

Abstract
Although cannabis has been used for pain management for millennia, very few approved cannabinoids are indicated for the treatment of pain and other medical symptoms. Cannabinoid therapy re-gained attention only after the discovery of endocannabinoids and fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), the enzymes playing a role in endocannabinoid metabolism. Nowadays, research has focused on the inhibition of these degradative enzymes and the elevation of endocannabinoid tonus locally; special emphasis is given on multi-target analgesia compounds, where one of the targets is the endocannabinoid degrading enzyme. In this review, I provide an overview of the current understanding about the processes accounting for the biosynthesis, transport and metabolism of endocannabinoids, and pharmacological approaches and potential therapeutic applications in this area, regarding the use of drugs elevating endocannabinoid levels in pain conditions.
Keywords: 2-AG, anandamide, endocannabinoids, FAAH, MAGL, pain
Cannabinoids are a group of chemical compounds that produce their effects via activating cannabinoid receptors; they include the phytocannabinoids (herbal cannabinoids/natural cannabinoids found in the cannabis plant), synthetic cannabinoids, and endogenous cannabinoids (endocannabinoids) (1, 2). Cannabis, also known as marijuana, has been used as both a recreational and medicinal drug for centuries. Pain management is the most important among the medicinal purposes, and has been drawing intense attention following the discovery of cannabinoid receptors and their endogenous ligands, endocannabinoids (3–5). Δ9-tetrahydrocannabinol (THC) and cannabidiol, the critical components of the cannabis plant, and the synthetic cannabinoids and their analogues have been shown to exert strong analgesic action both in preclinical and clinical studies (6–9). In this review, after a brief introduction to cannabinoid receptors, phytocannabinoids and synthetic cannabinoids, I provide an overview of what is currently known about the synthesis, release, degradation and biological actions of endocannabinoids, regarding their role in pain modulation, and describe the recent evidence of the promising results of augmentation of endogenous cannabinoid tonus for the treatment of pain.
CANNABINOID RECEPTORS AND THE SITE OF ACTION OF CANNABINOIDS
To date, two subtypes of cannabinoid receptors, termed cannabinoid-1 (CB1) and cannabinoid-2 (CB2) receptors, have been cloned (10, 11). CB1 receptors are most abundantly expressed in the central nervous system (CNS), most densely in motor and limbic regions, and in areas that are involved in pain transmission and modulation, such as periaqueductal grey (PAG), rostral ventromedial medulla (RVM), spinal cord dorsal horn, and in the periphery. CB1 receptors are generally located pre-synaptically on axons and terminals of neurons and mediate the inhibition of neurotransmitter release by the inhibition of adenylate cyclase, blockade of voltage-dependent calcium channels, and/or by the activation of potassium channels and mitogen-activated protein kinase. CB2 receptors, on the other hand, are found mainly, but not exclusively, outside the CNS, predominantly in peripheral tissues with immune functions, and most densely in the spleen. Similar to CB1 receptors, CB2 receptors are also G-protein coupled, inhibits adenylate cyclase and produce cellular inhibition, but neither blockade of calcium channels nor activation of potassium channels mediates this inhibitory effect (Figure 1) (1, 2, 12–14).
FIG. 1.
