology-modeling server. PDB files were also created by the SWISS-MODEL for each mutant protein. According to the Molsoft ICM-Pro output datas, amino acid substitutions did not completely change the conformation of the amino terminal end of the active site loop (Figure 4). Superimposition of the wild-type and mutant sequences showed that the amino terminal of the enzymes do not cause deflexion by means of carbon backbone at all. However new residues of the amino terminal end may be able to trigger some new connections with their microenvironments.
Figure 4.

a) Presentation of the active site of wild type PvLDH enzyme. Yellow; amino terminal end of active site loop (residues 100F, 103A, 105P and 108S are located at the amino terminal end of active site loop), green; The five residue insertion in the active site loop. b) Showing up every four different mutations at the amino terminal end of active site loop. (yellow; amino terminal end of active site loop, green; the five residue insertion in the active site loop of the PvLDH, mutated residues are shown in white)
Discussion
When comparing LDH enzymes from Plasmodiums and other Apicomplexan parasites, some amino acids are conserved between 100th and 110th residues where the active site is located (23). However the amino acids (100F, 101T, 102K, 103A, 105P, 106G, 107K, 108S) located in the terminal end of the acvite site loop region shows some degree of differences among Plasmodiums and some other Apicomplexans (Toxoplasma gondii LDH1 and LDH2, Eimeria tenella, Eimeria acervulina, Theileria parva) (17). In this study, kinetic analyses were conducted on previously mutated Plasmodium vivax LDH enzymes, that were obtained by mimicking Toxoplasma gondii I and II, Eimeria acervulina and Eimeria tenella LDH’s, to show the importance of N-terminal amino acids of the active site.
Previously mutated enyzmes were produced as recombinant in E. coli hosts and affinity purification were achieved before kinetic analysis (17). Results of SDS-PAGE analysis indicated that the produced mutant LDH enzyme was about 34 kDa and the amount of elutions were enough for a further kinetic analysis. Western blot of all LDH enzymes proved the cloned and purifed enzymes belonging to Plasmodium species (13). According to results taken from SDS-PAGE analysis and spectrophometric measurements, only the last 4 elutions had appropriate purity for kinetic studies.
Km and kcat values of the mutant enzymes obtained in this study were compared with the values of the wild type P. vivax LDH enzyme (23, 24). Km pyruvate value of PvLM3Tg1 mutant increased to 0.1294 mM and kcat value was same as the wild type enzyme. Increase of Km value shows decreasing substrate binding affinity of the enzyme considerably while doing catalysis function normally. The Km value of PvLM2Tg2 mutant increased about two folds compared to the wild type. On the other hand, the kcat value showed no distinctive change. Kinetic studies of PvLM2Ea mutant enzyme showed a dramatic increase in Km pyruvate value (0.1865 mM) but the kcat value was not so different compared to the wild type PvLDH. Increase of Km pyruvate value (0.1665 mM) of the PvLM3Et mutant was much higher than the same value of TgLDHs. However the kcat value was slightly higher than the other TgLDH mutant enzymes.
Comparison of the crystal structure of the Plasmodium LDH enzymes showed highly conserved structural models (10, 23). There were no significant alterations within the active site and cofactor binding regions (10, 23). Homology modelling of mutated sites also showed highly similar 3D structure among four enzymes (Toxoplasma gondii LDH1 and LDH2, Eimeria tenella LDH, Eimeria acervulina LDH). Results of superimpositon between wild and mutant PvLDHs indicated unchanged carbon backbone coordinates. However, mutated residues showed some micro changes on the amino terminal end of the active site loop backbone. This could cause some new connections with their microenvironments such as water molecules. This could explain the changes in the Km pyruvate and kcat values of mutant enzymes compared to the wild type enzyme.
Conclusion
The result of mimicking the active site terminal aminoacids of P. vivax LDH to other Apicomplexan LDHs shows the importance of this site as a drug target candidate. According to kinetic studies Km values of all mutant enzymes show some distinctive changes compared to the wild type LDH enzyme. Aminoacid changes in this region clearly cause decreasing enzyme affinity to its substrate. However kcat values of all mutant proteins are nearly the same and this shows that catalytic activity of enzymes were not disrupted completely. Sensitivity of the studied region emphasizes the significance of this site for drug design studies for both Plasmodium and some other Apicomplexans.
Acknowledgments
This project was supported by TUBITAK, The Scientific and Technological Research Council of Turkey, (Project No: 104T215).
This study was previously presented at the European Biotechnology Congress 2011, Istanbul, Turkey
Conflict of Interest
No conflict of interest was declared by the authors.