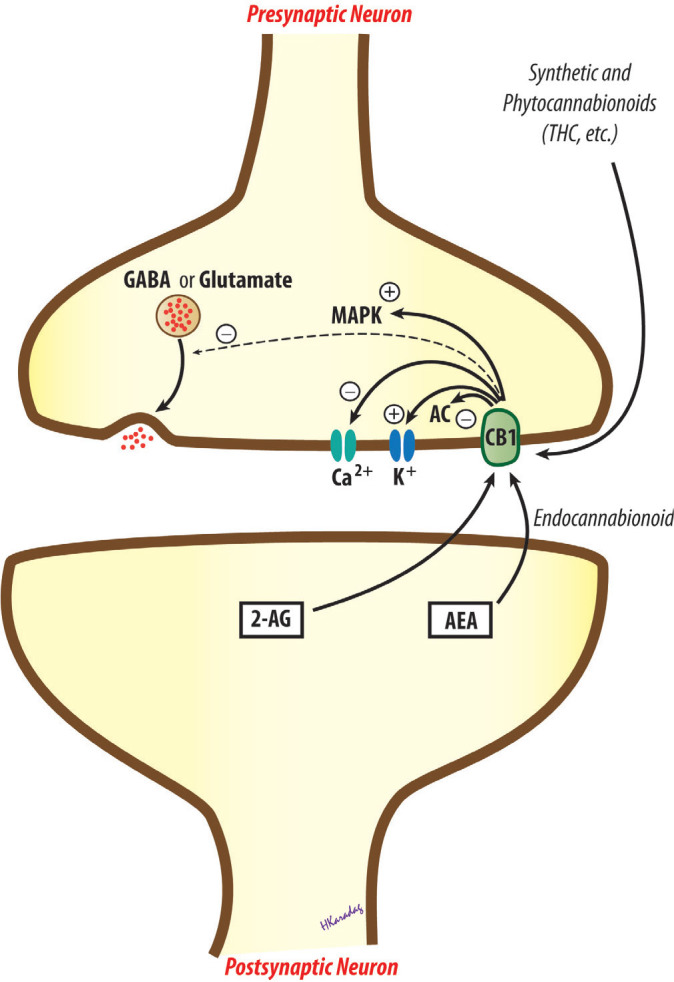
Transduction mechanisms for cannabinoid CB1 receptors. AC, adenylate cyclase; AEA, arachidonyl ethanolamide, anandamide; 2-AG, 2-arachidonylglycerol; CB1, cannabinoid receptor 1; GABA, gamma-aminobutyric acid; MAPK, mitogen-activated protein kinase; THC, tetrahydrocannabinol.
Phytocannabinoids, synthetic cannabinoids and endocannabinoids are thought to produce their anti-nociceptive action primarily through CB1 receptors, located at the supraspinal, spinal and peripheral levels (15, 16). Activation of descending inhibition by presynaptic inhibition of GABAergic and glutamatergic transmission in the PAG and modulation of on- and off-cells in the RVM seems to play pivotal roles in supraspinal analgesia (15, 17, 18). CB1 receptors found in presynaptic afferent terminals and on the terminals of intrinsic neurons and efferent supraspinal neurons are likely to mediate anti-nociception at the spinal level (15, 17). Topical cannabinoid anti-nociception and its synergy with spinal sites have also been suggested (19). CB2 receptors, although not as extensively studied as the CB1 receptors, are also proposed to play a role in the anti-nociceptive effects of cannabinoids (20).
PHYTOCANNABINOIDS AND SYNTHETIC CANNABINOIDS
Almost 80 of the chemical compounds in the cannabis plant, named phytocannabinoids, have the structure of a cannabinoid. Of these, THC is the best characterised and the primary psychoactive component of the plant. THC has an impact on many pathophysiological processes, including anti-nociception, through the activation of CB1 and CB2 receptors. However, its clinical utility is limited due to its unwanted CNS effects, which are mediated via brain CB1 receptors (5, 14). Cannabidiol, another important phytocannabinoid gaining attention recently, has a very low affinity at CB1 and CB2 receptors. Unlike THC, it does not cause any psychoactivity, but exerts many positive pharmacological effects, including anti-anxiety, anti-epileptic, anti-bacterial, anti-inflammatory, anti-cancer and anti-diabetic properties (14, 21, 22). In addition to its wide therapeutic spectrum, cannabidiol is proposed to reverse some of the central side effects of THC (21–23). Nabiximols (Sativex®), a herbal cannabis extract containing THC and cannabidiol at a 1:1 ratio in an oromucosal spray, has been approved for the treatment of neuropathic pain and spasticity associated with multiple sclerosis and intractable cancer pain (24).
Dronabinol (Marinol®), a synthetic THC, and its analogue nabilone (Cesamet®) are the currently available synthetic cannabinoids. Dronabinol and nabilone have been approved for chemotherapy-associated emesis in Canada and USA for many years. In addition, nabilone is indicated for anorexia associated with AIDS-related weight loss (14, 25). Recently, a clinical trial regarding the efficacy of nabilone in diabetic neuropathy has also ended in success (26). Rimonabant, a CB1 receptor antagonist/inverse agonist, was approved for obesity and smoking cessation, but withdrawn because of the increased incidence of depression.
ENDOCANNABINOIDS: SYNTHESIS, RELEASE, TRANSPORT, AND METABOLISM
The endocannabinoid system is comprised of cannabinoid CB1 and CB2 receptors, endogenous agonists of these receptors, “endocannabinoids”, and the processes playing a role in biosynthesis, release, transport and metabolism of these endogenous lipid-signalling molecules. Arachidonyl ethanolamide (AEA, anandamide), 2-arachidonylglycerol (2-AG), O-arachidonyl ethanolamine (virodhamine), N-arachidonyl dopamine (NADA), and 2-arachidonyl glyceryl ether (noladin ether) are the putative endocannabinoids. Of these endogenous metabolites of eicosanoid fatty acids, AEA and 2-AG are the best characterised (4, 5, 14). Endocannabinoids activate CB1 and/or CB2 receptors to modulate physiological and pathological conditions, including memory, appetite, immune function, sleep, stress response, thermoregulation, addiction, and no wonder analgesia. AEA and NADA are also shown to have an affinity to transient receptor potential vanilloid 1 (TRPV1) receptors (4, 14, 27).
Endocannabinoids are highly lipophilic compounds that are not stored in vesicles after production. Unlike classical cannabinoids, such as THC, they are substantially synthesised “on demand” from membrane phospholipids, and released immediately. Both AEA and 2-AG are produced at post-synaptic neurons. A two-step process is proposed as the main biosynthetic pathway for AEA. First, phosphatidylethanolamine, a membrane phospholipid, is converted to N-acyl-phosphatidylethanolamine (NAPE) by a calcium-dependent N-acyltransferase (NAT). Then, an NAPE-selective phospholipase D (NAPEPLD) catalyses NAPE to form N-acylethanolamines, such as AEA. For 2-AG, the major pathway also consists of a two-step process: diacylglycerol (DAG) is initially produced from inositol phospholipids by the phospholipase C enzyme, and subsequently hydrolysed to 2-AG by a diacylglycerol lipase (DAGL). Alternative enzymatic pathways also appear to be involved in the biosynthesis of both AEA and 2-AG. Following their release from depolarised post-synaptic neurons, endocannabinoids are regarded to act as retrograde messengers, and activate CB1 receptors on pre-synaptic terminals. Their overall effect may be excitatory or inhibitory, depending on a reduction in neurotransmitter release, mostly as the result of pre-synaptic inhibition of GABA or glutamatergic transmission (Figure 2) (4, 14, 27–29).
FIG. 2.
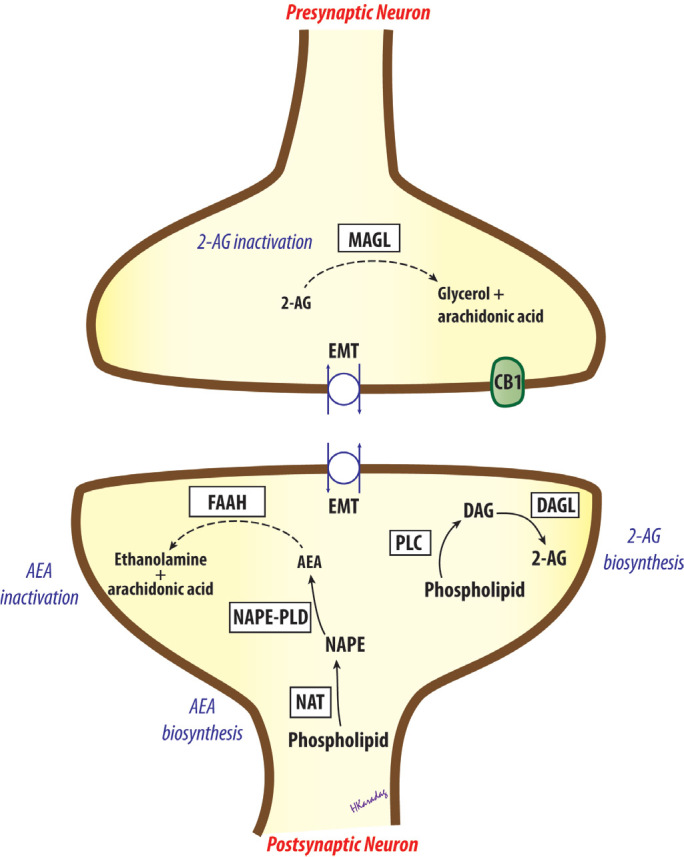
Biosynthesis and inactivation of endocannabinoids. AEA, arachidonyl ethanolamide, anandamide; 2-AG, 2-arachidonylglycerol; CB1, cannabinoid receptor 1; DAG, diacylglycerol; DAGL, diacylglycerol lipase; EMT, endocannabinoid membrane transporter; FAAH, fatty acid amide hydrolase; MAGL, monoacylglycerol lipase; NAPE, N-acylphosphatidylethanolamine; NAPE-PLD, NAPE-selective phospholipase D; NAT, N-acyltransferase; PLC, phospholipase C.