References
- 1.Knell AJ. Malaria. Oxford: Oxford University Press; 1991. [Google Scholar]
- 2.Cox-Singh J, Davis TM, Lee KS, Shamsul SS, Matusop A, Ratnam S, et al. Plasmodium knowlesi Malaria in humans is widely distributed and potentially life threatening. Clin Infect Dis. 2008;46:165–71. doi: 10.1086/524888. [CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization (WHO) The global burden of disease: 2004 update. Switzerland: WHO Press; 2008. [Google Scholar]
- 4.Patel AP, Staines HM, Krishna S. New antimalarial targets: The example of glucose transport. Travel Med Infect Dis. 2008;6:58–66. doi: 10.1016/j.tmaid.2008.01.005. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 5.Baird JK. Chloroquine resistance in Plasmodium vivax. Antimicrob Agents Chemother. 2004;48:4075–83. doi: 10.1128/AAC.48.11.4075-4083.2004. [CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sherman IW. Biochmesitry of Plasmodium (malarial parasites) Microbiol Rev. 1979;43:453–95. doi: 10.1128/mr.43.4.453-495.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basco LK, Marquet F, Makler MM, Le Bras J. Plasmodium falciparum and Plasmodium vivax: Lactate dehydrogenase activity and its application for in vitro drug susceptibility assay. Exp Parasitol. 1995;80:260–71. doi: 10.1006/expr.1995.1032. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 8.Royer RE, Deck LM, Campos NM, Hunsaker LA, Vander Jagt DL. Biologically active derivatives of gossypol: synthesis and anti-malarial activities of peri-acylated gossylic nitriles. J Med Chem. 1986;29:1799–801. doi: 10.1021/jm00159a043. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 9.Vivas L, Easton A, Kendrick H, Cameron A, Lavandera JL, Barros D, et al. Plasmodium falciparum: stage specific effects of a selective inhibitor of lactate dehydrogenase. Exp Parasitol. 2005;111:105–14. doi: 10.1016/j.exppara.2005.06.007. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 10.Dunn CR, Banfield MJ, Barker JJ, Higham CW, Moreton KM, Turgut-Balik D, et al. The structure of lactate dehydrogenase from Plasmodium falciparum reveals a new target for anti-malarial design. Nat Struct Bio. 1996;11:912–5. doi: 10.1038/nsb1196-912. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 11.Gomez MS, Piper RC, Hunsaker LA, Royer RE, Deck LM, Makler MT, et al. Substrate and cofactor specificity and selective inhibition of lactate dehydrogenase from the Malarial parasite P. falciparum. Mol Biochem Parasitol. 1997;90:235–46. doi: 10.1016/s0166-6851(97)00140-0. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 12.Turgut-Balık D, Holbrook JJ. Determination of the DNA and amino acid sequences of the lactate dehydrogenasegene from Plasmodium falciparum, strains K1 and PF FCBR: A route to the design of new antimalarials. Turk j Biol. 2001;25:241–50. [Google Scholar]
- 13.Turgut-Balik D, Shoemark DK, Sessions RB, Moreton KM, Holbrook JJ. Mutagenic exploration of the active site of lactate dehydrogenase from Plasmodium falciparum. Biotech Lett. 2001;23:923–7. [CrossRef] [Google Scholar]
- 14.Turgut-Balik D, Shoemark DK, Moreton KM, Sessions RB, Holbrook JJ. Over-production of lactate dehydrogenase from Plasmodium falciparum opens route to new antimalarials. Biotech Lett. 2001;23:917–21. [CrossRef] [Google Scholar]
- 15.Turgut-Balık D, Akbulut E, Shoemark DK, Celik V, Moreton KM, Sessions RB, et al. Cloning, sequence and expression of the lactate dehydrogenase gene from human malaria parasite Plasmodium vivax. Biotech Lett. 2004;26:1051–5. doi: 10.1023/B:BILE.0000032958.78158.10. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 16.Sambrook J, Russell DW. Molecular cloning a laboratory manual I-II-III. New York: CSHL Pres; 2001. [Google Scholar]
- 17.Atmıs B. Plasmodium vivax’ ın LDH enziminin aktif bölge halkasının amino terminal ucunun Analizi. 2007. Master of Science Thesis; Department of Biology; University of Firat. [Google Scholar]
- 18.Turgut-Balık D, Çelik V, Moreton KM, Brady RL. Overcoming cloning problems by staining agarose gels with crystal violet ınstead of ethidium bromide in lactate dehydrogenase gene from Plasmadium vivax and Plasmodium falciparum. Acta Biol Hung. 2005;56:389–97. doi: 10.1556/ABiol.56.2005.3-4.20. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 19.Shoemark DK. The kinetic characterization of the lactate dehydrogenase enzyme from Plasmodium falciparum. 2000. UK: Doctor of philosophy thesis; Department of biochemistry; University of Bristol. [DOI] [PubMed]
- 20.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;5259:680–5. doi: 10.1038/227680a0. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 21.Leatherbarrow RJ. (Grafit, Version 3.0.). Staines. UK: Erithracus Software Ltd; 1992. [Google Scholar]
- 22.Berwal R, Gopalan N, Chandel K, Prasad GB, Prakash S. Plasmodium falciparum: enhanced soluble expression, purification and biochemical characterization of lactate dehydrogenase. Exp Parasitol. 2008;120:135–41. doi: 10.1016/j.exppara.2008.06.006. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 23.Chaikuad A, Fairweather V, Conners R, Josep-Horne T, Turgut-Balik D, Brady RL. Structure of Lactate Dehydrogenase from Plasmodium vivax: Complexes with NADH and APADH. Biochemistry. 2005;44:16221–8. doi: 10.1021/bi051416y. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 24.Mutlu O. Plasmodium vivax yabanıl tip laktat dehidrogenaz enziminin saflaştırılmasının optimizasyonu ve yabanıl tip ile mutant enzimlerinin kinetik analizlerinin yapılması. 2009. Master of Science Thesis; Department of Bioengineering; Yildiz Technical University.