After activating cannabinoid receptors, endocannabinoids are removed from the extracellular space by a process of cellular uptake. Not much is known about the uptake of 2-AG, but a specific “endocannabinoid membrane transporter” appears to mediate the removal of AEA, although this has not yet been cloned (14, 28, 30, 31). Then, AEA is hydrolysed to arachidonic acid and ethanolamine in post-synaptic neurons, whereas 2-AG is hydrolysed to arachidonic acid and glycerol in pre-synaptic neurons, predominantly by the enzymes fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), respectively. Accordingly, FAAH appears to terminate endocannabinoid action during synthesis, while MAGL seems to play role after receptor activation. The endocannabinoids AEA and 2-AG are also metabolised via oxidation by the enzymes cyclooxygenase (COX), lipooxygenase, and cytochrome P-450 (4, 14, 28, 29).
MODULATION OF THE ENDOCANNABINOID SYSTEM
The central side effects of exogenous cannabinoids, most of which are related to CB1 receptors in the central nervous system (CNS), directed researchers to look for alternative strategies. Peripherally restricted CB1 agonists and/or selective CB2 receptor agonists deserve attention, since they are expected to minimise unwanted CNS effects mediated via CB1 receptors. There are reports indicating that selective peripheral CB1 agonists are effective in controlling conditions, such as chronic neuropathic pain and spasticity in multiple sclerosis (32, 33); however, no evidence of analgesic efficacy was observed for a peripherally acting CB1/CB2 receptor agonist in the human capsaicin pain model (34). Selective CB2 receptor agonists have also undergone clinical trials and found to be beneficial, but unwanted effects, such as immune depression, have prevented their use in the clinic (35, 36). Combining low doses of cannabinoids with other analgesics, such as opioids or NSAIDs, is also promising. Reducing the cannabinoid and opioid/NSAID dose needed may provide an advantageous treatment strategy by enhancing pain relief whilst minimising the incidence of adverse effects (6, 7, 37).
Elevating endocannabinoid levels appears to be the most striking strategy for developing analgesic drugs with cannabinoid properties but devoid of central psychotropic effects (38–40). During pain states, endocannabinoids are only synthesised and metabolised in the CNS structures involved in pain transmission and modulation. Thus, augmenting endocannabinoid levels in these structures will only effect CB receptors in these areas and possibly reduce central side effects. Inhibition of the degradative enzymes FAAH and MAGL, and inhibition of endocannabinoid cellular uptake are the pharmacological strategies used to modulate the endocannabinoid system and elevate endocannabinoid levels locally (3, 5, 22, 38, 40).
Fatty acid amide hydrolase inhibitors, such as URB597 and OL135, are shown to be effective at reducing sensitivity to pain in acute and chronic experimental pain models (41, 42). However, in a recent clinical trial, PF-04457845, an irreversible FAAH inhibitor, failed to induce effective analgesia in patients with knee osteoarthritis (43). It is noteworthy that AEA, at higher concentrations, activates TRPV1 channels, which are known to be involved in nociception, and that the COX pathway represents an alternative metabolic route for AEA. Thus, COX-1 and COX-2 inhibitors, and TRPV1 antagonists seem to be potential second targets to combine with FAAH inhibitors (22, 39, 44). Dual FAAH/TRPV1 blockers, such as N-arachidonoyl-serotonin (AA-5-HT) and OMDM198, are also efficacious in animal studies, but this multi-target strategy has not yet reached the clinic (45, 46).
Inhibition of MAGL using pharmacological agents, such as URB602, JZL184 and OMDM169, also elicits anti-nociceptive activity in experimental models, but tolerance may develop with fully effective doses of MAGL inhibitors (47). Dual FAAH/MAGL inhibition and using peripherally restricted inhibitors of FAAH and MAGL are also among the promising strategies (38, 39, 48). Moving to the inhibition of endocannabinoid cellular uptake, there are some potential therapeutic effects observed with the use of membrane transporter inhibitors in animal models (30, 38, 40). This approach has once again gained attention after the recent discovery of FAAH-like anandamide transporter (49), but clinical research remains to be determined.
It is worth mentioning that endocannabinoid system seems to mediate anti-nociceptive effects of 2 analgesics used worldwide, paracetamol and dipyrone. Paracetamol is metabolised in the brain by FAAH into AM404, which reinforces the activity of the descending serotonergic system through the endocannabinoid system via CB1 receptors (50, 51). FAAH also appears to be responsible for the formation of two novel arachidonyl-conjugated metabolites of dipyrone (52). Metabolites of these classical analgesics then possibly activate CB receptors or inhibit endocannabinoid metabolism (53–55), but clinical research is needed to determine how many of these findings also apply to humans.
CONCLUSION
The discovery of CB1 and CB2 receptors, their endogenous ligands (endocannabinoids), and the processes responsible for the biosynthesis, release, transport and metabolism of these compounds were a huge help in understanding the role of endocannabinoids in various physiological and pathological conditions, including pain modulation. Elevating endocannabinoid levels locally by the inhibition of endocannabinoid degrading enzymes, FAAH and MAGL, using pharmacological agents, and thereby reducing the unwanted central effects of exogenous cannabinoids, also made an additional contribution to this knowledge. Some of the most important future directions in this field are: (i) developing peripherally restricted CB1 agonists, (ii) inventing new CB2 agonists, (iii) combining cannabinoids with other analgesics, and (iv) elevating endocannabinoid levels using multi-target drugs such as FAAH/MAGL, FAAH (MAGL)/COX and FAAH (MAGL)/TRPV1 dual blockers. Clinical trials will demonstrate the value of these approaches.
Acknowledgments
I wish to thank Prof. Hakan Karadağ for drawing figures.
Footnotes
Ethics Committee Approval: N/A.
Informed Consent: N/A.
Peer-review: Internally peer-reviewed.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The author declared that this study has received no financial support.
REFERENCES
- 1.Pertwee RG. Cannabinoid receptors and pain. Prog Neurobiol. 2001;63:569–611. doi: 10.1016/s0301-0082(00)00031-9. [DOI] [PubMed] [Google Scholar]
- 2.Walker JM, Huang SM. Cannabinoid analgesia. Pharmacol Ther. 2002;95:127–35. doi: 10.1016/s0163-7258(02)00252-8. [DOI] [PubMed] [Google Scholar]
- 3.Di Marzo V. Targeting the endocannabinoid system: to enhance or reduce? Nat Rev Drug Discov. 2008;7:438–55. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- 4.Guindon J, Hohmann AG. The endocannabinoid system and pain. CNS Neurol Disord Drug Targets. 2009;8:403–21. doi: 10.2174/187152709789824660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pertwee RG. Targeting the endocannabinoid system with cannabinoid receptor agonists: pharmacological strategies and therapeutic possibilities. Philos Trans R Soc Lond B Biol Sci. 2012;367:3353–63. doi: 10.1098/rstb.2011.0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ulugol A, Ozyigit F, Yesilyurt O, Dogrul A. The additive antinociceptive interaction between WIN 55,212-2, a cannabinoid agonist, and ketorolac. Anesth Analg. 2006;102:443–7. doi: 10.1213/01.ane.0000194587.94260.1d. [DOI] [PubMed] [Google Scholar]
- 7.Gunduz O, Karadag HC, Ulugol A. Synergistic anti-allodynic effects of nociceptin/orphanin FQ and cannabinoid systems in neuropathic mice. Pharmacol Biochem Behav. 2011;99:540–4. doi: 10.1016/j.pbb.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 8.Dogrul A, Gul H, YildIz O, Bilgin F, Guzeldemir ME. Cannabinoids blocks tactile allodynia in diabetic mice without attenuation of its anti-nociceptive effect. Neurosci Lett. 2004;368:82–6. doi: 10.1016/j.neulet.2004.06.060. [DOI] [PubMed] [Google Scholar]
- 9.Ulugol A, Karadag HC, Ipci Y, Tamer M, Dokmeci I. The effect of WIN 55,212-2, a cannabinoid agonist, on tactile allodynia in diabetic rats. Neurosci Lett. 2004;371:167–70. doi: 10.1016/j.neulet.2004.08.061. [DOI] [PubMed] [Google Scholar]
- 10.Munro S, Thomas KL, Abushaar M. Molecular Characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–5. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 11.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned Cdna. Nature. 1990;346:561–4. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 12.Dogrul A, Seyrek M, Yalcin B, Ulugol A. Involvement of descending serotonergic and noradrenergic pathways in CB1 receptor-mediated anti-nociception. Prog Neuropsychopharmacol Biol Psychiatry. 2012;38:97–105. doi: 10.1016/j.pnpbp.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 13.Dogrul A, Seyrek M, Yalcin B, Ulugol A. Involvement of serotonergic system in cannabinoid analgesia. In: Van Bockstaele EJ, editor. Endocannabinoid regulation of monoamines in psychiatric and neurological disorders. New York: Springer; 2013. pp. 277–95. [Google Scholar]
- 14.Guindon J, Beaulieu P, Hohmann AG. Pharmacology of the cannabinoid system. In: Beaulieu P, Lussier D, Porreca F, Dickenson AH, editors. Pharmacology of pain. Seattle: IASP Press; 2010. p. 6. [Google Scholar]
- 15.Richardson JD. Cannabinoids modulate pain by multiple mechanisms of action. J Pain. 2000;1:2–14. [Google Scholar]
- 16.Manzanares J, Julian MD, Carrascosa A. Role of the cannabinoid system in pain control and therapeutic implications for the management of acute and chronic pain episodes. Curr Neuropharmacol. 2006;4:239–57. doi: 10.2174/157015906778019527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- 18.Meng ID, Manning BH, Martin WJ, Fields HL. An analgesia circuit activated by cannabinoids. Nature. 1998;395:381–3. doi: 10.1038/26481. [DOI] [PubMed] [Google Scholar]
- 19.Dogrul A, Gul H, Akar A, YildIz O, Bilgin F, Guzeldemir E. Topical cannabinoid antinociception: synergy with spinal sites. Pain. 2003;105:11–6. doi: 10.1016/s0304-3959(03)00068-x. [DOI] [PubMed] [Google Scholar]
- 20.Malan TP, Ibrahim MM, Vanderah TW, Makriyannis A, Porreca F. Inhibition of pain responses by activation of CB2 cannabinoid receptors. Chemistry and Physics of Lipids. 2002;121:191–200. doi: 10.1016/s0009-3084(02)00155-x. [DOI] [PubMed] [Google Scholar]
- 21.Izzo AA, Borrelli F, Capasso R, Di Marzo V, Mechoulam R. Non-psychotropic plant cannabinoids: new therapeutic opportunities from an ancient herb. Trends Pharmacol Sci. 2009;30:515–27. doi: 10.1016/j.tips.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 22.Starowicz K, Di Marzo V. Non-psychotropic analgesic drugs from the endocannabinoid system: “Magic bullet” or “multiple-target” strategies? Eur J Pharmacol. 2013;716:41–53. doi: 10.1016/j.ejphar.2013.01.075. [DOI] [PubMed] [Google Scholar]
- 23.Wright MJ, Vandewater SA, Taffe MA. Cannabidiol attenuates deficits of visuospatial associative memory induced by Delta 9tetrahydrocannabinol. Brit J Pharmacol. 2013;170:1365–73. doi: 10.1111/bph.12199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sastre-Garriga J, Vila C, Clissold S, Montalban X. THC and CBD oromucosal spray (Sativex (R)) in the management of spasticity associated with multiple sclerosis. Expert Rev Neurother. 2011;11:627–37. doi: 10.1586/ern.11.47. [DOI] [PubMed] [Google Scholar]
- 25.Wang T, Collet JP, Shapiro S, Ware MA. Adverse effects of medical cannabinoids: a systematic review. Can Med Assoc J. 2008;178:1669–78. doi: 10.1503/cmaj.071178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toth C, Mawani S, Brady S, Chan C, Liu CX, Mehina E, et al. An enriched-enrolment, randomized withdrawal, flexible-dose, double-blind, placebo-controlled, parallel assignment efficacy study of nabilone as adjuvant in the treatment of diabetic peripheral neuropathic pain. Pain. 2012;153:2073–82. doi: 10.1016/j.pain.2012.06.024. [DOI] [PubMed] [Google Scholar]
- 27.Zogopoulos P, Vasileiou I, Patsouris E, Theocharis SE. The role of endocannabinoids in pain modulation. Fund Clin Pharmacol. 2013;27:64–80. doi: 10.1111/fcp.12008. [DOI] [PubMed] [Google Scholar]
- 28.Cascio MG. PUFA-derived endocannabinoids: an overview. P Nutr Soc. 2013;72:451–9. doi: 10.1017/S0029665113003418. [DOI] [PubMed] [Google Scholar]
- 29.Di Marzo V. Endocannabinoids: synthesis and degradation. Rev Physiol Biochem Pharmacol. 2008;160:1–24. doi: 10.1007/112_0505. [DOI] [PubMed] [Google Scholar]
- 30.Fowler CJ. Anandamide uptake explained? Trends Pharmacol Sci. 2012;33:181–5. doi: 10.1016/j.tips.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 31.Jhaveri MD, Richardson D, Chapman V. Endocannabinoid metabolism and uptake: novel targets for neuropathic and inflammatory pain. Brit J Pharmacol. 2007;152:624–32. doi: 10.1038/sj.bjp.0707433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karst M, Salim K, Burstein S, Conrad I, Hoy L, Schneider U. Analgesic effect of the synthetic cannabinoid CT-3 on chronic neuropathic pain: a randomized controlled trial. JAMA. 2003;290:1757–62. doi: 10.1001/jama.290.13.1757. [DOI] [PubMed] [Google Scholar]
- 33.Pryce G, Visintin C, Ramagopalan SV, Al-Izki S, De Faveri LE, Nuamah RA, et al. Control of spasticity in a multiple sclerosis model using central nervous system-excluded CB1 cannabinoid receptor agonists. FASEB J. 2014;28:117–30. doi: 10.1096/fj.13-239442. [DOI] [PubMed] [Google Scholar]
- 34.Kalliomaki J, Annas P, Huizar K, Clarke C, Zettergren A, Karlsten R, et al. Evaluation of the analgesic efficacy and psychoactive effects of AZD1940, a novel peripherally acting cannabinoid agonist, in human capsaicin-induced pain and hyperalgesia. Clin Exp Pharmacol P. 2013;40:212–8. doi: 10.1111/1440-1681.12051. [DOI] [PubMed] [Google Scholar]
- 35.Ostenfeld T, Price J, Albanese M, Bullman J, Guillard F, Meyer I, et al. A randomized, controlled study to investigate the analgesic efficacy of single doses of the cannabinoid receptor-2 agonist GW842166, ibuprofen or placebo in patients with acute pain following third molar tooth extraction. Clin J Pain. 2011;27:668–76. doi: 10.1097/AJP.0b013e318219799a. [DOI] [PubMed] [Google Scholar]
- 36.Atwood BK, Straiker A, Mackie K. CB2: therapeutic target-in-waiting. Prog Neuropsychopharmacol Biol Psychiatry. 2012;38:16–20. doi: 10.1016/j.pnpbp.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith PA, Selley DE, Sim-Selley LJ, Welch SP. Low dose combination of morphine and [Delta]9-tetrahydrocannabinol circumvents antinociceptive tolerance and apparent desensitization of receptors. Eur J Pharmacol. 2007;571:129–37. doi: 10.1016/j.ejphar.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pertwee RG. Elevating endocannabinoid levels: pharmacological strategies and potential therapeutic applications. Proc Nutr Soc. 2014;73:96–105. doi: 10.1017/S0029665113003649. [DOI] [PubMed] [Google Scholar]
- 39.Maione S, Costa B, Di Marzo V. Endocannabinoids: a unique opportunity to develop multitarget analgesics. Pain. 2013;154:S87–93. doi: 10.1016/j.pain.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 40.Piscitelli F, Di Marzo V. “Redundancy” of Endocannabinoid Inactivation: New Challenges and Opportunities for Pain Control. ACS Chem Neurosci. 2012;3:356–63. doi: 10.1021/cn300015x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naidu PS, Kinsey SG, Guo TL, Cravatt BF, Lichtman AH. Regulation of inflammatory pain by inhibition of fatty acid amide hydrolase. J Pharmacol Exp Ther. 2010;334:182–90. doi: 10.1124/jpet.109.164806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caprioli A, Coccurello R, Rapino C, Di Serio S, Di Tommaso M, Vertechy M, et al. The novel reversible fatty acid amide hydrolase inhibitor ST4070 increases endocannabinoid brain levels and counteracts neuropathic pain in different animal models. J Pharmacol Exp Ther. 2012;342:188–95. doi: 10.1124/jpet.111.191403. [DOI] [PubMed] [Google Scholar]
- 43.Huggins JP, Smart TS, Langman S, Taylor L, Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153:1837–46. doi: 10.1016/j.pain.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 44.Fowler CJ, Naidu PS, Lichtman A, Onnis V. The case for the development of novel analgesic agents targeting both fatty acid amide hydrolase and either cyclooxygenase or TRPV1. Brit J Pharmacol. 2009;156:412–9. doi: 10.1111/j.1476-5381.2008.00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morera E, De Petrocellis L, Morera L, Moriello AS, Ligresti A, Nalli M, et al. Synthesis and biological evaluation of piperazinyl carbamates and ureas as fatty acid amide hydrolase (FAAH) and transient receptor potential (TRP) channel dual ligands. Bioorg Med Chem Lett. 2009;19:6806–9. doi: 10.1016/j.bmcl.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 46.Costa B, Bettoni I, Petrosino S, Comelli F, Giagnoni G, Di Marzo V. The dual fatty acid amide hydrolase/TRPV1 blocker, N-arachidonoylserotonin, relieves carrageenan-induced inflammation and hyperalgesia in mice. Pharmacol Res. 2010;61:537–46. doi: 10.1016/j.phrs.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 47.Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci. 2010;13:1113–U111. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Niphakis MJ, Johnson DS, Ballard TE, Stiff C, Cravatt BF. O-hydroxyacetamide carbamates as a highly potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem Neurosci. 2012;3:418–26. doi: 10.1021/cn200089j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fu J, Bottegoni G, Sasso O, Bertorelli R, Rocchia W, Masetti M, et al. A catalytically silent FAAH-1 variant drives anandamide transport in neurons. Nat Neurosci. 2012;15:64–U82. doi: 10.1038/nn.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hogestatt ED, Jonsson BAG, Ermund A, Andersson DA, Bjork H, Alexander JP, et al. Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J Biol Chem. 2005;280:31405–12. doi: 10.1074/jbc.M501489200. [DOI] [PubMed] [Google Scholar]
- 51.Mallet C, Daulhac L, Bonnefont J, Ledent C, Etienne M, Chapuy E, et al. Endocannabinoid and serotonergic systems are needed for acetaminophen-induced analgesia. Pain. 2008;139:190–200. doi: 10.1016/j.pain.2008.03.030. [DOI] [PubMed] [Google Scholar]
- 52.Rogosch T, Sinning C, Podlewski A, Watzer B, Schlosburg J, Lichtman AH, et al. Novel bioactive metabolites of dipyrone (metamizol) Bioorg Med Chem. 2012;20:101–7. doi: 10.1016/j.bmc.2011.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elmas P, Ulugol A. Involvement of cannabinoid CB1 receptors in the antinociceptive effect of dipyrone. J Neural Transm. 2013;120:1533–8. doi: 10.1007/s00702-013-1052-7. [DOI] [PubMed] [Google Scholar]
- 54.Ottani A, Leone S, Sandrini M, Ferrari A, Bertolini A. The analgesic activity of paracetamol is prevented by the blockade of cannabinoid CB1 receptors. Eur J Pharmacol. 2006;531:280–1. doi: 10.1016/j.ejphar.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 55.Escobar W, Ramirez K, Avila C, Limongi R, Vanegas H, Vazquez E. Metamizol, a non-opioid analgesic, acts via endocannabinoids in the PAG-RVM axis during inflammation in rats. Eur J Pain. 2012;16:676–89. doi: 10.1002/j.1532-2149.2011.00057.x. [DOI] [PubMed] [Google Scholar]